

Abstract
Advanced paternal age can have deleterious effects on various traits in the next generation. Here, we establish a paternal‐aging model in mice to understand the molecular mechanisms of transgenerational epigenetics. Whole‐genome target DNA methylome analyses of sperm from aged mice reveal more hypo‐methylated genomic regions enriched in REST/NRSF binding motifs. Gene set enrichment analyses also reveal the upregulation of REST/NRSF target genes in the forebrain of embryos from aged fathers. Offspring derived from young mice administrated with a DNA de‐methylation drug phenocopy the abnormal vocal communication of pups derived from aged fathers. In conclusion, hypo‐methylation of sperm DNA can be a key molecular feature modulating neurodevelopmental programs in offspring by causing fluctuations in the expression of REST/NRSF target genes.
Keywords: DNA hypo‐methylation, paternal aging, REST/NRSF, transgenerational epigenetic inheritance, ultrasonic vocalization
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Neuroscience
Paternal aging affects health and disease of the next generation, but the causative mechanisms are largely unknown. This study identifies hypo‐methylated sites enriched in REST/NRSF motifs in sperm DNA as epigenetic key features for inheritance.

Introduction
The Developmental Origins of Health and Disease (DOHaD) concept, which links health status and risk of disease in later childhood and adult life with environmental conditions in early life, has been proposed since the 1980s. The concept generally reflects the maternal influence on offspring, although it has recently been expanded to include paternal influence as well (Paternal Origin of Health and Disease, POHaD) (Soubry, 2018). Besides paternal exposure to environmental contaminants, advanced paternal age also has deleterious effects on the health of the next generation, including low birth weight (Reichman & Teitler, 2006; Alio et al, 2012) and psychiatric disorders such as schizophrenia (Malaspina et al, 2001) and autism spectrum disorder (ASD) (Tsuchiya et al, 2008; Lundström et al, 2010; Sandin et al, 2016). The latter has been modeled in rodents—learning deficits, impaired social behavior, and hyper‐anxiety are observed in offspring derived from aged fathers (Smith et al, 2009; Foldi et al, 2010; Sampino et al, 2014; Milekic et al, 2015). Exome analyses have also revealed more paternally derived de novo mutations in ASD‐offspring of aged fathers (Kong et al, 2012; O’Roak et al, 2012), although some studies have contradicted their results (Gratten et al, 2016; Taylor et al, 2019). Moreover, sperm DNA methylation levels show age‐related changes in humans (Jenkin et al, 2014), and a longitudinal analysis of ASD‐high‐risk children has indeed shown a correlation between sperm DNA methylation and risk of ASD in offspring (Feinberg et al, 2015). However, the transgenerational impact of sperm DNA hypo‐methylation remains unclear.
In the present study, we first established a paternal‐aging mouse model characterized by vocal communication deficits in offspring at infancy. We then conducted whole‐genome target methylome analyses using sperm DNA from young and aged mice and found more hypo‐methylated regions in the genome of the latter. Motif analysis identified enrichment of binding motifs for the RE1‐silencing transcription factor (REST), also known as neuron‐restrictive silencer factor (NRSF), in hypo‐methylated regions of sperm DNA from aged fathers. Likewise, comprehensive gene expression analysis using gene set enrichment analyses (GSEA) also revealed enrichment of REST/NRSF target genes in upregulated transcripts in the developing brain of offspring derived from aged fathers. Consistently, the motor cortex, which is responsible for vocal communication deficits, was specifically thinner in pups derived from aged fathers, especially in the deeper layers. Finally, we replicated the effects of paternal aging on behavior in offspring derived from young male mice administrated with a DNA de‐methylation drug. These findings suggest that DNA hypo‐methylation with the REST/NRSF binding motif can be a key to understanding the molecular neuropathogenesis of offspring born to aged fathers.
Results
Establishment of a paternal‐aging model in mice
Since the risk of offspring mental illness increases from the paternal age of 40–50 years in humans (Malaspina et al, 2001; Tsuchiya et al, 2008; Lundström et al, 2010; Sandin et al, 2016), we designated wild‐type C57BL/6J male mice over 12 months of age as “aged” mice. Young (3‐month‐old wild‐type) and aged male mice were housed with wild‐type virgin female mice for mating and separated from pregnant mice to avoid any possible effects on maternal separation‐induced ultrasonic vocalization (USV) in their pups. Per litter, the number of offspring from young fathers (YFO, 5.7 ± 0.7) and those from aged fathers (AFO, 6.5 ± 0.6) (P = 0.441) were comparable.
Maternal separation‐induced USV is an early infancy social‐communicative behavior. We recorded the USV of YFO and AFO and measured their body weights at postnatal day 6 (PND6) (Fig 1A). YFO and AFO had comparable body weights (3.22 ± 0.07 g: YFO vs. 3.07 ± 0.07 g: AFO, P = 0.17, Fig 1B); USV syllables, which are the sound elements of USV, were reduced by 36.1% in AFO compared with YFO (189.1 ± 22.8 calls: YFO vs. 120.7 ± 16.1 calls: AFO, P = 0.018, Fig 1C), although the average duration, maximum frequency (kHz), and maximum amplitude (−dB) of overall syllables were not significantly different between the two groups (Fig 1D–F). To better understand the USV composition, syllables were classified into 12 types according to Scattoni’s classification, with some modifications (Fig 1G) (Scattoni et al, 2008). The percentage of each syllable type was drastically different between YFO and AFO. For AFO, the percentage of “Downward” syllables was increased, while the percentages of “One jump”, “Chevron”, “Upward”, “More jump”, “Wave”, and “More jump + Harmonics” syllables were decreased (Fig 1H). In addition, AFO emitted significantly fewer types of syllables (Fig 1I) with lower entropy scores (Fig 1J), showing less diversity in the syllable types. These findings suggest that paternal aging led to abnormal vocal behavior in offspring in the early postnatal period.
Figure 1. Advanced paternal age caused vocal communication deficits in offspring.
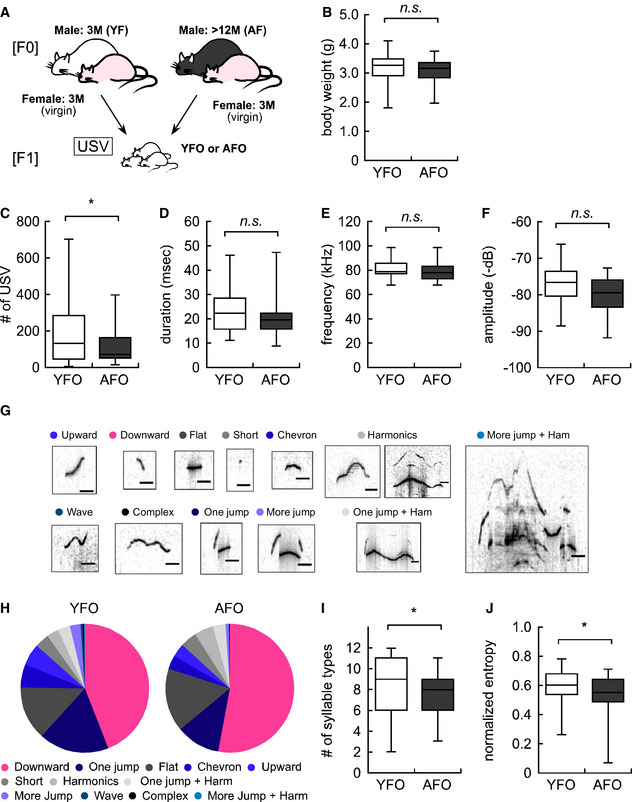
- Experimental design for F1 offspring derived from either young or aged fathers (YFO: n = 57, AFO: n = 39).
- Body weight in YFO and AFO.
- Total USV number in YFO and AFO.
- Average duration (msec) of each syllable in YFO and AFO.
- Maximum frequency (kHz) of each syllable in YFO and AFO.
- Maximum amplitude (−dB) of each syllable in YFO and AFO.
- Individual syllable categories. The black line indicates 20 msec.
- Ratio of each syllable type in YFO and AFO. Each color corresponds to the individual syllable categories shown in Fig 1G. Red and blue colors indicate a statistically significant increase and decrease, respectively, while gray indicates statistically ‘not significant’ changes.
- Number of syllable types in YFO and AFO.
- Normalized entropy in YFO and AFO.
Data information: Data are presented in the box plot or pie chart format. The central band in each box represents the median; boxes indicate the middle quartiles, and whiskers extend to the minimum and maximum values. *P < 0.05, determined using Student’s or Welch t‐test.
Target methylome analysis of sperm from aged mice
To uncover the molecular basis of the transgenerational influence of paternal aging, we first examined the epigenetic changes in sperm DNA. Since longitudinal cohort analyses suggest that paternal sperm DNA methylation is associated with early signs of autism risk (Feinberg et al, 2015), we conducted target methylome analyses using SureSelect Methyl‐Seq technique from Agilent Technology (Koike et al, 2016) and sperm samples from four young and nine aged mice (Fig 2A). After checking the sequencing data quality of sperm DNA samples (Fig 2B and C), we analyzed differentially methylated regions (DMRs) in sperm DNA using the model based analysis of bisulfite sequence data (MOABS) tool (Method2 and minDepthForComp = 10) (Sun et al, 2014). Using × 10 ≧ depth CpG site data, 16 hyper‐ and 96 hypo‐DMRs in sperm DNA from aged mice were identified (Fig 2D, Datasets EV1 and EV2). Half to two‐thirds of hyper‐DMRs were in intergenic and intron regions, and the remaining half to one‐third were in exon regions. In contrast, almost all hypo‐DMRs were in intergenic and intron regions, and the remaining 20% or less were in exon and promoter regions (Fig 2E). This suggests that aging induces hypo‐methylation, rather than hyper‐methylation, of sperm chromosomal DNA. We should keep in mind that the DMRs were found across all chromosomes, except for sex chromosomes. This might be because the SureSelect target regions specific to X and Y chromosomes are extremely small in number (7,347 base pairs (bps) and 114 bps, respectively) compared with the size of the X and Y chromosomes (171,031,299 bps and 91,744,698 bps, respectively).
Figure 2. Comprehensive target methylome analysis identified more hypo‐methylated genomic regions in sperm from aged mice.
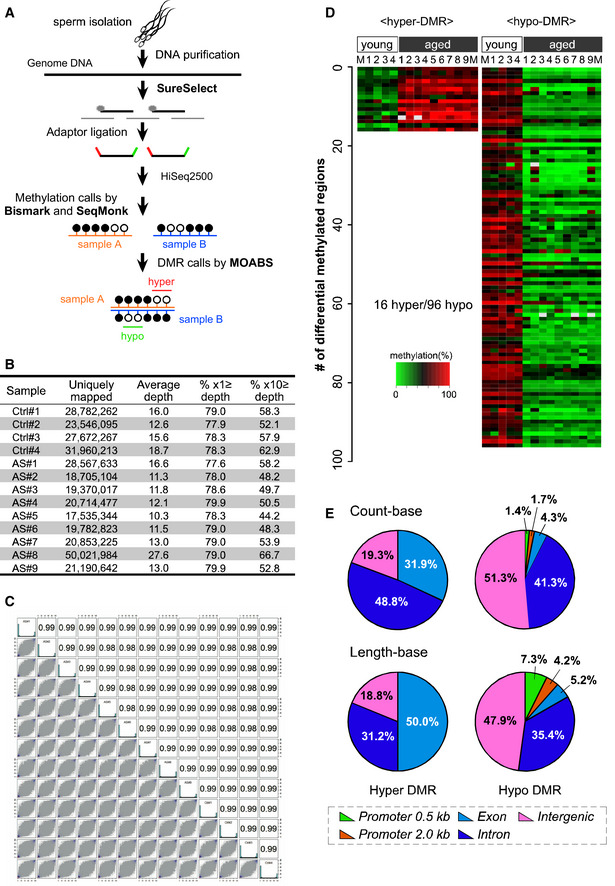
- Strategy for DNA target methylome analyses of sperm from young (3‐month‐old, n = 4) and aged (18‐month‐old, n = 9) mice.
