

Abstract
Phosphorylation is one of the most dynamic and widespread post‐translational modifications regulating virtually every aspect of eukaryotic cell biology. Here, we assemble a dataset from 75 independent phosphoproteomic experiments performed in our laboratory using Saccharomyces cerevisiae. We report 30,902 phosphosites identified from cells cultured in a range of DNA damage conditions and/or arrested in distinct cell cycle stages. To generate a comprehensive resource for the budding yeast community, we aggregate our dataset with the Saccharomyces Genome Database and another recently published study, resulting in over 46,000 budding yeast phosphosites. With the goal of enhancing the identification of functional phosphorylation events, we perform computational positioning of phosphorylation sites on available 3D protein structures and systematically identify events predicted to regulate protein complex architecture. Results reveal hundreds of phosphorylation sites mapping to or near protein interaction interfaces, many of which result in steric or electrostatic “clashes” predicted to disrupt the interaction. With the advancement of Cryo‐EM and the increasing number of available structures, our approach should help drive the functional and spatial exploration of the phosphoproteome.
Keywords: budding yeast, phosphorylation, protein–protein interaction, Rad23, TRAPP
Subject Categories: Methods & Resources; Post-translational Modifications, Proteolysis & Proteomics;
This study compiles 75 independent SILAC‐based experiments into a comprehensive phosphoproteome resource for budding yeast. 3D analysis of protein interaction interfaces and other strategies are used to predict functionality amongst the ≥ 40,000 reported phosphorylation events.
Introduction
Post‐translational modification of proteins by phosphorylation controls virtually every cellular process. Regulatory mechanisms based on phosphorylation have been widely explored and characterized. In classical approaches, phosphorylation sites are often biochemically identified on substrate proteins of interest and then mutated to either prevent or constitutively mimic a phosphorylation event. The phenotypes associated with these “phosphomutant” proteins inform on the biological purpose of phosphorylation at that site. In the last 15 years, technological advances in mass spectrometry, along with the development of enrichment methods for phosphorylated peptides (Ficarro et al, 2002; Gruhler et al, 2005; Larsen et al, 2005; Bodenmiller et al, 2007), have greatly expanded our ability to identify phosphorylation events, leading to large phosphoproteomic datasets (Aebersold & Mann, 2003; Olsen et al, 2006; Olsen et al, 2010; Swaney et al, 2013; Sharma et al, 2014; Bastos de Oliveira et al, 2015; Hu et al, 2019). However, our ability to probe the biological relevance of the identified phosphorylation events still relies on low‐throughput methods. As a consequence, the functional importance of most cataloged phosphorylation events has not yet been determined (Needham et al, 2019). Notably, given the overwhelming number of identified phosphorylation events, over 100,000 in the case of a human cell (Hornbeck et al, 2019; Ochoa et al, 2020), and the likely promiscuity in kinase actions, it is debatable whether all of these events are functionally relevant (Lienhard, 2008; Landry et al, 2009). In many cases where attempts have been made to investigate the role of specific phosphorylation events, the results are often negative (Dephoure et al, 2013), consistent with the notion that many phosphorylation events may be extensively redundant in nature or, perhaps, not functional (Landry et al, 2009; Levy et al, 2012). These issues highlight the necessity for strategies to predict functional phosphorylation sites from large phosphoproteome datasets. While guidelines for interpreting phosphoproteomic datasets to identify candidate sites for mutational analysis are available (Dephoure et al, 2013), strategies to efficiently and systematically identify functional phosphorylation events are lacking, especially in the case of budding yeast.
Here, we present an in‐depth phosphoproteome for budding yeast that constitutes the largest collection of phosphorylation sites for this organism. Over 10.6 million high‐resolution MS/MS spectra were acquired in our mass spectrometer. In addition, we utilized two independent methods for scoring phosphosite localization and employed an in‐house algorithm to capture ambiguous phosphosites that fall within clusters of consecutive, phosphorylate‐able residues. When considering the new phosphorylation events identified by this study, the aggregated budding yeast phosphoproteome currently constitutes over 46,000 phosphosites. In addition to performing cell cycle‐ and DNA damage‐related analyses, we computationally positioned phosphorylation onto all available 3D protein structures in order to systematically identify potentially functional phosphorylation events. Results reveal many phosphorylation sites that map to or near protein interaction interfaces, some of which result in steric or electrostatic “clashes” predicted to disrupt the interaction. Phosphorylation site mutants experimentally validate our predictions and establish roles for phosphorylation in negatively regulating protein–protein interactions. We have compiled our in‐depth phosphoproteome into an online database open to the budding yeast community. This resource should help drive the functional and spatial exploration of the yeast phosphoproteome.
Results
In‐depth phosphoproteome of budding yeast
We sought to generate an in‐depth phosphoproteomic database for the model system budding yeast. The spectra used to assemble the dataset were generated from 75 independent SILAC‐based experiments conducted in our laboratory. These experiments were originally performed for various unrelated biological inquiries and explored a range of conditions, including distinct cell cycle stages, DNA damage treatment, and carbon deprivation (Fig 1A, Dataset EV1). Phosphopeptides enriched from proteolytically digested whole cell lysates were subsequently pre‐fractionated by HILIC chromatography (Fig 1A). Our data are sourced strictly from high‐resolution spectral data acquired using a single, in‐house mass spectrometer and processed through a unified data processing pipeline. A fraction of this dataset was previously published (See Dataset EV1 (Lanz et al, 2018)). In all, the dataset consists of fragmentation spectra acquired from over 825 LC‐MS/MS injections (1,500+ h of data‐dependent acquisition time). To identify peptide spectrum matches (PSMs) from our library of fragmentation spectra, we utilized two prominent search engines—Sequest and Andromeda (MaxQuant; Appendix Fig S1, Datasets EV2 and EV3; see details under Materials and Methods). The search parameters for the Andromeda and Sequest searches were mostly the same, the primary difference being the semi‐specificity for tryptic ends in the Sequest search (we were unable to search large amounts of data with semi‐specific digestion using our MaxQuant platform). Nearly 2,300 sites were identified with highest confidence in semi‐tryptic phosphopeptides (Dataset EV4), demonstrating the utility of the secondary search. We performed one phosphoproteomic experiment using chymotrypsin as the digestive enzyme (Dataset EV5 and Appendix Fig S1). In all, our dataset consists of ~ 3.5 million PSMs, representing ~ 45,000 called phosphosites detected within ~ 4,000 proteins (Fig 1A).
Figure 1. In‐depth analysis of the budding yeast phosphoproteome.
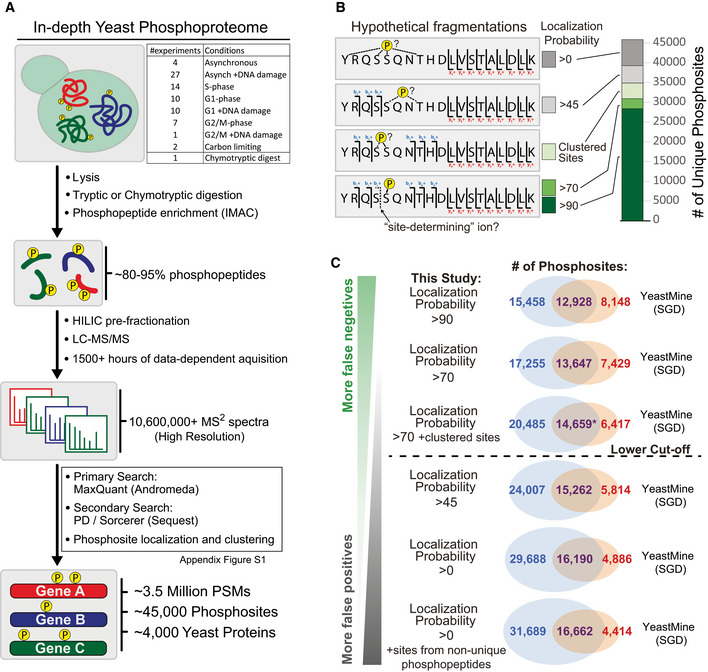
- A generalized workflow for mapping the budding yeast phosphoproteome using mass spectrometry. Dataset EV1 contains detailed information on the experiments included in this dataset. Appendix Fig S1 contains a decision tree that describes how the primary and secondary searches were compiled into a final dataset.
- Rationale for phosphosite localization analysis. Site localization probabilities were determined using MaxQuant and the PhosphoRS node within Proteome Discoverer. Hypothetical fragmentations illustrate how fragment ion information impacts the ability to resolve phosphorylated residues within phosphopeptides. Clustered sites were identified using an in‐house algorithm.
- Overlay of this study with YeastMine, the public repository for phosphosites utilized by the Saccharomyces Genome Database (SGD). Localization criteria for phosphosites identified by this study are relaxed in descending Venn diagrams. Our lower cutoff reflects a balance between confidence in localization and the prevention of false negatives. In the comparison marked with the asterisk, phosphosites that we derived from non‐unique phosphopeptides were included in the overlay if already present in YeastMine. These non‐unique sites were excluded from the analysis if not present in YeastMine.
A critical challenge in the analysis of peptide‐centric phosphoproteomic workflows is the need to properly assign the phosphorylated STY residue within a fragmented phosphopeptide (Thompson et al, 2012). The use of multiple search engines allowed us to employ two prominent algorithms for determining phosphosite localization, PhosphoRS and PTM‐Score, each of which utilize distinct methods to identify “site‐determining” ions (Taus et al, 2011; Sharma et al, 2014). We present our dataset with a range of cutoffs for localization probability (Fig 1B, Dataset EV2). We also implemented an in‐house clustering algorithm to capture several thousand “phosphosites” whose site localization probabilities were distributed within consecutive STY residues (Fig 1B illustrates how clustered phosphosites differ from other phosphosites with ambiguous localization). As phosphosite localization confidence decreases, the total number reported phosphosites is inflated by false positives because, in some cases, multiple ambiguous phosphosites are called from phosphopeptides that may only harbor a single phosphorylated residue. With this fact in mind, we overlaid our dataset with the primary public repository for budding yeast phosphosites, YeastMine. YeastMine contains over 21,000 “unique” phosphosites (Balakrishnan et al, 2012) and is the contributing repository for the Saccharomyces Genome Database (SGD), an online resource used by nearly all budding yeast biologists. YeastMine is comprised of phosphosites identified from high‐throughput MS‐based studies in addition to phosphosites identified from low‐throughput investigations of individual proteins or protein complexes. When considering only the phosphosites we identified with greater than 90 localization probability, we detect over 60% of the sites contained within YeastMine. As the stringency for phosphosite localization is relaxed, the overlap of our dataset with YeastMine increases. This is true even as poorly localized sites are considered, as well as phosphosites identified within non‐unique phosphopeptides that map to multiple different proteins (Fig 1C). This comparative analysis suggests that public phosphosite repositories may contain many mis‐localized phosphorylation sites that, although originating from a true PSM, result from differences in how individual contributors account for phosphosite localization. Our suggested cutoff (Fig 1C; dashed line) aims to strike a balance between the false positives associated with poorly localized sites and the false negatives resulting from a strict reliance on highly localized sites.
