

Abstract
Differentiation of naïve peripheral B cells into terminally differentiated plasma cells is characterized by epigenetic alterations, yet the epigenetic mechanisms that control B‐cell fate remain unclear. Here, we identified a role for the histone H3K79 methyltransferase DOT1L in controlling B‐cell differentiation. Mouse B cells lacking Dot1L failed to establish germinal centers (GC) and normal humoral immune responses in vivo. In vitro, activated B cells in which Dot1L was deleted showed aberrant differentiation and prematurely acquired plasma cell characteristics. Similar results were obtained when DOT1L was chemically inhibited in mature B cells in vitro. Mechanistically, combined epigenomics and transcriptomics analysis revealed that DOT1L promotes expression of a pro‐proliferative, pro‐GC program. In addition, DOT1L indirectly supports the repression of an anti‐proliferative plasma cell differentiation program by maintaining the repression of Polycomb Repressor Complex 2 (PRC2) targets. Our findings show that DOT1L is a key modulator of the core transcriptional and epigenetic landscape in B cells, establishing an epigenetic barrier that warrants B‐cell naivety and GC B‐cell differentiation.
Keywords: B‐cell differentiation, DOT1L, germinal center B cell, plasma cell, PRC2
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology; Post-translational Modifications, Proteolysis & Proteomics
The histone H3K79 methyltransferase DOT1L plays a central role in B cell development and differentiation. DOT1L maintains B cells naivety by orchestrating critical transcriptional and epigenetic regulators.

Introduction
B lymphocytes are key cellular components of the adaptive immune system, and their functional deregulation is associated with immune deficiencies and autoimmunity (Rajewsky, 1996; LeBien & Tedder, 2008). Several well‐coordinated processes during B‐cell differentiation coincide with specific adaptations of the epigenome (Parra, 2009; Busslinger & Tarakhovsky, 2014; Bao & Cao, 2016). Given the critical contribution of B cells to the immune system, it is important to understand the molecular mechanisms underlying the epigenetic programming during their differentiation.
We previously identified DOT1L as a conserved epigenetic writer that catalyzes mono‐, di‐, or tri‐methylation of lysine 79 of histone H3 (H3K79me) (van Leeuwen et al, 2002; Frederiks et al, 2008). DOT1L‐mediated H3K79me resides on the nucleosome core surface away from histone tails (Vlaming et al, 2014). H3K79me is associated with active transcription, but its function in gene regulation remains unclear (Steger et al, 2008; Vlaming & van Leeuwen, 2016; Wood et al, 2018). DOT1L has gained wide attention as a specific drug target in the treatment of Mixed Lineage Leukemia (MLL) characterized by rearrangements of the MLL gene. Oncogenic MLL‐fusion proteins recruit DOT1L, leading to hypermethylation of H3K79 and increased expression of MLL‐target genes, thereby introducing a druggable dependency on DOT1L activity (Okada et al, 2006; Bernt et al, 2011; Daigle et al, 2011; McLean et al, 2014; Chen et al, 2015; Wang et al, 2016; Stein et al, 2018). In addition, we recently observed a similar dependency in a mouse model of thymic lymphoma caused by loss of the histone deacetylase HDAC1 (Vlaming et al, 2019). While DOT1L is emerging as a drug target in leukemia and lymphoma, the role of DOT1L in gene regulation during normal B lymphocyte development and differentiation is not known. Analysis of publicly available RNA‐sequencing data shows that Dot1L expression is regulated during B‐cell development (see below).
An active humoral immune response is characterized by the activation of clonally selected, antigen‐primed B cells within secondary lymphoid organs. This results in the formation of a specific micro‐environment, known as the germinal center (GC) (MacLennan, 1994, 2005). Rapidly proliferating GC B cells pass through the process of somatic hypermutation that lays the molecular basis of antibody affinity maturation (Odegard & Schatz, 2006; Di Noia & Neuberger, 2007; Calado et al, 2012; Victora & Nussenzweig, 2012; Pilzecker & Jacobs, 2019). Ultimately, B cells selected on the basis of antibody affinity may differentiate either into memory B cells to establish long‐term immunological memory or via a plasma‐blast stage into terminally differentiated, antibody‐secreting plasma cells. Development and functionality of B lymphocytes are associated with dynamic changes in the epigenetic landscape (Martin‐Subero & Oakes, 2018). Recent studies indicate that specific alterations in B‐cell function and identity are intimately linked with well‐established histone modifications such H3K4 tri‐methylation (H3K4me3) related with active gene promoters (Li et al, 2007; Zhang et al, 2015) and H3K27me3 associated with gene repression (Beguelin et al, 2016). Furthermore, the H3K27 methyltransferase EZH2, the catalytic component of the Polycomb repressive complex 2 (PRC2), has been shown to have an essential role in establishing GC B cells (Béguelin et al, 2013).
Here, we determined the role of the H3K79 methyltransferase DOT1L in normal mouse B‐cell development by deleting Dot1L early in the B‐cell lineage and investigating specific dependencies of B‐lineage cells on DOT1L. Our findings, corroborated by in vitro DOT1L inhibition studies, show that DOT1L fine‐tunes the core transcriptional and epigenetic landscape of B cells and in doing so establishes a critical epigenetic barrier coordinating the stepwise transitions toward terminally differentiated plasma cells.
Results
Effective deletion of Dot1L in B‐cell lineage cells
Given the DOT1L dependencies in leukemia (Daigle et al, 2013; Shukla et al, 2016; Wang et al, 2016; Stein et al, 2018) and lymphoma (Vlaming et al, 2019), we quantified the expression of Dot1L during normal B‐cell development using publicly available data (Shi et al, 2015). We observed that Dot1L is transcriptionally regulated in B‐cell subsets and more highly expressed in GC B cells (Fig 1A). To determine the relevance of this regulation in controlling the development and differentiation of B lineage cells, we inactivated Dot1L during early B‐cell development by crossing the Mb1‐Cre knockin allele into a Dot1Lfl/fl background. DOT1L is the sole enzyme responsible for H3K79me; knockout of Dot1L has been shown to lead to complete loss of H3K79me (Jones et al, 2008; Feng et al, 2010; Vlaming & van Leeuwen, 2016; Vlaming et al, 2019). However, loss of H3K79 methylation requires dilution of modified histones by replication‐dependent and replication‐independent mechanisms of histone exchange (De Vos et al, 2011; Radman‐Livaja et al, 2011; Chory et al, 2018). Mb1‐Cre was chosen because it leads to deletion of Dot1L at an early stage in B‐cell development that is followed by successive rounds of replication. This ensures complete loss of H3K79me in all subsequent B‐cell subsets. Dot1L was specifically and efficiently deleted in B cells, as confirmed at the transcript level (Fig EV1A and Appendix Fig S1), as well as by intracellular staining for the DOT1L product H3K79me2 in proB, preB, immature B cells, and mature B cells. While some proB cells retained H3K79me2, preB cells and all stages beyond lacked detectable levels of H3K79me2 (Fig 1B). As a control, the methylation mark remained unchanged in mature T cells from Mb1‐Cre+/− ;Dot1Lfl/fl and Mb1‐Cre+/− ;Dot1L wt/wt mice (Fig 1B). We refer to Mb1‐Cre+/− ;Dot1Lfl/fl and Mb1‐Cre+/− ;Dot1L wt/wt B cells as Dot1L KO and WT cells, respectively. To study the impact of Dot1L ablation on the development of B cells in the bone marrow, we determined the cellularity of specific developmental subsets in the DOT1L‐proficient and DOT1L‐deficient settings. Early ablation of Dot1L resulted in an overall 1.6‐fold reduction of bone marrow B lineage cells. This reduction appeared to be caused primarily by an early differentiation block at the proB to preB cell stage; preB cell were reduced 1.8‐fold in the Dot1L KO as compared to WT and proB cells were increased by 2.0‐fold. In line with this partial developmental inhibition, the cellularity of all subsequent stages of development including immature B and mature B cells was significantly reduced in the bone marrow (Figs 1C and EV1B).
Figure 1. Expression levels, efficient deletion of Dot1L in B cells, and its effect on cellularity of B lineage subsets in the bone marrow.
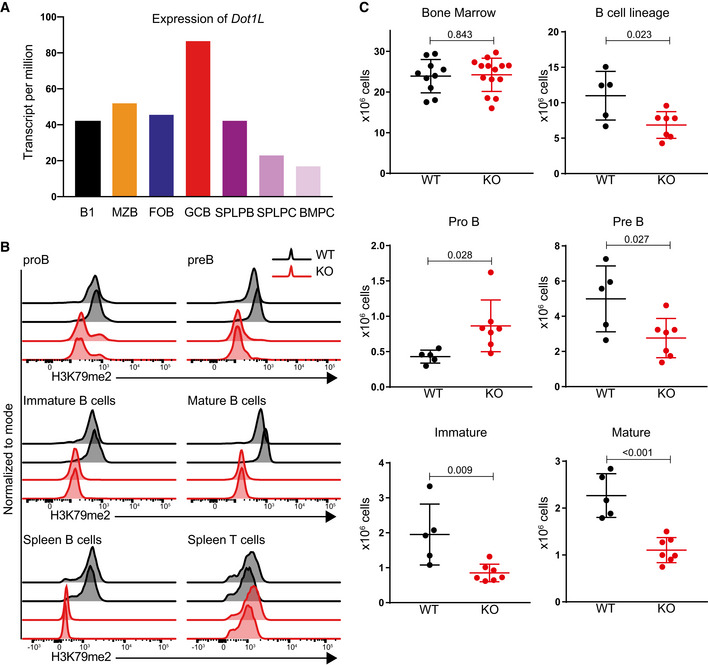
- Expression of Dot1L in different B‐cell populations (Shi et al, 2015). B1: B1 cells, MZB: marginal zone B, FOB: follicular B, GCB: germinal center B, SPLPB: spleen plasma blast, SPLPC: spleen plasma cells, and BMPC: Bone marrow plasma cells. Expression is shown as transcript per million (TPM). Results represent the data from two biological replicates except SPLPB and BMPC which lack replicates (WT; n = 2). Bars indicate mean values.
- Intracellular flow cytometry staining for H3K79me2 in bone marrow B‐cell subsets as well as splenic B and T cells from MB1Cre+/−; Dot1Lwt/wt (WT) and MB1Cre+/−; Dot1Lfl/fl (KO) mice. Results represent the data from two independent experiments.
- Statistical analysis of the absolute number of total nucleated cells from bone marrow B‐cell subsets. Results represent the data pooled from three independent experiments, and numbers represent biological replicates for each group (WT; n = 5, KO; n = 7), except bone marrow cellularity (WT; n = 10, KO; n = 14). Bars and error bars indicate mean ± SD.
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value of less than 0.05 was considered as significant.
Figure EV1. Validation of the experimental system, flow cytometer gating strategy to identify and compare different bone marrow B lineage subsets and ex vivo and in situ analysis of cell viability in WT and KO mice.
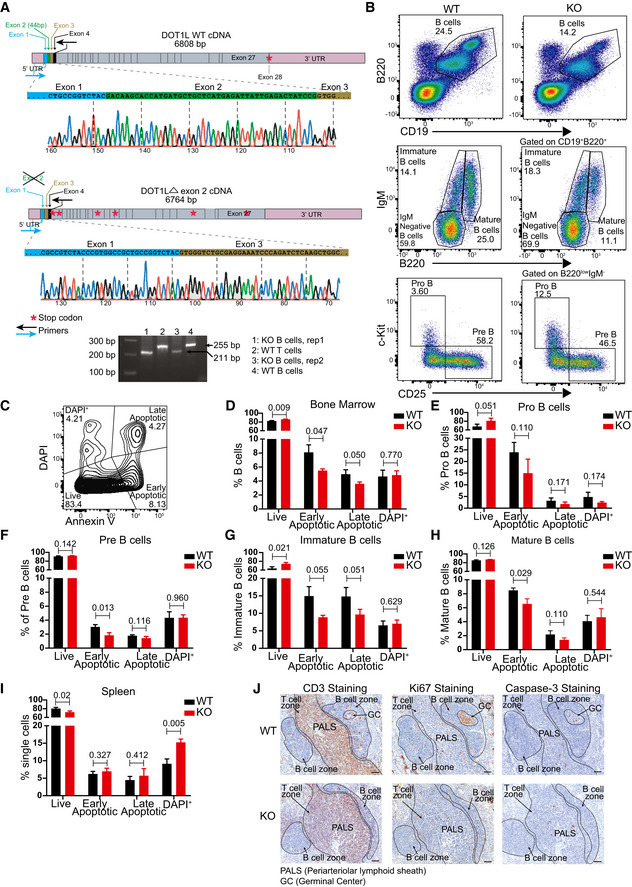
-
ASchematic view of WT and KO (Δ exon 2) cDNA of Dot1L showing UTRs, exons, physiological translational stop codon for WT, multiple translational stop codons (position not to scale) generated after frameshifting induced in KO and location of primers used in RT–PCR to amplify the region flanking exon 2. Chromatograph generated by sanger sequencing showing Cre‐mediated deletion of exon 2 leading to joining of exon 1 to exon 3 in KO. Agarose gel picture showing the deletion of 44 bp corresponding to exon 2 specifically in KO‐B cells. Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 1, KO; n = 2).
-
BRepresentative flow cytometry plots showing gating strategy to identify and compare relative frequency of different B lineage precursor subsets in the bone marrow from WT and KO mice.
-
CRepresentative flow cytometry plots showing gating strategy to identify and compare relative frequency of live, early apoptotic, late apoptotic, and DAPI+ cells in bone marrow.
-
D–HStatistical analysis of the relative frequency of live, early apoptotic, late apoptotic, and DAPI+ cells from the total B lineage and its subsets in the bone marrow from WT and KO mice. Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 3, KO; n = 3).
-
IStatistical analysis of the relative frequency of live, early apoptotic, late apoptotic and DAPI+ cells from mature spleen B cells from WT and KO mice. Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 3, KO; n = 3).
-
JImmunohistochemistry staining for CD3, Ki67 and cleaved caspase‐3 on spleen sections from WT and KO mice. Results represent the data from at one experiment, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 4).
Data information: Statistical analysis were performed using Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating P‐value. P‐value less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
The reduction in preB cell numbers and subsequent B‐cell subsets did not relate to a compromised cell viability (Fig EV1, EV2, EV3, EV4, EV5). This suggests that impaired VDJ recombination in the absence of DOT1L‐dependent pro‐recombinogenic H3K79me marks (Xu et al, 2012; Deng et al, 2015) underlies the reduced B‐cell cellularity in the Dot1L‐KO setting. However, other causative factors cannot be excluded. Regardless of this partial developmental block, B cells could mature in the absence of DOT1L and H3K79 methylation, providing a system to study the role of this epigenetic mark in B‐cell differentiation.