- The average level of CpG methylation at the targeted regions was 33.3–34.6% and 27.7–36.2% in sperm datasets from young and aged mice, respectively. The average depth of each sample was 11.3–27.6, which covered 44.2–62.9% of the target regions at a depth of 10.
- Correlation coefficients were also high (i.e., 0.98–0.99), indicating that the obtained sequence data were sufficient for further analysis of age‐specific methylation changes.
- A heat map of the 16 hyper‐ and 96 hypo‐methylated DMRs in sperm from aged mice ordered by their chromosomal positions. M indicates the average.
- A majority of hyper‐methylated genomic regions were within the genes (31.9% in exons and 48.8% in introns in count‐base analysis and 50.0% in exons and 31.2% in introns in length‐base analysis). In contrast, almost half (47.9%) of the hypo‐methylated regions were in intergenic regions (51.3% in count‐base analysis and 47.9% in length‐base analysis); only 4.3% of the regions in count‐base analysis and 5.2% in length‐base analysis were in exons, and 3.1% in count‐base analysis and 11.5% in length‐base analysis were in promoter regions (0.5 kb or 2.0 kb upstream of the transcription starting site).
Data information: Data are presented in the volcano plot format. Statistical significance was determined using Fisher’s exact test.
It is of note that gene ontology (GO) analyses using genes within or neighboring the DMRs further revealed that hypo‐DMRs in sperm DNA from aged mice were highly and significantly enriched with genes related to “cell projection”, “learning”, “learning or memory”, “neuron part”, and “connexin complex” (Figs 3A and EV1A). Gene lists such as “abnormal nervous system physiology”, “nervous system phenotype”, and “abnormal long‐term depression” were found in genes related to hypo‐DMRs in GO analysis of mouse genome informatics (MGI) as well (Figs 3B and EV1B).
Figure 3. The REST/NRSF binding motif was enriched in DNA hypo‐methylation regions.
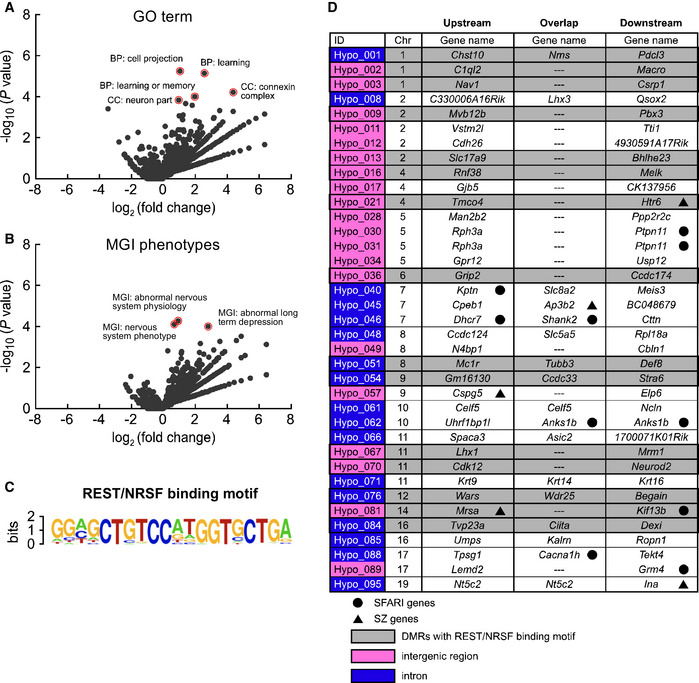
- Volcano plots showing GO terms enriched for genes near hypo‐methylated regions relative to other RefSeq genes.
- Volcano plots showing MGI phenotypes enriched for genes near hypo‐methylated regions relative to other RefSeq genes.
- Enrichment of the REST/NRSF binding motif in hypo‐methylated DMRs (19/96) in aged mice.
- Gene lists near the 37 hypo‐methylated genomic regions with REST/NRSF binding confirmed using the ChIP‐Atlas database. Blue highlights indicate DMRs in the intron of overlap genes. Magenta highlights indicate DMRs in intergenic regions between upstream and downstream genes. Gray highlights indicate DMRs with the REST/NRSF binding motif. The black circle indicates ASD‐related genes (SFARI genes), and the black triangle indicates schizophrenia‐related genes (SZ genes).
Data information: Data are presented in the volcano plot format. Statistical significance was determined using Fisher’s exact test.
Figure EV1. Gene ontology analysis of hypo‐methylated DMRs.
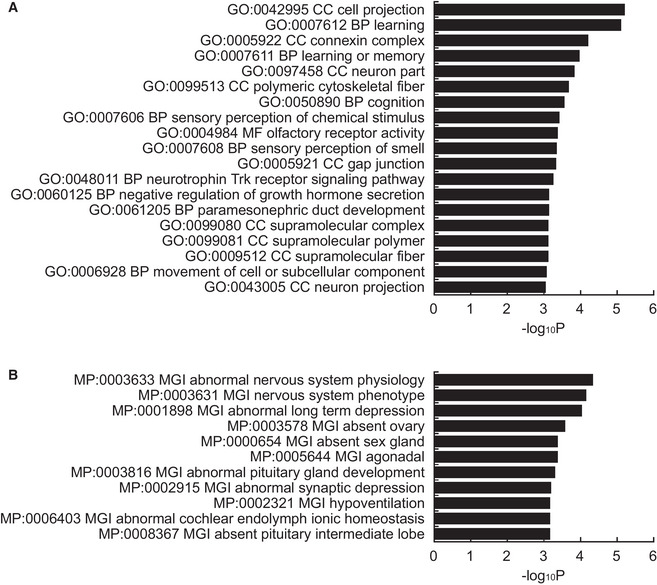
- Top 19 GO terms (P < 0.001) associated with the biological process (BP), cellular component (CC), and molecular function (MF).
- Top 11 MGI phenotypes (P < 0.001) of genes near hypo‐methylated DMRs in sperm from aged mice. Both indicate that genes associated with neuronal function are enriched.
Data information: Data are presented in the bar graph format. Statistical significance was determined using Fisher’s exact test.
Motif analysis using hypergeometric optimization of motif enrichment (Tatsumi et al, 2018) revealed significant enrichment (P = 1e‐25) of a unique consensus sequence, GGAGCTGTCCATGGTGCTGA, which indicates potential binding to REST/NRSF (Fig 3C), which is a pivotal regulator of brain development (Paquette et al, 2000; Mandel et al, 2011). Strikingly, these REST/NRSF binding motifs were detected only in the hypo‐DMRs (19/96) of sperm from aged mice. As an alternative approach, we used ChIP‐Atlas (http://chip‐atlas.org), an online database integrating published ChIP‐seq data (Oki et al, 2018), to find transcription factors with enriched binding for hypo‐DMRs. Surprisingly, the most significant was the ChIP‐seq data of REST/NRSF in embryonic stem (ES) cell‐derived neuronal progenitor cells (GSM671100) (Arnold et al, 2013); these REST/NRSF‐bound regions were detected in 37/96 of the hypo‐DMRs (Fig 3D, Dataset EV3). When the 19 sequences from the motif analysis and 37 sequences from the ChIP‐Atlas analysis were compared, 15 were found to be common (shown in gray in Fig 3D). Interestingly, some of the 37 REST/NRSF‐bound hypo‐DMRs were found to be close to genes associated with autism archived in Simons Foundation Autism Research Initiative (SFARI genes) (Banerjee‐Basu & Packer, 2010) and/or with schizophrenia (SZ genes) (Allen et al, 2008), including typical risk genes such as Shank2 and Htr6 (Tsai, 1999; Monteiro & Feng, 2017). These results suggest that DNA hypo‐methylation due to paternal aging did not occur randomly but was enriched within REST/NRSF binding motifs in the hypo‐DMRs, which may implicate deficits in neuronal development.
Transcriptome analyses in the embryonic brain
To examine whether paternal aging indeed affected neural development, we conducted transcriptome analyses in the forebrain at embryonic day 14.5 (E14.5), the peak stage of neuronal production. A volcano plot of RNA‐seq data indicated no genes that were significantly up‐ or downregulated (by fold changes > 2.0 or < 0.5, respectively, and at a significance level of P < 0.01), in the forebrain of AFO compared with YFO (Fig 4A). Nevertheless, principal components (PC) analysis (PCA) indicated differences in global expression profiles between YFO and AFO at E11.5 (PC1 vs. PC2 and PC2 vs. PC3 in Fig EV2A and B, respectively) and E14.5 (similarly in Fig EV2C and D). Therefore, we applied GSEA, which is suitable for detecting global changes in gene expression sets (Mootha et al, 2003), and revealed significant enrichment of gene sets with GO terms related to neuronal differentiation/function (e.g., voltage‐gated cation channel activity, cation channel complex, and synapse membrane) in genes upregulated in the forebrain of AFO (Fig 4B). In contrast, gene sets downregulated in the forebrain of AFO had GO terms related to DNA replication and extracellular structure among their top five GO terms (Fig 4C). It should be noted that the REST/NRSF target gene set was also enriched in the upregulated genes in AFO (Fig 4D and E; plotted as red dots in Fig 4A). Interestingly, we noticed a significant enrichment of the early‐fetal gene set (Kang et al, 2011; Katayama et al, 2016) in the upregulated genes in the embryonic brain of YFO (normalized enrichment score (NES) = −3.112, P < 0.001, Fig 4F). Conversely, a significant enrichment of the late‐fetal gene set (Kang et al, 2011; Katayama et al, 2016) (NES = 3.370, P < 0.001, Fig 4G) was observed in the upregulated genes in AFO. It is thus reasonable to assume that precocious neurogenesis occurs in the embryonic brain of AFO. Furthermore, the SFARI gene set was also enriched in the embryonic brain of AFO (NES = 1.781, P < 0.001, Fig 4H).
Figure 4. Comprehensive transcriptome analysis also identified REST/NRSF target genes in upregulated genes in offspring derived from aged fathers.