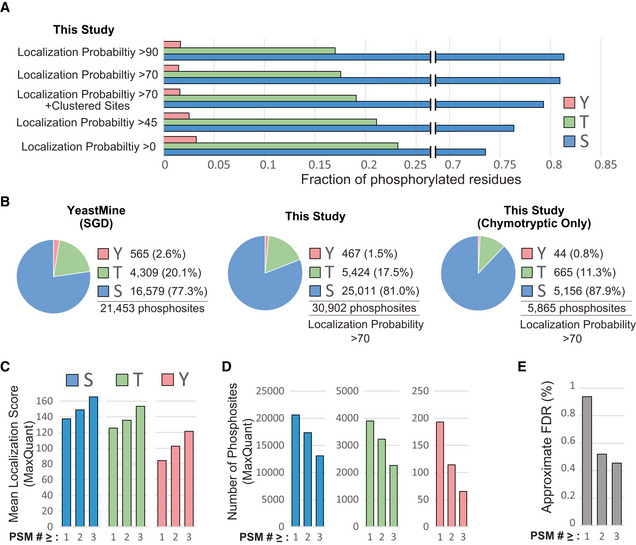
We found that phosphosite localization probability tolerance also impacted the proportionality of STY phosphorylation within our dataset and that the fraction of phospho‐tyrosine residues doubles as the threshold for site localization probability is relaxed (Fig 2A). This observation could in part explain the slightly higher proportion of tyrosine phosphorylation reported on YeastMine (Fig 2B). In fact, the average quality of phospho‐tryosine sites identified in our dataset is lower than that of phospho‐serine or phospho‐threonine (Fig 2C). Because sites identified as phospho‐tyrosine in our study (and possibly YeastMine) are prone to represent mis‐localized phospho‐serine or phospho‐threonine, we encourage the careful consideration of the PSM quality metrics when investigating tyrosine phosphorylation. We found that filtering based on the number of phosphosite identifications (PSMs) increases overall data quality (Fig 2C and D) and reduces the false discovery rate (Fig 2E, Appendix Fig S2).
Figure 2. S, T, Y phosphorylation proportionality and quality control in the budding yeast phosphoproteome.
-
AProportionality of S, T, Y phosphorylation as a function of phosphosite localization probability. Clustered sites are excluded.
-
BProportionality of S, T, Y phosphorylation in the indicated datasets. Clustered sites are excluded.
-
C, D(C) MaxQuant's site localization scores for and (D) total number of S, T, and Y phosphosites with at least one, two, or three identifications (PSMs). Only sites identified by the primary search (Maxquant) were included.
-
EFDR approximation for the final dataset (Dataset EV2) when considering phosphosites with at least one, two, or three identifications (PSMs). FDR was estimated by a Target/Decoy analysis designed to track the Target/Decoy ratio at each step of the analysis pipeline, both before and after combining results from the primary and secondary search engines (Appendix Fig S2).
To contextualize the depth of our study, we plotted our identified phosphosites as a function of protein abundance (Ho et al, 2018). Despite the fact that the enrichment of phosphopeptides directly from cell lysates can hinder the detection of phosphorylation events that occur in low abundant proteins (Solari et al, 2015), we readily identified novel phosphorylation events in very low abundant yeast proteins, and the distribution of phosphosite discovery was mostly independent of the estimated protein abundance (Fig 3A). The scale of our analysis (825 independent MS runs) produced a large dynamic range of phosphopeptide identifications (i.e. PSMs) per phosphorylation site. By plotting the number of PSMs for each phosphosite as a function of the harboring protein's estimated copy number (Fig 3B), we highlighted ~ 500 phosphosites with highest PSM# ‐to‐ protein abundance ratios, which could potentially serve as crude indicators of high stoichiometry phosphorylation within low abundance proteins, despite the noted caveats of using number of PSMs and generalizing estimated protein abundances for such types of inferences (Fig 3B, see “#identifications” and “ProteinAbundance” columns in Dataset EV2).
Figure 3. Contextualizing the depth of the budding yeast phosphoproteome.
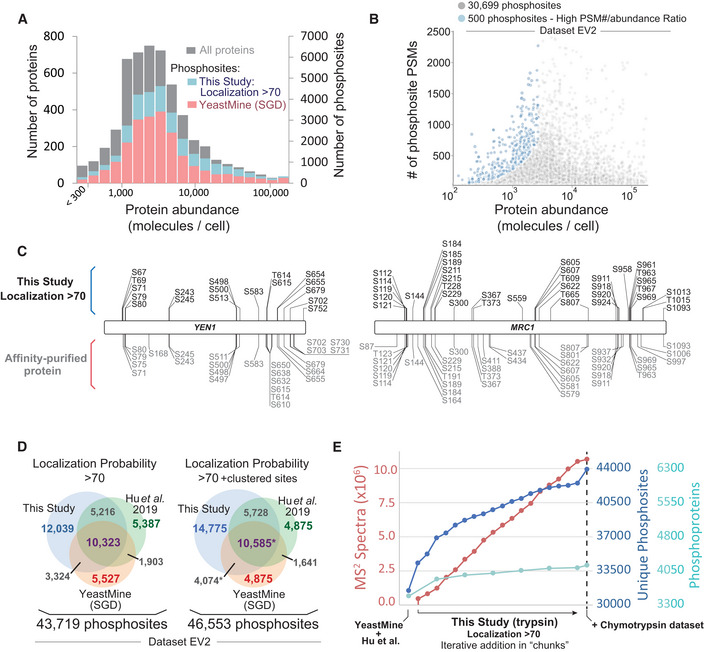
- Histogram depicting the distribution of identified phosphosites as a function of protein copy number (estimated from Ho et al, 2018). Bars representing the number of phosphosites identified in this study are plotted behind (not on top of) the bars representing YeastMine.
- The PSM count for identified phosphosites as a function of protein copy number (estimated from Ho et al, 2018). Only phosphosites within proteins with copy number estimations are depicted. Blue dots highlight a small subset of sites with a high PSM#/protein copy number ratio.
- Coverage maps comparing the Yen1 and Mrc1 phosphosites identified in this study (above, in black) with the sites identified in low‐throughput studies (below, in gray). For the low‐throughput MS analyses, phosphopeptides were enriched after affinity purification of Yen1 or Mrc1 from yeast lysates.
- The current state of the budding yeast phosphoproteome (Dataset EV2). This study is combined with YeastMine and another recent large‐scale analysis, Hu et al, 2019, which used a localization probability cutoff of 75 for their dataset. See Dataset EV3 for a dataset sourced exclusively from the Sequest searches. In the comparison marked with the asterisk, phosphosites that we derived from non‐unique phosphopeptides were included in the overlay if already present in YeastMine. These non‐unique sites were excluded from the analysis if not present in YeastMine.
- Dot graph examining saturation in the ability to identify novel phosphoproteins and phosphosites from the budding yeast phosphoproteome. Unique, non‐redundant phosphosites from this study were iteratively added to an aggregate set (left–right) in randomized chunks (localization probability of > 70, clustered sites are excluded). Dataset generated using chymotrypsin (Dataset EV5) is the ultimate addition to the plot.
Because it was previously demonstrated that the extent of phosphorylation identified in high‐throughput studies is less than that which can be detected from affinity‐purified proteins (Albuquerque et al, 2008), we next compared our coverage to various low‐throughput MS analyses. One such low‐throughput study identified 25 phosphosites in Yen1 (Blanco et al, 2014), a nuclease regulated by cyclin‐dependent kinase. We were able to detect 18 phosphosites in Yen1 (Fig 3C), nearly all of which were identified by Blanco and colleagues. Only four Yen1 phosphosites are contained within YeastMine. Another study identified 39 phosphosites in the replisome protein Mrc1 (Albuquerque et al, 2008), a number comparable to the 36 sites identified in our study (Fig 3C). Together, these examples illustrate that, in some cases, our depth of coverage compares to the depth achieved in the analysis of affinity‐purified proteins. In addition, we note that our analysis confirmed the presence of phosphorylation at putative phosphosites, whose mutation was previously shown to preclude phosphorylation‐dependent mobility shifts and disrupt genuine phospho‐mediated regulation (Appendix Fig S3, bolded dark blue sites with asterisk) (Kono et al, 2008; Rossi et al, 2015).
We next sought to define the current state of the budding yeast phosphoproteome by aggregating our dataset with two prominent publicly available datasets. In addition to YeastMine, we also incorporated a recent large‐scale phosphoproteomic screen performed by Hu et al (2019). Unlike our study, Hu et al, utilized high‐pH reverse phase chromatography for phosphopeptide prefractionation (Batth et al, 2014). Aggregation of these three datasets (our dataset, Hu et al, and YeastMine) resulted in a composite dataset containing 46,553 phosphosites (Fig 3D, Dataset EV2). Similar to what has been done previously (Amoutzias et al, 2012), and using the YeastMine and Hu et al, datasets as a foundation, we iteratively incorporated our dataset in a randomized “chunk”‐wise manner. As the final portions of our tryptic dataset were considered, our ability to detect new phosphoproteins and phosphosites was approaching saturation (Fig 3E). However, the ultimate addition of a dataset derived from phosphopeptides generated by chymotryptic digestion broke the plateau of the saturation curve (Fig 3E), suggesting that the size of the yeast phosphoproteome can still be significantly expanded using alternative digestive enzymes. It is also likely that the phosphoproteome can also be expanded by exploring a more diverse set of cellular states. For example, our dataset lacks spectra acquired from meiotic conditions and, therefore, may not contain phosphorylation events mediated by meiosis‐specific kinases, like Ime2 (Foiani et al, 1996; Guttmann‐Raviv et al, 2002). Moreover, our search pipeline does not capture phosphorylation that occurs on non‐canonical residues, which has recently been identified in other eukaryotes (Hardman et al, 2019).