Figure EV2. Gating strategy to identify and compare the spleen B cells, different B‐cell subsets, and germinal center B cells from Peyer’s patches of WT and KO conditions.
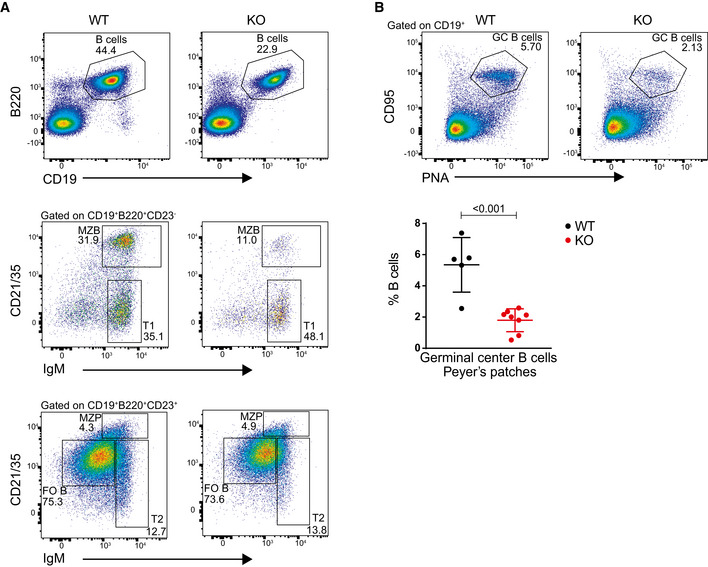
- Representative flow cytometry plots showing gating strategy to identify and compare relative frequency of mature B cells and different B‐cell subsets from the spleen of WT and KO mice.
- Representative flow cytometry plots showing gating strategy to identify and compare germinal center B cells from Peyer’s patches from WT and KO mice (upper panel). Statistical analysis of the percentage of germinal center B cells of total B from Peyer’s patches (lower panel). Results represent the data from three independent experiments, and numbers represent biological replicates for each group (WT; n = 5, KO; n = 8).
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. P‐value less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Figure EV3. Dot1L activity in B cells controls proliferation in response to in vitro T cell‐dependent stimuli, class switch recombination in response to in vitro T‐independent stimuli, and viability in response to both the stimuli.
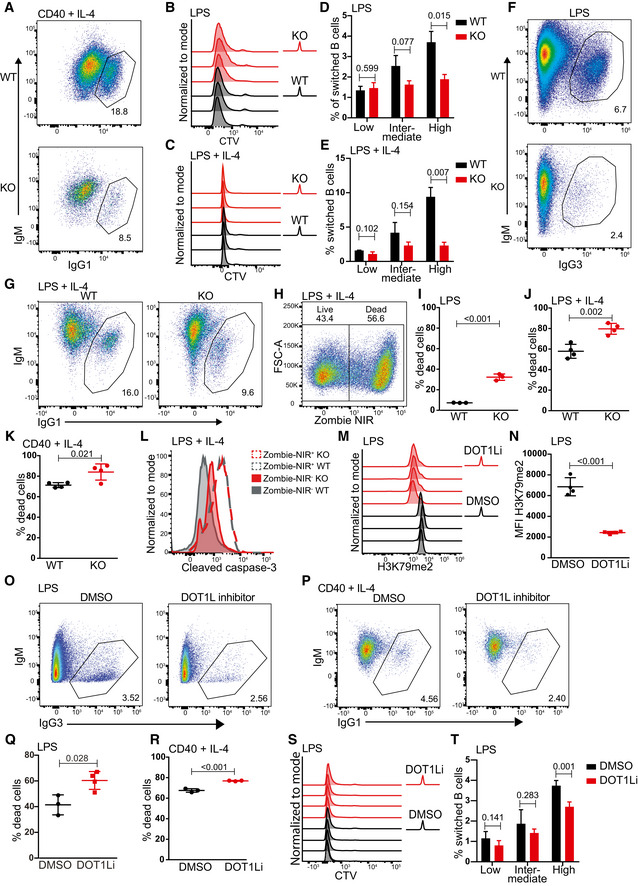
-
ARepresentative flow cytometry plots showing gating strategy to identify and compare relative frequency of IgG1 switched cells from WT and KO mice after 4 days of activation with anti CD40 + IL‐4.
-
B, CNumber of cell divisions traced by CTV dilution of B cells after 4 days of stimulation with LPS (B) or LPS + IL‐4 (C). Data represent three biological replicates for each genotype.
-
D, EStatistical analysis of the percentage of IgG1 switched cells per generation of proliferating B cells after 4 days of stimulation with LPS (D) or LPS + IL‐4 (E). Results represent the data from one experiment and numbers represent biological replicates for each group (WT; n = 3, KO; n = 3).
-
F, GRepresentative flow cytometry plots showing gating strategy to identify and compare relative frequency of IgG1 switched cells from WT and KO mice after 4 days of activation with LPS (F) or LPS + IL‐4 (G).
-
HRepresentative flow cytometry plot from single cells showing gating strategy to identify live (Zombie NIR−) and dead cells (Zombie NIR+) after 4 days of stimulation with LPS + IL‐4.
-
I–KStatistical analysis of dead cells indicated as the percentage of total single cells after 4 days of stimulation with LPS (I), LPS + IL‐4 (J) or anti‐CD40 + IL‐4 (K). Results represent the data from one experiment and numbers represent biological replicates for each group (I: (WT) n = 3, (KO) n = 3; J: (WT) n = 4, (KO) n = 4; K: (WT) n = 4, (KO) n = 4).
-
LRepresentative histograms indicating the levels of cleaved caspase‐3 in live (Zombie NIR−) after 4 days of stimulation with LPS + IL‐4. Cleaved caspase‐3 levels on dead (Zombie NIR+) population shown, as positive control.
-
MIntracellular flow cytometry staining for H3K79me2 in activated B‐cell after 4 days of stimulation with LPS either in the presence of DOT1L inhibitor, Pinometostat or DMSO as a control. Data represent three biological replicates for each treatment.
-
NStatistical analysis of Median Fluorescence Intensity (MFI) of H3K79me after 4 days of stimulation with LPS either in the presence of DOT1L inhibitor, Pinometostat or DMSO as a control is shown. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
-
O, PRepresentative flow cytometry plots showing gating strategy to identify and compare relative frequency of switched cells after 4 days of stimulation of naive B cells with LPS (O) or LPS + IL‐4 (P) either in the presence of DOT1L inhibitor, Pinometostat, or DMSO as a control.
-
Q, RStatistical analysis of dead cells indicated as the percentage of total single cells after 4 days of stimulation with LPS (Q) or LPS + IL‐4 (R) either in the presence of DOT1L inhibitor, Pinometostat, or DMSO as a control. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
-
SNumber of cell divisions traced by CTV dilution of B cells after 4 days of stimulation with LPS either in the presence of the DOT1L inhibitor Pinometostat or DMSO as a control. Data represent four biological replicates for each treatment.
-
TStatistical analysis of the percentage of IgG3 switched cells per generation of proliferating B cells after 4 days of stimulation with LPS either in the presence of the DOT1L inhibitor Pinometostat or DMSO as a control. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
Data information: Statistical analyses were performed using Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating P‐value. P‐value less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Figure EV4. Dot1L‐deficient B cells show reduced serum Ig titers and aberrant plasma cells formation.
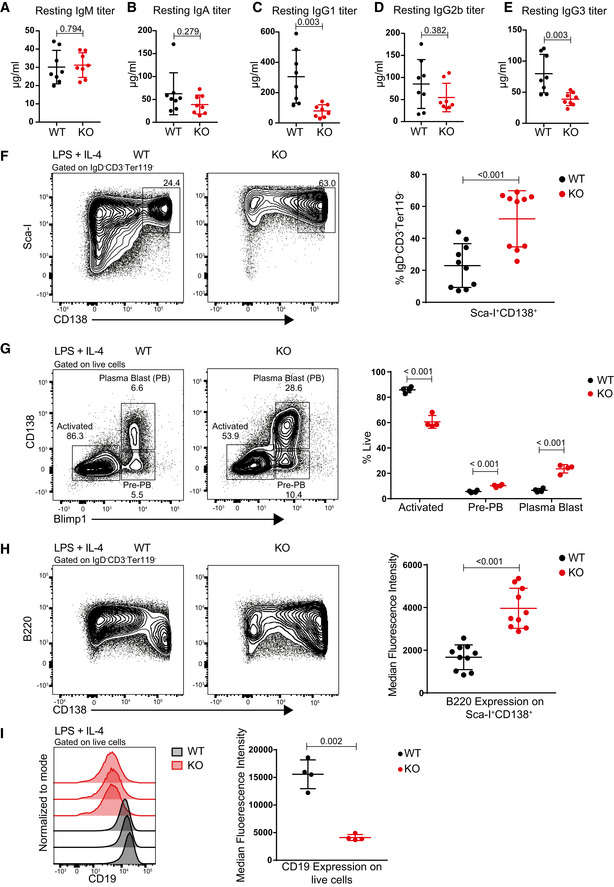
-
A–EStatistical analysis of resting serum titers of IgM (A), IgA (B), IgG1 (C), IgG2b (D), and IgG3 (E) from WT and KO mice. P‐value for IgA (B) was calculated by Mann–Whitney test. Results represent the data from two independent experiments and numbers represent biological replicates for each group (WT; n = 8, KO; n = 8).
-
FRepresentative flow cytometry plots showing gating strategy to identify and compare plasma cells (Sca‐I+CD138+) between WT and KO cells after 4 days of stimulation with LPS + IL‐4 (left panel) and statistical analyses of their relative numbers (right panel). Results represent the data from three independent experiments, and numbers represent biological replicates for each group (WT; n = 10, KO; n = 10).
-
GRepresentative flow cytometry plots showing gating strategy to identify and compare activated B cells (CD138+ Blimp1+), Pre‐PB cells (CD138− Blimp1+) and plasma blast (PB) cells between WT and KO cells after 4 days of stimulation with LPS + IL‐4 (left panel) and statistical analyses of their relative numbers (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 4).
-
HRepresentative flow cytometry plots showing the relative surface density for B220 (CD45R) of WT and KO cells after 4 days of stimulation with LPS + IL‐4 (left panel) and statistical analysis of MFI of B220 for each group (right panel). Results represent the data from three independent experiments, and numbers represent biological replicates for each group (WT; n = 10, KO; n = 10).
-
IRepresentative histograms showing the relative surface density for CD19 on total live WT and KO cells after 4 days of stimulation with LPS + IL‐4 (left panel) and statistical analysis of MFI of CD19 for each group (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 4).
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Figure EV5. Dot1L ablation in B cells results in transcriptional biasness, premature acquisition of plasma cell markers in naïve B cells, and represents transcriptional resemblance with Ezh2‐KO plasma cells.
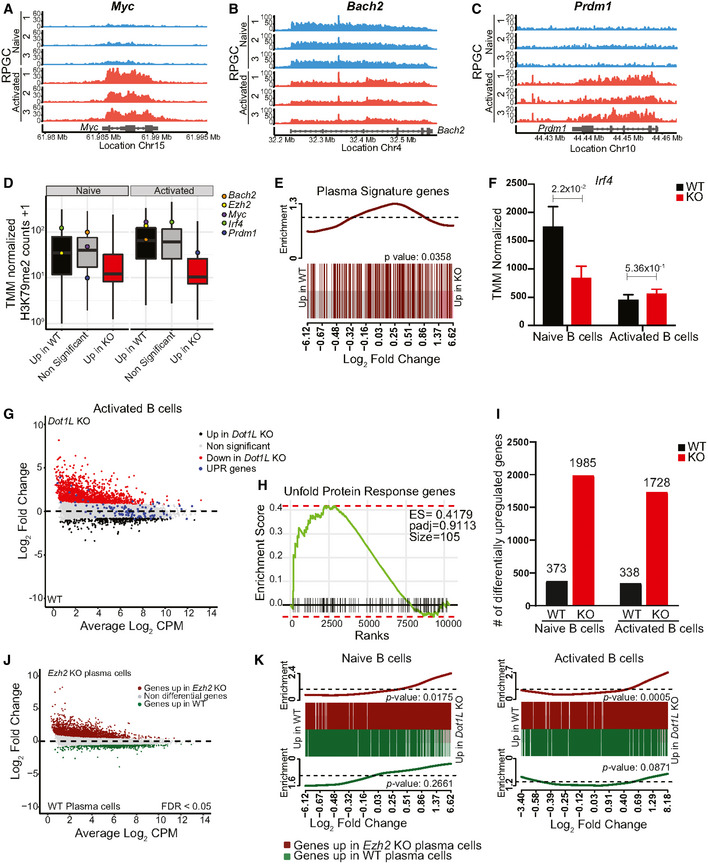
-
A–CH3K79me2 methylation at the Myc (A), Bach2 (B), and Prdm1 (C) loci from WT activated and naïve B cells, as determined by ChIP‐seq reads per genomic content (RPGC). Data represent three independent biological replicates for each condition.
-
DBox plot showing the distribution of mean WT H3K79me2 level for Bach2, Ezh2, Myc, Irf4, and Prdm1 among different indicated gene sets from activated and naïve B cells. Boxes in Box plot indicate Inter quartile range (IQR) and whiskers show 1.5 IQR of highest and lowest quartile. Central horizontal line within the bars represent median of the TMM normalized H3K79me2 counts + 1 values of the respective genes for each condition. Results represent the data generated from three biological replicates for each group.
-
ERelative distribution of plasma cell signature genes between WT and Dot1L‐KO naïve B cells depicted by BARCODE plot. The P‐values calculated via FRY test show the statistical significance of enrichment of each gene set. P‐value less than 0.05 was considered as significant.
-
FDifferential (FDR < 0.05) expression of Irf4 transcripts as indicated by counts per million after TMM normalization from naïve and activated B cells from WT and KO mice. Data were generated from three independent biological replicates for each genotype. Bars and error bars indicate mean ± SD. Statistical significance is indicated by FDR after the Benjamini–Hochberg multiple testing correction performed by edgeR package using R language.
-
GMA‐Plot of normalized RNA‐Seq data generated from three independent biological replicates for each genotype showing relative distribution of Unfold protein response (UPR) genes between WT and Dot1L‐KO Activated B cells.