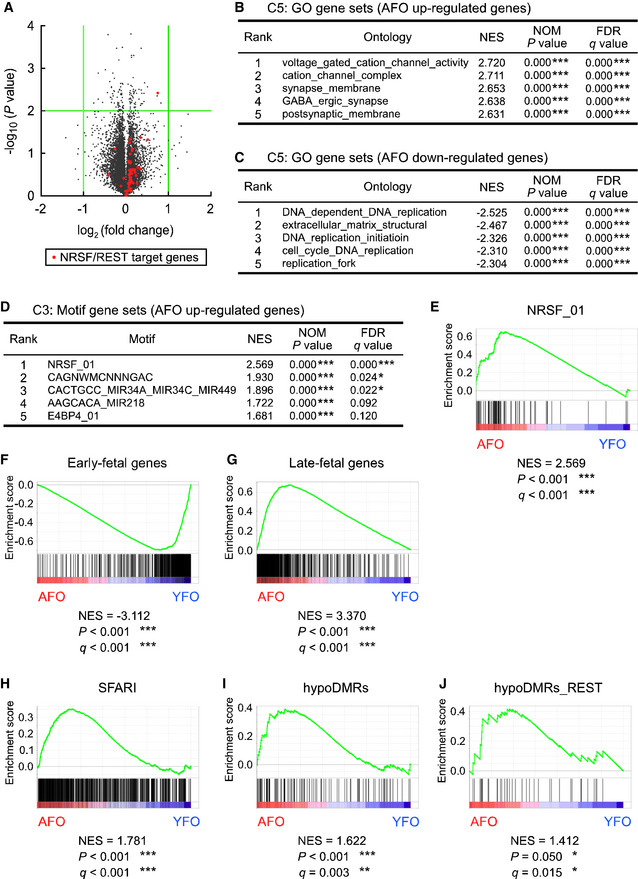
- Volcano plot for DEGs in the embryonic brain of YFO and AFO at E14.5 (n = 3 in each group). Green lines indicate a twofold change and P = 0.01. Orange dots indicate REST/NRSF target genes.
- GO gene sets in AFO upregulated genes.
- GO gene sets in AFO downregulated genes.
- Motif gene sets in AFO upregulated genes.
- GSEA plots of DEGs in NRSF/REST target gene sets.
- GSEA plots of DEGs for early‐fetal gene sets.
- GSEA plots of DEGs for late‐fetal gene sets.
- GSEA plots of DEGs for SFARI‐related gene sets.
- GSEA plots of DEGs for DNA hypo‐methylation neighboring gene sets.
- GSEA plots of DEGs for DNA hypo‐methylation neighboring gene sets with REST/NRSF binding motifs.
Data information: Data are presented in the volcano plot or line chart format. *P < 0.05, **P < 0.01, ***P < 0.001, determined by nominal GSEA P‐value. NES, normalized enrichment score.
Figure EV2. PCA of the comprehensive transcriptome of offspring derived from young and aged fathers.
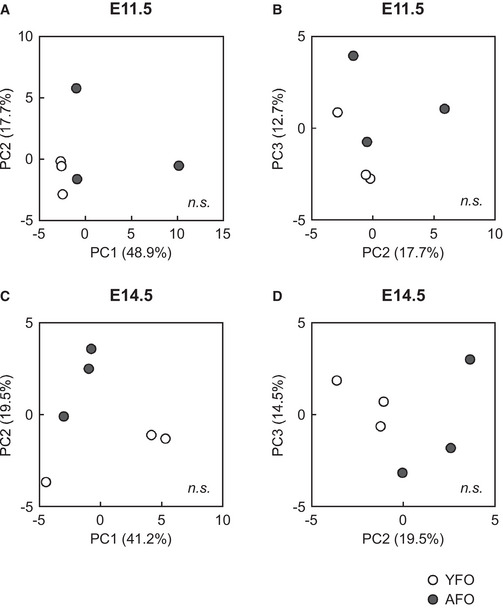
- A two‐dimensional scatter plot of the PCA analysis of gene expression at E11.5. PC1 and PC2 accounted for 48.9 and 17.7% of the contribution of the variance, respectively.
- A two‐dimensional scatter plot of the PCA analysis of gene expression at E11.5. PC2 and PC3 accounted for 17.7 and 12.7% of the contribution of the variance, respectively.
- A two‐dimensional scatter plot of the PCA analysis of gene expression at E14.5. PC1 and PC2 accounted for 41.2 and 19.5% of the contribution of the variance, respectively.
- A two‐dimensional scatter plot of the PCA analysis of gene expression at E14.5. PC2 and PC3 accounted for 19.5 and 14.5% of the contribution of the variance, respectively.
Data information: Data are presented in the scatter plot format. The open circle indicates an individual YFO pattern, and a dark circle denotes an individual AFO pattern. PC1, principal component 1. PC2, principal component 2. PC3, principal component 3. Statistical significance was determined using the prcomp R package.
At the earlier stage E11.5, distortions of gene expression in the AFO brain were not detected (Fig 5A). Genes slightly up‐ and downregulated in the forebrain of AFO were found to have GO terms related to the centromere, chromatin remodeling, and histone exchange (Fig 5B) and to extracellular matrix and collagen (Fig 5C), respectively, in which neuronal differentiation/function (e.g., voltage‐gated cation channel activity, cation channel complex, and synapse membrane) were not listed. Importantly, the REST/NRSF target gene set was also not enriched in the genes that were upregulated in AFO (Fig 5D and E; plotted as red dots in Fig 5A). The early‐fetal gene set was slightly enriched in genes that were upregulated in YFO (NES = −1.427, P < 0.001, Fig 5F), a tendency similar to that at E14.5. Unlike in the case of E14.5, the late‐fetal gene set was enriched in the genes that were upregulated at E11.5 in YFO (NES = −1.410, P = 0.001, Fig 5G), but not AFO. Furthermore, significant enrichment of the SFARI gene set was also not detected in embryonic AFO brains at E11.5. It could be speculated that precocious neurogenic gene expression found in the brain of AFO at E14.5 was not detected at E11.5. This suggests that paternal aging might disrupt the harmonious expression of multiple genes in a stage‐specific manner in the embryonic brain.
Figure 5. REST/NRSF target genes were not enriched at E11.5 in offspring embryonic brains derived from aged fathers.
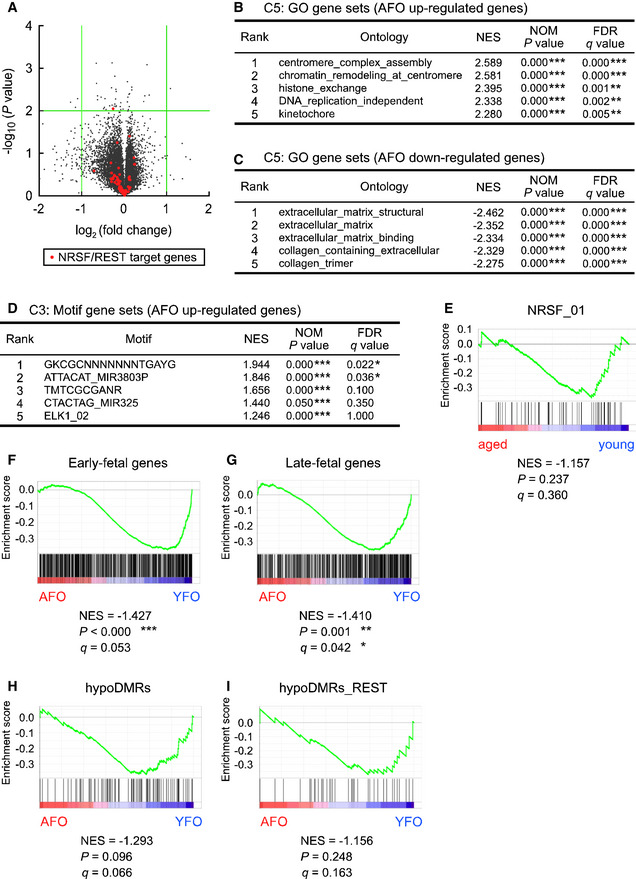
- Volcano plot for DEGs in the embryonic brain of YFO and AFO at E11.5 (n = 3 in each group). Green lines indicate a twofold change and P = 0.01. Orange dots indicate REST/NRSF target genes.
- GO gene sets in AFO upregulated genes.
- GO gene sets in AFO downregulated genes.
- Motif gene sets in AFO upregulated genes.
- GSEA plots of DEGs for REST/NRSF target gene sets.
- GSEA plots of DEGs for early‐fetal gene sets.
- GSEA plots of DEGs for late‐fetal gene sets.
- GSEA plots of DEGs for DNA hypo‐methylation neighboring gene sets.
- GSEA plots of DEGs for DNA hypo‐methylation neighboring gene sets with REST/NRSF binding motifs.
Data information: Data are presented in the volcano plot or line chart format. *P < 0.05, **P < 0.01, ***P < 0.001, determined by volcano or nominal GSEA P‐value. NES, normalized enrichment score.
To examine whether altered DNA methylation in the sperm of aged mice impacts gene expression in the embryonic brain of AFO, we also conducted GSEA and revealed a significant enrichment of the gene set near the hypo‐DMRs in the upregulated genes (NES = 1.622, P < 0.001, Fig 4I). Furthermore, a set of genes near the hypo‐DMRs with the REST/NRSF binding motif was enriched in the upregulated genes (NES = 1.412, P = 0.050, Fig 4J). In contrast, the gene set near neither hypo‐DMRs (NES = −1.293, P > 0.05, Fig 5H) nor hypo‐DMRs with the REST/NRSF binding motif (NES = −1.156, P > 0.05, Fig 5I) was enriched in the upregulated genes in the E11.5 embryonic brain of AFO. Our results suggest that DNA hypo‐methylation in aged mouse sperm upregulates the expression of REST/NRSF target genes in a stage‐specific manner in the embryonic brain.
Based on these gene expression data of the embryonic brain, we next addressed the brain structure of pups at PND6, when an impairment in vocal behavior was observed. We noticed that the cortical thickness of the primary motor cortex (M1) was reduced by 18.0% in AFO (P = 0.011, Fig 6A and B). Using layer‐specific markers (Fig 6C and D) (Arlotta et al, 2005; Alcamo et al, 2008; Britanova et al, 2008; Bedogni et al, 2010), reduction of thickness was specifically observed in Tbr1+/Ctip2− layer 6 neurons (by 63.0%, P = 0.033), but not in Satb2+/Ctip2− layer 2–4 neurons nor in Ctip2+ layer 5 neurons, in AFO (Fig 6E). Correspondingly, a 39.8% reduction in the number of neurons was confirmed in layer 6 neurons in the M1 of AFO (P = 0.007, Fig 6F). We also noticed that cell density in layer 6 was reduced to 59.6% in the M1 of AFO (P = 0.015), but not in layer 5 nor in layers 2–4 (Fig 6G). These findings imply that early neurogenesis producing deep layer neurons is impaired in the neocortex of the AFO. Smaller brains are phenotypes in both humans and mice that have impairments in the REST pathway (Yang et al, 2012; Nechiporuk et al, 2016). It has also been reported that lesions of the motor cortex in humans severely impair or eliminate the ability to produce vocalization (Jurgens, 2002; Jarvis, 2004). The thinner motor cortex in the present study, particularly in the deeper layers that are specifically related to motor control, can be considered to be associated with the vocal deficiencies in the AFO.
Figure 6. Abnormal cortical structures in offspring brains derived from aged fathers in the early postnatal period.