Functional and regulatory exploration of the budding yeast phosphoproteome
Despite the large quantity of phosphorylation revealed by mass spectrometry, the inability to distinguish meaningful phosphorylation events from “noise” within the phosphoproteome represents a fundamental limitation of the technology. To address this limitation, we employed a variety of strategies to systematically reveal potentially meaningful phosphorylation events. First, we took advantage of an extensive compilation of temperature sensitive (ts) budding yeast mutants (Li et al, 2011). We reasoned that, since ts mutations fall within chemically sensitive regions of a protein's structure, phosphorylation events which occur at or near these ts residues are more likely to be impactful. Our analysis revealed 50 phosphorylation events that occur in immediate proximity (± 3 a.a.) to residues that harbor ts mutations (Dataset EV6), and in several cases the ts residue is itself phosphorylated. In one such case, rsp5‐T104A, the sensitizing mutation, is the substitution of a threonine to alanine, which suggests that the phosphorylation of the Rsp5 ubiquitin ligase at T104 is somehow important for its function.
Because phosphorylation that is subjected to dynamic regulation is more likely to be functionally important (Kanshin et al, 2015), we next aimed to extract regulatory information for the phosphorylation events we identified. A unique feature of our dataset is the multitude of analyses performed on yeast treated with DNA damaging agents, synchronized to distinct cell cycle stages, or both. We selected from our dataset a set of 11 independent SILAC experiments that probe the response to DNA damage in different stages of the cell cycle. This curation contains measurements for the behavior of over 23,000 phosphosites (Dataset EV7). In budding yeast, DNA damage signaling is mediated by the sensor kinases Mec1 and Tel1 and the downstream checkpoint kinase Rad53 (see recent reviews (Giannattasio & Branzei, 2017; Pardo et al, 2017; Cussiol et al, 2019; Lanz et al, 2019)), while the cyclin‐dependent kinase orders the progression of the budding yeast cell cycle. Phosphopeptides whose abundance is dependent on the action of these kinases have been identified previously (Holt et al, 2009; Bastos de Oliveira et al, 2015), and the behavior of these “substrate” phosphopeptides can be used to track kinase activity (Fig 4A) (Hustedt et al, 2015; Bastos de Oliveira et al, 2018; Lanz et al, 2018). We assessed changes in the activity of these kinases, along with changes to the rest of the phosphoproteome, in response to DNA damage and cell cycle progression (Fig 4B–D, Appendix Fig S4). The treatment of G1‐arrested cells with a UV‐mimicking drug, 4‐Nitroquinoline N‐oxide (4NQO), results in short tracts of ssDNA exposure (as a byproduct of nucleotide excision repair pathway (Giannattasio et al, 2004)) and is sufficient for the activation of both the apical and downstream checkpoint kinases (Fig 4B). We found that signaling from the DNA damage response kinases uncouples during unperturbed S phase, where Mec1 and Tel1 exhibit an activity independent of Rad53 (Fig 4C), a finding consistent with previous work (Bastos de Oliveira et al, 2015; Lanz et al, 2018). However, replication in the presence of a DNA alkylating agent, MMS, strongly induces Rad53 activity during S phase, revealing more efficient signal transduction from Mec1 to Rad53. Strikingly, in addition to the established targets of Mec1, Tel1, and Rad53 previously reported, the data presented here highlight many unexplored DNA damage‐ and cell cycle‐regulated phosphorylation events.
Figure 4. Probing regulation: DNA damage and cell cycle‐induced changes to the phosphoproteome.
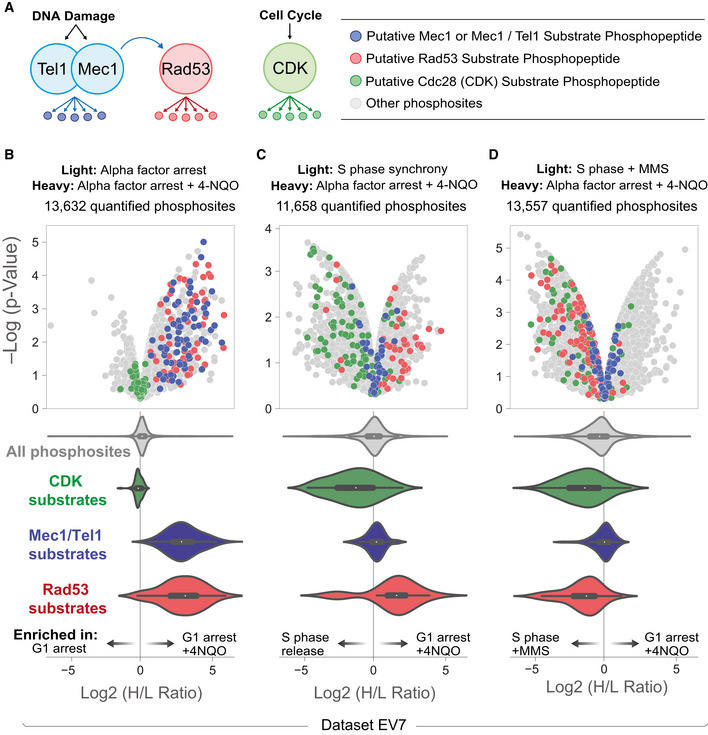
- Tracking the activity of DNA damage and cyclin‐dependent kinase signaling by monitoring the behavior of substrate phosphopeptides. In brief, “substrate” sites are kinase‐dependent phosphopeptides and harbor phosphorylated residues that lie in consensus target sequence of the indicated kinase. These putative substrate sites were defined previously by (Holt et al, 2009; Bastos de Oliveira et al, 2015).
- SILAC quantitation for the indicated experiment in the form of a volcano plot (Dataset EV7). Alpha factor arrest was for 3 h. 4NQO was added 2 h and 20 min after G1 arrest. Mec1/Tel1, Rad53, and CDK substrate phosphopeptides are highlighted in blue, red, and green, respectively. The violin plots represent the distribution of SILAC ratios for putative kinase substrate phosphopeptides (a proxy for kinase activity in the given experiment). Central dot and boxes represent the median and interquartile range, respectively. P‐value was calculated via t‐test of four independent biological replicates.
- (C) As in (B), S phase synchrony was achieved by releasing alpha factor arrested cells for 40 min. Central dot and boxes represent the median and interquartile range, respectively. P‐value was calculated via t‐test of two independent biological replicates.
- (D) As in (B), S phase synchrony was achieved by treating asynchronous cultures with 0.02 % MMS for 2 h. Central dot and boxes represent the median and interquartile range, respectively. P‐value was calculated via t‐test of four independent biological replicates.
We used another curation of experiments from our dataset (see Dataset EV1) to obtain cell cycle‐related regulatory information for ~ 11,000 phosphorylation events. While the experiments that comprise this resource were not originally designed to precisely and systematically monitor phosphorylation dynamics across the cell cycle, we compared the relative prevalence of phosphopeptides identified in bulk from yeast that were synchronized within three distinct cell cycle stages (G1 arrested, S phase enriched, and G2M arrested). Approximately 20% of the 11,000 phosphorylation events were either enriched or depleted in one stage of the cell cycle (Appendix Fig S5; Dataset EV8). Many phosphorylation events that were subjected to cell cycle regulation were either established cell stage‐specific events or occurred within proteins with cell cycle‐related functions (highlighted in Appendix Fig S5). Though our cell cycle analysis constitutes a large catalog of phosphorylation events, we caution that, due to technical and experimental limitations, our approach using “spectral counting” for this dataset lacks the quantitative accuracy and temporal resolution achieved by more focused investigations of cell cycle phosphorylation dynamics in yeast (Swaffer et al, 2018; Touati et al, 2018).
The 3D budding yeast phosphoproteome
In addition to probing the regulation of phosphorylation, we performed structural analysis of phosphorylation site position with the goal of improving the systematic prediction of functional phosphorylation events. Because phosphorylation events occurring at or near protein–protein interfaces may also be more likely to impact protein function, we first plotted the phosphoproteome within the context of protein–protein interactions. We utilized Interactome INSIDER (Meyer et al, 2018), a tool that has systematically identified, proteome‐wide, every amino acid residue found at the interface between proteins in a crystal structure or homology model (interactome3D is the source for our homology models (Mosca et al, 2013)). In total, we identified 646 phosphosites that lie at the surface between interacting proteins and many more sites that fall within 5 a.a. residues of an interface (Dataset EV9). An additional feature of Interactome INSIDER is its ability to predict interface residues for known protein–protein interactions that do not currently have any structural information (Meyer et al, 2018); we found 1,932 phosphorylation events that occur on these predicted protein‐protein interface residues (Dataset EV9). This information has been compiled into the SuperPhos online database (superphos.yulab.org), which lists all identified phosphorylation sites in budding yeast and displays their proximity to known or predicted protein interaction interfaces (Fig 5A).
Figure 5. 3D analysis of budding yeast phosphoproteome reveals potential regulatory phosphorylation at protein interaction interfaces.
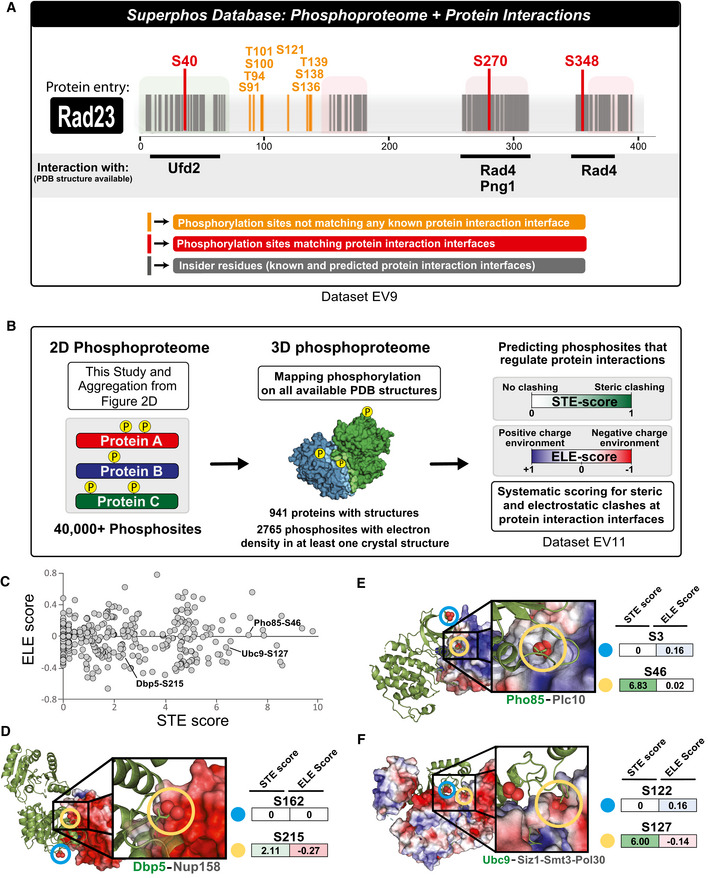
-
AThe SuperPhos database (beta version accessible at: http://superphos.yulab.org). Representative example of data display for a protein entry. The database merges an updated version of the budding yeast phosphoproteome with the protein interface calculations made by interactome INSIDER. In addition to indicating interactions for which PDB structures are available (as shown in this example), the SuperPhos database also provides information on all predicted interaction interfaces from INSIDER (not shown in this example). INSIDER mapping and phosphosite positional information used for the website can be found within Dataset EV9.