-
HLack of significant enrichment of UPR gene set between WT and Dot1L‐KO activated B cells shown by Fast gene set enrichment analysis.
-
INumber of differentially (FDR < 0.05) expressed genes from naïve and activated WT and Dot1L‐KO B cells. Results represent the data generated from three biological replicates for each group and condition.
- J
-
KRelative distribution of up‐ (dark‐red) and downregulated (green) genes from Ezh2‐KO plasma cells between WT and Dot1L‐KO naïve and activated B cells depicted by BARCODE plot. The P‐values calculated via FRY test show the statistical significance of enrichment of each gene set. A P‐value of less than 0.05 was considered as significant.
Source data are available online for this figure.
Lack of DOT1L prohibits differentiation of germinal center B cells
In the spleen of Mb1‐Cre+/− ;Dot1Lfl/fl mice, B‐cell cellularity decreased 3.5‐fold while T‐cell numbers remained unaffected, resulting in a 2.0‐fold decreased overall cellularity (Fig 2A and B). Similar to the results from the bone marrow B‐cell subsets, the frequency of late apoptotic cells as measured by annexin V and DAPI staining of spleen B cells (ex vivo) remained unaltered in the absence of DOT1L (Fig EV1I). This was further corroborated in situ by immunohistochemistry for the apoptosis marker cleaved caspase‐3 (Fig EV1J). Among the various peripheral B‐cell subsets, the strongest reduction was found in the number of marginal zone B cells and GC B cells to the extent that they were nearly absent (Figs 2C–H and EV2A). The reduction of GC B cells in Dot1L KO was of particular interest given the highest expression of Dot1L mRNA in this subset (Figs 1A and 2H), suggesting that the formation of germinal centers critically depends on DOT1L. Indeed, in situ histological analyses also revealed the absence of GCs in the spleen of Dot1L‐KO mice (Fig 2I and J). Similarly, in Peyer’s patches, lack of DOT1L resulted in a marked reduction of GC B cells (Fig EV2B).
Figure 2. Dot1L ablation affects the cellularity of mature peripheral B‐cell populations with a strong reduction in Marginal Zone and Germinal Center B cells.
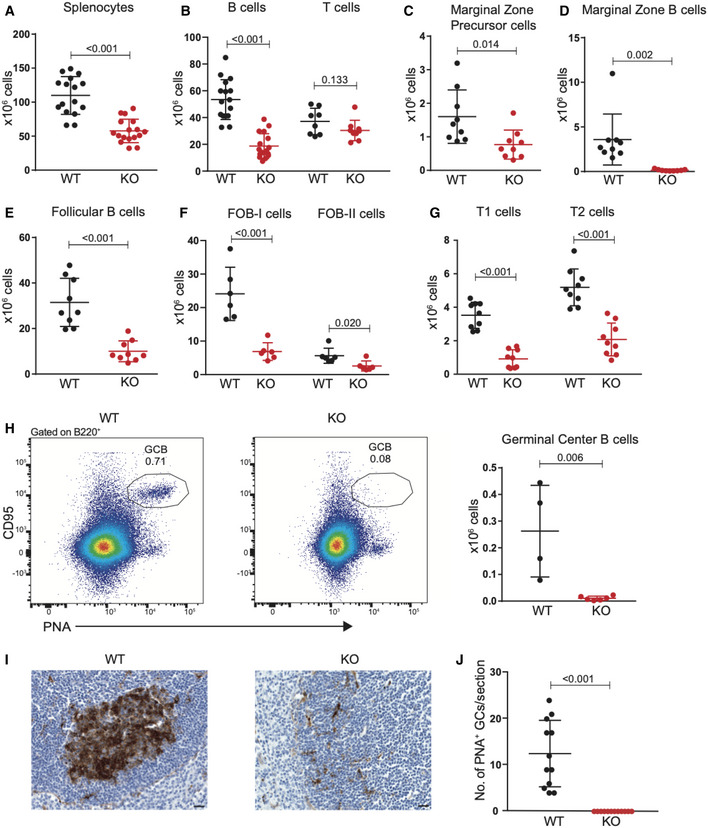
-
A–GStatistical analysis of absolute number of total nucleated splenocytes, splenic B‐ and T‐cells, and indicated mature B‐cell subsets in WT and KO mice. Results represent the data pooled from at least two independent experiments and numbers represent biological replicates for each group (A: (WT) n = 16, (KO) n = 17; B (B‐cells): (WT) n = 16, (KO) n = 17; B (T‐cells): (WT) n = 8, (KO) n = 9; C–E: (WT) n = 9, (KO) n = 9; F: (WT) n = 6, (KO) n = 6; G: (WT) n = 9, (KO) n = 9).
-
HRepresentative flow cytometry plots showing gating strategy (left panel) to identify germinal center B cells (PNAhigh, CD95+) from the spleen of unchallenged WT and KO mice and statistical analysis of their absolute number (right panel). Results represent the data pooled from two independent experiments, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 6).
-
IIdentification of germinal centers by lectin histochemistry of Peanut agglutinin (PNA) in spleens from WT and KO mice. The scale bar: 20 μm.
-
JQuantification of germinal centers identified in spleen sections as shown in (I). Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 6, KO; n = 6).
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value of less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Dot1L deletion impairs proliferation in response to T cell‐dependent stimuli and reduces class switch recombination in response to T cell‐independent stimuli in vitro
Following B‐cell priming, B cells undergo class switch recombination (CSR) and form GCs. CSR is an important feature of humoral immunity and is linked to B‐cell activation and initiation of proliferation. Given the critical role of DOT1L in GC B‐cell differentiation, we here examined the CSR potential and proliferative capacity of naïve splenic B cells stimulated in vitro. Upon the T cell‐dependent stimulus mimetic anti‐CD40 + IL‐4, the lack of Dot1L was found associated with a reduced frequency of class‐switched cells (Figs 3A and EV3A). The reduction in CSR might relate to a defect in the CSR machinery or impaired proliferation. Upon anti‐CD40 + IL‐4 stimulation, the proliferative response of Dot1L‐KO B cells was strongly impaired, as revealed by tracing the dilution of a fluorescent label (Fig 3B). Further analysis based on frequency of switched cells (IgG1+) among different generations of cell divisions revealed that apparently, the overall reduction in frequency of switched cells in KO as compared to WT B cells was associated with impaired proliferation rather than a defect in CSR (Fig 3C). The reduced proliferation in response to T‐dependent stimulation suggested that Dot1L‐KO B cells are compromised in mounting T cell‐dependent immune responses, which is in line with the lack of GCs in Dot1L‐KO mice.
Figure 3. Class switch recombination and proliferative potential of Dot1L‐deficient B cells in response to T cell‐dependent and T cell‐independent stimuli in vitro .
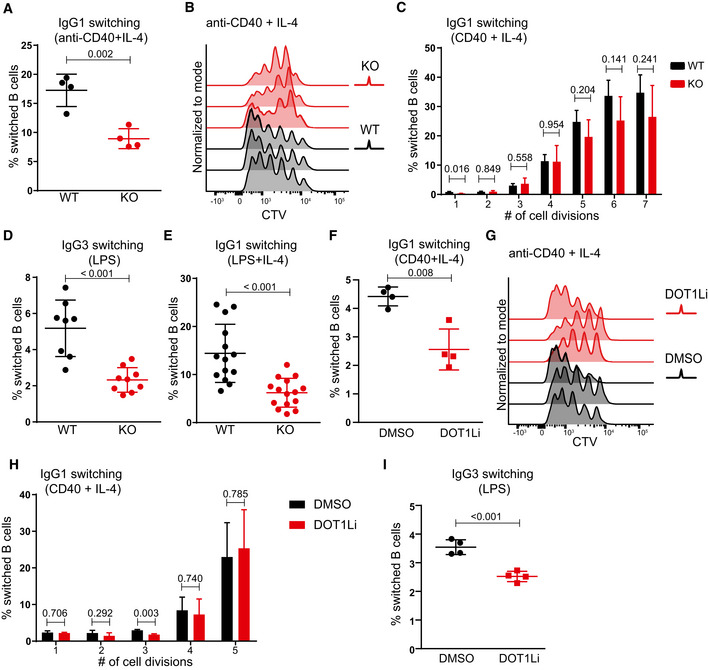
-
AStatistical analysis of IgG1 switching of Dot1L‐proficient (WT) and Dot1L‐deficient (KO) B cells after 4 days of activation with anti‐CD40 + IL‐4. Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 4).
-
BNumber of cell divisions traced by CTV dilution of B cells stimulated for 4 days with anti‐CD40 + IL‐4. Data represent three biological replicates for each genotype.
-
CStatistical analysis of the percentage of IgG1 switched cells per generation of proliferating WT and KO B cells after 4 days of stimulation with anti‐CD40 + IL‐4. Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 4, KO; n = 4).
-
D, EStatistical analysis of switching of WT and KO naïve B cells activated for 4 days with LPS alone (IgG3 switching) (D) or with LPS + IL‐4 (IgG1 switching) (E). Results represent the data pooled from at least four independent experiments, and numbers represent biological replicates for each group (D: (WT) n = 8, (KO) n = 9; E: (WT) n = 11, (KO) n = 13).
-
FStatistical analysis of IgG1 switching of naïve B cells after 4 days of activation with anti‐CD40 + IL‐4 in the presence of the DOT1L inhibitor Pinometostat or DMSO as a control. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
-
GNumber of cell divisions traced by CTV dilution of B cells stimulated for 4 days with anti‐CD40 + IL‐4 either in the presence of DOT1L inhibitor Pinometostat or DMSO as a control. Data represents three biological replicates for each treatment.
-
HStatistical analysis of the percentage of IgG1 switched cells per generation of proliferating WT B cells after 4 days of stimulation with anti‐CD40 + IL‐4 either in the presence of DOT1L inhibitor Pinometostat or DMSO as a control. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 3).
-
IStatistical analysis of IgG3 switching of naïve B cells after 4 days of activation with LPS alone in the presence of the DOT1L inhibitor Pinometostat or DMSO as a control. Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value of less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Unlike stimulation with anti‐CD40 + IL‐4, mimicking T cell‐dependent B‐cell activation, the proliferative capacity of mature B cells was found indistinguishable in response to T cell‐independent activation stimuli using the strong B‐cell mitogen lipopolysaccharide (LPS) (Dziarski, 1982) or LPS + IL‐4. These findings indicate that Dot1L‐KO B cells are not intrinsically impaired in their proliferative potential (Fig EV3B and C). However, despite their proficiency in proliferation, the frequency of switched cells in Dot1L‐KO B cells was found reduced in response to both LPS alone (inducing a switch to IgG3) or LPS + IL‐4 (inducing a switch to IgG1) (Figs 3D and E, and EV3, EV4, EV5). The underlying mechanism by which DOT1L might affect CSR under these conditions of strong activation warrants further investigation. RNA‐Seq analysis of KO and WT B cells activated with LPS + IL‐4 did not show defects in the mRNA expression of the known trans elements involved in CSR in Dot1L‐KO B cells, (Appendix Table S1). However, under LPS conditions, DOT1L may affect these factors by unknown post‐transcriptional mechanisms or by altering cis acting features associated with CSR, like switch region accessibility. Alternatively, the switching cells may be prone to die in the absence of DOT1L, as DOT1L has been associated with DNA damage responses and induction of apoptosis (Huyen et al, 2004; Nguyen & Zhang, 2011; Kari et al, 2019; Bian et al, 2020). In vitro activation of naïve B cells with anti‐CD40 + IL‐4, LPS, and LPS + IL‐4 led to a higher proportion of dead cells (Zombie NIR positive) in Dot1 L‐KO compared to WT (Fig EV3, EV4, EV5). In addition, the viable (Zombie NIR negative) subset in activated (LPS + IL‐4 mediated) KO B cells consistently showed higher levels of cleaved caspase‐3, a marker of apoptosis (Fig EV3L). Apparently, upon in vitro activation the viability of Dot1L‐deficient B cells is compromised.
In vitro inhibition of DOT1L activity in mature B‐cell phenocopies early Dot1L deletion
The pleiotropic changes associated with Dot1L ablation in developing B cells led us to investigate whether the effects observed in mature Dot1L‐KO B cells in vitro could be recapitulated by inhibiting the methyltransferase activity of DOT1L. For this purpose, we employed the highly specific DOT1L inhibitor Pinometostat during T cell‐dependent and T cell‐independent activation of mature splenic B cells isolated from wild‐type mice. Treatment with Pinometostat closely mimicked our observations regarding B‐cell activation, CSR, proliferation, and viability in Dot1L‐KO mice (Figs 3F–I and EV3, EV4, EV5). These data highlight the relevance of DOT1L activity in controlling normal B‐cell differentiation and proliferation induced by T‐dependent stimuli in vitro. Importantly, these insights exclude potential indirect effects associated with the early ablation of Dot1L during B‐cell ontogeny.
Dot1L‐deficient B cells fail to mount efficient immune responses in vivo and acquire plasma cell features in vitro
The virtual absence of GC B cells, the impaired proliferative response to T cell‐dependent stimulation, and the decreased viability of Dot1L‐deficient B cells upon activation implicated a severe defect of Mb1‐Cre+/− ;Dot1Lfl/fl mice in establishing effective humoral immunity. In unchallenged Dot1L‐KO mice, serum IgM titers appeared normal but IgG1 and IgG3 titers were decreased, while IgG2b and IgA titers were not significantly affected (Fig EV4, EV5). To determine the immune responsiveness of Dot1L‐deficient B cells, Mb1‐Cre+/− ;Dot1Lfl/fl and Mb1‐Cre+/− ;Dot1Lwt/wt mice were challenged with an acute lymphocytic choriomeningitis virus (LCMV) infection. During the LCMV infection, Dot1L‐deficient B cells failed to establish GC B cells and generate plasma cells (Fig 4A and B). In accordance, these mice also failed to mount normal IgM serum titers against LCMV (Fig 4C). The failure to establish GCs in response to LCMV is also in line with the very low LCMV‐specific IgG serum titers (Fig 4C). The inability of Mb1‐Cre+/− ;Dot1Lfl/fl mice to mount efficient antibody responses to T cell‐dependent antigen was confirmed using 4‐hydroxy‐3‐nitrophenylacetyl conjugated to chicken gamma globulin (NP‐CGG) in alum as immunogen (Fig 4D). Together, these data suggest that DOT1L is essential for establishing a normal humoral immune response. Upon in vitro activation of naïve Dot1L‐KO B cells with LPS + IL‐4, a significantly increased frequency of cells expressing the plasma cell marker CD138 (Sanderson et al, 1989; McCarron et al, 2017) was observed (Fig EV4F). This observation was supported by the increased proportion of cells expressing the pan‐plasma cell transcription factor BLIMP1 (Tellier et al, 2016; Fig EV4G) in Dot1L‐KO versus WT. However, these cells failed to downregulate B220, also known as CD45R (Fig EV4H). Downregulation of B220 is considered as a hallmark of post‐mitotic plasma cells (Kallies et al, 2004). In addition, in vitro‐activated Dot1L‐KO B cells failed to differentiate into CD19 high‐activated B‐cell blasts or CD19‐negative plasma cells, but instead they remained at a transitional state, expressing intermediate levels of CD19 on cell surface (Fig EV4I). Of note, these results were reproduced using the DOT1L inhibitor Pinometostat during in vitro stimulation of naïve WT B cells with LPS (Fig 5A–D). Together, these phenotypes observed upon in vitro activation suggest that in the absence of DOT1L‐mediated H3K79 methylation, naïve B cells prematurely gain some plasma cell features which may form the basis of skipping the GC stage. However, these cells do not accomplish a complete plasma cell differentiation program.