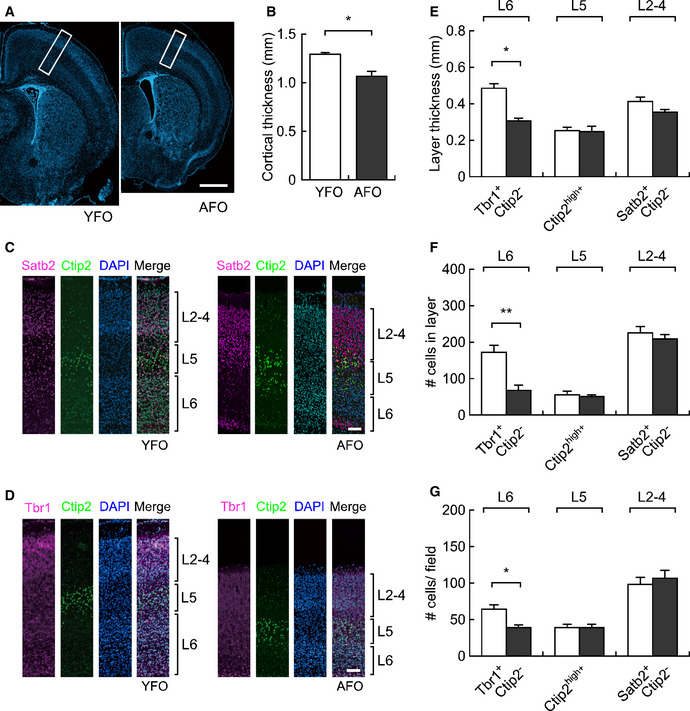
- Representative images of the cerebral cortex of YFO (left) and AFO (right) at postnatal day 6. Scale bar: 1 mm.
- Cortical thickness in the postnatal brain of YFO (n = 4, from different mothers) and AFO (n = 5, from different mothers).
- Immunostaining for cortical layer markers in the postnatal brain of YFO and AFO (representative magnified images of the white boxes in panel A). Satb2+/Ctip2−: layer 2–4, Ctip2high: layer 5, Satb2−/Ctip2−: layer 6. Scale bar: 100 μm.
- Immunostaining for another cortical layer markers in the postnatal brain of YFO and AFO (representative magnified images of the white boxes in panel A). Tbr1−/Ctip2−: layer 2–4, Ctip2high: layer 5, Tbr1+/Ctip2−: layer 6. Scale bar: 100 μm.
- Layer thickness (mm) in each cortical layer in the postnatal brain of YFO (n = 4, from different mothers) and AFO (n = 5, from different mothers).
- Cell number in each cortical layer in the postnatal brain of YFO and AFO.
- Cell density in each cortical layer in the postnatal brain of YFO and AFO.
Data information: All data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, determined using Student t‐test.
Impact of treatment with a de‐methylation drug
To replicate the DNA hypo‐methylation in sperm from aged mice, we injected young (3‐month‐old) male mice with saline or 5‐aza‐2′‐deoxycytidine (5‐Aza, 0.05 mg/kg body weight), an inhibitor of DNA methyltransferase, once every 2 days for 7 weeks (Fig 7A). Consistent with a previous study (Kelly et al, 2003), 5‐Aza injection in mice decreased testis weight (Fig 7B). In addition, we found a decreased level of 5‐methylcytosine in sperm (Fig 7C and D) and an increased level of 5‐hydroxymethylcytosine in the testis (Fig EV3A and B).
Figure 7. DNA hypo‐methylation in sperm from 5‐Aza‐treated mice caused vocal communication deficits in offspring.
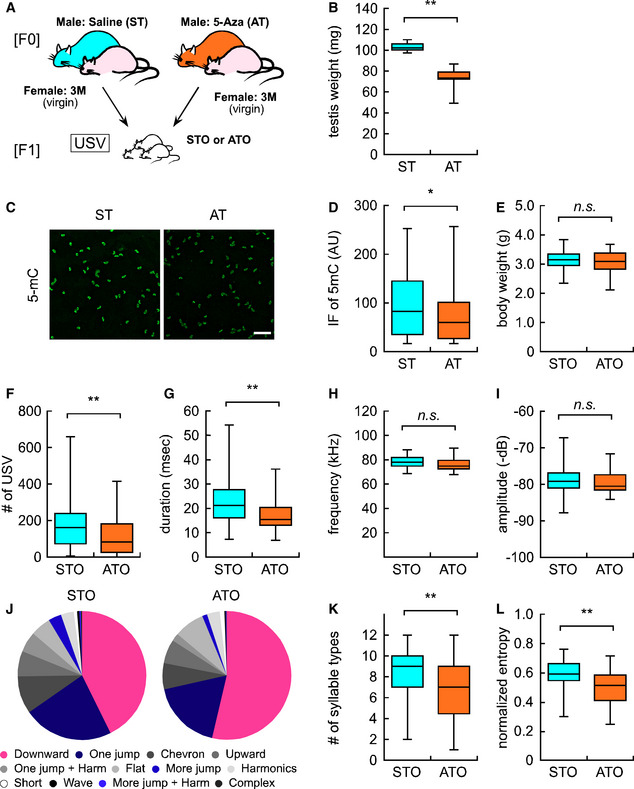
- Experimental design for F1 offspring derived from either saline or 5‐Aza‐treated fathers (ST and AT, respectively).
- Testis weight in ST (n = 6, from different male mice) and AT (n = 6, from different male mice).
- Representative image of 5‐mC immunofluorescence in the sperm of saline‐ and 5‐Aza‐treated mice (ST and AT, respectively). Scale bar: 100 μm.
- Quantification analysis of 5‐mC immunofluorescence in individual sperm cells from STO (n = 1,673 sperm from 4 male mice) and ATO (n = 2,675 sperm from 4 male mice).
- Bodyweight in STO (n = 49) and ATO (n = 36).
- Overall USV number in STO and ATO.
- Average duration (msec) of each syllable in STO and ATO.
- Maximum frequency (kHz) of each syllable in STO and ATO.
- Maximum amplitude (−dB) of each syllable in STO and ATO.
- Ratio of each syllable type in STO and ATO. Each color corresponds to individual syllable categories shown in Fig 1G.
- Number of syllable types in STO and ATO.
- Normalized entropy in STO and ATO.
Data information: All data are presented in the box plot format. The central band in each box represents the median, boxes indicate the middle quartiles, whiskers extended to the minimum and maximum values. *P < 0.05 and **P < 0.01, determined using Student’s or Welch t‐test.
Figure EV3. 5‐Aza treatment globally increased 5‐hydroxymethylcytosine levels in the testes.
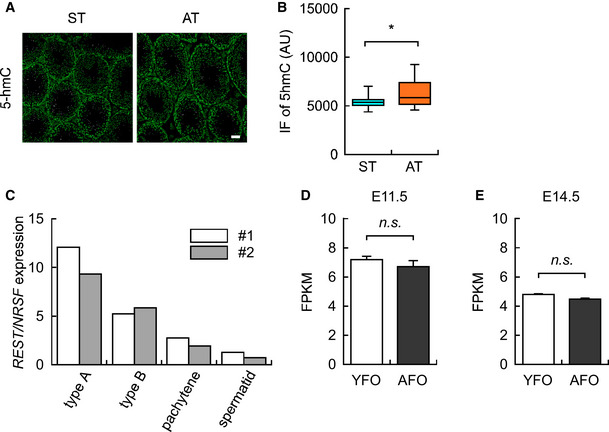
- Representative images of 5‐hydroxymethylcytosine (5‐hmC) in the testis of saline‐ and 5‐Aza‐treated mice (ST and AT, respectively). Scale bar: 50 μm.
- Quantification of 5‐hmC immunofluorescence in the testis of ST (n = 798 spermatogonia from 4 male mice) and AT (n = 1,031 spermatogonia from 4 male mice).
- Reanalysis of REST/NRSF expression in the germline cells (i.e., type A spermatogonia, type B spermatogonia, pachytene spermatocytes, and spermatid) in a public database of the testis (n = 2) (Namekawa et al, 2006). The open and light bars indicate samples #1 and #2, respectively.
- REST/NRSF expression in E11.5 brains from YFO (n = 3, from different mothers) and AFO (n = 3, from different mothers) based on RNA‐seq data.
- REST/NRSF expression in the E14.5 brains from YFO (n = 3, from different mothers) and AFO (n = 3, from different mothers) based on RNA‐seq data.
Data information: Data are presented in the box plot (the central band in each box represents the median, boxes indicate the middle quartiles, and whiskers extended to the minimum and maximum values) or bar graph (mean ± SEM) format. *P < 0.05, determined using Student’s t‐test.
We further assessed the DNA methylation status in the selected hypo‐DMRs by targeted bisulfite sequencing. Among the 96 hypo‐DMRs, we chose 11 hypo‐DMRs that were enriched for REST/NRSF binding based on ChIP‐Atlas analysis (see Fig 3D), and their nearby genes were upregulated in the embryonic AFO brain (Dataset EV4). Indeed, we confirmed a significant reduction in the DNA methylation ratio at CpG sites of M2_hypo_003 and M2_hypo_046 in the autism‐related genes Nav1 (Scn2a) (Fig EV4A) and Shank2 (Fig EV4B), respectively. In Nav1, altered methylation of the CpG site was in a binding motif of specificity protein 1 (Sp1) (Philipsen & Suske, 1999). The Sp1 site is located between two adjacent REST/NRSF binding motifs, and it has been reported that REST directly inhibits Sp1‐mediated transcription in the SYN1 promoter region (Paonessa et al, 2013). In Shank2, on the other hand, reduced methylation of CpG sites was located on both sides of the REST/NRSF binding motif within a few base pairs. In contrast, we did not detect significant DNA hypo‐methylation at the CpG sites of M2_hypo028 (Fig EV5B), M2_hypo030 (Fig EV5C), M2_hypo034 (Fig EV5D), M2_hypo036 (Fig EV5E), M2_hypo040 (Fig EV5F), M2_hypo045 (Fig EV5G), M2_hypo066 (Fig EV5H), M2_hypo085 (Fig EV5I), and M2_hypo088 (Fig EV5J) or non‐hypo‐methylated DNA regions in aged sperm (Fig EV5A). These results suggest that 5‐Aza injection could affect USV phenotypes in the offspring, at least, through local DNA hypo‐methylation in these genes during paternal spermatogenesis, which might lead to alterations in neuronal or synaptic activity by changing the expression of Nav1 or Shank2, or both.
Figure EV4. 5‐Aza treatment significantly induced DNA hypo‐methylation in sperm genomic loci.

- Percentages of DNA methylation in DMRs (M2_hypo_003) near the Nav1 gene (38.0% vs. 29.0% in Saline vs. 5‐Aza, respectively; P < 0.05) in sperm from young father with Saline (blue, n = 29 from different 3 male) and 5‐Aza treatment (orange, n = 29 from different 3 male).
- Percentage of DNA methylation of DMRs (M2_hypo_046) near Shank2 gene (49.7% vs. 42.8% in Saline vs. 5‐Aza, respectively; P < 0.05) in sperm from young father with Saline (blue, n = 28 from different 3 male) and 5‐Aza treatment (orange, n = 29 from different 3 male).
Data information: Horizontal red and blue bars indicate REST/NRSF and Sp1 binding motifs, respectively (for more detail, see Discussion). Black and open circles indicate methylated and unmethylated CpG sites, respectively. Statistical significance was determined by Fisher’s exact test.
