

Antigen-driven immune synaptic T cell contacts reprogram gene expression, epigenetics, and functions of dendritic cells.
Abstract
Understanding the fate of dendritic cells (DCs) after productive immune synapses (postsynaptic DCs) with T cells during antigen presentation has been largely neglected in favor of deciphering the nuances of T cell activation and memory generation. Here, we describe that postsynaptic DCs switch their transcriptomic signature, correlating with epigenomic changes including DNA accessibility and histone methylation. We focus on the chemokine receptor Ccr7 as a proof-of-concept gene that is increased in postsynaptic DCs. Consistent with our epigenomic observations, postsynaptic DCs migrate more efficiently toward CCL19 in vitro and display enhanced homing to draining lymph nodes in vivo. This work describes a previously unknown DC population whose transcriptomics, epigenomics, and migratory capacity change in response to their cognate contact with T cells.
INTRODUCTION
Interactions between naïve T cells and primed antigen-presenting cells, mostly dendritic cells (DCs), are the cornerstone of adaptive immunity (1). These contacts (“immune synapses”, IS) mainly occur in peripheral lymph nodes, where mature DCs travel from infected tissues and establish exploratory contacts until found by antigen-specific T cell clones, which undergo subsequent activation and amplification (2, 3). Most studies have thoroughly analyzed the IS as a platform that drives T cell activation, differentiation, and effector function (1), but fewer studies have focused on the DC side (4–6). During IS formation, DCs rearrange some of the antigen-presenting machinery to enhance their function (7, 8). An active exchange of information ensues, with DCs communicating antigenic information to T cells. In turn, T cells instruct DCs through membrane receptor interactions and T cell–derived exosomes that contain genetic information, mainly microRNAs and mitochondrial DNA (9, 10). Such genetic information transferred from T cells primes DCs to boost their antiviral capabilities (9). In addition, some studies suggest that antigen presentation to CD4+ T cells leads to DC “licensing” and priming, enhancing cross-presentation to CD8+ T cells (cross-priming) (11). In addition, DCs receive anti-apoptotic and survival signals from the T cell through the IS (4). Moreover, the IS locally promotes autophagy and mitophagy and reduces cell death (6). DCs can stay alive for up to 15 days in the lymph node after productive IS formation (12). Some cytokines, for example, type I interferon, increase the life span of CD8α+ DCs, enhancing cross-presentation and priming of CD8+ T cells during bacterial responses (13). Overall, this evidence suggests that some DCs could become “memory-like” innate cells after cognate immune contact, with enhanced ability to respond to subsequent immune challenges. Other cells such as monocytes and natural killer cells have been described to confer a nonspecific protection upon direct infection with a specific pathogen (14, 15). These cells display epigenomic (14) and metabolic changes (16), sustaining the expression of host defense–related genes that would enhance potential responses against future infections. In this study, we have analyzed the epigenetic and functional changes induced in CD11c+ bone marrow–derived DCs (BMDCs) upon being instructed by antigen cognate interaction with T cells.