Figure 4. DOT1L‐deficient B cells fail to mount an efficient immune response.
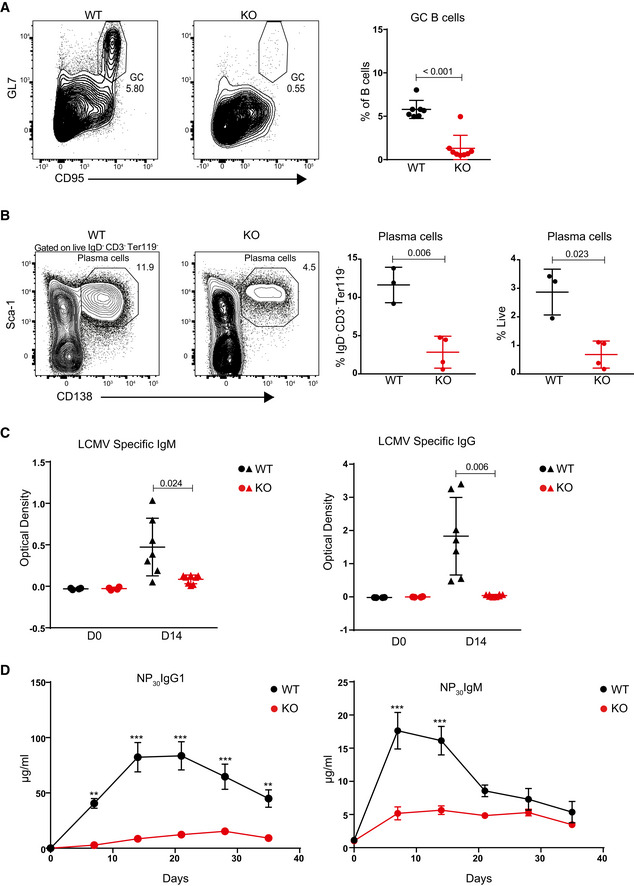
- Representative flow cytometry plots showing gating strategy to identify splenic GC B cells 14 days after LCMV Armstrong infection in WT and Dot1L‐KO mice (left panel) and statistical analysis of their relative number. Results represent the data from two experiments, and numbers represent biological replicates for each group (WT; n = 7, KO; n = 8).
- Representative flow cytometry plots showing gating strategy to identify plasma cells from the spleen 14 days after LCMV Armstrong infection (left panel) and statistical analysis of their relative number (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 3, KO; n = 4).
- Statistical analysis of serum titers of LCMV‐specific IgM and IgG of WT and KO mice before (D0) and 14 days after (D14) LCMV Armstrong infection. Results represent the data from two experiments, and numbers represent biological replicates for each group (WT; n = 7, KO; n = 8).
- Statistical analysis of serum titers of NP‐specific IgG1 and IgM quantified by ELISA from WT and KO mice at the indicated days following immune challenge. Adjusted P‐value were calculated using two‐way ANOVA. Error bars indicate mean ± standard error of mean (SEM). Results represent the data from one experiment, and numbers represent biological replicates for each group (WT; n = 6, KO; n = 6).***P‐value < 0.0001 and **P‐value < 0.001.
Data information: Except for Fig 4D, statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value of less than 0.05 was considered as significant. Bars and error bars except Fig 4D indicate mean ± SD.
Figure 5. Chemical inhibition of DOT1L results in aberrant plasma cell differentiation in response to in vitro stimulation.
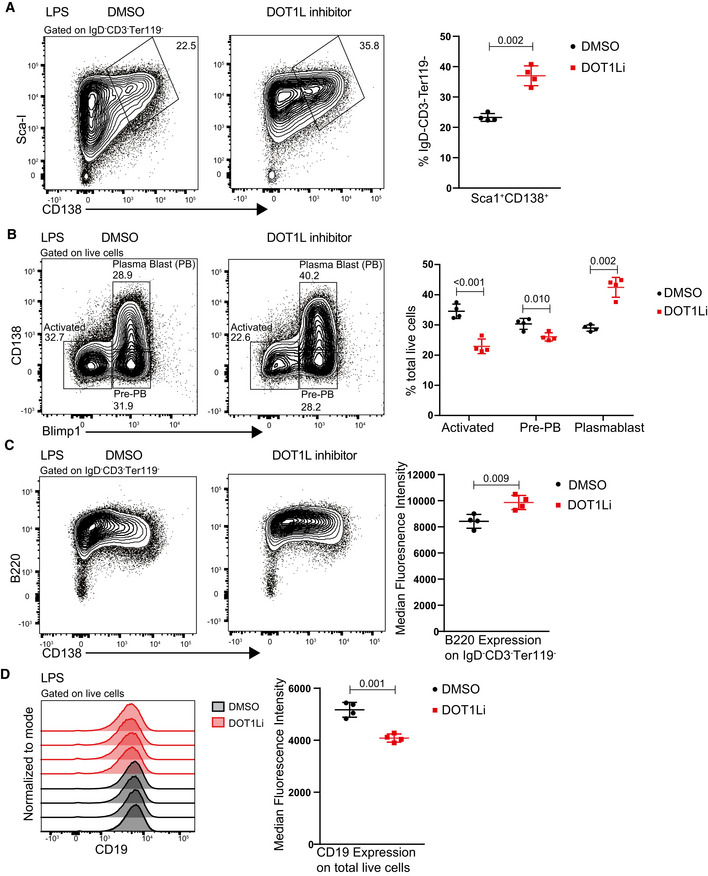
- Representative flow cytometry plots showing gating strategy to identify and compare plasma cells (Sca‐I+CD138+) after 4 days of stimulation with LPS either in the presence of DOT1L inhibitor, Pinometostat, or DMSO as a control (left panel) and statistical analyses of their relative numbers (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
- Representative flow cytometry plots showing gating strategy to identify and compare activated B cells (CD138+ Blimp1+), Pre‐PB cells (CD138− Blimp1+) and plasma blast (PB) cells after 4 days of stimulation with LPS either in the presence of DOT1L inhibitor, Pinometostat, or DMSO as a control (left panel) and statistical analyses of their relative numbers (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
- Representative flow cytometry plots showing the relative surface density for B220 (CD45R) after 4 days of stimulation with LPS either in the presence of DOT1L inhibitor, Pinometostat or DMSO as a control (left panel) and statistical analysis of MFI of B220 for each treatment (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
- Representative histograms showing the relative surface density for CD19 on total live cells after 4 days of stimulation with LPS (left panel) and statistical analysis of MFI of CD19 for each treatment (right panel). Results represent the data from one experiment, and numbers represent biological replicates for each treatment (n = 4).
Data information: Statistical analyses were performed using the Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating the P‐value. A P‐value less than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
DOT1L supports a pro‐proliferative, MYC‐high GC stage and prohibits premature differentiation toward plasma cells in vitro
To unravel the underlying molecular mechanisms that prohibit Dot1L‐KO GC B‐cell differentiation and stimulate partial differentiation toward plasma cells, we performed RNA‐Seq analyses of in vitro‐activated B cells under Dot1L‐proficient and Dot1L‐deficient conditions (Fig 6A). To avoid confounding issues associated with impaired replication upon in vitro stimulation with anti‐CD40 + IL‐4, naïve B cells were stimulated in vitro with LPS + IL‐4 as an activating condition that facilitates replication of both WT and KO B cells. Among the differentially expressed genes, the genes encoding the pro‐GC transcription factor BACH2 and the pro‐proliferative transcription factor MYC (Muto et al, 2004; Muto et al, 2010; Calado et al, 2012) were H3K79me2 methylated in WT cells (see below) and transcriptionally downregulated in activated DOT1L‐deficient B cells (Fig EV5A, B and D). In agreement with this, Prdm1, repressed by BACH2 and encoding the pan‐plasma cell transcription factor BLIMP1 (Martins & Calame, 2008; Tellier et al, 2016), was found upregulated (Figs 6B and EV5C). These transcriptional changes are in line with the increased formation of cells with plasma cell features in KO cells. In addition, analysis of a published plasma cell gene signature (Shi et al, 2015) revealed that the transcriptome of activated Dot1L‐deficient B cells was indeed strongly enriched for plasma cell‐associated transcripts (Fig 6C). Simultaneously, MYC‐target gene transcripts were strongly reduced (Fig 6D). Together, these observations indicate that DOT1L‐mediated H3K79 methylation licenses a transient entrance into a pro‐proliferative, MYC‐high GC stage and is required to prevent premature differentiation of activated B cells toward non‐proliferative terminally differentiated plasma cells. Importantly, transcriptomic data from ex vivo isolated naïve Dot1L‐deficient B cells also revealed enrichment of some plasma cell‐associated genes, suggesting that antigen‐inexperienced B cells are prematurely differentiated to some extent in the absence of DOT1L (Fig EV5E). However, upon activation, Irf4, a factor essential for plasma cell differentiation (Klein et al, 2006), was not differentially expressed between WT and KO (Fig EV5F). This dichotomy, among other factors, likely contributes to the failure to adopt an identity of terminally differentiated plasma cells. Plasma cells strongly depend on the endoplasmic reticulum (ER) stress pathway (Gass et al, 2004), and recently, DOT1L has been connected to the expression of unfolded protein response (UPR) genes involved in this pathway (Dafflon et al, 2020). However, our data from activated B cells revealed that the expression of most UPR genes remained unchanged in the absence of DOT1L. This suggests that at least from the transcriptional point of view an altered ER stress response is not the cause of the aberrant differentiation in the absence of DOT1L (Fig EV5G and H, Appendix Table S2). Together, our results indicate that DOT1L modulates the expression of several key transcriptional regulators essential in controlling stepwise B‐cell differentiation.
Figure 6. Transcriptome analysis of in vitro‐activated B cells shows accelerated plasma cell differentiation and compromised activation of MYC‐target genes in the absence of DOT1L.
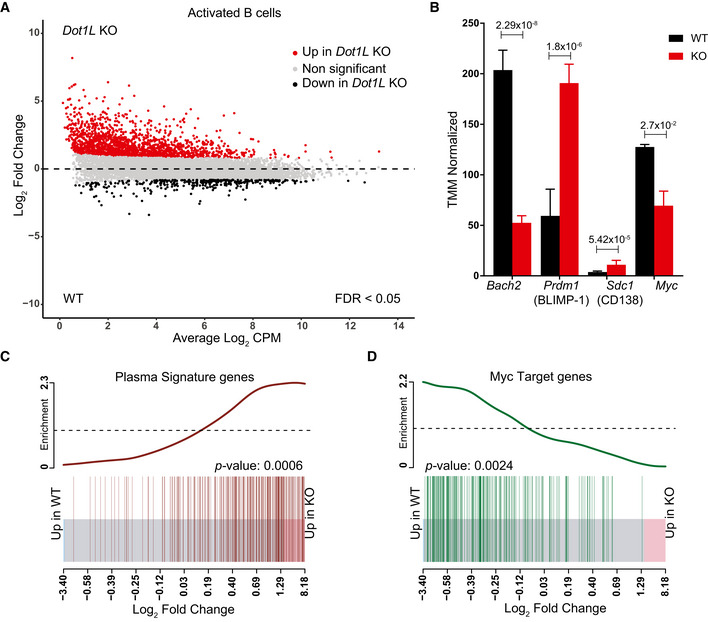
-
AMA‐Plot of normalized RNA‐Seq data generated from three independent biological replicates for each genotype showing differential (false discovery rate (FDR) < 0.05) expression of genes between Dot1L‐KO and WT B cells after 2 days of in vitro activation with LPS + IL‐4.
-
BDifferential (FDR < 0.05) expression of Bach2, Prdm1 (encoding BLIMP‐1), Cd138 (a plasma cell marker), and Myc transcripts as indicated by counts per million after TMM normalization from WT and KO. Data were generated from three independent biological replicates for each genotype. Bars and error bars indicate mean ± SD. Statistical significance is indicated by FDR after the Benjamini–Hochberg multiple testing correction performed by edgeR package using R language.
-
C, DEnrichment of plasma cell signature genes in KO as compared to WT‐activated B cells (C) and Enrichment of MYC‐target genes in WT as compared to KO‐activated B cells (D). Enrichment of gene sets is depicted by a BARCODE plot; P‐value calculated via FRY test show the statistical significance of enrichment of each gene set. A P‐value less than 0.05 was considered as significant.
Source data are available online for this figure.