-
BMapping the yeast phosphoproteome to all available PDB structures and systematic prediction of phosphorylation that regulates protein–protein interactions. For each phosphosite that mapped to a structured region, ELE and STE scores were systematically calculated based on the proximity and charge of atoms from neighboring proteins. See Materials and Methods for a detailed explanation of how the scores were calculated. Phosphosites that map to more than one crystal structure or to multiple chains within a single crystal structure were assigned multiple STE and ELE scores (Dataset EV11).
-
CDistribution of STE and ELE scores assigned to phosphosites in the 3D budding yeast phosphoproteome. Labeled dots are highlighted in panels D–F.
-
D–FRepresentative examples of the mapping and scoring of phosphosites within the structural context of protein complexes. The phosphoprotein is displayed as a green ribbon cartoon; the electron density of the surrounding protein(s) is colored based on the electrostatic environment (as calculated by default using APBS: Adaptive Poison‐Boltzmann Solver). Blue coloring and red coloring represent more positive and negative charge environment, respectively. We note that, due to technical limitations related to size of the Ypt1‐TRAPP structure, it is not present in INSIDER and thus not searchable in our webtool.
To further exploit available structural information in our effort to systematically identify functional phosphorylation sites, we computationally positioned the budding yeast phosphoproteome onto all available 3D protein structures within the Protein Data Bank (PDB; Fig 5B). When considering 941 yeast proteins with structural information in PDB with resolution better than 4 Å, we found that the majority of phosphorylation occurs only within in regions with no structural information (5,943 of 8,708 phosphosites), a finding consistent with the importance of intrinsic disorder for protein phosphorylation (Iakoucheva et al, 2004). Despite this, we were still able to map 2,765 phosphorylation events onto structured regions (Fig 5B). Because most crystal structures are prepared under conditions in which the crystalized proteins would not be phosphorylated (e.g., protein purified from bacteria), phosphorylation sites that map to solvent‐inaccessible regions within these protein structures would have a higher likely‐hood of being impactful. We distinguished between two types of solvent‐inaccessible residues: (i) residues buried within the core of a single polypeptide chain and (ii) residues that lie at the interface between interacting proteins (similar to our INSIDER approach). We identified 539 phosphorylation sites that mapped to a buried, solvent‐inaccessible region within a single protein (Dataset EV10). However, we opted to focus more on phosphorylation sites found at the interfaces where proteins interact, since these sites potentially play key regulatory roles.
Mapping phosphorylation sites to protein interaction interfaces reveals phosphorylation events that regulate protein–protein interactions
Due to the potentially disruptive nature of adding a bulky and negatively charged phosphate group to an S/T/Y residue near a protein interaction surface, the presence of phosphorylation at a protein‐protein interface could result in a steric or electrostatic clash. In these instances, we predict that interface phosphorylation would disrupt or prevent protein–protein interactions and therefore reflect a potentially important regulatory mechanism. This concept was previously explored in budding yeast by Studer et al, albeit on a smaller scale (Studer et al, 2016). To systematically identify phosphorylation that causes “clashes” between interacting proteins, we devised a minimal scoring system based on the steric and electrostatic environment surrounding phosphosites near a protein interface region (see Materials and Methods for detailed explanation of how the scores were calculated). Our method is similar to the approach employed by Studer et al (2016). In brief, our method utilizes the per‐atom charge calculated by employing PDB2PQR pipeline (Dolinsky et al, 2004). Here, steric clash (STE) and electrostatic (ELE) scores for a given phosphosite are calculated based on the distance and charge of atoms from neighboring proteins (Fig 5C, Dataset EV11). Manual inspections of several phosphorylation events within their 3D context found that a phosphosite's STE and ELE scores accurately represent the surrounding steric and electrostatic environment (Fig 5D–F). We caution that the quality of our predictions is dependent on the quality and content of the structural information deposited on PDB, which can vary from structure to structure. Studer et al also reported that phosphorylation sites that lie at interface residues tend to exhibit more conservation throughout multiple fungal species, a finding supported by our own investigation of phosphosite conservation in our dataset (see Materials and Methods for full description). We distilled our conservation analysis into a single score and incorporated into Dataset EV9.
Using our scoring system, we extracted from our dataset hundreds of sites that, if phosphorylated in the context of the crystal structure, would cause steric clashing, occur within a negatively charged environment, or both (Fig 5C, Dataset EV11). We hypothesized that phosphorylation events with high STE scores and low ELE scores may disrupt protein–protein interactions. To test this hypothesis, we constitutively mimicked phosphorylation by mutating a phosphosite residue from serine to aspartic acid and determined how that mutation impacted a predicted set of protein–protein interactions. We performed proof‐of‐principle experiments in Rad23, an evolutionarily conserved protein with dual roles in nucleotide excision repair and proteolysis (Schauber et al, 1998). For its DNA repair functions, Rad23, together with Rad4, recognizes damaged DNA (de Laat et al, 1999). For its role in protein degradation, Rad23 interacts with several proteins in the ubiquitin pathway (Schauber et al, 1998). One of these proteins is Png1, a protein involved in the degradation of misfolded ER proteins (Kim et al, 2006). We chose Rad23 for our proof‐of‐principle experiments because it harbored a phosphosite (serine 270; S270) that mapped to an interface that binds two different proteins, Rad4 and Png1 (Fig 6A). Based on its STE and ELE scores, we anticipated that phosphorylation at S270 might significantly impact Rad23's interaction with Png1 while having a milder effect on its interaction with Rad4. To test our prediction, we generated a phospho‐mimetic RAD23 mutant (Rad23S270D) and performed IP‐MS to quantitatively compare interacting proteins pulled down with Rad23WT vs Rad23S270D. Consistent with our prediction, we found that Rad23's interaction with Png1 was specifically disrupted when phosphorylation was mimicked at S270 (greater that 25‐fold change in the average SILAC ratios for Png1 peptides; Fig 6B and C). Importantly, the ability of Rad23 to bind to its other interacting proteins, including Rad4, was not disrupted by the mutation of serine 270. A Rad23S270N substitution resulted in a less dramatic disruption of the Rad23‐Rad4 interaction (Appendix Fig S6B), suggesting that in this case electrostatics plays a more important role in impairing the interaction as compared to steric constraints.
Figure 6. Validation of the predicted effects of phosphorylation of Rad23 and Ypt1 in disrupting specific protein–protein interactions.
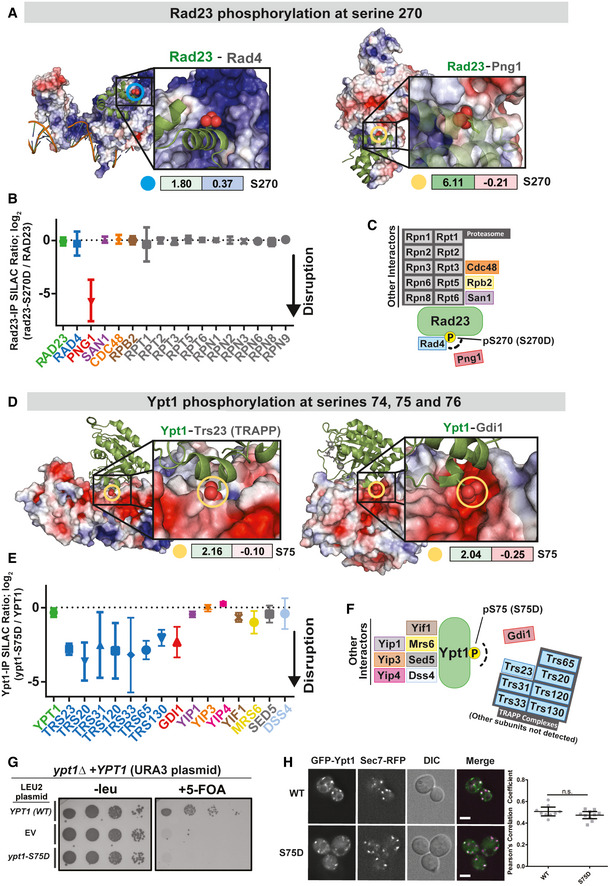
- Rad23 in complex with Rad4 (left) and Png1 (right). STE (left box) and ELE (right box) scores are displayed for the phosphorylation of Rad23 at serine 270 (circled in both structures). The electron density of the surrounding protein(s) is colored based on the electrostatic environment (as calculated by default using APBS). Blue coloring and red coloring represent more positive and negative charge environment, respectively.
- Quantitative mass spectrometry analysis of the Rad23 interaction network and the effect of a phospho‐mimetic mutation at serine 270. SILAC‐labeled yeast cultures expressing Rad23‐FLAG or an empty vector were subjected to anti‐FLAG IP to pre‐define the list of specific Rad23 interacting proteins shown in the graph. The average SILAC ratios represent fold changes for each of the Rad23 interactions in an IP using wild‐type Rad23 vs Rad23‐S270D mutant as bait. The average SILAC ratio for each protein (central icon) is derived from the measurement of multiple unique peptides (Dataset EV10). Error bars represent the standard deviation between independent peptide measurements. Data shown are representative of two independent experiments (data from the replicate experiment can be found in Appendix Fig S6A).
- Schematics depicting Rad23's protein interaction network and the impact of a phospho‐mimetic mutation at serine 270, as defined by SILAC IP‐MS.