RESULTS AND DISCUSSION
To assess the transcriptomic changes displayed by postsynaptic DCs, BMDCs were incubated with lipopolysaccharide (LPS) and pulsed (psDCs) or not (nsDCs) with the ovalbumin (OVA) peptide OVA323–339. Then, cells were allowed to conjugate with resting CD4+ OT-II T cells for 24 hours. CD11c+ MHCIIhigh DCs were sorted, and their RNA was isolated and analyzed by RNA sequencing (RNA-seq). Differential expression testing revealed 768 genes up-regulated in psDCs compared to nsDCs, with a fold increase (log2) of more than 1. On the other hand, 340 genes were down-regulated, with a fold reduction of below −1 (log2). Data analysis showed tight clustering among replicates of both conditions, confirming the robustness of the transcriptomic signature changes observed in both populations (fig. S1, A and B). Within the 60 most significantly changed genes, some are expressed in nsDCs, e.g., Cd40, Isg15, or Ccr7 [this is likely due to LPS maturation (17)]; however, their expression increases one to five times (in log2) in psDCs (Fig. 1A). To provide a more general overview of the pathways modified by productive interaction with T cells, differentially expressed genes were analyzed using Ingenuity Pathway Analysis (Qiagen) and clusterProfiler. The former approach revealed a clear activation of pathways including DC maturation, T helper 1 cell differentiation, and actin cytoskeleton reorganization (Fig. 1B). By comparing some known biological functions with the fold change of their associated genes, we identified some Gene Ontology (GO) terms corresponding to the inflammatory response, including response to cytokines or innate immune defense, which were globally activated, while cell adhesion was mostly repressed (Fig. 1C). A detailed analysis of antiviral response and virus clearance ontologies showed that many genes that participate in these pathways were also up-regulated in psDCs (Fig. 1D). These results indicate that establishment of the IS drives DCs toward a more mature, migratory, and inflammatory phenotype that serves the cell to better perform its function upon successive triggers with different stimuli. To validate the RNA-seq results, we performed real-time quantitative polymerase chain reaction (qPCR) of some genes identified in the RNA-seq screening in nsDC and psDC samples. psDCs up-regulated genes such as Ccr7, Tlr3, Fscn1, Cd40, Isg15, Ifit1, and Cxcl10 but not DCs pulsed with a control peptide lymphocytic choriomeningitis virus (LCMV) GP61–80 which, similar to OVA323–339, also binds H-I-Ab (Fig. 1E) (18). Incubation with OVA323–339, but not with LCMV GP61–80 peptide, leads to a productive IS as indicated by CD4+ T cell activation (fig. S2A). Moreover, when DCs were pulsed with whole OVA protein, we observed the up-regulation of these genes and CD4+ T cell activation (fig. S2, A and B). These data confirmed the findings revealed by the RNA-seq screening and indicated that the transcriptomic changes observed in psDCs are mostly driven by the cognate IS between DC and CD4+ T cells, although a minor contribution of antigen processing to this effect (19) cannot be totally ruled out.
Fig. 1. psDCs enhance the expression of genes related with inflammation, cytokine response, and antiviral immunity.
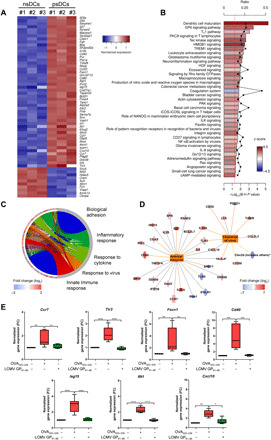
(A) Heatmap showing normalized gene expression values for a selection of 60 genes detected as differentially expressed [Benjamini and Hochberg (B-H)–adjusted P value < 0.05; abs(log2FC) > 1]. RNA-seq–based transcriptomic profile comparison of psDCs versus nsDCs (n = 3). (B) Enriched canonical pathways detected with IPA on the complete set of 1108 differentially expressed genes, after filtering with B-H–adjusted P value < 0.05 and abs(z score) > 1. The length of the bars represents enrichment significance [−log10(B-H P value)], and the red dashed line indicates the significance threshold. Bar colors represent “z-score” values (predicted activation state of the pathway, IPA); positive values (dark red) indicate higher activity in psDCs. Connected black dots represent parameter “ratio,” this is, the fraction of genes associated with any given pathway included in the collection of differentially expressed genes. NF-κB, nuclear factor κB; IL-8, interleukin-8. (C) Enriched biological process GO terms detected with clusterProfiler on the complete set of 1108 differentially expressed genes (right side of the circular plot). Only five selected terms are shown (B-H–adjusted P value < 1 × 10−8) and are connected to small rectangles representing differentially expressed genes associated to those terms (left side of the circular plot), colored according to the logFC value. (D) IPA network connecting functional terms “virus clearance” and “antiviral response” to associated differentially expressed genes. Genes up- or down-regulated in psDCs relative to nsDCs are colored in red and blue, respectively. (E) qPCR analysis of the gene expression levels indicated in psDCs versus nsDCs. Pulsed with or without OVA323–339 cognate OT-II peptide or LCMV GP61–80 control peptide [n = 7; Kruskal-Wallis test with Dunn’s posttest or one-way analysis of variance (ANOVA) test with Tukey’s posttest; *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001).