DOT1L‐mediated H3K79 methylation is associated with gene activity in B cells
To link the phenotypes of loss of DOT1L to its role as an epigenetic regulator in B cells, we generated genome‐wide maps of DOT1L‐mediated H3K79me2 in naïve and activated WT B cells by ChIP‐Seq. H3K79me2 is known to mark the region downstream of the transcription start site of transcribed genes and positively correlate with gene activity (Steger et al, 2008; Huff et al, 2010; Vlaming & van Leeuwen, 2016; Godfrey et al, 2019). While H3K79me1 shows the same trends, it has a broader distribution, and H3K79me3 is detectable only at limited levels (Steger et al, 2008; Huff et al, 2010; Vlaming et al, 2019). Here, we analyzed how the gene expression changes in Dot1L‐KO versus WT B cells related to genes being marked by H3K79me2 in WT cells. Both in naïve and activated B cells more than 83% of the differentially expressed genes was found upregulated in the absence of DOT1L; only a small subset was downregulated (Figs 7A and EV5I). The upregulation was biased toward more lowly expressed genes. The observed upregulation of genes in Dot1L‐KO B cells was unexpected given the fact that H3K79me2 generally correlates with transcriptional activity (Schubeler et al, 2004; Steger et al, 2008; Wang et al, 2008; Bernt et al, 2011; Cano‐Rodriguez et al, 2016; Yang et al, 2016; Wood et al, 2018; Godfrey et al, 2019). However, repressive functions of DOT1L have been proposed as well (Zhang et al, 2006a; Zhang et al, 2006b; Cecere et al, 2013; Xiao et al, 2016). Comparing H3K79me2 ChIP values in WT B cells with the gene expression changes caused by loss of DOT1L revealed that the genes upregulated in Dot1L‐KO B cells were mostly hypomethylated in WT cells, indicating that they are likely indirectly affected by the loss of DOT1L (Fig 7A and B). In contrast, genes downregulated in Dot1L‐KO B cells were generally highly expressed and H3K79 methylated in WT B cells, indicating that this gene set harbors the genes directly dependent on DOT1L. This set of candidate direct targets of DOT1L includes the previously mentioned pro‐proliferative factor Myc, the pro‐GC transcription factor Bach2, and Prdm1, a target of BACH2 (Fig EV5A–D). These findings show that DOT1L‐mediated H3K79me2 is a mark of many active genes in B cells, but suggest that only a small fraction of these genes requires H3K79me2 for maintenance of gene expression, since only a subset of the active genes was downregulated in Dot1L‐KO B cells. Similar observations in transcriptome changes were also made in CD8+ T cells lacking DOT1L as well as upon DOT1L inhibition in mouse ES cells and during human cellular reprogramming (Kwesi‐Maliepaard et al, 2020; Ferrari et al, 2020; Kim et al, 2021).
Figure 7. DOT1L‐mediated H3K79 methylation is associated with gene activity in B cells and indirectly promotes repression of PRC2 target genes.
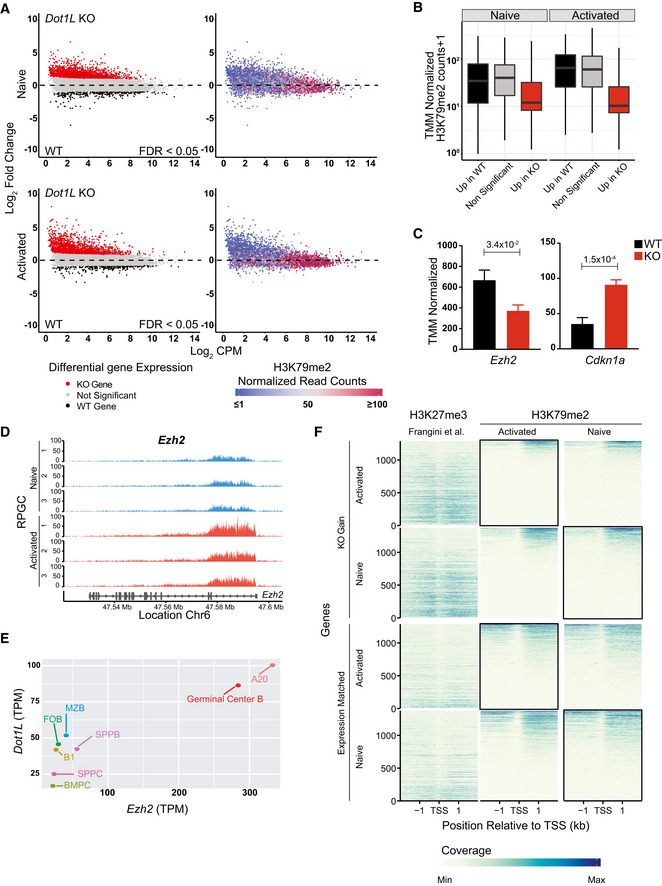
- Integrative analyses of differentially (FDR < 0.05) expressed transcripts from naïve and activated Dot1L‐deficient B cells (left panel) with H3K79me2 ChIP values from WT naïve and activated B cells (right panel).
- The distribution of mean H3K79me2 among different gene sets (6A, left panel) from activated and naïve B cells depicted by box plots. Boxes in Box plot indicate Inter quartile range (IQR) and whiskers show 1.5 IQR of highest and lowest quartile. Central horizontal line within the bars represent median of the TMM normalized H3K79me2 counts + 1 values of the respective genes for each condition. Results represent the data generated from three biological replicates for each group.
- Differential (FDR < 0.05) expression of Ezh2 and Cdkn1a as indicated by counts per million after TMM normalization from WT and Dot1L‐KO‐activated B cells. Data were generated from three independent biological replicates for each genotype. Bars and error bars indicate mean ± SD. Statistical significance is indicated by FDR after the Benjamini–Hochberg multiple testing correction performed by edgeR package using R language.
- H3K79me2 methylation at the Ezh2 locus from WT activated and naïve B cells, as determined by reads per genomic content (RPGC). Data represent three independent biological replicates.
- Correlation between expression of Ezh2 and Dot1L as depicted by TPM in different mature B‐cell subsets; B1: B1 cells, MZB: marginal Zone B, FOB: follicular B, GCB: germinal Center B, SPLPB: spleen plasma blast, SPLPC: spleen plasma cells) and Bone Marrow (BMPC: Bone marrow plasma cells) A20: Germinal Center like cell lymphoma cell line. Correlation shown by scatter plot.
- Coverage plot of H3K27me3 from naïve B cells (Frangini et al, 2013) and H3K79me2 (from WT‐activated and naïve B cells) flanking 4 kb around transcriptional start sites (TSS) for genes upregulated in KO (KO Gain) or non‐differential Expression‐matched genes. Coverage was calculated as reads per genomic content cutoff at the 0.995th quantile and rescaled to a maximum of 1. Black boxes indicate the relavant B‐cell population for comparing the distribution of H3K27me3 and H3K79me2 for the indicated gene sets obtained from the differential gene expression analysis of Dot1L‐proficient and deficient B cells.
Data information: Statistical analysis were performed using Student’s two‐tailed unpaired t‐test. Statistical significance was determined by calculating P‐value. P‐value lesser than 0.05 was considered as significant. Bars and error bars indicate mean ± SD.
Source data are available online for this figure.
DOT1L supports repression of PRC2 target genes
The large number of genes found upregulated in Dot1L‐KO B cells indicated that DOT1L positively regulates the expression of a transcriptional repressor whose target genes are de‐repressed in the absence of DOT1L. To identify such candidate repressors, we investigated the relatively small fraction of genes downregulated in naïve and activated Dot1L‐KO B cells. Unbiased identification of upstream transcriptional regulators by Ingenuity Pathway analyses (IPA) pointed toward potential regulators that were differentially upregulated in WT B cells (Tables 1 and 2). To further narrow down the list, we filtered for genes that are (i) H3K79‐dimethylated by DOT1L, (ii) encode transcriptional repressors, and (iii) play a role in B‐cell differentiation/proliferation and GC formation. This led to the identification of the histone H3K27 methyltransferase EZH2 as a candidate factor (Tables 1 and 2). We verified that Ezh2 expression was reduced in Dot1L KO B cells (Fig 7C) and that the gene is H3K79‐dimethylated in WT cells (Fig 7D), indicating that the expression of Ezh2 might be directly promoted by DOT1L activity (Fig 7C and D). Further supporting this notion, the expression of Ezh2 and Dot1L was found co‐regulated in B‐cell subsets (Fig 7E).
Table 1.
Ingenuity pathway analysis of upstream transcriptional regulators that are differentially expressed higher in WT than Dot1L knockout naïve B cells (FDR < 0.05).
| Upstream transcriptional regulator | Expression Log ratio (KO/WT) | Activation z‐score | P‐value of overlap | Ensemble gene id | TMM normalized H3K79me2 |
|---|---|---|---|---|---|
| HIF1A a | −1.534 | 5.138 | 3.22E‐10 | ENSMUSG00000021109 | 8.44 |
| EZH2 | −1.195 | 0.27 | 0.000133 | ENSMUSG00000029687 | 33.46 |
| REL | −1.146 | 1.685 | 9.25E‐05 | ENSMUSG00000020275 | 41.44 |
| TAF4B | −1.042 | 0.0396 | ENSMUSG00000054321 | 23.16 | |
| IRF4 | −1.038 | −3.202 | 0.00193 | ENSMUSG00000021356 | 120.62 |
| MED13 | −0.978 | −2.143 | 0.0067 | ENSMUSG00000034297 | 153.69 |
| SIRT1 a | −0.973 | −3.106 | 7.11E‐09 | ENSMUSG00000020063 | 7.74 |
| PURA | −0.964 | 1.067 | 0.00223 | ENSMUSG00000043991 | 47.75 |
| EBF1 | −0.95 | 2.612 | 7.55E‐08 | ENSMUSG00000057098 | 112.63 |
Genes with TMM normalized H3K79me2 score lesser than 10 were not considered.
Table 2.
Ingenuity pathway analysis of upstream transcriptional regulators that are differentially expressed higher in WT than Dot1L knockout activated B cells (LPS + IL‐4, Day 2; FDR < 0.05).
| Upstream transcriptional regulator | Expression Log ratio (KO/WT) | Activation z‐score | P‐value of overlap | Ensemble gene id | TMM normalized H3K79me2 |
|---|---|---|---|---|---|
| BACH2 | −1.956 | −3.367 | 0.00186 | ENSMUSG00000040270 | 66.81 |
| EGR1 a | −1.678 | 3.22 | 6.76E‐15 | ENSMUSG00000038418 | 10.41 |
| TAF4B | −1.436 | 0.00102 | ENSMUSG00000054321 | 53.19 | |
| PURA | −1.392 | 0.00633 | ENSMUSG00000043991 | 49.35 | |
| HIF1A a | −1.269 | 4.754 | 7.83E‐10 | ENSMUSG00000021109 | 6.13 |
| CREBZF | −1.233 | −1.994 | 0.0409 | ENSMUSG00000051451 | 206.73 |
| SMAD2 | −1.178 | 2.439 | 0.0061 | ENSMUSG00000024563 | 94.23 |
| EBF1 | −1.15 | 1.207 | 0.000138 | ENSMUSG00000057098 | 155.78 |
| HHEX | −1.059 | −0.538 | 0.00453 | ENSMUSG00000024986 | 41.60 |
| EGR2 a | −1.018 | 1.566 | 1.16E‐06 | ENSMUSG00000037868 | 8.55 |
| PDLIM1 | −0.967 | 0.0383 | ENSMUSG00000055044 | 16.33 | |
| SATB1 | −0.962 | −1.998 | 4.34E‐07 | ENSMUSG00000023927 | 116.85 |
| CREB1 | −0.949 | 3.751 | 6.80E‐13 | ENSMUSG00000025958 | 92.51 |
| PAX5 | −0.917 | −1.807 | 0.00318 | ENSMUSG00000014030 | 330.58 |
| KLF2 | −0.912 | 1.144 | 1.98E‐15 | ENSMUSG00000055148 | 148.02 |
| NPM1 | −0.881 | −0.509 | 0.00309 | ENSMUSG00000057113 | 177.98 |
| MYC | −0.878 | −3.848 | 2.74E‐14 | ENSMUSG00000022346 | 163.26 |
| REL | −0.866 | 1.473 | 1.58E‐10 | ENSMUSG00000020275 | 102.56 |
| EZH2 | −0.846 | −0.678 | 2.11E‐06 | ENSMUSG00000029687 | 135.73 |
| CBFB a | −0.814 | 0.538 | 2.45E‐05 | ENSMUSG00000031885 | 6.84 |
| NFKB1 | −0.813 | 2.31 | 6.65E‐12 | ENSMUSG00000028163 | 176.38 |
Genes with TMM normalized H3K79me2 score less than 10 were not considered.
We next investigated the physiological relevance of the connection between DOT1L and EZH2. First, Ezh2‐KO and Dot1L‐KO B cells have overlapping phenotypes (Su et al, 2003; Béguelin et al, 2013; Caganova et al, 2013; Beguelin et al, 2017; Guo et al, 2018). Second, we took advantage of publicly available RNA‐Seq data (Guo et al, 2018) from Ezh2‐KO plasma cells to identify genes that require EZH2 for their repression (Fig EV5J). Using this as a signature of EZH2‐dependent genes, we found that many of the genes de‐repressed in Ezh2‐KO cells were also found de‐repressed in activated Dot1L‐KO B cells (Fig EV5K). Many of these genes were found de‐repressed also in naïve Dot1L‐KO B cells (Fig EV5K). As an independent validation, we evaluated the expression of a known PRC2‐target gene Cdkn1a (p21) (Fan et al, 2011; Sato et al, 2013; Beguelin et al, 2017) and found that it was upregulated in activated Dot1L‐KO B cells (Fig 6C). Third, we analyzed the level of H3K27me3 and H3K79me2 in the set of genes that was de‐repressed in Dot1L KO using our H3K79me2 ChIP‐Seq data and publicly available H3K27me3 ChIP‐Seq data from naïve B cells (Frangini et al, 2013). Further analysis revealed that this gene set was enriched for H3K27me3 in WT cells compared to expression‐matched non‐differentially expressed genes (Fig 7F). Lastly, chemical inhibition of both DOT1L (this study) and EZH2 (Scharer et al, 2018) upon in vitro stimulation led to enhanced dysfunctional plasma cell formation. Together, these findings suggest that in B cells, DOT1L supports the repression of PRC2 target genes, thus uncovering a previously unknown connection between two conserved histone methyltransferases associated with activation and repression, respectively. Together, our findings place H3K79 methylation by DOT1L at the heart of maintaining epigenetic identity of B cells, by orchestrating the activity of central transcriptional and epigenetic regulators such as BACH2, MYC, EZH2, and their target genes.