Figure EV5. 5‐Aza treatment‐induced DNA hypo‐methylation in sperm genomic loci at non‐significant levels.
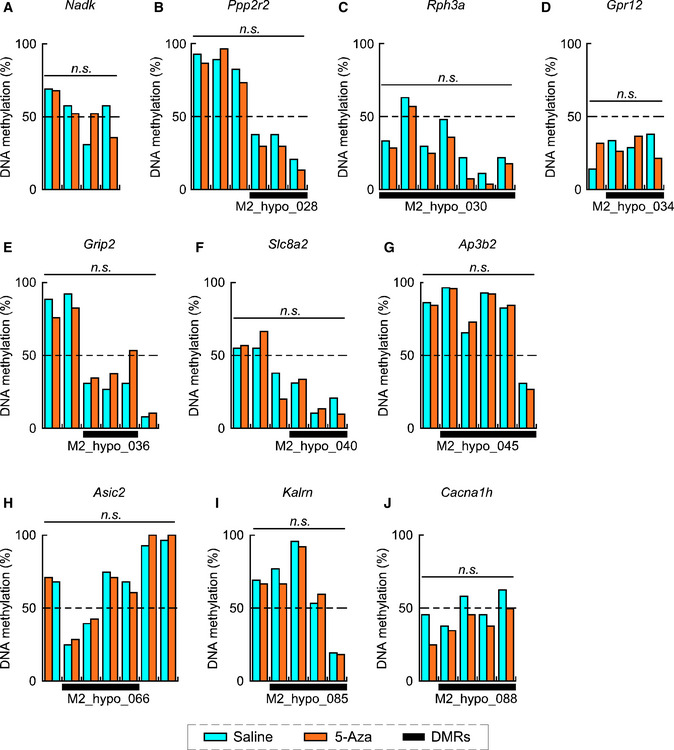
- Percentages of DNA methylation of CpG sites near Nadk (a control) (53.3% vs. 50.8% in saline vs. 5‐Aza, respectively; P = 0.744) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_028) near Ppp2r2 (33.3% vs. 22.6% in saline vs. 5‐Aza, respectively; P = 0.275) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_030) near Rph3a (29.6% vs. 21.9% in saline vs. 5‐Aza, respectively; P = 0.334) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_034) near Gpr12 (30.0% vs. 23.0% in saline vs. 5‐Aza, respectively; P = 0.869) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_036) near Grip2 (29.6% vs. 40.7% in saline vs. 5‐Aza, respectively; P = 0.654) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_040) near Slc8a2 (53.0% vs. 50.6% in saline vs. 5‐Aza, respectively; P = 0.882) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_045) near Ap3b2 (69.1% vs. 69.6% in saline vs. 5‐Aza, respectively; P = 0.578) in sperm from young fathers with saline (blue; n = 30, from different 3 males) or 5‐Aza treatment (orange; n = 29, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_066) near Asic2 (51.8% vs. 49.2% in saline vs. 5‐Aza, respectively; P = 0.812) in sperm from young fathers with saline (blue; n = 28, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_085) near Kalrn (55.6% vs. 56.3% in saline vs. 5‐Aza, respectively; P = 0.821) in sperm from young fathers with saline (blue; n = 29, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
- Percentages of DNA methylation of DMRs (M2_hypo_088) near Cacna1h (52.5% vs. 39.5% in saline vs. 5‐Aza, respectively; P = 0.075) in sperm from young fathers with saline (blue; n = 29, from different 3 males) or 5‐Aza treatment (orange; n = 30, from different 3 males).
Data information: Data are presented in the bar graph format. Statistical significance was determined using Fisher’s exact test.
Male mice treated with either saline or 5‐Aza were mated with young (3‐month‐old) female mice to produce offspring. The litter size derived from the mice treated with saline (STO) and 5‐Aza (ATO) was comparable (8.2 ± 0.3 for STO and 7.2 ± 0.8 for ATO, P = 0.31). Body weight did not differ between STO and ATO (Fig 7E); however, the number of USV syllables in ATO significantly decreased to 58.9% compared with those in STO (198.8 ± 23.4 calls in STO vs. 117.1 ± 18.2 calls in ATO, P = 0.010, Fig 7F). Detailed syllable analyses revealed that duration, but not maximum frequency nor amplitude, was significantly decreased in ATO (Fig 7G–I). Furthermore, the syllable types differed between STO and ATO; the percentage of “Downward” syllables was significantly increased, while the percentages of “One jump”, “More jump”, and “More jump + Harmonic” syllables were decreased in ATO (Fig 7J). In addition, the number of syllable types and entropy scores of syllables were also significantly decreased in ATO (Fig 7K and L). As expected, the syllable deficiency in ATO was similar to those in AFO (Fig 1I and J). This suggests that DNA hypo‐methylation in sperm led to USV deficits, but not to low body weight in offspring at the early postnatal stage.
Discussion
We showed here that paternal aging indeed results in vocal communication deficits in pups, as observed in genetic models of ASD (Hiramoto et al, 2011); USV syllables and syllable types, especially complex syllables (“One Jump”, “More jump”, and “More jump + Harmonics”), were significantly decreased in AFO. Our entropy analyses showed reduced diversity (as indicated by a lower entropy score) in vocal communication in AFO. Therefore, we propose that paternal aging in mice is a suitable model for further analyzing epigenetic mechanisms in ASD.
We also performed comprehensive whole‐genome target methylome analyses of sperm DNA obtained from young and aged mice. Consistent with previous studies in mice and humans (Feinberg et al, 2015; Milekic et al, 2015; Xie et al, 2018), we found higher frequencies of DNA hypo‐methylation in sperm from aged mice. Furthermore, whole‐genome level analysis comparing lists of genes near the hypo‐DMRs with those of hypo‐methylated DNA promoters (Xie et al, 2018) did not reveal any genes in common with those in previous studies in the vicinity of our hypo‐DMRs. One possible explanation is a methodological difference; the method used in the previous studies was reduced representation bisulfite sequencing (RRBS), while we used SureSelect Methyl‐Seq from Agilent Technology (Koike et al, 2016), which can cover a wider range of genome sequences than RRBS (Miura & Ito, 2015). The motif analysis and ChIP‐Atlas analyses revealed a common enrichment of REST/NRSF binding motifs in hypo‐, but not in hyper‐, DMRs of aged mouse sperm. Therefore, our study goes a step further than previous studies, and we believe that we have gained a deeper understanding of the molecular mechanisms underlying the influence of paternal aging on brain development in the next generation.
How could REST/NRSF contribute to DNA methylation and subsequent gene expression? There can be several alternative scenarios. First, since DNA methyltransferase 3b is known to promote REST/NRSF occupancy in the developing heart (Zhang et al, 2017), REST/NRSF cannot bind to hypo‐DMRs, which might subsequently lead to leaky expression of the target genes. Alternatively, it has been reported that REST/NRSF binding induces DNA hypo‐methylation in the neighboring genomic region in ES cells (Stadler et al, 2011). By reanalyzing a publicly available data for the testes, we found that Rest expression was highest in testicular stem cells (i.e., type A spermatogonia, Fig EV3C; Namekawa et al, 2006). Analysis of a single‐cell RNA sequencing dataset from testicular cells also confirmed a distinctly high expression of Rest in the most immature sub‐population of spermatogonia (Green et al, 2018). Thus, the hypo‐DMRs in aged mouse sperm may be a result of REST/NRSF binding in testicular stem cells.
We also revealed that gene sets near the DMRs were enriched in genes that were upregulated in the embryonic AFO brain (Fig 4I and J). Since most of the DMRs were located in intergenic regions, it would be interesting to know how DNA hypo‐methylation at REST/NRSF binding sites affects gene expression. Recently published interactome studies have shown that risk variants in non‐coding regions of the genome identified by genome‐wide association studies may influence phenotypes by causing perturbations in promoters and enhancers (Gallagher & Chen‐Plotkin, 2018; Nott et al, 2019). In addition, many functional cis‐elements are reported to be located hundreds of kilobases away from the coding regions of target genes (West & Fraser, 2005; Bulger & Groudine, 2011; Sanyal et al, 2012). Indeed, in our RNA‐seq data, we found that gene sets for REST/NRSF target genes and late‐fetal genes were upregulated in the brain of AFO at E14.5, while those for early‐fetal genes were downregulated (Fig 4A, F and G); moreover, the difference was not attributed to REST/NRSF expression itself (Fig EV3D and E). In contrast, distortions in gene expression were not observed in the brain of AFO at E11.5 (Fig 5). Given the fact that REST/NRSF represses the expression of neuronal genes in neural stem cells (Ballas et al, 2005), enrichment of late‐fetal gene sets in the forebrain of AFO could be attributed to increased expression of REST/NRSF target genes.
A previous study did not find a significant relationship between DNA methylation and gene expression in the brain of offspring from aged mice (Milekic et al, 2015). This may be due to a difference in the analytical methods; we adopted a comprehensive GSEA that is suitable for detecting slight gene expression changes in gene sets (Mootha et al, 2003), while they used a conventional Pearson correlation analysis between DNA methylation and expression levels of limited genes (Milekic et al, 2015). Using the more advanced technique, we first investigated the possibility that DNA hypo‐methylation due to advanced paternal age does indeed have an impact on gene expression in the offspring brain. Although it is unclear why hypo‐methylation of the genomic regions is related to leaky expression of the target genes for REST/NRSF, which functions as a transcriptional repressor, a recent study has shown that REST/NRSF can bind to hyper‐methylated regions (Lunyak & Rosenfeld, 2005); that is, aging‐induced hypo‐methylation at the REST/NRSF binding sites in the sperm genome might be involved in causing de‐repression of inadequate gene expression in the embryonic brain derived from an aged father.
To understand the global effects on sperm DNA methylation due to 5‐Aza treatment, we reanalyzed publicly available RRBS data from three 5‐Aza‐treated and two control mice (Kläver et al, 2015). Among the 96 hypo‐DMRs we identified, 21 regions were calculatable, of which 12 regions (including Nav1, which we validated by BS Sanger sequencing) were significantly DNA hypo‐methylated in 5‐Aza‐treated mice. As RRBS enriches for CpG‐rich regions of the genome, we are unable to estimate methylation of cytosines outside CpG‐rich regions. We imagine that this is the main reason why these RRBS datasets seldom cover the regions we focused on (e.g., REST binding sites).
In another model of ASD, which was based on a Chd8+/ΔL mutant, GSEA was applied because the expression of genes remained almost unchanged in the embryonic brain (Katayama et al, 2016). Intriguingly, REST/NRSF target genes were downregulated, and consistently early‐fetal genes were upregulated in the embryonic brain of Chd8+/ΔL mutant mice, which is in contrast to our results, where REST/NRSF target genes were upregulated, and consistently early‐fetal genes were downregulated in the embryonic brain of AFO. Likewise, the results related to brain structure also seems to be contrasting—Chd8+/ΔL mutants had larger brains, while AFO pups exhibited a reduction in cortical thickness. These results suggest that both up‐ and downregulation of REST/NRSF target genes during the neurogenic period can be a risk for ASD due to genetic and/or epigenetic factors.