To assess whether the observed gene expression reprogramming of psDCs was specific of CD4 T-IS, the previous strategy was similarly applied to psDCs that have been pulsed with the peptide OVA257–264 and allowed to interact with CD8+ OT-I cells. The same previously validated genes (Ccr7, Tlr3, Fscn1, Cd40, Isg15, Ifit1, and Cxcl10) were up-regulated in psDC that had undergone an OVA257–264 peptide–specific IS with CD8+ T cells. In contrast, DCs pulsed with the control peptide vaccinia virus (VACV) B8R20–27, which, similar to OVA257–264, binds H-2kb, did not show such response (Fig. 2) (20). OVA257–264 but not VACV B8R20–27 resulted in a productive synapse as indicated by CD8+ T cell state of activation after the IS (fig. S3). The fold change of these genes in psDCs was in the same range as the one observed for IS with CD4+ T cells (Figs. 1E and 2).
Fig. 2. Gene expression of psDCs upon CD8+ T cell IS.
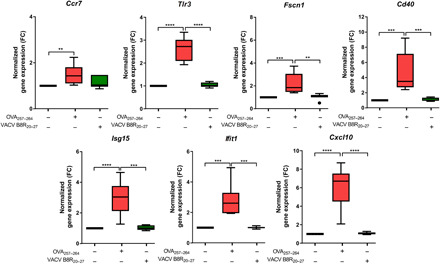
qPCR analysis of the expression levels of the indicated genes in DCs cocultured with CD8+ OT-I T cells under the presence or absence of OVA257–264 cognate OT-I peptide or VACV B8R20–27 control peptide (n = 7; Kruskal-Wallis test and Dunn’s posttest or one-way ANOVA with Tukey’s posttest; **P < 0.01, ***P < 0.001, and ****P < 0.0001).
While some genes up-regulated in psDCs were common to those observed in DCs treated with T cell–derived extracellular vesicles (EVs) (9), some others differ, suggesting that other mechanisms besides EVs transmission are involved. It is likely that membrane-bound receptor-ligand interactions, for example, CD40-CD40L (21), may be required in the context of the intimate membrane-to-membrane juxtaposition that occurs during IS formation. In addition, it is interesting that bacterially derived LPS triggering directs DCs to better respond to viral infections, suggesting that priming against a specific stimulus may generate a state of “general alert” that enhances immune protection against a variety of aggressors.
Next, we assessed whether modifications in the transcriptomic signature of psDCs reflect the acquisition of an innate immune memory-like phenotype. Changes in the accessibility of chromatin of psDCs were evaluated using ATAC-seq (assay for transposase-accessible chromatin with high-throughput sequencing). psDCs and nsDCs were isolated and treated with transposase 5 (Tn5) to detect regions undergoing active chromatin remodeling. This analysis revealed that around 25,000 regions were differentially accessible when comparing both conditions [false discovery rate (FDR) < 0.05; 15,062 with an abs(log2FC) > 1] (Fig. 3A). Most of the differential peaks were located in intronic or intergenic areas, at distances ranging 1 to 100 kb from transcription starting sites, suggesting a link with regulatory regions (fig. S4, A and B).
Fig. 3. Epigenomic signature of psDCs is modified upon T cell contact.
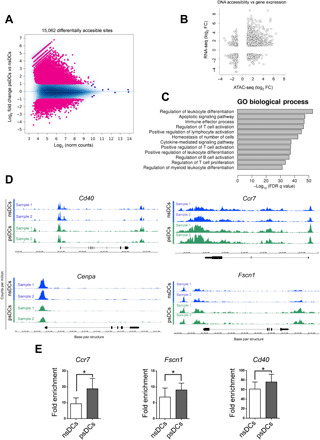
(A) MA plot representing differential accessibility analysis results for ATAC-seq data. Differentially accessible sites (15,062) [with FDR < 0.05 and abs(log2FC) > 1] are symbolized as pink dots over a blue cloud that represents the complete consensus set of 98,731 ATAC-seq sites. X-axis values (“log concentration”) represent logarithmically transformed, normalized counts, averaged for all samples, for each site. Y-axis values represent log2(fold change) values for ATAC-seq counts in psDCs relative to nsDCs. Positive log2FC values indicate higher accessibility in psDCs. (B) Scatterplot relating log2(fold change) values for ATAC-seq counts and log2(fold change) values for RNA-seq counts. Dots represent the association between 1634 differentially accessible genomic sites and 632 differentially expressed genes that were located at the shortest distance of a differentially accessible site. (C) Enriched biological processes GO terms, detected with GREAT, were associated to differentially accessible site proximal genes. Only the top 12 terms, sorted by significance, are presented. (D) Peak representation of the DNA accessibility in psDCs (green) versus nsDCs (blue) within regions proximal to locus for the indicated genes. The height of the peak represents the counts per million from the ATAC-seq data (n = 2). (E) ChIP-qPCR assay of the coimmunoprecipitation H3K4me3 with the promoter regions of the indicated genes. Results are expressed as fold enrichment relative to a glyceraldehyde-3-phosphate dehydrogenase promoter region (n = 5, paired t test; *P < 0.05).