Discussion
Given the critical contribution of B cells to the immune system, it is important to understand the molecular mechanisms underlying the epigenetic programming during their differentiation. Taking advantage of a B cell‐specific mouse knockout model, we observed that the H3K79 methyltransferase DOT1L has a central role in B‐cell physiology. Among mature B cells, GC B cells express the highest levels of Dot1L and they were strongly reduced in Dot1L‐KO mice. Indeed, GC B‐cell differentiation was found to be critically dependent on DOT1L. Upon in vitro activation, Dot1L‐KO B cells failed to proliferate and after an in vivo immune challenge these cells failed to differentiate into GC B cells and establish an effective immune response. In addition, we observed an accelerated but incomplete plasma cell differentiation in Dot1L‐KO B cells in vitro. RNA‐Seq data generated from naïve and in vitro‐activated Dot1L‐KO and WT B cells demonstrated a strong enrichment of genes associated with plasma cell differentiation among the genes upregulated in Dot1L KO. However, in vitro‐activated Dot1L KO B cells failed to downregulate B220, indicating an incomplete differentiation into plasma cells (Kallies et al, 2004), which is in agreement with the failure of these cells to upregulate Irf4, a factor essential for plasma cell differentiation (Klein et al, 2006). Our results also revealed that DOT1L supports MYC and BACH2 activity, which B cells depend on to effectively differentiate into pro‐proliferative GC B cells and maintain that state (Calado et al, 2012). Recent studies have shown that inhibition of DOT1L also leads to reduced Myc expression in multiple myeloma (Ishiguro et al, 2019), in MYC‐driven B‐cell lymphoma (Deshpande et al, 2018), and in androgen‐dependent prostate cancers (Vatapalli et al, 2020) indicating that the connection between DOT1L and MYC has broad implications in B‐cell physiology as well as other cell types. Furthermore, in neuroblastoma H3K79me2 methylation has been shown to be a strict prerequisite for MYC‐induced transcriptional activation, indicating a mutual interaction between DOT1L and MYC (Wong et al, 2017). Interestingly, this novel interaction has also been reported recently in colorectal cancer (Yang et al, 2019). In addition to the crucial role in GC formation, we also identify DOT1L as a critical factor in maintaining MZ B cells. Further exploring the strong reduction of MZ B cells upon loss of Dot1L should provide additional insights regarding the contribution of DOT1L in orchestrating normal B‐cell physiology.
In addition to supporting MYC and BACH2 activity, our findings also suggest that DOT1L supports the repression of target genes of PRC2. Regarding influence of EZH2 and DOT1L on GC B cells, it was found that in both spleen and Peyer’s patches, GC B cells were drastically reduced in the absence of EZH2 (Caganova et al, 2013) and DOT1L (this study). Phenotypically, this connection between DOT1L and PRC2 targets is further supported by overlap in transcriptome changes between Ezh2 KO and Dot1L KO B cells. The mechanism explaining the connection between DOT1L and PRC2 targets is unknown and needs further investigation. The observation that Ezh2 is normally H3K79‐dimethylated and downregulated in Dot1L‐KO B cells, and that Dot1L and Ezh2 are co‐regulated in B cells indicates that DOT1L might promote repression of PRC2 targets by maintaining expression of Ezh2. Alternatively, DOT1L might affect a subset of PRC2 targets by altered activity of co‐repressor complexes, as has been suggested for DOT1L in mouse ES cell differentiation (Ferrari et al, 2020). Finally, additional factors controlled by DOT1L may impact differentiation of B cells (e.g. see Tables 1 and 2). A direct stimulatory effect of H3K79me on H3K27me3 synthesis is not likely to be involved since these modifications occur at distinct locations in the genome and are associated with opposite transcriptional states. The link between DOT1L and multiple transcriptional regulators implies the existence of a complex regulatory network in B cells that warrants further investigation.
Understanding the critical role of DOT1L in B‐cell physiology might uncover important implications in B cell‐related pathologies. Considering the requirement for DOT1L in supporting humoral immune responses that we show here, targeting DOT1L may offer an opportunity for immune suppression. Given the strong dependency of GC B cells on DOT1L and the role of DOT1L in controlling PRC2 targets and the oncogenic factor MYC, DOT1L inhibition may also offer a novel therapeutic angle in the treatment of diffuse large B‐cell lymphoma of the GCB type. In summary, in B cells, DOT1L has a central role in guiding dynamic epigenetic states controlling differentiation and ensuring functional immune responses, with a potential for clinical exploitation.
Materials and Methods
Mice
Mb1‐Cre+/− ;Dot1Lfl/fl mice were derived by crossing the Dot1Ltm1a(KOMP)Wtsi line—generated by the Wellcome Trust Sanger Institute (WTSI) and obtained from the KOMP Repository (www.komp.org)—with the MB1‐Cre strain kindly provided by M. Reth (Hobeika et al, 2006). Mice from this newly created Mb1‐Cre+/− ;Dot1L strain were maintained under specific pathogen‐free (SPF) conditions at the animal laboratory facility of the Netherlands Cancer Institute (NKI; Amsterdam, Netherlands). Mice used for experiments were between 6–8 weeks old and of both genders unless stated otherwise. All experiments were approved by the Animal Ethics Committee of the NKI and performed in accordance with the Dutch Experiments on Animals Act and the Council of Europe.
Genotyping PCR
Mice were genotyped for Dot1L using the forward primer (Dot1L: FWD, 5′‐GCAAGCCTACAGCCTTCATC‐3′) and reverse primer 1 (Dot1L:REV1, 5′‐CACCGGATAGTCTCAATAATCTCA‐3′) to identify WT allele; Dot1L wt (517 bp) while the floxed allele; Dot1L fl (335 bp) was identified by using Dot1L: FWD and reverse primer 2 (Dot1L:REV2, 5′‐GAACCACAGGATGCTTCAG‐3′). The region flanking floxed exon 2 of Dot1L was amplified by using My Taq DNA polymerase (meridian BIOSCIENCE, catalog number BIO‐21107) and the following thermocycler conditions: Initial denaturation 95°C for 5 min, followed by 35 cycles of denaturation 95°C for 30 s, annealing 60°C for 30 s and extension 72°C for 1 min. Final extension was done at 72°C for 5 min. The WT allele (418 bp) for Mb1 was detected by using forward primer (Mb1‐FWD1: 5′‐CTGCGGGTAGAAGGGGGTC‐3′) and reverse primer (Mb1‐REV1: 5′‐CCTTGCGAGGTCAGGGAGCC‐3′) while Cre (219 bp) was detected by using forward primer (Mb1‐FWD2: 5′‐GTGCAAGCTGAACAACAGGA‐3′) and reverse primer (Mb1‐REV2: 5′‐AAGGAGAATGTGGATGCTGG‐3′). PCR to amplify the region of the MB1 locus was performed by using My Taq DNA polymerase (meridian BIOSCIENCE, catalog number BIO‐21107) and the thermocycler conditions used to amplify region of MB1 locus consists of one cycle of Initial denaturation: 95°C for 3 min, annealing: 75°C for 5 min and extension: 72°C for 90 s followed by 34 cycles of denaturation: 94°C for 1 min, annealing: 63°C for 1 min, extension: 72°C for 90 s. Final extension was done at 72°C for 10 min.
Reverse transcriptase (RT)–PCR
Total RNA from FACS sorted mature B and T lymphocytes from the spleen of mice was isolated using RNeasy mini (Qiagen, cat. no. 74106) according to the manufacturer instruction. The cDNA libraries were prepared using Invitrogen Superscript III kit and Random hexamer primers (catalog number 18080051) according to the manufacturer instructions. Deletion of exon 2 (44 bp) from Dot1l transcript was detected by amplifying the portion of Dot1L cDNA with Taq polymerase (Invitrogen, catalog number 10342053) using forward primer (P1:FWD, 5′‐CGAGAAGCTGGAGCTGAG‐3′) and reverse primer (P2:REV, 5′‐TGGTGGATGCTGTCGATG‐3′). Thermocycler conditions used for amplification consists of Initial denaturation: 95°C for 3 min, followed by 15 cycles of denaturation: 95°C for 30 s, annealing: 65.7°C for 45 s and extension: 72°C for 1 min. This was followed by 25 cycles of denaturation: 95°C for 30 s, annealing: 60°C for 45 s, extension: 72°C for 1 min. Final extension was done at 72°C for 5 min. PCR fragments of 255 and 211 bp, for WT and KO, respectively, were extracted from 2% Precast Agarose E‐gel (catalog number G401002) using E‐gel Agarose electrophoresis system (Invitrogen) and subjected to sequencing using the same set of individual primers. Sequenced products were analyzed by SnapGene (version 4.1.6).
In silico analysis
The predicated translated product from the KO Dot1L transcript missing exon 2 was generated using SnapGene (version 4.1.6). Alignments of the open reading frame from the WT Dot1L transcript and the predicted translated product from the KO Dot1L transcript were performed by ClustalW under default settings.
Flow cytometry
Single cell suspensions were made from bone marrow, spleen, and Peyer’s patches. Bone marrow, spleen, and blood samples were subjected to erythrocyte lysis. Distinct cellular populations were identified using a combination of fluor‐conjugated antibodies against surface markers (Appendix Table S3). Cells were stained with fluorescently labeled antibodies (Appendix Table S4). For intracellular staining, cells were fixed and permeabilized using the Transcription Factor Buffer set (Becton Dickinson, BD, catalog number 562574). Antibodies for intracellular staining were diluted in Perm/Wash buffer except for cleaved caspase‐3. For H3K79me2 staining, cells were first stained with surface markers and fixed and permeabilized as described before. After fixation and permeabilization cells were washed with Perm/Wash containing 0.25% SDS. H3K79me2‐specific antibody (Millipore, clone NL59, catalog number 04‐835) was diluted 1:200 into Perm/Wash + 0.25% SDS and cells were incubated for 30 min. Cells were washed with Perm/Wash and incubated with the secondary antibodies Donkey anti‐Rabbit AF555 (Thermo Scientific, catalog number A‐31572) or Goat anti‐Rabbit AF488 (Invitrogen, catalog number A‐11034) 1:1,000 in Perm/Wash. For cleaved caspase‐3 staining, the cells were first stained with surface markers and then fixed with 4% formaldehyde for 15 min at room temperature. After washing with 0.25% tween buffer, the cells were permeabilized with 0.25% tween buffer for 15 min at room temperature. Following permeabilization, the cells were stained with cleaved caspase‐3 antibody as 1:50 diluted in 0.25% tween buffer for 45 min at room temperature under dark condition. For annexin V staining, following surface marker stainings, the cells were washed once with PBS and later with Binding Buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Cells were stained with annexin V stain mix prepared as 1:20 in Binding Buffer and incubated for 20 min at room temperature in the dark. Following annexin V (Biolegend, catalog number 640920) staining, the cells were washed once with Binding buffer and resuspended in Binding buffer containing DAPI (1:100). Flow cytometry was performed using the LSR Fortessa (BD Biosciences) and data were analyzed with FlowJo software (Tree Star Inc.). Histograms were smoothed.
Immunization
Adult mice were inoculated intravenously with sub‐lethal dose 2×105 PFU (Plaque forming units) of lymphocytic choriomeningitis virus strain Armstrong. Serum was collected prior to immunization and 14 days post‐immune challenge. For NP‐CGG immunization, mice were injected intraperitoneally with 100 μg of alumprecipitated NP‐CGG [(4‐hydroxy‐3‐nitrophenyl) acetyl coupled to chicken γ–globulin, BIOSEARCHTM TECHNOLOGIES] in a 200 μl of NP‐CGG alum solution. To determine the serum titers of NP‐specific IgM and IgG1, mice were bled from the tail vein on days 0, 7, 14, 21, 28, and 35.
In vitro stimulation: class switch recombination, proliferation, and plasma cell formation
Single cells suspensions were prepared from the spleen of 6‐ to 8‐week‐old mice. Following erythrocyte lysis, naïve splenic B cells were enriched by the depletion of CD43‐expressing cell using biotinylated anti‐CD43 antibody (Clone S7, BD Biosciences), BD IMag Streptavidin Particles Plus, and the IMag® system (BD Biosciences), as described by the manufacturer. To measure their proliferative capacity, naïve B cells (CD43−) were labeled for 10 min at 37°C with 5 μM Cell Trace Violet (CTV, Life Technologies, Invitrogen™) in IMDM medium containing 2% FCS, with pen/strep, and 100 µM β‐mercaptoethanol. After washing, cells were cultured in complete IMDM medium (IMDM supplemented with 8% FCS, with pen/strep, and 100 µM β‐mercaptoethanol) at a density of 2 × 105 cells/well in 24‐well plates. CSR to IgG3 and IgG1 was induced in T cell‐independent manner by exposure to 5 μg/ml of Lipopolysaccharide (Escherichia coli LPS, 055:B5, Sigma) or LPS + IL‐4. IL4 was used at a concentration of 10 ng/ml. Cells were exposed to anti‐CD40 (1 µg/ml, BD Clone HM40‐3) and IL‐4 (10 ng/ml) to induce IgG1 switching in a T cell‐dependent manner. Four days later, the cells were harvested and either stained to determine CSR frequency or plasma cells formation. CTV dilution as an indicator of cell multiplication. In order to determine the effect of chemical inhibition of DOT1L on CSR and plasma cell formation under in vitro stimulation (LPS or anti‐CD40 + IL‐4), naïve B cells isolated from WT spleen were labeled with CTV and stimulated (LPS or anti‐CD40 + IL‐4) under the above‐mentioned conditions either in the presence of DOT1L inhibitor Pinometostat (EPZ‐5676, catalog number S7062, Selleckchem) at a concentration of 10 μM or 0.1% dimethyl sulfoxide (DMSO) as a control.
Enzyme‐linked Immunosorbent Assay (ELISA)
LCMV‐specific serum IgM and IgG levels were measured by ELISA. In short, Nunc‐Immuno Maxisorp plates (Fisher Scientific) were coated overnight at 4°C with virus in bicarbonate buffer. Plates were subsequently incubated for 1h with blocking buffer (PBS/5% milk powder (Fluka Biochemika)). Sera from mice were diluted in PBS/1% milk powder and incubated for 1h at 37°C. Next, HRP‐conjugated IgG and IgM antibodies (Southern Biotech) were diluted 1:4,000 in PBS/1% milk powder and incubated 1 h at 37°C. Plates were developed with TMB substrate (Sigma Aldrich), and the color reaction was stopped by the addition of 1 M H2SO4. Optical density was read at 450 nm (OD450) using a Microplate reader (Model 680, Bio‐Rad).
To quantify NP‐specific serum antibodies, plates were coated with 2 μg/ml NP30‐BSA. Serum was added at a starting dilution of 1:100 followed by threefold serial dilutions and incubated for 2 h at room temperature. Bound serum antibodies were detected with polyclonal biotinylated goat anti‐mouse IgM or anti‐IgG1 (Southern Biotech, catalog numbers 1020‐08 and 1071‐08, respectively), streptavidin‐alkaline phosphatase conjugate (Roche, catalog number 11089161001) and chromogenic substrate 4‐nitrophenyl phosphate (Sigma, catalog number N2765‐100TAB) in diethanolamine buffer. Purified monoclonal antibodies (B1‐8μ and 18‐1‐16y1, home‐made) were used as standards for quantification.