According to the DOHaD theory, low body weight at birth or at the early postnatal stage can be a risk factor for many diseases, such as cardiovascular diseases and metabolic syndrome, in adults (Barker & Osmond, 1986; Eriksson, 2016). As with previous studies in humans (Reichman & Teitler, 2006; Alio et al, 2012), we found that AFO tended to have lower body weight, although the difference was not statistically significant. Low body weight was not observed in ATO (Fig 7E), suggesting the possible involvement of other mechanisms related to paternal aging in determining offspring body weight.
In conclusion, we found that paternal aging induced predominant DNA hypo‐methylation in sperm and abnormal behavior in offspring, which was phenocopied in offspring derived from young mice administrated with a DNA de‐methylation drug. We also revealed enriched REST/NRSF binding motifs in hypo‐methylated regions of sperm DNA, as well as significant upregulation of REST/NRSF target genes in the developing AFO brain. On the basis of these results, we propose a molecular scenario in which paternal aging may induce leaky expression of REST/NRSF target genes that have been marked with hypo‐methylation within sperm cells, causing precocious neurogenesis and thereby resulting in abnormalities in brain structures and neuronal activities, which may cause behavioral phenotypes corresponding to neurodevelopmental disorders (see Synopsis). Due to the current technical limitations of target‐specific epigenome editing, further proof is currently limited. The next challenge will be to elucidate whether DNA hypo‐methylation of REST/NRSF binding motifs in sperm impacts the expression of REST/NRSF target genes in the embryonic brain and subsequent behavioral abnormalities. A similar mechanism could underlie the scenario of paternal exposure to environmental contaminants in rats, where the effect on the next generation can be partially mitigated by folic acid (Lessard et al, 2019). Although it remains unclear how de‐methylation marks in the sperm genome can influence gene expression in the offspring brain, recent epidemiological data suggest a possible involvement of methylation levels with regard to the risk for ASD (Levine et al, 2018; Raghavan et al, 2018).
Materials and Methods
Animals
All experimental procedures were approved by the Ethics Committee for Animal Experiments of Tohoku University Graduate School of Medicine (#2014‐112) and of Tokyo University of Agriculture, and animals were treated according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Three‐month‐old (young) or > 12‐month‐old (aged) male C57BL6/J mice were crossed with young virgin female mice to obtain offspring for this study (Fig 1A). All animals were housed in standard cages in a temperature‐ and humidity‐controlled room with a 12‐h light/dark cycle (light on at 8:00) and had free access to standard lab chow and tap water. For DNA methylation analyses, sperm was obtained from 3‐month or 18‐month‐old male mice.
USV measurement
Each pup was assessed for USV on PND6 according to previously described protocols (Scattoni et al, 2008; Yoshizaki et al, 2016; Yoshizaki et al, 2017). Pups were separated from their mother and littermates, one at a time, and placed in a plastic dish with floor cloth in a soundproof chamber, and USV calls were recorded for 5 min using a microphone connected to the UltraSound Gate 416H detector (Avisoft Bioacoustics, Germany) set at 25–125 kHz to measure the number of USV calls and latency until first USV call. For syllable analyses, MATLAB scripts (USVSEG) were applied to convert audio files to the spectrogram, and USV syllables were automatically segmented (Tachibana et al, 2020). Each segmented syllable was manually inspected to exclude contaminating noise and error. Syllables longer than 2 ms and with frequencies between 60 and 120 kHz were subjected to further analyses of duration, maximum frequency (peak frequency at maximum amplitude), and maximum amplitude. Syllables were manually classified into 12 types according to a previous report (Scattoni et al, 2008). Briefly, (i) “Upward” was characterized by upward modulation in pitch with a terminal frequency change ≥ 6.25 kHz than the beginning of the syllable. (ii) “Downward” was characterized by downward modulation in pitch with a terminal frequency change ≥ 6.25 kHz than the beginning of the syllable. (iii) “Flat” was continuous with a frequency modification ≤ 3 kHz. (iv) “Short” was displayed as a dot and shorter than or equal to 5 ms. (v) “Chevron” formed a U or an inverted U. (vi) “Wave” was regulated with two‐directional changes in frequency > 6.25 kHz. (vii) “Complex” was regulated with three or more directional changes in frequency > 6.25 kHz. (viii) “One jump” contained two components, in which the second component was changed ≥ 10 kHz frequency compared with the first component, and there was no time interval between the two components. (ix) “More jump” contained three or more components, in which the second component was changed ≥ 10 kHz frequency compared with the first and third components and there was a time interval between adjacent components. (x) “Harmonics” was displayed as one main component stacking with other harmonic components of different frequencies. (xi) “One jump + harmonics” contained one jump and harmonics syllables together, with no time interval between them. (xii) “More jump + harmonics” contained more jumps and harmonics syllables together, with no time interval between them. Variations in emitted syllables from individual mouse pups were examined based on normalized entropy scores calculated using the following equation:
Here, T indicates the number of syllable types and pt indicates the production rate of a specific syllable type t. Pups that had fewer than 10 calls during the 5 min isolation were excluded from this analysis.
Target methylome analysis
DNA preparation
Sperm DNA was isolated by a standard phenol‐chloroform extraction procedure using dithiothreitol. One microgram of DNA was dissolved in 130 μl of 10 mM Tris–HCl (pH 8.0) and sheared using an S220 focused ultrasonicator (Covaris, Woburn, MA, USA) to yield 500‐bp fragments. The AMPure XP system (Agilent Technologies, Santa Clara, CA, USA) was used to purify the fragmented DNA. Briefly, sheared DNA (130 μl) was mixed with 1.8× volume (234 μl) of AMPure XP reagent and allowed to stand for 15 min at room temperature. Beads were collected using a magnetic stand, the supernatant was removed, and the resulting pelleted beads were rinsed with 70% ethanol and dried by incubation at 37°C for 5 min. DNA was then eluted from the beads in 20 μl of RNase‐free water. The eluted DNA was then dried in vacuum and dissolved in 7 μl of RNase‐free water.
Target enrichment
Target enrichment by liquid‐phase hybridization capture was performed using the SureSelect Mouse Methyl‐Seq kit (Agilent Technologies) (Sun et al, 2014). Genomic DNA (7 μl) fragmented and purified as described above was supplemented with 3 μl of formamide (biochemistry grade; Wako Pure Chemical Industries, Osaka, Japan) and overlaid with 80 μl of mineral oil (Sigma‐Aldrich, St. Louis, MO, USA). DNA was completely denatured by incubating the tube at 99°C for 10 min; then, the tube was cooled to 65°C and maintained at that temperature for at least 5 min before adding the following reagents. Hybridization buffer and capture probe mix were prepared according to the manufacturer’s protocol, overlaid with 80 μl of mineral oil each, and incubated at 65°C for 10 min. The two solutions were then combined and mixed thoroughly by pipetting. The combined solution was transferred to a tube containing the denatured input DNA (maintained at 65°C as described above), and the solution was thoroughly mixed by pipetting. The tube was incubated at 65°C for 24 h to allow hybridization between probes and targets. A 50 μl volume of well‐suspended DynaBeads MyOne Streptavidin T1 solution (Life Technologies, Carlsbad, CA, USA) was placed in a 1.5 ml tube, and the beads were washed twice with 200 μl of binding buffer. The hybridization reaction, supplemented with 200 μl of binding buffer, was added to the pelleted beads and thoroughly mixed. After incubation at room temperature for 30 min with agitation, the beads were collected using a magnetic stand and washed with 500 μl of wash buffer 1, subjected to three rounds of washing and re‐suspension in pre‐warmed buffer 2, and then incubated at 65°C for 10 min. After removing the washing solution from the tube, enriched DNA was eluted by incubating the beads in 20 μl of elution buffer at room temperature for 20 min. The elution fraction was immediately subjected to bisulfite treatment.
Bisulfite treatment
The EZ DNA Methylation‐Gold kit (Zymo Research, Irvine, CA, USA) was used for bisulfite treatment of target‐enriched DNA according to the manufacturer’s instructions. The enriched DNA solution (20 μl) was mixed with 130 μl of freshly prepared cytosine‐thymine conversion reagent, and the mixture was incubated at 64°C for 2.5 h. The 10 min incubation step at 98°C was omitted, since the target‐enriched DNA was already denatured. After purification and desulfonation, bisulfite‐treated DNA was eluted with 20 μl of M‐elution buffer.
Post‐bisulfite adapter tagging (PBAT) library construction and Illumina sequencing
We used bisulfite‐treated DNA for library preparation according to the PBAT protocol (available from http://crest‐ihec.jp/english/epigenome/index.html), except that the following primers were used: the primer used for the first‐strand synthesis was 5′‐biotin ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT WWW WNN NN‐3′ (W = A or T) and the indexed primer used for second‐strand synthesis was 5′‐CAA GCA GAA GAC GGC ATA CGA GAT XXX XXX GTA AAA CGA CGG CCA GCA GGA AAC AGC TAT GAC WWW WNN NN‐3′, where XXX XXX represents the index sequence of each primer. Constructed SureSelect Methyl‐Seq (SSM)‐PBAT libraries were sequenced as previously described (Kobayashi et al, 2013; Miura & Ito, 2015; Koike et al, 2016) using the Illumina HiSeq2500 system (San Diego, CA, USA) to generate 100‐nt single‐end sequence reads. Before alignment, each random sequence was trimmed from the sequence datasets. These trimmed data (93‐nt) are deposited in the DNA Data Bank of Japan (DDBJ) (accession number DRA007933).
Target methylome sequence alignment and statistical analysis
SSM‐PBAT reads were aligned to the mouse genome (mm10, Genome Reference Consortium Mouse Build 38) using the Bismark tool (v.0.10.0; http://www.bioinformatics.babraham.ac.uk/projects/bismark/) with the specific options: −q −n 2 –l 93 –pbat. Statistical significance of DNA methylation at each CpG site or CpG island was evaluated using MOABS. Composite profiles were generated using SeqMonk (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/).
Gene expression analyses of embryonic brains
RNA extraction for RNA sequencing
Total RNA was extracted from embryonic forebrains of pups from different mothers at E11.5 and E14.5 using the RNeasy Micro kit (Qiagen, Germany) according to the manufacturer's protocol. The libraries were obtained using 2.0 μg of total RNA and the TruSeq® Stranded mRNA Library Prep Kit (Illumina, USA) following the manufacturer’s guidelines. The quality of the libraries was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Germany). Library cluster generation was performed on a cBot with the Illumina HiSeq PE Cluster Kit v4 – cBot – HS (Illumina, USA). The libraries were sequenced using a paired‐end method (100 base pair reads) using the HiSeq 2500 system and HiSeq Control Software v2.2.58/Real‐Time Analysis v1.18.64 (Illumina, USA). BCL files were converted to the FASTQ file format by using bcl2fastq2 (Illumina, USA).
Analysis of RNA sequencing data
A series of RNA‐seq data analyses were performed according to the Genedata Profiler Genome (Genedata) instructions. Reads in FASTQ format were aligned to the reference mouse genome (GRCm38/mm10, Dec. 2011) using the TopHat v2.0.14, Bowtie, and SAM tools (Li et al, 2009; Trapnell et al, 2009; Langmead & Salzberg, 2012; Kim et al, 2013); these filtered data have been deposited in the DDBJ (accession number DRA008023). To remove unreliable data, the genes were selected by filtering according to the following criteria: the dispersion was not zero in fragments per kilobase of transcript per million mapped reads, and the number of counts was 16 or more. PCA was performed using the prcomp R package.