- Ypt1 in complex with the TRAPP (left) and Gdi1 (right). We detected phosphorylation at S74, S75, and S76 of Ypt1 (all with high‐confidence localization in singly phosphorylated phosphopeptides). STE (left) and ELE (right) scores for the monophosphorylation of Ypt1 at serine 75 are displayed and modeled into both structures (circled). The electron density of the surrounding protein(s) is colored based on the electrostatic environment (as calculated by default using APBS). Blue coloring and red coloring represent more positive and negative charge environment, respectively. We note that, due to technical limitations related to size of the Ypt1‐TRAPP structure, it is not present in INSIDER and thus not searchable in our webtool.
- Quantitative mass spectrometry analysis of the Ypt1 interaction network and the effect of a phospho‐mimetic mutation at serine 75. SILAC‐labeled yeast cultures expressing Ypt1‐FLAG or an empty vector were subjected to anti‐FLAG IP to pre‐define the list of specific Ypt1‐interacting proteins shown in the graph. The average SILAC ratios represent fold changes for each of the Ypt1 interactions in IP using wild‐type Ypt1 vs Ypt1‐S75D mutant as bait. The average SILAC ratio for each protein (central icon) is derived from the measurement of multiple unique peptides (Dataset EV10). Error bars represent the standard deviation. Data shown are representative of two independent experiments.
- Schematics depicting Ypt1's protein interaction network and the impact of a phospho‐mimetic mutation at serine 75, as defined by SILAC IP‐MS.
- Plasmid shuffling assay to test whether ypt1‐S75D can fulfill Ypt1's essential functions. Yeast were spotted onto plates containing 5‐Fluoroorotic acid (5‐FOA) to relinquish the URA plasmid.
- Fluorescent and differential interference contrast (DIC) microscopy to determine whether ypt1‐S75D retains proper localization to the Golgi complex. Plasmids expressing either wild‐type GFP‐Ypt1 or GFP‐Ypt1‐S75D were transformed into yeast cells. Single focal planes are shown of live‐cell fluorescence microscopy images under normal growth conditions. Cells are expressing an endogenously tagged Golgi marker, Sec7‐DsRed. Scale bar is 2 µm. A total of 105 cells for WT and S75D. The amount of overlap between Ypt1 and Sec7 colocalization was quantified using the Pearson's correlation coefficient (PCC). An unpaired t‐test with Welch's correction was used to compare WT and mutant PCC data points (see Materials and Methods). The PCC for the WT and S75D mutant is not significantly different (P = 0.1667). WT Mean = 0.5068, S75D Mean = 0.4732, error bars represent 95% CIs.
We performed a similar analysis for the Golgi‐resident Rab family GTPase protein, Ypt1. Ypt1 is an essential Rab1 homolog that regulates ER‐to‐Golgi membrane trafficking by recruiting effectors to the membrane surfaces of ER‐derived vesicles and the Golgi complex (Hutagalung & Novick, 2011; Yang et al, 2016). The primary guanine‐nucleotide exchange factor (GEF) that activates Ypt1 in vivo is the TRAPPIII complex (Wang et al, 2000; Thomas et al, 2018); the related TRAPPII complex is also capable of promoting Ypt1 activation (Thomas & Fromme, 2016). Ypt1 interacts with the TRAPP complexes, in part, by binding to the Trs23 subunit. The inactive (GDP‐bound) form of Ypt1 is kept soluble in the cytoplasm by binding to Gdi1 (Garrett et al, 1994). Gdi1 therefore prevents inactive Ypt1 from accumulating on the membrane of the Golgi complex or other organelles. Interestingly, we found three consecutive phosphosites in Ypt1 (S74, S75, and S76) that lie at its interface with both Trs23 and Gdi1, potentially disrupting these interactions when phosphorylated (Fig 6D). A phospho‐mimetic mutation of just one of these residues, S75D, was sufficient to disrupt the interactions of Ypt1 with subunits of the TRAPP complexes (including Trs23) and with Gdi1, without disrupting other interactions (Fig 6E and F). We also tested whether the S75D mutation retained the essential function of Ypt1. We observed that ypt1‐S75D was unable to support viability in the absence of endogenous Ypt1 (Fig 6G). However, the GFP‐Ypt1‐S75D mutant protein appeared to retain normal Golgi localization, measured by colocalization with the Golgi‐resident protein Sec7 (Fig 6H), suggesting that S75D is a separation‐of‐function mutation that will be useful to dissect distinct mechanisms of Ypt1 regulation. Overall, together with previous work (Studer et al, 2016), these examples reinforce the concept that it is possible to systematically predict the impact of phosphorylation on the regulation of protein–protein interactions based on the structural context of its occurrence.
Discussion
Here, we conducted the most in‐depth analysis of the phosphoproteome in budding yeast to date. As it currently stands, if considering both our current analysis and previous reports, the budding yeast phosphoproteome presently consists of approximately 45,000 phosphorylation sites, though this number varies greatly depending on thresholds set for phosphosite localization probability. Our analysis of phosphosites identified over a range of localization probabilities should help inform the yeast community on the limitations inherent to the mass spectral identification of phosphosites and illustrate the pros and cons of both laxity and stringency in setting cutoffs for localization probability. Importantly, the saturation analysis in Fig 3E suggests that the size of the phosphoproteome can be expanded further using alternative digestive enzymes.
The biological significance of nearly all these identified phosphorylation events remains unknown. The extensive scope of the phosphoproteome raises the question as to whether many of the identified phosphorylation events have tangible biological significance. While the quantity of phosphosites identified and phosphoproteome coverage achieved in our study has inherent value, the ability to distinguish functional phosphorylation from what could potentially be “off‐target” or promiscuous kinase action represents the primary challenge in dealing with large‐scale phosphoproteomic datasets. Importantly, while the “detectability” of a particular phosphorylation event is impacted by factors other than its abundance (e.g., peptide solubility or ionization and accessibility to tryptic digestion), an argument could be made that many of the phosphorylation events that are buried deep within the phosphoproteome are low abundant and therefore are likely less important than the events which are readily detectable. If true, by expanding the depth with which the phosphoproteome is profiled, to what extent is our dataset revealing functional phosphorylation sites? While this question is difficult to address, the examples presented in Appendix Fig S3 indicate that even phosphorylation that exists near the threshold of MS detection can be biologically meaningful. In addition, the relative prevalence of a particular phosphorylation event does not predetermine its importance. Although specialized MS applications can assess the stoichiometry of protein phosphorylation (Wu et al, 2011; Lim et al, 2017), general MS‐based phosphoproteomics does not inherently inform phosphorylation stoichiometry. Therefore, low abundant phosphoproteins that are stoichiometrically phosphorylated are often indistinguishable from very abundant proteins whose phosphorylated form represents only a small fraction of their total protein.
Given the challenges highlighted above, how can one distinguish biologically meaningful phosphorylation from what might just be the “noise” of the phosphoproteome? For one, it is clear that the evolutionary conservation of a phosphorylated residue does not dictate the relevancy of the event, as many functional phosphorylation events occur on poorly conserved residues (Holt et al, 2009; Amoutzias et al, 2012). One clever way to distinguish a “deliberate” phosphorylation event from promiscuous ones may be to measure how dynamically it changes. With the assumption that functional kinase–substrate interactions are better optimized for binding than promiscuous interactions, Kanshin et al (2015) demonstrated that changes in phosphorylation occur faster on functional vs promiscuous substrates. Nevertheless, the current standard for determining the functionality of a phosphorylation event requires the generation of mutant yeast strains that either lack or constitutively mimic the phosphorylated residues in a substrate protein, with the ultimate goal of phenocopying the effects of a kinase's action or inaction. However, generating phosphosite mutations is labor intensive and often times, due to the recessive nature of phosphomutant phenotypes, requires genetic manipulation at the endogenous locus. Efforts to elicit phenotypes from phosphosite mutants can be further complicated by functional redundancy, which in some cases can be found in serines or threonines neighboring the identified phosphorylation event or in another substrate whose phosphorylation results in a redundant effect. The phospho‐regulation of Slx4 and Sld3/Dbf4 exemplifies the extensive redundancy that must be overcome when making phosphosite mutants (Ohouo et al, 2010; Zegerman & Diffley, 2010).
The challenges that hinder the interpretation of large phosphoproteomic datasets are, in some ways, similar to those faced in the field of human genomics. As mass spectrometry has exponentially expanded the catalog of phosphorylation events, genomics has similarly revealed tens of thousands of disease‐associated mutations (Stenson et al, 2003; Landrum et al, 2014). Akin to the biologically impactful phosphorylation events in our dataset, impactful mutations exist amidst many less meaningful polymorphisms (Tennessen et al, 2012; The, 1000 Genomes Project Consortium, 2012; The Genome of the Netherlands Consortium, 2014). Recent efforts to identify the key mutations that underpin human disease phenotypes have utilized the expanding collection of protein structural information (Wang et al, 2012; Wei et al, 2014; Meyer et al, 2016; Chen et al, 2018), with the logic being that mutations that occur at or near the interfaces where proteins interact will have a higher likelihood of impacting protein function. Building on this logic, here we streamlined the identification of the functional phosphosites by identifying those located at or near protein‐interaction interfaces. In addition, we were able to make systematic predictions about the impact of a phosphorylation event occurring near protein‐protein interfaces and its potential for causing steric clashes or an electrostatic environment incompatible with the crystal structure. Our minimalistic approach to predicting regulatory phosphorylation based on available structural information, though simpler than methods employed previously (Nishi et al, 2011; Beltrao et al, 2012; Betts et al, 2017; Ochoa et al, 2020), successfully predicted disruptive phosphorylation (Fig 6). For example, we demonstrated that phosphorylation‐mimicking mutation of Rad23 at S270 specifically disrupts the interaction between Rad23 and Png1. The formation of the Rad23‐Png1 complex is critical for the efficient degradation of glycosylated ER‐associated proteins (Kim et al, 2006). Thus, while the kinase responsible for the phosphorylation of S270 and the biological context in which this phosphorylation occurs remain unclear, the phosphorylation of Rad23 at S270 could act as a switch to inhibit the degradation of glycosylated ER proteins. Moreover, we found that phospho‐mimetic mutation of S75 in the Rab GTPase Ypt1 disrupts its interactions with both Gdi1 and the TRAPP GEF complexes. This suggests that phosphorylation of S75 would result in decreased activation of Ypt1 but persistence of inactive Ypt1 on the Golgi membrane. Correspondingly, the Ypt1 phosphomutant retained its localization to the Golgi yet was unable to provide the essential function of Ypt1. Previous studies have demonstrated that phosphorylation of another Rab GTPase, Sec4, is a negative regulatory mechanism coupled to the cell cycle (Heger et al, 2011; Lepore et al, 2016) and that Rab family proteins are regulated by phosphorylation in humans (Steger et al, 2017). Therefore, although it remains to be determined how phosphorylation of Ypt1 is regulated, our results suggest that phosphorylation of Ypt1 is a plausible regulatory mechanism for controlling when and where it is activated and that the ypt1‐S75D separation‐of‐function mutation will be useful to further dissect distinct mechanisms of regulation.