To quantify resting serum Ig titers, plates were coated with 1 μg/ml of goat anti‐mouse Kappa and Lambda prepared in BBS (Borate Buffered Saline). Serum was diluted with 0.05% Tween in PBS and added at a starting dilution of 1:100, followed by fourfold serial dilutions. The plates were incubated overnight at 4°C. Bound serum antibodies were detected with biotinylated goat anti‐mouse IgM, IgG1, IgG2b, IgG3, and IgA (Southern Biotech, catalog numbers 1020‐08, 1071‐08, 1090‐08, 1100‐08, and 1040‐08, respectively), streptavidin‐alkaline phosphatase conjugate (Roche, catalog number 11089161001), and chromogenic substrate 4‐nitrophenyl phosphate (Sigma, catalog number N2765‐100TAB) in diethanolamine buffer. Mouse IgM, IgG1, IgG2b, IgG3, and IgA antibodies (Southern Biotech, catalog numbers 0101‐01, 0102‐01, 0104‐01, 0105‐01, and 0106‐01, respectively) were used as standards for quantification.
Lectin histochemistry
Lymphoid tissues such as spleens were fixed in EAF (ethanol, acetic acid, formaldehyde, saline) for 24 h and subsequently embedded in paraffin. 4‐µm‐thick sections were stained with the lectin Peanut Agglutinin (PNA, Vector Laboratories) at 1:1,500 dilution to reveal germinal centers. The sections were counterstained with hematoxyline.
Immunohistochemistry
Spleens were collected and fixed in EAF fixative (ethanol/acetic acid/formaldehyde/saline at 40:5:10:45 v/v). Fixed spleens were embedded in paraffin and sections of 4 µm thickness were made. Immunohistochemistry (IHC) was conducted for CD3 (Thermo Fischer Scientific, RM‐9107‐S1, at a dilution of 1:600), cleaved caspase‐3 (Cell Signaling, catalog number 9661, at a dilution of 1:400), and Ki67 (Abcam, catalog number ab15580, at a dilution of 1:3000). The sections were reviewed with a Zeiss Axioskop2 Plus microscope (Carl Zeiss Microscopy, Jena, Germany), and images were captured with a Zeiss AxioCam HRc digital camera and processed with AxioVision 4 software (both from Carl Zeiss Vision, München, Germany). The scale bars were set at 50 μm.
Sorting and in vitro activation for RNA and ChIP‐Seq
For RNA sequencing, cells were first depleted for CD43+ cells and either subjected to MACS sorting for CD19+ cells as naïve B cells or activated for 2 days with LPS + IL‐4. Following activation, the cells were enriched for CD19+ by MACS according to the manufacturer instructions. For ChIP‐Seq, CD43− cells were either FACS sorted for CD19+ as naïve B‐cell pool or activated for 3 days with LPS + IL‐4 and subjected to FACS sorting for CD19+ expression.
RNA‐Seq sample preparation
MACS sorted CD19+ cells were resuspended in TRIzol (Ambion Life Technologies), and total RNA was extracted according to the manufacturer’s protocol. Quality and quantity of the total RNA were assessed by the 2100 Bioanalyzer using a Nano chip (Agilent). Only RNA samples having an RNA Integrity Number (RIN) > 8 were subjected to library generation.
RNA‐Seq library preparation
Strand‐specific cDNA libraries were generated using the TruSeq Stranded mRNA sample preparation kit (Illumina) according to the manufacturer’s protocol. The libraries were analyzed for size and quantity of cDNAs on a 2100 Bioanalyzer using a DNA 7500 chip (Agilent), diluted, and pooled in multiplex sequencing pools. The libraries were sequenced as 65 base single reads on a HiSeq2500 (Illumina).
RNA‐Seq preprocessing
Strand‐specific RNA reads (11–33 million reads per sample), 65 bp single‐end, were aligned against the mouse reference genome (Ensembl build 38) using Tophat (version 2.1, bowtie version 1.1). Tophat was supplied with a Gene Transfer File (GTF, Ensembl version 77) and was supplied with the following parameters: “‐‐prefilter‐multihits –no‐coverage‐search –bowtie1 –library‐type fr‐firststrand”. In order to count the number of reads per gene, a custom script which is based on the same ideas as HTSeq‐count has been used. A list of the total number of uniquely mapped reads for each gene that is present in the provided Gene Transfer Format (GTF) file was generated. Genes that have no expression across all samples within the dataset were removed. Analysis was restricted to genes that have least two counts per million (CPM) value in all samples in specific contrasts, to exclude very low abundance genes. Differential expression analysis was performed in R language (version 3.5.1) on only relevant samples using edgeR package and default arguments with the design set to either Dot1LKO status, Ezh2KO status or cell type. Genes were considered to be differentially expressed when the False discovery rate (FDR) was below 0.05 after the Benjamini–Hochberg multiple testing correction. Sets of differentially expressed genes in indicated conditions were called “gene signatures”. MA plots were generated after differential expression analysis carried by edgeR package (Robinson et al, 2010; McCarthy et al, 2012). Read counts were corrected for gene length based on the longest transcript of the gene followed by normalization for the library size and shown as transcript per million (TPM). Counts are shown as counts per million after trimmed mean of M‐values (TMM) normalization using the edgeR R package. For analyses where we performed expression matching, we chose genes with an absolute log2 fold changes less than 0.1 and false discovery rate corrected P‐values above 0.05 that were closest in mean expression to each of the genes being matched without replacement. The RNA‐Seq datasets reported in this article have been deposited at the National Center for Biotechnology Information under the accession number GSE138909.
Ingenuity pathway analysis
Lists of differentially expressed genes (FDR < 0.05) between WT and KO B cells both from naïve and activated conditions were submitted to IPA using default settings to identify potential upstream regulators.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) was carried out after differential expression analysis and was shown as barcode plot where the genes were ranked according to the log2 fold change between the compared conditions. Statistical significance for the enrichment of gene set was determined by Fast approximation to mroast (FRY) gene set test (Wu et al, 2010) from limma package (version 3.44.3) (Ritchie et al, 2015), and two‐sided directional P‐value less than 0.05 was considered significant. For Fig EV5H, GSEA was performed by Fast Gene Set Enrichment Analysis (fgsea) Bioconductor package (version 1.14.0) (preprint: Korotkevich et al, 2019) using ranked, shrunken fold changes from the differential expression analysis as input.
ChIP‐Seq sample preparation
Sorted cells were centrifuged at 500 rcf. The pellet was resuspended in IMDM containing 2% FCS and formaldehyde (Sigma) was added to a final concentration of 1%. After 10‐min incubation at RT, glycine (final concentration 125 mM) was added and incubated for 5 min. Cells were washed twice with ice‐cold PBS containing Complete, EDTA‐free, protein inhibitor cocktail (PIC) (Roche). Cross‐linked cell pellets were stored at −80°C. Pellets were resuspended in cold Nuclei lysis buffer (50 mM Tris–HCl pH 8.0, 10 mM EDTA pH 8.0, 1% SDS) + PIC and incubated for at least 10 min. Cells were sonicated with a PICO sonicator to an average length of 200–500 bp using 30 s on/30 s off for 3 min. After centrifugation at high speed, debris was removed and 9× volume of ChIP dilution buffer (50 mM Tris–HCl pH 8, 0.167 M NaCl, 1.1% Triton X‐100, 0.11% sodium deoxycholate) + PIC and 5× volume of RIPA‐150 (50 mM Tris–HCl pH 8, 0.15 M NaCl, 1 mM EDTA pH 8, 0.1% SDS, 1% Triton X‐100, 0.1% sodium deoxycholate) + PIC was added. Shearing efficiency was confirmed by reverse crosslinking the chromatin and checking the size on agarose gel. Chromatin was pre‐cleared by adding ProteinG Dynabeads (Life Technologies) and rotation for 1 h at 4°C. After the beads were removed 2 μl H3K79me1, 2 μl H3K79me2 (NL59, Merck Millipore, catalog number 04‐835) and 1 μl H3K4me3 (Abcam, catalog number ab8580) were added and incubated overnight at 4°C. ProteinG Dynabeads were added to the IP and incubated for 3 h at 4°C. Beads with bound immune complexes were subsequently washed with RIPA‐150, two times RIPA‐500 (50 mM Tris–HCl pH 8, 0.5 M NaCl, 1 mM EDTA pH 8, 0.1% SDS, 1% Triton X‐100, 0.1% sodium deoxycholate), two times RIPA‐LiCl (50 mM Tris–HCl pH 8, 1 mM EDTA pH 8, 1% Nonidet P‐40, 0.7% sodium deoxycholate, 0.5 M LiCl2) and TE. Beads were resuspended in 150 μl Direct elution buffer (10 mM Tris–HCl pH 8, 0.3 M NaCl, 5 mM EDTA pH 8, 0.5% SDS) and incubated overnight at 65°C, and input samples were included. Supernatant was transferred to a new tube, and 1 μl RNase A (Sigma) and 3 μl ProtK (Sigma) were added per sample and incubated at 55°C for 1 h. DNA was purified using Qiagen purification columns.
ChIP‐Seq library preparation
Library preparation was done using KAPA LTP Library preparation kit using the manufacturer’s protocol with slight modifications. Briefly, after end‐repair and A‐tailing adaptor were ligated followed by Solid Phase Reversible Immobilization (SPRI) clean‐up. Libraries were amplified by PCR and fragments between 250 and 450 bp were selected using AMPure XP beads (Beckman Coulter, catalog number A63881). The libraries were analyzed for size and quantity of DNAs on a 2100 Bioanalyzer using a High Sensitivity DNA kit (Agilent), diluted and pooled in multiplex sequencing pools. The libraries were sequenced as 65 base single reads on a HiSeq2500 (Illumina).
ChIP‐Seq preprocessing
ChIP‐Seq samples were mapped to mm10 (Ensembl GRCm38) using BWA‐MEM with the option “‐M”. Duplicate reads were removed using MarkDuplicates from the Picard toolset with “VALIDATION_STRINGENCY = LENIENT” and “REMOVE_DUPLICATES = true” as arguments. Bigwig tracks were generated from these bam files by using bamCoverage from deepTools using the following arguments: “‐of bigwig –binsize 25 –normalizeUsing RPGC –ignoreForNormalization chrM –effectiveGenomeSize 2652783500”. Bigwig files were loaded into R using the “import.bw()” function from the rtracklayer R package for visualization of heatmaps and genomic tracks. TSSs for heatmaps were taken from Ensembl GRCm38.77 gene models by taking the first base pair of the 5′ UTR of transcripts. When such annotation was missing, the most 5′ position of the first exon was taken.
Statistical analyses
Statistical analyses were performed using Prism 7 (GraphPad). Data are presented as mean ± SD except for Fig 4D where it is presented as mean ± SEM. Unless stated otherwise, the unpaired Student’s t‐test with two‐tailed distributions was used to calculate the P‐value. A P‐value < 0.05 was considered statistically significant.
Author contributions
Conception and design, MAA, MFA, EMK‐M, FvL, and HJ. Acquisition of data, MAA, MFA, EMK‐M, FIM, MC, INP, TvW, and J‐YS. Analysis and interpretation of data, MAA, MFA, EMK‐M, RA, EdW, KR, FvL, and HJ. Bioinformatics Analysis, MAA, TvdB, MFA, EdW, and IdR. Writing of the manuscript, MAA, MFA, EMK‐M, FvL, and HJ.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We highly appreciate the support from Michael Reth in providing his Mb1‐Cre mouse model system. We acknowledge Paul van den Berk for advice and Muddassir Malik for critically reading the manuscript. We thank the NKI animal pathology facility for performing histology and immunohistochemistry, as well as advice; the NKI Genomics Core Facility for library preparations and sequencing; the NKI Research High Performance Computing (RHPC) facility for providing computational resources; the NKI Flow Cytometry facility for assistance; and the caretakers of the NKI Animal Laboratory facility for assistance and excellent animal care. This work would not have been possible without the generous support kindly provided by the Netherlands Organization for Scientific Research (NWO‐VICI‐016.130.627 to FvL; ZonMW Top 91213018 to HJ; ZonMW Top91218022 to FvL and HJ) and the Dutch Cancer Society (NKI 2014‐7232 to FvL and HJ and NKI 2019‐2/12825 to HJ and FvL). T.v.d.B. and E.d.W. are part of the Oncode Institute which is partly financed by the Dutch Cancer Society. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
EMBO reports (2021) 22: e51184.
Contributor Information
Heinz Jacobs, Email: h.jacobs@nki.nl.
Fred van Leeuwen, Email: fred.v.leeuwen@nki.nl.
Data availability
The RNA‐Seq and ChIP‐Seq datasets reported in this article have been deposited at the National Center for Biotechnology Information under the accession number GSE138909 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138909) and GSE138906 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138906), respectively.