Enrichment analysis
Genome coordinates of DMRs and other SureSelect target regions were converted from the mm10 to the mm9 assembly using the UCSC liftOver tool (https://genome.ucsc.edu/cgi‐bin/hgLiftOver) before enrichment analyses based on the ChIP‐Atlas. Enrichment analyses of GO terms (http://geneontology.org) and MGI phenotypes (http://www.informatics.jax.org) were performed for genes located near DMRs against the other RefSeq coding genes. GSEA was performed as previously described (Katayama et al, 2016), and SFARI (Banerjee‐Basu & Packer, 2010) and SZ gene sets (Allen et al, 2008) were obtained from https://www.sfari.org and http://www.szgene.org, respectively.
Immunohistochemistry
Procedures for immunohistochemistry were according to previous studies (Kikkawa et al, 2013; Kimura et al, 2015). Coronal serial sections (16 μm thick) were prepared using a cryostat, washed with Tris‐based saline containing 0.1% Tween20 (TBST), and treated with 3% bovine serum albumin and 0.3% TritonX‐100 in PBS solution (blocking solution). The sections were then incubated with primary antibodies against Tbr1 (AB10554; Millipore, Morocco; dilution, 1: 200), Satb2 (ab51502; Abcam, Cambridge, UK; dilution, 1:1,000), Ctip2 (ab18465; Abcam, Cambridge, UK; dilution, 1:1,000), Cux1 (sc‐13024; Santacruz Biotechnology, Dallas, US; dilution, 1:500) in blocking solution overnight at 4°C. Subsequently, the sections were incubated with Cy3 or Alexa488 conjugated secondary antibodies (Jackson ImmunoResearch Inc.; dilution, 1:500) and 4′, 6‐diamino‐2‐phenylindole (DAPI; dilution, 1:1,000) for 1 h at room temperature. Images were obtained using a fluorescent microscopic system (BZ‐X, Keyence, Osaka, Japan); cortical thickness was measured using an analyzing tool on the system.
De‐methylation drug treatment
A 5 mg/ ml stock solution of 5‐Aza (A3656, Sigma) in DMSO was prepared and stored at −20°C until further use. As 5‐Aza is unstable in aqueous solution, a working solution was prepared at the time of use by diluting the stock solution with saline (Otsuka). Male mice (12‐week‐old) were weighed and intraperitoneally injected with the working solution of 5‐Aza (0.05 mg/kg) or saline between 13:00 and 17:00 every 2 days for 7 weeks according to a previous protocol (Kelly et al, 2003). Mice showing a significant reduction in body weight during the regimen were excluded from the experiment. After completion of the regimen, male mice treated with saline or 5‐Aza were mated with virgin female wild‐type mice (10‐week‐old) to obtain the offspring. For methylation status detection, sperm was squeezed out from the cauda epididymis of the mice and smeared on glass slides. Sperm was decondensed for 60 min at room temperature in a decondensation solution (10 mM EDTA, 10 mM DTT, 0.25 mg/ml heparin, 0.3% Triton X‐100 in PBS) and fixed with 1% PFA for 10 min at room temperature. The slides were further treated with 1 N HCl at 37°C for 30 min and then with blocking solution for 60 min at room temperature. Diluted antibody against 5‐methylcytosine (5mC) (dilution, 1:300; clone 33D3, Calbiochem) was applied on the glass slides and incubated overnight at 4°C. After washing, the slides were incubated with an Alexa488 conjugated antibody (Jackson ImmunoResearch Inc.; dilution, 1:500) for 1 h at room temperature. Images were then obtained using a confocal microscope (LSM 800, Zeiss). Contours of sperm heads were detected using the U‐Net software (Falk et al, 2019), and sperm head immunofluorescence was measured using ImageJ software (National Institute of Health, Bethesda, Maryland, USA).
Sperm DNA methylation analysis
Mouse sperm genomic DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen, Germany). Sperm were suspended in 300 μl of a lysis buffer containing 100 mM Tris–HCl (pH 8.0), 10 mM EDTA, 500 mM NaCl, 1% SDS, and 2% 2‐ME. Proteinase K solution was added to the suspension and incubated for 2 h at 55°C on a rocking platform. Another 20 μl of Proteinase K solution was added before further incubation overnight. After adding 400 μl of Buffer AL and 400 μl of ethanol, samples were applied to the columns provided and processed according to the manufacturer’s protocol. The EZ DNA Methylation‐Direct Kit (Zymo Research, California) was used to convert unmethylated sperm DNA cytosine to uracil. Freshly prepared CT conversion reagent was mixed with 350 ng of purified genomic DNA, incubated for 8 min at 98°C, and then for 3.5 h at 64°C. Bisulfite‐converted DNA was eluted with 12 μl of M‐elution buffer and stored at −20°C until further use. To assess the DNA methylation status, target genomic regions were amplified from bisulfite‐converted sperm DNA using nested PCR. Primers were designed using the MethPrimer tool (Li & Dahiya, 2002). See Dataset EV5 (BS_seq_primer.xls) for the list of primers. Second PCR products were subcloned into the pGEM‐T easy vector (Promega, Madison) and transformed into DH5α (Takara, Japan). The sequence of each PCR product was determined by sequencing plasmids from 8–10 different colonies; sequence data were processed and analyzed using the QUMA tool (Kumaki et al, 2008).
Statistical analyses
All data are presented in the dot plot, box plot (centerline, median, box limits, upper and lower quartiles), or mean ± standard error of the mean (SEM) format. The two‐side Student test, Welch’s t‐test, Fisher’s exact test, and Mann–Whitney U‐test were used for comparisons. JMP®, version 14 (SAS Institute Inc., Cary, NC) was used for all statistical analyses, and P‐values less than 0.05 were considered statistically significant.
Conflict of interest
The authors declare that they have no conflict of interest.
Author contributions
KY: Conceptualization, Data curation, Formal analysis, Funding acquisition, Validation, Visualization, Writing—original draft, Writing—review and editing. RK: Conceptualization, Data curation, Formal analysis, Validation, Writing—review and editing. HK: Data curation, Formal analysis, Validation, Writing—review and editing. SO: Formal analysis, Validation, Writing—review and editing. TK: Data curation, Formal analysis, Validation, Writing—review and editing. LM: Data curation, Formal analysis, Validation. KK: Data curation, Formal analysis. KM: Formal analysis, Validation. HI: Formal analysis, Validation. YM: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing—review and editing. TK: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing—review and editing. NO: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing—review and editing.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Acknowledgements
The authors thank Mr. Tasuku Koike and Mr. Tomoya Takashima for their contributions to the sperm methylome and neuronal activity analyses, respectively. We are also grateful to Drs. Keiichi Nakayama and Masaaki Nishiyama for their advice on gene set enrichment analyses (GSEA), and to Drs. Kouta Kanno, Takeru Matsuda, and Fumiyasu Komaki for suggestions regarding mathematical analyses for USV. This research was supported in part by CREST (to Y.M, T.K, and N.O), the MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (to T.K), as well as JSPS KAKENHI Grants # 25640002, #15K12764, #16H06530 (to N.O.), #15K06694, and #19K08060 (to K.Y.).
EMBO reports (2021) 22: e51524.
Contributor Information
Yasuhisa Matsui, Email: yasuhisa.matsui.d3@tohoku.ac.jp.
Tomohiro Kono, Email: tomohiro@nodai.ac.jp.
Noriko Osumi, Email: osumi@med.tohoku.ac.jp.
Data availability
DNA methylome data of sperm from young and aged mice that support the findings of this study have been deposited in the DNA Data Bank of Japan (DDBJ) (accession number DRA007933). URL: https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA007933
Embryonic brain RNA‐sequence data derived from young and aged fathers that support the findings of this study have been deposited in DDBJ (accession number DRA008023). URL: https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008023
References
- Alio AP, Salihu HM, McIntosh C, August EM, Weldeselasse H, Sanchez E, Mbah AK (2012) The effect of paternal age on fetal birth outcomes. Am J Mens Health 6: 427–435 [DOI] [PubMed] [Google Scholar]
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Fariñas I, Grosschedl R, McConnell SK (2008) Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron 57: 364–377 [DOI] [PubMed] [Google Scholar]
- Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L (2008) Systematic meta‐analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 40: 827–834 [DOI] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD (2005) Neuronal subtype‐specific genes that control corticospinal motor neuron development in vivo . Neuron 45: 207–221 [DOI] [PubMed] [Google Scholar]
- Arnold P, Schöler A, Pachkov M, Balwierz PJ, Jørgensen H, Stadler MB, van Nimwegen E, Schübeler D (2013) Modeling of epigenome dynamics identifies transcription factors that mediate Polycomb targeting. Genome Res 23: 60–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121: 645–657 [DOI] [PubMed] [Google Scholar]
- Banerjee‐Basu S, Packer A (2010) SFARI Gene: an evolving database for the autism research community. Dis Model Mech 3: 133–135 [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C (1986) Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1: 1077–1081 [DOI] [PubMed] [Google Scholar]
- Bedogni F, Hodge RD, Nelson BR, Frederick EA, Shiba N, Daza RA, Hevner RF (2010) Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc Natl Acad Sci USA 107: 13129–13134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, de Juan RC, Cheung A, Kwan KY, Schwark M, Gyorgy A, Vogel T, Akopov S, Mitkovski M, Agoston D et al (2008) Satb2 is a postmitotic determinant for upper‐layer neuron specification in the neocortex. Neuron 57: 378–392 [DOI] [PubMed] [Google Scholar]
- Bulger M, Groudine M (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG (2016) Developmental Origins of Health and Disease – from a small body size at birth to epigenetics. Ann Med 48: 456–467 [DOI] [PubMed] [Google Scholar]
- Falk T, Mai D, Bensch R, Çiçek Ö, Abdulkadir A, Marrakchi Y, Böhm A, Deubner J, Jäckel Z, Seiwald K et al (2019) U‐Net: deep learning for cell counting, detection, and morphometry. Nat Methods 16: 67–70 [DOI] [PubMed] [Google Scholar]
- Feinberg JI, Bakulski KM, Jaffe AE, Tryggvadottir R, Brown SC, Goldman LR, Croen LA, Hertz‐Picciotto I, Newschaffer CJ, Fallin MD et al (2015) Paternal sperm DNA methylation associated with early signs of autism risk in an autism‐enriched cohort. Int J Epidemiol 44: 1199–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldi CJ, Eyles DW, McGrath JJ, Burne TH (2010) Advanced paternal age is associated with alterations in discrete behavioural domains and cortical neuroanatomy of C57BL/6J mice. Eur J Neurosci 31: 556–564 [DOI] [PubMed] [Google Scholar]
- Gallagher MD, Chen‐Plotkin AS (2018) The Post‐GWAS Era: from association to function. Am J Hum Genet 102: 717–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratten J, Wray NR, Peyrot WJ, McGrath JJ, Visscher PM, Goddard ME (2016) Risk of psychiatric illness from advanced paternal age is not predominantly from de novo mutations. Nat Genet 48: 718–724 [DOI] [PubMed] [Google Scholar]