A recent study explored in‐depth the functional landscape of the human phosphoproteome (Ochoa et al, 2020), curating the massive amount of raw mass spectral data gathered on the human cell lines and processing it through a unified pipeline (requiring 2 months of dedicated PSM search time). Taking advantage of the wealth of structural and mutational information available in human systems, the authors assigned a predicted functional “score” for each phosphosite. Unfortunately, relative to the human phosphoproteome, the budding yeast phosphoproteome has currently been profiled to a far lesser extent and much less structural information is available for this organism. Moving forward, the ever‐expanding repository of structural information represents an invaluable resource in the study of post‐translational modifications and their functionality. Elucidation of more protein structures, particularly through the use of emerging technologies like Cyro‐EM, will expand the 3D characterization of the phosphoproteome and support the approaches presented here.
Materials and Methods
Protein extraction and sample preparation for phosphoproteome analysis
The phosphoproteomic experiments used as the source for the database were performed for a variety of focused biological investigations. In almost all cases, these experiments were performed with the intention of quantifying changes in phosphopeptide abundance and thus relied on a two‐channel SILAC‐based workflow. "Light" and "heavy"‐labeled cultures ("light" version complemented with normal arginine and lysine; "heavy" version complemented with l‐Lysine 13C6, 15N2.HCl and l‐Arginine 13C6, 15N4.HCl) were combined, harvested by centrifugation in TE buffer pH 8.0 containing protease inhibitors and stored frozen at −80°C until cell lysis. Approximately 0.3 g of yeast cell pellet (in three separate 2 ml screwcap tubes) was lysed by bead beating at 4°C in 3 ml of lysis buffer (1 ml per tube) containing 50 mM Tris–HCl, pH 8.0, 0.2% Tergitol, 150 mM NaCl, 5 mM EDTA, complete EDTA‐free protease inhibitor cocktail (Roche), 5 mM sodium fluoride and 10 mM β‐glycerophosphate. Lysates of light and heavy conditions were mixed together (approximately 6 mg of protein from each condition). The mixed lysate was then denatured in 1% SDS, reduced with DTT, alkylated with iodoacetamide and then precipitated with three volumes of a solution containing 50% acetone and 50% ethanol. Proteins were solubilized in a solution of 2 M urea, 50 mM Tris–HCl, pH 8.0, and 150 mM NaCl, and then TPCK‐treated trypsin was added. Digestion was performed overnight at 37°C, and then trifluoroacetic acid and formic acid were added to a final concentration of 0.2%. For the digestion using chymotrypsin, the urea concentration was reduced to 1 M. Peptides were desalted with Sep‐Pak C18 column (Waters). C18 column was conditioned with five column volumes of 80% acetonitrile and 0.1% acetic acid and washed with five column volumes of 0.1% trifluoroacetic acid. After samples were loaded, column was washed with five column volumes of 0.1% acetic acid followed by elution with four column volumes of 80% acetonitrile and 0.1% acetic acid. Elution was dried in a SpeedVac evaporator and resuspended in 1% acetic acid.
Phosphopeptide enrichment
After protein extraction and trypsin digestion, desalted peptides were resuspended in 1% acetic acid and loaded in a tip column containing ~ 22 µl of immobilized metal affinity chromatography (IMAC) resin prepared as previously described (Bastos de Oliveira et al, 2018). After loading, the IMAC resin was washed with one column volume of 25% acetonitrile, 100 mM NaCl, and 0.1% acetic acid solution followed by two column volumes of 1% acetic acid, one column volume of deionized water and finally, eluted with three column volumes of 12% ammonia and 10% acetonitrile solution. The elutions were then dried and resuspended in 16.5 μl H2O. 1.5 μls (10% of the sample) was diluted to 10 μl of 0.1% TFA and ran as input samples to assess phosphopeptide purity.
HILIC fractionation
After phosphopeptide enrichment, samples were dried in a SpeedVac, reconstituted in 80% acetonitrile and 1% formic acid, and fractionated by hydrophilic interaction liquid chromatography (HILIC) with TSK gel Amide‐80 column (2 mm × 150 mm, 5 µm; Tosoh Bioscience). 90 s fractions were collected between 10 and 25 min of the gradient. Three solvents were used for the gradient: buffer A (90% acetonitrile); buffer B (80% acetonitrile and 0.005% trifluoroacetic acid), and buffer C (0.025% trifluoroacetic acid). The gradient used consists of a 100% buffer A at time = 0 min; 88% of buffer B and 12% of buffer C at time = 5 min; 60% of buffer B and 40% of buffer C at time = 30 min; and 5% of buffer B and 95% of buffer C from time = 35 to 45 min in a flow of 150 µl/min.
Mass spectrometry analysis and data acquisition
Hydrophilic interaction liquid chromatography fractions were dried in a SpeedVac, reconstituted in 0.1% trifluoroacetic acid, and subjected to LC‐MS/MS analysis using a 20‐cm‐long 125‐µm inner diameter column packed in‐house with 3 µm C18 reversed‐phase particles (Magic C18 AQ beads, Bruker). Separated phosphopeptides were electrosprayed into a QExactive Orbitrap mass spectrometer (Thermo Fisher Scientific). Xcalibur software (Thermo Fischer Scientific) was used for the data acquisition, and the Q Exactive was operated in data‐dependent mode. Survey scans were acquired in the Orbitrap mass analyzer over the range of 380 to 1,800 m/z with a mass resolution of 70,000 (at m/z 200). MS/MS spectra were performed selecting up to the 10 most abundant ions with a charge state using of 2, 3, or 4 within an isolation window of 2.0 m/z. Selected ions were fragmented by higher‐energy collisional dissociation (HCD) with normalized collision energies of 27, and the tandem mass spectra were acquired in the Orbitrap mass analyzer with a mass resolution of 17,500 (at m/z 200). Repeated sequencing of peptides was kept to a minimum by dynamic exclusion of the sequenced peptides for 30 s. For MS/MS, AGC target was set to 1e5 and max injection time was set to 120 ms.
Phosphopeptide and phosphorylation site identification: primary search using Andromeda
Three separate search engines were used to search the raw MS/MS spectra. All searches were performed on 19 separate “chunks”, with each chunk containing an average of 500,000 MS/MS spectra. The primary search engine used was Andromeda, as part of the MaxQuant software package (version 1.6.5.0). Searching parameters for MaxQuant included a fully‐tryptic requirement. After a “first search” at 20 ppm, the precursor match tolerance was set to 4.5 ppm. Differential modifications were 8.0142 daltons for lysine, 10.00827 daltons for arginine, 79.966331 daltons for phosphorylation of serine, threonine and tyrosine, and a static mass modification of 57.021465 daltons for alkylated cysteine residues. N‐terminal acetylation was also set as a variable modification, but only for peptides that correspond to the N‐terminus of protein. A complete list of searching parameters can be found in Dataset EV13. The primary source for the phosphosite identification was the “Phospho STY” output table in MQ (Sharma et al, 2014). The quality threshold for a PSM to be considered for phosphosite identification was an Andromeda score greater than 40 and a delta score of greater than 6 (similar to criteria used previously (Sharma et al, 2014)). The 19 “Phospho STY” files (from each of the 19 chunks) were concatenated and redundancy eliminated by retaining the PSM entry with best phosphosite localization score for every identified phosphosite. The primary dataset contains only phosphosites with high‐confidence localization, which we considered as having a MaxQuant localization probability of > 0.70.
Phosphopeptide and phosphorylation identification: secondary search using Sequest
All spectra were also searched using two Sequest‐based engines, Proteome Discoverer (Thermo) and SORCERER (Sage N Research, Inc.). For PD and SORCERER, we used similar search parameters as MaxQuant, with the exception that we permitted semi‐tryptic digestion, rather than require fully typtic. Precursor match tolerance for both Sequest searches was set to 10 ppm. We considered only high‐confidence PSMs for the pipeline, filtered to < 1% FDR using percolator and sorcererscore for PD and SORCERER, respectively. To increase the confidence in our Sequest searches further, we only considered phosphopeptides whose backbone sequence appeared in both the PD and SORCERER PSM searches. For phosphopeptides that passed this backbone requirement, we then retained the PSM information acquired using Proteome Discoverer. The phosphorylation localization probabilities were determined using ptmRS (PhosphoRS) within Proteome Discoverer (Taus et al, 2011). The threshold for “high‐confidence” phosphosite localization was a phosphoRS percentage of > 70%.
Inclusion of phosphosite clusters
Phosphopeptides that did not contain phosphosite localization scores that met our “high‐confidence” threshold (MQ: localization probability above 0.70; PD/SORCERER: phosphoRS percentage above 70%) were subsequently searched for phosphosite “clusters”. This involved identifying consecutive S/T/Y residues that were assigned localization probabilities which sum to > 0.9 (MQ) or 90% (phosphoRS).
SILAC quantitation
Maxquant was used to determine the SILAC ratios for the experiments related to Fig 4B–D and Dataset EV7. These 11 experiments were subjected to an independent Maxquant search (using the same PSM search parameters described above (Dataset EV13)). Phospho STY output file from Maxquant was used to assemble Dataset EV7.
Analysis of cell cycle phosphorylation dynamics
The spectral counts used to perform the cell cycle analysis were extracted from the primary search outlined in Appendix Fig S1. A curated set of runs were given either a G1, S phase, or G2M annotation. For every phosphosite (specifically, its best corresponding phosphopeptide), the number of identifications (i.e., spectral counts, PSMs) within runs with cell cycle annotation was tallied. Only phosphopeptides that were detected more than five times in the annotated runs (G1 + G2M + MMS) were retained. The stringency for enrichment or depletion in a particular cell cycle state was a five‐fold difference in the number of identifications for one cell cycle stage vs the other two.