References
- Bao Y, Cao X (2016) Epigenetic control of B cell development and B‐cell‐related immune disorders. Clin Rev Allergy Immunol 50: 301–311 [DOI] [PubMed] [Google Scholar]
- Béguelin W, Popovic R, Teater M, Jiang Y, Bunting Karen L, Rosen M, Shen H, Yang Shao N, Wang L, Ezponda T et al (2013) EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23: 677–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM et al (2016) EZH2 and BCL6 cooperate to assemble CBX8‐BCOR complex to repress bivalent promoters, mediate germinal center formation and lymphomagenesis. Cancer Cell 30: 197–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, Shen H, Challman MF, Elemento O, Singh A et al (2017) EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb‐E2F1 feedback loop. Nat Commun 8: 877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L et al (2011) MLL‐rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20: 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, Nwosu ZC, Zhang L, Czerwonka A, Pawlowska A et al (2020) Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature 585: 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger M, Tarakhovsky A (2014) Epigenetic control of immunity. Cold Spring Harb Perspect Biol 6: a024174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK et al (2013) Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest 123: 5009–5022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, de Alboran IM, Janz M, Rodig S, Rajewsky K (2012) The cell‐cycle regulator c‐Myc is essential for the formation and maintenance of germinal centers. Nat Immunol 13: 1092–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano‐Rodriguez D, Gjaltema RA, Jilderda LJ, Jellema P, Dokter‐Fokkens J, Ruiters MH, Rots MG (2016) Writing of H3K4Me3 overcomes epigenetic silencing in a sustained but context‐dependent manner. Nat Commun 7: 12284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecere G, Hoersch S, Jensen MB, Dixit S, Grishok A (2013) The ZFP‐1(AF10)/DOT‐1 complex opposes H2B ubiquitination to reduce Pol II transcription. Mol Cell 50: 894–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C‐W, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J et al (2015) DOT1L inhibits SIRT1‐mediated epigenetic silencing to maintain leukemic gene expression in MLL‐rearranged leukemia. Nat Med 21: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chory EJ, Calarco JP, Hathaway NA, Bell O, Neel DS, Crabtree GR (2018) Nucleosome turnover regulates histone methylation patterns over the genome. Mol Cell 73: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafflon C, Gaulis S, Barys L, Kapur K, Cornacchione V, Schukur L, Bergling S, Traggiai E, Jansky S, Hellmann L et al (2020) DOT1L inhibition is lethal for multiple myeloma due to perturbation of the endoplasmic reticulum stress pathway. Oncotarget 11: 56–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y et al (2011) Selective killing of mixed lineage leukemia cells by a potent small‐molecule DOT1L inhibitor. Cancer Cell 20: 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack‐Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP et al (2013) Potent inhibition of DOT1L as treatment of MLL‐fusion leukemia. Blood 122: 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos D, Frederiks F, Terweij M, van Welsem T, Verzijlbergen KF, Iachina E, de Graaf EL, Maarten Altelaar AF, Oudgenoeg G, Heck AJR et al (2011) Progressive methylation of ageing histones by Dot1 functions as a timer. EMBO Rep 12: 956–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Liu H, Liu X (2015) RAG1‐mediated ubiquitylation of histone H3 is required for chromosomal V(D)J recombination. Cell Res 25: 181–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Chen B, Ramezani‐Rad P, Pastore A, Zhao L, Weigert O, Armstrong S, Rickert R, Ren B, Deshpande A (2018) Targeting MYC‐driven B‐cell lymphoma by inhibition of the histone methyltransferase DOT1L. Blood 132: 2839 [Google Scholar]
- Di Noia JM, Neuberger MS (2007) Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76: 1–22 [DOI] [PubMed] [Google Scholar]
- Dziarski R (1982) Studies on the mechanism of peptidoglycan‐ and lipopolysaccharide‐induced polyclonal activation. Infect Immun 35: 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, Hornyak TJ (2011) EZH2‐dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/CDKN1A expression. Mol Cancer Res 9: 418–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, Fields TA, Vivian JL, Fields PE (2010) Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 116: 4483–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Arrigoni L, Franz H, Izzo A, Butenko L, Trompouki E, Vogel T, Manke T (2020) DOT1L‐mediated murine neuronal differentiation associates with H3K79me2 accumulation and preserves SOX2‐enhancer accessibility. Nat Commun 11: 5200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangini A, Sjoberg M, Roman‐Trufero M, Dharmalingam G, Haberle V, Bartke T, Lenhard B, Malumbres M, Vidal M, Dillon N (2013) The aurora B kinase and the polycomb protein ring1B combine to regulate active promoters in quiescent lymphocytes. Mol Cell 51: 647–661 [DOI] [PubMed] [Google Scholar]
- Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, van Leeuwen F (2008) Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol 15: 550–557 [DOI] [PubMed] [Google Scholar]
- Gass JN, Gunn KE, Sriburi R, Brewer JW (2004) Stressed‐out B cells? Plasma‐cell differentiation and the unfolded protein response. Trends Immunol 25: 17–24 [DOI] [PubMed] [Google Scholar]
- Godfrey L, Crump NT, Thorne R, Lau IJ, Repapi E, Dimou D, Smith AL, Harman JR, Telenius JM, Oudelaar AM et al (2019) DOT1L inhibition reveals a distinct subset of enhancers dependent on H3K79 methylation. Nat Commun 10: 2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD (2018) EZH2 Represses the B cell transcriptional program and regulates antibody‐secreting cell metabolism and antibody production. J Immunol 200: 1039–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M (2006) Testing gene function early in the B cell lineage in mb1‐cre mice. Proc Natl Acad Sci USA 103: 13789–13794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, Plocik AM, Guthrie C, Yamamoto KR (2010) Reciprocal intronic and exonic histone modification regions in humans. Nat Struct Mol Biol 17: 1495–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, DiTullio RA, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double‐strand breaks. Nature 432: 406–411 [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Kitajima H, Niinuma T, Ishida T, Maruyama R, Ikeda H, Hayashi T, Sasaki H, Wakasugi H, Nishiyama K et al (2019) DOT1L inhibition blocks multiple myeloma cell proliferation by suppressing IRF4‐MYC signaling. Haematologica 104: 155–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones B, Su H, Bhat A, Lei H, Bajko J, Hevi S, Baltus GA, Kadam S, Zhai H, Valdez R et al (2008) The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet 4: e1000190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies A, Hasbold J, Tarlinton DM, Dietrich W, Corcoran LM, Hodgkin PD, Nutt SL (2004) Plasma cell ontogeny defined by quantitative changes in blimp‐1 expression. J Exp Med 200: 967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kari V, Raul SK, Henck JM, Kitz J, Kramer F, Kosinsky RL, Übelmesser N, Mansour WY, Eggert J, Spitzner M et al (2019) The histone methyltransferase DOT1L is required for proper DNA damage response, DNA repair, and modulates chemotherapy responsiveness. Clin Epigenet 11: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KP, Choi J, Yoon J, Bruder JM, Shin B, Kim J, Arauzo‐Bravo MJ, Han D, Wu G, Han DW et al (2021) Permissive epigenomes endow reprogramming competence to transcriptional regulators. Nat Chem Biol 17: 47–56 [DOI] [PubMed] [Google Scholar]
- Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, Ludwig T, Rajewsky K, Dalla‐Favera R (2006) Transcription factor IRF4 controls plasma cell differentiation and class‐switch recombination. Nat Immunol 7: 773–782 [DOI] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Sergushichev A (2019) Fast gene set enrichment analysis. bioRxiv 10.1101/060012 [PREPRINT] [DOI] [Google Scholar]
- Kwesi‐Maliepaard EM, Aslam MA, Alemdehy MF, van den Brand T, McLean C, Vlaming H, van Welsem T, Korthout T, Lancini C, Hendriks S et al (2020) The histone methyltransferase DOT1L prevents antigen‐independent differentiation and safeguards epigenetic identity of CD8+ T cells. Proc Natl Acad Sci USA 117: 201920372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBien TW, Tedder TF (2008) B lymphocytes: how they develop and function. Blood 112: 1570–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109: 745–756 [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128: 707–719 [DOI] [PubMed] [Google Scholar]
- MacLennan IC (1994) Germinal centers. Annu Rev Immunol 12: 117–139 [DOI] [PubMed] [Google Scholar]
- MacLennan IC (2005) Germinal centers still hold secrets. Immunity 22: 656–657 [DOI] [PubMed] [Google Scholar]
- Martins G, Calame K (2008) Regulation and functions of Blimp‐1 in T and B lymphocytes. Annu Rev Immunol 26: 133–169 [DOI] [PubMed] [Google Scholar]
- Martin‐Subero JI, Oakes CC (2018) Charting the dynamic epigenome during B‐cell development. Semin Cancer Biol 51: 139–148 [DOI] [PubMed] [Google Scholar]
- McCarron MJ, Park PW, Fooksman DR (2017) CD138 mediates selection of mature plasma cells by regulating their survival. Blood 129: 2749–2759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 40: 4288–4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CM, Karemaker ID, van Leeuwen F (2014) The emerging roles of DOT1L in leukemia and normal development. Leukemia 28: 2131–2138 [DOI] [PubMed] [Google Scholar]
- Muto A, Tashiro S, Nakajima O, Hoshino H, Takahashi S, Sakoda E, Ikebe D, Yamamoto M, Igarashi K (2004) The transcriptional programme of antibody class switching involves the repressor Bach2. Nature 429: 566–571 [DOI] [PubMed] [Google Scholar]
- Muto A, Ochiai K, Kimura Y, Itoh‐Nakadai A, Calame KL, Ikebe D, Tashiro S, Igarashi K (2010) Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J 29: 4048–4061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen AT, Zhang Y (2011) The diverse functions of Dot1 and H3K79 methylationanh. Genes Dev 25: 1345–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard VH, Schatz DG (2006) Targeting of somatic hypermutation. Nat Rev Immunol 6: 573–583 [DOI] [PubMed] [Google Scholar]
- Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y (2006) Leukaemic transformation by CALM–AF10 involves upregulation of Hoxa5 by hDOT1L. Nat Cell Biol 8: 1017–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M (2009) Epigenetic events during B lymphocyte development. Epigenetics 4: 462–468 [DOI] [PubMed] [Google Scholar]
- Pilzecker B, Jacobs H (2019) Mutating for good: DNA damage responses during somatic hypermutation. Front Immunol 10: 438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman‐Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F (2011) Patterns and mechanisms of ancestral histone protein inheritance in budding yeast. PLoS Biol 9: e1001075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky K (1996) Clonal selection and learning in the antibody system. Nature 381: 751–758 [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucl Acids Res 43: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson RD, Lalor P, Bernfield M (1989) B lymphocytes express and lose syndecan at specific stages of differentiation. Cell Regul 1: 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, Yamanaka R, Tanaka Y, Nukiwa T, Marquez VE et al (2013) PRC2 overexpression and PRC2‐target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep 3: 1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer CD, Barwick BG, Guo M, Bally APR, Boss JM (2018) Plasma cell differentiation is controlled by multiple cell division‐coupled epigenetic programs. Nat Commun 9: 1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill LP, Turner BM, Delrow J et al (2004) The histone modification pattern of active genes revealed through genome‐wide chromatin analysis of a higher eukaryote. Genes Dev 18: 1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, Corcoran LM (2015) Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody‐secreting plasma cells. Nat Immunol 16: 663–673 [DOI] [PubMed] [Google Scholar]
- Shukla N, Wetmore C, O'Brien MM, Silverman LB, Brown P, Cooper TM, Thomson B, Blakemore SJ, Daigle S, Suttle B et al (2016) Final report of Phase 1 Study of the DOT1L inhibitor, pinometostat (EPZ‐5676), in children with relapsed or refractory MLL‐r acute leukemia. Blood 128: 2780 [Google Scholar]
- Steger DJ, Lefterova MI, Ying L, Stonestrom AJ, Schupp M, Zhuo D, Vakoc AL, Kim JE, Chen J, Lazar MA et al (2008) DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol 28: 2825–2839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EM, Garcia‐Manero G, Rizzieri DA, Tibes R, Berdeja JG, Savona MR, Jongen‐Lavrenic M, Altman JK, Thomson B, Blakemore SJ et al (2018) The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 131: 2661–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A (2003) Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4: 124–131 [DOI] [PubMed] [Google Scholar]
- Tellier J, Shi W, Minnich M, Liao Y, Crawford S, Smyth GK, Kallies A, Busslinger M, Nutt SL (2016) Blimp‐1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 17: 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatapalli R, Sagar V, Rodriguez Y, Zhao JC, Unno K, Pamarthy S, Lysy B, Anker J, Han H, Yoo YA et al (2020) Histone methyltransferase DOT1L coordinates AR and MYC stability in prostate cancer. Nat Comm 11: 4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Nussenzweig MC (2012) Germinal centers. Annu Rev Immunol 30: 429–457 [DOI] [PubMed] [Google Scholar]
- Vlaming H, van Welsem T, de Graaf EL, Ontoso D, Altelaar AF, San‐Segundo PA, Heck AJ, van Leeuwen F (2014) Flexibility in crosstalk between H2B ubiquitination and H3 methylation in vivo . EMBO Rep 15: 1077–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlaming H, van Leeuwen F (2016) The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 125: 593–605 [DOI] [PubMed] [Google Scholar]
- Vlaming H, McLean CM, Korthout T, Alemdehy MF, Hendriks S, Lancini C, Palit S, Klarenbeek S, Kwesi‐Maliepaard EM, Molenaar TM et al (2019) Conserved crosstalk between histone deacetylation and H3K79 methylation generates DOT1L‐dose dependency in HDAC1‐deficient thymic lymphoma. EMBO J 38: e101564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ et al (2008) Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 40: 897–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen CW, Armstrong SA (2016) The role of DOT1L in the maintenance of leukemia gene expression. Curr Opin Genet Dev 36: 68–72 [DOI] [PubMed] [Google Scholar]
- Wong M, Tee AEL, Milazzo G, Bell JL, Poulos RC, Atmadibrata B, Sun Y, Jing D, Ho N, Ling D et al (2017) The histone methyltransferase DOT1L promotes neuroblastoma by regulating gene transcription. Cancer Res 77: 2522–2533 [DOI] [PubMed] [Google Scholar]
- Wood K, Tellier M, Murphy S (2018) DOT1L and H3K79 methylation in transcription and genomic stability. Biomolecules 8: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Lim E, Vaillant F, Asselin‐Labat ML, Visvader JE, Smyth GK (2010) ROAST: rotation gene set tests for complex microarray experiments. Bioinformatics 26: 2176–2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Chen L, Zhou Q, Zhang W (2016) Dot1l deficiency leads to increased intercalated cells and upregulation of V‐ATPase B1 in mice. Exp Cell Res 344: 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zan H, Pone EJ, Mai T, Casali P (2012) Immunoglobulin class‐switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 12: 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Ha S, Ahn J, Kim R, Kim S, Lee Y, Kim J, Soll D, Lee HY, Park HS (2016) A chemical biology route to site‐specific authentic protein modifications. Science 354: 623–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Lei Q, Li L, Yang J, Dong Z, Cui H (2019) Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c‐Myc expression in colorectal cancer. Clin Epigenet 11: 199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia X, Jalal DI, Kuncewicz T, Xu W, Lesage GD, Kone BC (2006a) Aldosterone‐sensitive repression of ENaCalpha transcription by a histone H3 lysine‐79 methyltransferase. Am J Physiol Cell Physiol 290: C936–C946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC (2006b) Dot1a‐AF9 complex mediates histone H3 Lys‐79 hypermethylation and repression of ENaCalpha in an aldosterone‐sensitive manner. J Biol Chem 281: 18059–18068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dominguez‐Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, Vlasevska S, Mo T, Tang H, Basso K et al (2015) Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med 21: 1190–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The RNA‐Seq and ChIP‐Seq datasets reported in this article have been deposited at the National Center for Biotechnology Information under the accession number GSE138909 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138909) and GSE138906 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138906), respectively.