- Green CD, Ma Q, Manske GL, Shami AN, Zheng X, Marini S, Moritz L, Sultan C, Gurczynski SJ, Moore BB et al (2018) A comprehensive roadmap of murine spermatogenesis defined by single‐cell RNA‐Seq. Dev Cell 46: 651–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiramoto T, Kang G, Suzuki G, Satoh Y, Kucherlapati R, Watanabe Y, Hiroi N (2011) Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum Mol Genet 20: 4775–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis ED (2004) Learned birdsong and the neurobiology of human language. Ann N Y Acad Sci 1016: 749–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins TG, Aston KI, Pflueger C, Cairns BR, Carrell DT (2014) Age‐associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet 10: e1004458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens U (2002) Neural pathways underlying vocal control. Neurosci Biobehav Rev 26: 235–258 [DOI] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, Sousa AM, Pletikos M, Meyer KA, Sedmak G et al (2011) Spatio‐temporal transcriptome of the human brain. Nature 478: 483–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Nishi yama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, Suyama M, Takumi T, Miyakawa T, Nakayama KI (2016) CHD8 haploinsufficiency results in autistic‐like phenotypes in mice. Nature 537: 675–679 [DOI] [PubMed] [Google Scholar]
- Kelly TL, Li E, Trasler JM (2003) 5‐aza‐2'‐deoxycytidine induces alterations in murine spermatogenesis and pregnancy outcome. J Androl 24: 822–830 [DOI] [PubMed] [Google Scholar]
- Kikkawa T, Obayashi T, Takahashi M, Fukuzaki‐Dohi U, Numayama‐Tsuruta K, Osumi N (2013) Dmrta1 regulates proneural gene expression downstream of Pax6 in the mammalian telencephalon. Genes Cells 18: 636–649 [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Yoshizaki K, Osumi N (2015) Dynamic expression patterns of Pax6 during spermatogenesis in the mouse. J Anat 227: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kläver R, Sánchez V, Damm OS, Redmann K, Lahrmann E, Sandhowe‐Klaverkamp R, Rohde C, Wistuba J, Ehmcke J, Schlatt S et al (2015) Direct but no transgenerational effects of decitabine and vorinostat on male fertility. PLoS One 10: e0117839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Sakurai T, Miura F, Imai M, Mochiduki K, Yanagisawa E, Sakashita A, Wakai T, Suzuki Y, Ito T et al (2013) High‐resolution DNA methylome analysis of primordial germ cells identifies gender‐specific reprogramming in mice. Genome Res 23: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T, Wakai T, Jincho Y, Sakashita A, Kobayashi H, Mizutani E, Wakayama S, Miura F, Ito T, Kono T (2016) DNA methylation errors in cloned mouse sperm by germ line barrier evasion. Biol Reprod 94: 128 [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Jonasdottir A et al (2012) Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y, Oda M, Okano M (2008) QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36(Web Server): W170–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard M, Herst PM, Charest PL, Navarro P, Joly‐Beauparlant C, Droit A, Kimmins S, Trasler J, Benoit‐Biancamano MO, MacFarlane AJ et al (2019) Prenatal exposure to environmentally‐relevant contaminations perturbs male reproductive parameters across multiple generations that are partially protected by folic acid supplementation. Sci Rep 9: 13829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine SZ, Kodesh A, Viktorin A, Smith L, Uher R, Reichenberg A, Sandin S (2018) Association of maternal use of folic acid and multivitamin supplements in the periods before and during pregnancy with the risk of autism spectrum disorder in offspring. JAMA Psychiatry 75: 176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LC, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18: 1427–1431 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundström S, Haworth CM, Carlström E, Gillberg C, Mill J, Råstam M, Hultman CM, Ronald A, Anckarsäter H, Plomin R et al (2010) Trajectories leading to autism spectrum disorders are affected by paternal age: findings from two nationally representative twin studies. J Child Psychol Psychiatry 51: 850–856 [DOI] [PubMed] [Google Scholar]
- Lunyak VV, Rosenfeld MG (2005) No rest for REST: REST/NRSF regulation of neurogenesis. Cell 121: 499–501 [DOI] [PubMed] [Google Scholar]
- Malaspina D, Harlap S, Fennig S, Heiman D, Nahon D, Feldman D, Susser ES (2001) Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry 58: 361–367 [DOI] [PubMed] [Google Scholar]
- Mandel G, Fiondella CG, Covey MV, Lu DD, Loturco JJ, Ballas N (2011) Repressor element 1 silencing transcription factor (REST) controls radial migration and temporal neuronal specification during neocortical development. Proc Natl Acad Sci USA 108: 16789–16794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milekic MH, Xin Y, O'Donnell A, Kumar KK, Bradley‐Moore M, Malaspina D, Moore H, Brunner D, Ge Y, Edwards J et al (2015) Age‐related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol Psychiatry 20: 995–1001 [DOI] [PubMed] [Google Scholar]
- Miura F, Ito T (2015) Highly sensitive targeted methylome sequencing by post‐bisulfite adaptor tagging. DNA Res 22: 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P, Feng G (2017) SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci 18: 147–157 [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E et al (2003) PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Namekawa SH, Park PJ, Zhang LF, Shima JE, McCarrey JR, Griswold MD, Lee JT (2006) Postmeiotic sex chromatin in the male germline of mice. Curr Biol 16: 660–667 [DOI] [PubMed] [Google Scholar]
- Nechiporuk T, McGann J, Mullendorff K, Hsieh J, Wurst W, Floss T, Mandel G (2016) The REST remodeling complex protects genomic integrity during embryonic neurogenesis. Elife 5: e09584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y et al (2019) Brain cell type‐specific enhancer‐promoter interactome maps and disease‐risk association. Science 366: 1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, Kawaji H, Nakaki R, Sese J, Meno C (2018) ChIP‐Atlas: a data‐mining suite powered by full integration of public ChIP‐seq data. EMBO Rep 19: e46255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD et al (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paonessa F, Latifi S, Scarongella H, Cesca F, Benfenati F (2013) Specificity protein 1 (Sp1)‐dependent activation of the synapsin I gene (SYN1) is modulated by RE1‐silencing transcription factor (REST) and 5'‐cytosine‐phosphoguanine (CpG) methylation. J Biol Chem 288: 3227–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette AJ, Perez SE, Anderson DJ (2000) Constitutive expression of the neuron‐restrictive silencer factor (NRSF)/REST in differentiating neurons disrupts neuronal gene expression and causes axon pathfinding errors in vivo. Proc Natl Acad Sci USA 97: 12318–12323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipsen S, Suske G (1999) A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res 27: 2991–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan R, Riley AW, Volk H, Caruso D, Hironaka L, Sices L, Hong X, Wang G, Ji Y, Brucato M et al (2018) Maternal multivitamin intake, plasma folate and vitamin B12 levels and autism spectrum disorder risk in offspring. Paediatr Perinat Epidemiol 32: 100–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman NE, Teitler JO (2006) Paternal age as a risk factor for low birthweight. Am J Public Health 96: 862–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampino S, Juszczak GR, Zacchini F, Swiergiel AH, Modlinski JA, Loi P, Ptak GE (2014) Grand‐paternal age and the development of autism‐like symptoms in mice progeny. Transl Psychiatry 4: e386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Schendel D, Magnusson P, Hultman C, Surén P, Susser E, Grønborg T, Gissler M, Gunnes N, Gross R et al (2016) Autism risk associated with parental age and with increasing difference in age between the parents. Mol Psychiatry 21: 693–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J (2012) The long‐range interaction landscape of gene promoters. Nature 489: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Crawley J, Ricceri L (2008) Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PLoS One 3: e3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RG, Kember RL, Mill J, Fernandes C, Schalkwyk LC, Buxbaum JD, Reichenberg A (2009) Advancing paternal age is associated with deficits in social and exploratory behaviors in the offspring: a mouse model. PLoS One 4: e8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubry A (2018) POHaD: why we should study future fathers. Environ Epigenet 4: dvy007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D et al (2011) DNA‐binding factors shape the mouse methylome at distal regulatory regions. Nature 480: 490–495 [DOI] [PubMed] [Google Scholar]
- Sun D, Xi Y, Rodriguez B, Park HJ, Tong P, Meong M, Goodell MA, Li W (2014) MOABS: model based analysis of bisulfite sequencing data. Genome Biol 15: R38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana RO, Kanno K, Okabe S, Kobayasi KI, Okanoya K (2020) USVSEG: a robust method for segmentation of ultrasonic vocalizations in rodents. PLoS One 15: e0228907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumi D, Hayashi Y, Endo M, Kobayashi H, Yoshioka T, Kiso K, Kanno S, Nakai Y, Maeda I, Mochizuki K et al (2018) DNMTs and SETDB1 function as co‐repressors in MAX‐mediated repression of germ cell‐related genes in mouse embryonic stem cells. PLoS One 13: e0205969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JL, Debost JPG, Morton SU, Wigdor EM, Heyne HO, Lal D, Howrigan DP, Bloemendal A, Larsen JT, Kosmicki JA et al (2019) Paternal‐age‐related de novo mutations and risk for five disorders. Nat Commun 10: 3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LY (1999) Psychopharmacology in autism. Psychosom Med 61: 651–665 [DOI] [PubMed] [Google Scholar]
- Tsuchiya KJ, Matsumoto K, Miyachi T, Tsujii M, Nakamura K, Takagai S, Kawai M, Yagi A, Iwaki K, Suda S et al (2008) Paternal age at birth and high‐functioning autistic‐spectrum disorder in offspring. Br J Psychiatry 193: 316–321 [DOI] [PubMed] [Google Scholar]
- West AG, Fraser P (2005) Remote control of gene transcription. Hum Mol Genet 14(Spec No 1): R101–R111 [DOI] [PubMed] [Google Scholar]
- Xie K, Ryan DP, Pearson BL, Henzel KS, Neff F, Vidal RO, Hennion M, Lehmann I, Schleif M, Schröder S et al (2018) Epigenetic alterations in longevity regulators, reduced life span, and exacerbated aging‐related pathology in old father offspring mice. Proc Natl Acad Sci USA 115: E2348–E2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall‐Walker CA, Barry BJ, Murn J et al (2012) Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151: 1097–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki K, Furuse T, Kimura R, Tucci V, Kaneda H, Wakana S, Osumi N (2016) Paternal aging affects behavior in Pax6 mutant mice: a gene/environment interaction in understanding neurodevelopmental disorders. PLoS One 11: e0166665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki K, Koike K, Kimura R, Osumi N (2017) Early postnatal vocalizations predict sociability and spatial memory in C57BL/6J mice: individual differences in behavioral traits emerge early in development. PLoS One 12: e0186798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Wu B, Wang P, Wang Y, Lu P, Nechiporuk T, Floss T, Greally JM, Zheng D, Zhou B (2017) Non‐CpG methylation by DNMT3B facilitates REST binding and gene silencing in developing mouse hearts. Nucleic Acids Res 45: 3102–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Review Process File
Data Availability Statement
DNA methylome data of sperm from young and aged mice that support the findings of this study have been deposited in the DNA Data Bank of Japan (DDBJ) (accession number DRA007933). URL: https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA007933
Embryonic brain RNA‐sequence data derived from young and aged fathers that support the findings of this study have been deposited in DDBJ (accession number DRA008023). URL: https://ddbj.nig.ac.jp/DRASearch/submission?acc=DRA008023