G1 synchrony was achieved through alpha factor arrest. S phase synchrony was primarily achieved via 2 h MMS treatment (in some cases, 40 min release from alpha factor arrest). G2/M synchrony was achieved via 2.5 h of nocodozole treatment. The use of DNA damaging agents to synchronize cells in S phase was counterbalanced by the inclusion of multiple experiments involving the addition of 4NQO to both G1‐ and G2/M‐arrested cell. Thus, each cell cycle stage contains analyses done with and without the presence of a DNA damage.
Conservation analysis
Fungal protein sequences were downloaded from Uniprot database (UniProt, 2019) (Taxonomy id: 4751; downloaded on 10th September 2020). Homologous fungal proteins for all the Saccharomyces cerevisiae phosphoproteins in our dataset were identified using BLASTP (Camacho et al, 2009). From the BLASTP output, for the phosphoproteins that had at least three homologs (percent identity ≥ 30%; E‐value cutoff: 0.05), multiple sequence alignment (MSA) was performed using MAFFT3 (in clustal format) (Katoh & Standley, 2013). Further, with the MSAs as input, conservation score was generated for all the aligned positions in phosphoproteins utilizing AL2CO (Pei & Grishin, 2001).
Calculation of ELE and STE scores for phosphosites near protein–protein interaction interfaces
All the phosphosites were mapped onto the available 3D structures in PDB (Gilliland et al, 2000) using residue‐level mapping information obtained from SIFTS database (Gutmanas et al, 2018). The sites that were mapped to structures with more than one chain were further considered for calculation of ELE and STE scores. For each site, neighboring atoms with in a distance of 10 Å were identified (excluding the atoms present in the same chain as the phosphosite) using an in‐house python script (Atoms chosen to be the “phosphosite atoms”: “OG” for serine, “OG1” for threonine and “OH” for tyrosine). Next, the charge for all the individual neighboring atoms was obtained using a command line version of PDB2PQR pipeline (Dolinsky et al, 2004) (with amber as the force field and using “‐‐nodebump” option). Finally, the STE and ELE scores were calculated using the following equations:
where, Chargei is the charge of neighboring atom i and Distancei is the distance between atom i and the phosphosite atom.
Quantitative MS analysis pull‐down protein complexes
Yeast carrying either GFP‐YPT1 or Rad23‐FLAG were grown to an O.D.600 of 0.4 in 200 ml of ‐Arg ‐Lys dropout media ("light" version complemented with normal arginine and lysine; "heavy" version complemented with l‐Lysine 13C6, 15N2.HCl and l‐Arginine 13C6, 15N4.HCl). After centrifugation, pellets were kept at −80°C prior to cell lysis. Approximately 0.3 g of cell pellet of each strain was lysed by bead beating at 4°C in 3 ml of lysis buffer (50 mM Tris–HCl pH 7.5, 0.2% Tergitol, 150 mM NaCl, 5 mM EDTA, Complete EDTA‐free protease inhibitor cocktail (Roche), 5 mM sodium fluoride, 10 mM β‐glycerol‐phosphate). Lysates were incubated with GFP‐TRAP (in‐house) or anti‐FLAG agarose resin (Sigma) for 4 h at 4°C. After three washes with lysis buffer, bound proteins were eluted with 90 μls of elution buffer (100 mM Tris–HCl pH 8.0, 1% SDS). Eluted proteins from normal or heavy media grown cells were mixed together, reduced, alkylated and then precipitated with three volumes of a solution containing 50% acetone and 50% ethanol. Proteins were solubilized in a solution of 2 M urea, 50 mM Tris–HCl, pH 8.0, and 150 mM NaCl, and then Trypsin Gold was added. Digestion was performed overnight at 37°C, and then trifluoroacetic acid and formic acid were added to a final concentration of 0.2%. Peptides were desalted with Sep‐Pak C18 column (Waters). Elution from C18 column was dried in a SpeedVac evaporator and resuspended in 0.1% trifluoroacetic acid.
Fluorescent microscopy
Overnight cultures were grown to an OD600 between 0.1 and 0.8, then imaged. Single focal planes are shown of live‐cell fluorescence microscopy images under normal growth conditions. Cells are expressing endogenously tagged Sec7‐6xDsRed. Scale bar is 2 um. The amount of overlap between Ypt1 and Sec7 was quantified using the Pearson's Correlation Coefficient (PCC). A region of interest was selected surrounding 1–4 cells and propagated to all of the focal planes containing those cells for correlation analysis. Each data point represents the PCC for an image (WT = 9 images, S75D = 10 images) containing several regions of interest totaling 4–21 cells. A total of 105 cells for WT and S75D. An unpaired two‐tailed t‐test with Welch's correction was used to analyze the data points.
Author contributions
MCL performed all phosphoproteomic experiments. MCL, EJS, AMNJ and SV performed experiments. MCL, KY, MBS, JCF, VF, and HY conceived the data processing pipeline. KY and SG wrote various scripts for the consolidation and interpretation of the dataset and designed the website. KY wrote the site clustering and clash scoring algorithms. MCL wrote and MBS edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Dataset EV13
Review Process File
Acknowledgements
We would like to thank Bik Tye and Yuanliang Zhai for critical reading of the manuscript and Josh Chappie and Yuxin Mao for useful discussions. Mimi Xie assisted in making the ternary plot. Kiran Madura graciously provided Rad23‐related plasmids. Adm Chrysler and Ellen Miller provided invaluable IT assistance. Finally, we thank members of the Smolka and Yu Labs for useful discussions and constructive comments. K.Y. thanks the Sam and Nancy Fleming Research Fellowship. This work was supported by grants from the National Institutes of Health to M.B.S. (R01GM097272, R01GM123018, and R01HD095296; Equipment Supplement R01GM097272‐07S1), H.Y. (R01GM124559 and R01GM125639), and J.C.F. (R35GM136258). Additional funding was provided through the Alfred P. Sloan Foundation Fellowship and a National Science Foundation Graduate Research Fellowship (grant DGE‐1144153) to A.M.N.J.
EMBO reports (2021) 22: e51121.
Contributor Information
Haiyuan Yu, Email: haiyuan.yu@cornell.edu.
Marcus B Smolka, Email: mbs266@cornell.edu.
Data availability
The phosphoproteomic datasets produced in this study have been deposited to the PRIDE database (https://www.ebi.ac.uk/pride/archive/) and assigned the identifier PXD012395 (https://www.ebi.ac.uk/pride/archive/projects/PXD012395). Some data from PXD012395 were previously published in PXD009734 (https://www.ebi.ac.uk/pride/archive/projects/PXD009734).
References
- Aebersold R, Mann M (2003) Mass spectrometry‐based proteomics. Nature 422: 198–207 [DOI] [PubMed] [Google Scholar]
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H (2008) A multidimensional chromatography technology for in‐depth phosphoproteome analysis. Mol Cell Proteomics 7: 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoutzias GD, He Y, Lilley KS, Van de Peer Y, Oliver SG (2012) Evaluation and properties of the budding yeast phosphoproteome. Mol Cell Proteomics 11(M111): 009555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan R, Park J, Karra K, Hitz BC, Binkley G, Hong EL, Sullivan J, Micklem G, Cherry JM (2012) YeastMine–an integrated data warehouse for Saccharomyces cerevisiae data as a multipurpose tool‐kit. Database (Oxford) 2012: bar062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Cussiol JR, Das J, Jeong MC, Doerfler L, Schmidt KH, Yu H, Smolka MB (2015) Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell 57: 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos de Oliveira FM, Kim D, Lanz M, Smolka MB (2018) Quantitative analysis of DNA damage signaling responses to chemical and genetic perturbations. Methods Mol Biol 1672: 645–660 [DOI] [PubMed] [Google Scholar]
- Batth TS, Francavilla C, Olsen JV (2014) Off‐line high‐pH reversed‐phase fractionation for in‐depth phosphoproteomics. J Proteome Res 13: 6176–6186 [DOI] [PubMed] [Google Scholar]
- Beltrao P, Albanese V, Kenner LR, Swaney DL, Burlingame A, Villen J, Lim WA, Fraser JS, Frydman J, Krogan NJ (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150: 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts MJ, Wichmann O, Utz M, Andre T, Petsalaki E, Minguez P, Parca L, Roth FP, Gavin AC, Bork P et al (2017) Systematic identification of phosphorylation‐mediated protein interaction switches. PLoS Comput Biol 13: e1005462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco MG, Matos J, West SC (2014) Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54: 94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R (2007) Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods 4: 231–237 [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009) BLAST+: architecture and applications. BMC Bioinformatics 10: 421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Fragoza R, Klei L, Liu Y, Wang J, Roeder K, Devlin B, Yu H (2018) An interactome perturbation framework prioritizes damaging missense mutations for developmental disorders. Nat Genet 50: 1032–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cussiol JRR, Soares BL, Oliveira FMB (2019) From yeast to humans: understanding the biology of DNA damage response (DDR) kinases. Genet Mol Biol 43: e20190071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Gould KL, Gygi SP, Kellogg DR (2013) Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol Biol Cell 24: 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA (2004) PDB2PQR: an automated pipeline for the setup of Poisson‐Boltzmann electrostatics calculations. Nucleic Acids Res 32: W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae . Nat Biotechnol 20: 301–305 [DOI] [PubMed] [Google Scholar]
- Foiani M, Nadjar‐Boger E, Capone R, Sagee S, Hashimshoni T, Kassir Y (1996) A meiosis‐specific protein kinase, Ime2, is required for the correct timing of DNA replication and for spore formation in yeast meiosis. Mol Gen Genet 253: 278–288 [DOI] [PubMed] [Google Scholar]
- Garrett MD, Zahner JE, Cheney CM, Novick PJ (1994) GDI1 encodes a GDP dissociation inhibitor that plays an essential role in the yeast secretory pathway. EMBO J 13: 1718–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi‐Falconi M (2004) Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J 23: 429–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Branzei D (2017) S‐phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci 74: 2361–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland G, Berman HM, Weissig H, Shindyalov IN, Westbrook J, Bourne PE, Bhat TN, Feng Z (2000) The protein data bank. Nucleic Acids Res 28: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruhler A, Olsen JV, Mohammed S, Mortensen P, Faergeman NJ, Mann M, Jensen ON (2005) Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol Cell Proteomics 4: 310–327 [DOI] [PubMed] [Google Scholar]
- Guttmann‐Raviv N, Martin S, Kassir Y (2002) Ime2, a meiosis‐specific kinase in yeast, is required for destabilization of its transcriptional activator, Ime1. Mol Cell Biol 22: 2047–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmanas A, Dana JM, Velankar S, Qi G, Martin M, Tyagi N, O’Donovan C (2018) SIFTS: updated structure integration with function, taxonomy and sequences resource allows 40‐fold increase in coverage of structure‐based annotations for proteins. Nucleic Acids Res 47: D482–D489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardman G, Perkins S, Brownridge PJ, Clarke CJ, Byrne DP, Campbell AE, Kalyuzhnyy A, Myall A, Eyers PA, Jones AR et al (2019) Strong anion exchange‐mediated phosphoproteomics reveals extensive human non‐canonical phosphorylation. EMBO J 38: e100847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger CD, Wrann CD, Collins RN (2011) Phosphorylation provides a negative mode of regulation for the yeast Rab GTPase Sec4p. PLoS One 6: e24332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho B, Baryshnikova A, Brown GW (2018) Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst 6: 192–205 [DOI] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Latham V, Murray B, Nandhikonda V, Nord A, Skrzypek E, Wheeler T, Zhang B, Gnad F (2019) 15 years of PhosphoSitePlus(R): integrating post‐translationally modified sites, disease variants and isoforms. Nucleic Acids Res 47: D433–D441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Raucci S, Jaquenoud M, Hatakeyama R, Stumpe M, Rohr R, Reggiori F, De Virgilio C, Dengjel J (2019) Multilayered control of protein turnover by TORC1 and Atg1. Cell Rep 28: 3486–3496 [DOI] [PubMed] [Google Scholar]
- Hustedt N, Seeber A, Sack R, Tsai‐Pflugfelder M, Bhullar B, Vlaming H, van Leeuwen F, Guenole A, van Attikum H, Srivas R et al (2015) Yeast PP4 interacts with ATR homolog Ddc2‐Mec1 and regulates checkpoint signaling. Mol Cell 57: 273–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutagalung AH, Novick PJ (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev 91: 119–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK (2004) The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res 32: 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanshin E, Bergeron‐Sandoval LP, Isik SS, Thibault P, Michnick SW (2015) A cell‐signaling network temporally resolves specific versus promiscuous phosphorylation. Cell Rep 10: 1202–1214 [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Ahn J, Liu C, Tanabe K, Apodaca J, Suzuki T, Rao H (2006) The Png1‐Rad23 complex regulates glycoprotein turnover. J Cell Biol 172: 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono K, Nogami S, Abe M, Nishizawa M, Morishita S, Pellman D, Ohya Y (2008) G1/S cyclin‐dependent kinase regulates small GTPase Rho1p through phosphorylation of RhoGEF Tus1p in Saccharomyces cerevisiae . Mol Biol Cell 19: 1763–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH (1999) Molecular mechanism of nucleotide excision repair. Genes Dev 13: 768–785 [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR (2014) ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry CR, Levy ED, Michnick SW (2009) Weak functional constraints on phosphoproteomes. Trends Genet 25: 193–197 [DOI] [PubMed] [Google Scholar]
- Lanz MC, Oberly S, Sanford EJ, Sharma S, Chabes A, Smolka MB (2018) Separable roles for Mec1/ATR in genome maintenance, DNA replication, and checkpoint signaling. Genes Dev 32: 822–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Dibitetto D, Smolka MB (2019) DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J 38: e101801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics 4: 873–886 [DOI] [PubMed] [Google Scholar]
- Lepore D, Spassibojko O, Pinto G, Collins RN (2016) Cell cycle‐dependent phosphorylation of Sec4p controls membrane deposition during cytokinesis. J Cell Biol 214: 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED, Michnick SW, Landry CR (2012) Protein abundance is key to distinguish promiscuous from functional phosphorylation based on evolutionary information. Philos Trans R Soc Lond B Biol Sci 367: 2594–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, Vandersluis B, Bellay J, Devit M et al (2011) Systematic exploration of essential yeast gene function with temperature‐sensitive mutants. Nat Biotechnol 29: 361–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lienhard GE (2008) Non‐functional phosphorylations? Trends Biochem Sci 33: 351–352 [DOI] [PubMed] [Google Scholar]
- Lim MY, O'Brien J, Paulo JA, Gygi SP (2017) Improved method for determining absolute phosphorylation stoichiometry using bayesian statistics and isobaric labeling. J Proteome Res 16: 4217–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MJ, Lapcevic R, Romero AE, Yoon M, Das J, Beltran JF, Mort M, Stenson PD, Cooper DN, Paccanaro A et al (2016) Mutation3D: cancer gene prediction through atomic clustering of coding variants in the structural proteome. Hum Mutat 37: 447–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MJ, Beltran JF, Liang S, Fragoza R, Rumack A, Liang J, Wei X, Yu H (2018) Interactome INSIDER: a structural interactome browser for genomic studies. Nat Methods 15: 107–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosca R, Ceol A, Aloy P (2013) Interactome3D: adding structural details to protein networks. Nat Methods 10: 47–53 [DOI] [PubMed] [Google Scholar]
- Needham EJ, Parker BL, Burykin T, James DE, Humphrey SJ (2019) Illuminating the dark phosphoproteome. Sci Signal 12: eaau8645 [DOI] [PubMed] [Google Scholar]
- Nishi H, Hashimoto K, Panchenko AR (2011) Phosphorylation in protein‐protein binding: effect on stability and function. Structure 19: 1807–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa D, Jarnuczak AF, Vieitez C, Gehre M, Soucheray M, Mateus A, Kleefeldt AA, Hill A, Garcia‐Alonso L, Stein F et al (2020) The functional landscape of the human phosphoproteome. Nat Biotechnol 38: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107‐Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306 [DOI] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M (2006) Global, in vivo, and site‐specific phosphorylation dynamics in signaling networks. Cell 127: 635–648 [DOI] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA et al (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3: ra3 [DOI] [PubMed] [Google Scholar]
- Pardo B, Crabbé L, Pasero P (2017) Signaling pathways of replication stress in yeast. FEMS Yeast Res 17: fow101 [DOI] [PubMed] [Google Scholar]
- Pei J, Grishin NV (2001) AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17: 700–712 [DOI] [PubMed] [Google Scholar]
- Rossi SE, Ajazi A, Carotenuto W, Foiani M, Giannattasio M (2015) Rad53‐mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep 13: 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauber C, Chen L, Tongaonkar P, Vega I, Lambertson D, Potts W, Madura K (1998) Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature 391: 715–718 [DOI] [PubMed] [Google Scholar]
- Sharma K, D'Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr‐based signaling. Cell Rep 8: 1583–1594 [DOI] [PubMed] [Google Scholar]
- Solari FA, Dell'Aica M, Sickmann A, Zahedi RP (2015) Why phosphoproteomics is still a challenge. Mol Biosyst 11: 1487–1493 [DOI] [PubMed] [Google Scholar]
- Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F, Martinez TN, Lorentzen E, Pfeffer SR et al (2017) Systematic proteomic analysis of LRRK2‐mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 6: e31012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN (2003) Human gene mutation database (HGMD): 2003 update. Hum Mutat 21: 577–581 [DOI] [PubMed] [Google Scholar]
- Studer RA, Rodriguez‐Mias RA, Haas KM, Hsu JI, Vieitez C, Sole C, Swaney DL, Stanford LB, Liachko I, Bottcher R et al (2016) Evolution of protein phosphorylation across 18 fungal species. Science 354: 229–232 [DOI] [PubMed] [Google Scholar]
- Swaffer MP, Jones AW, Flynn HR, Snijders AP, Nurse P (2018) Quantitative phosphoproteomics reveals the signaling dynamics of cell‐cycle kinases in the fission yeast Schizosaccharomyces pombe . Cell Rep 24: 503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney DL, Beltrao P, Starita L, Guo A, Rush J, Fields S, Krogan NJ, Villen J (2013) Global analysis of phosphorylation and ubiquitylation cross‐talk in protein degradation. Nat Methods 10: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K (2011) Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10: 5354–5362 [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G et al (2012) Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Genome of the Netherlands Consortium (2014) Whole‐genome sequence variation, population structure and demographic history of the Dutch population. Nat Genet 46: 818–825 [DOI] [PubMed] [Google Scholar]
- Thomas LL, Fromme JC (2016) GTPase cross talk regulates TRAPPII activation of Rab11 homologues during vesicle biogenesis. J Cell Biol 215: 499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LL, Joiner AMN, Fromme JC (2018) The TRAPPIII complex activates the GTPase Ypt1 (Rab1) in the secretory pathway. J Cell Biol 217: 283–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Abu M, Hanger DP (2012) Key issues in the acquisition and analysis of qualitative and quantitative mass spectrometry data for peptide‐centric proteomic experiments. Amino Acids 43: 1075–1085 [DOI] [PubMed] [Google Scholar]
- Touati SA, Kataria M, Jones AW, Snijders AP, Uhlmann F (2018) Phosphoproteome dynamics during mitotic exit in budding yeast. EMBO J 37: e98745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt C (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Sacher M, Ferro‐Novick S (2000) TRAPP stimulates guanine nucleotide exchange on Ypt1p. J Cell Biol 151: 289–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wei X, Thijssen B, Das J, Lipkin SM, Yu H (2012) Three‐dimensional reconstruction of protein networks provides insight into human genetic disease. Nat Biotechnol 30: 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Das J, Fragoza R, Liang J, Bastos de Oliveira FM, Lee HR, Wang X, Mort M, Stenson PD, Cooper DN et al (2014) A massively parallel pipeline to clone DNA variants and examine molecular phenotypes of human disease mutations. PLoS Genet 10: e1004819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Haas W, Dephoure N, Huttlin EL, Zhai B, Sowa ME, Gygi SP (2011) A large‐scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods 8: 677–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XZ, Li XX, Zhang YJ, Rodriguez‐Rodriguez L, Xiang MQ, Wang HY, Zheng XF (2016) Rab1 in cell signaling, cancer and other diseases. Oncogene 35: 5699–5704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Diffley JF (2010) Checkpoint‐dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 467: 474–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Dataset EV13
Review Process File
Data Availability Statement
The phosphoproteomic datasets produced in this study have been deposited to the PRIDE database (https://www.ebi.ac.uk/pride/archive/) and assigned the identifier PXD012395 (https://www.ebi.ac.uk/pride/archive/projects/PXD012395). Some data from PXD012395 were previously published in PXD009734 (https://www.ebi.ac.uk/pride/archive/projects/PXD009734).