RNA-seq and ATAC-seq data seem to concur with more than 1600 differentially accessible sites located near a differentially expressed gene (Fig. 3B). Also, functional enrichment analyses for all the genes associated to differentially accessible sites revealed biological processes that were compatible with those associated with the set of differentially expressed genes, particularly those related with adaptive immunity and cytokine signaling (Fig. 3C). We then verified that some up-regulated gene products, such as Cd40, Ccr7, and Fscn1, also presented higher chromatin accessibility in psDCs. Conversely, Cenpa, a gene that was down-regulated in the psDCs RNA-seq comparison, displayed less chromatin accessibility (Fig. 3D). We then undertook a larger-scale effort by performing a chromatin immunoprecipitation (ChIP)–qPCR analysis of some of the genes of interest. Sheared chromatin from isolated psDCs versus nsDCs was immunoprecipitated with an anti-H4K3me3 antibody [H4K3me3 is an activating and priming histone marker (22)]. qPCR of the open regions showed that Ccr7, Fscn1, or Cd40 associated peaks were enriched in psDCs in this assay, confirming the correlation between the transcriptomic and epigenetic data (Fig. 3E).
These changes in DNA accessibility and the epigenetic marks of the psDCs reflect a contact-dependent priming and reprogramming. This priming could reflect an “alert” state. Hence, psDCs could activate faster the expression of genes such as Ccr7 or Cd40 against future challenges with enhanced subsequent responses.
To functionally validate some of the candidate genes detected in the global genome studies, we addressed the migratory capacity of psDCs as they display increased levels of Ccr7. Ccr7 belongs to the DC gene signature when differentiating with granulocyte-macrophage colony-stimulating factor (GM-CSF) and stimulating with LPS (23). Moreover, GM-CSF–derived DCs have been described to cluster more closely to migratory DCs (24). Thus, to study the migratory properties of psDCs, we performed experiments in Boyden-modified chambers toward the CCR7 ligand CCL19. We found that psDC migrate more efficiently than nsDC (Fig. 4A). Furthermore, psDCs migrated faster and more directionally toward CCL19 in under-agarose assays. Both parameters together reflect an enhanced chemotactic movement of psDCs in response to CCL19 (Fig. 4, B and C, and movie S1). To extend these observations to a more physiological context, psDCs and nsDCs from CD45.1 mice were injected in the footpad of the two limbs of wild-type mice (CD45.2). After 24 hours of migration, popliteal, inguinal, and axillary lymph nodes were extracted, processed, and analyzed by flow cytometry. An increased homing of psDCs to the draining lymph node (popliteal) was observed, in agreement with an enhanced CCR7-dependent migration (Fig. 4, D and E). These results validate the outcome of the RNA-seq and ATAC-seq assays at a functional level, indicating that Ccr7 up-regulation after a productive synaptic contact could be one of the reasons of the improved maturation (25) in addition to more efficient cell migration (26). In turn, our data indicate that DCs undergo a transcriptomic, epigenomic, and functional reprogramming upon a productive IS. Our findings have been described on an IS model using BMDC and OT-II or OT-I T cells. We therefore cannot unravel whether other factors, not specifically tested in our experimental system, could additionally influence postsynaptic reprogramming of DC. For instance, T cell receptor (TCR) affinity for peptide–major histocompatibility complex (pMHC) complexes functionally influences T cells (27, 28). In this regard, it would be of interest to determine whether (i) the affinity of different TCRs for their cognate pMHC complexes or (ii) the heterogeneity of peptide binding affinity to MHC molecules could differentially affect the reprogramming of psDCs.
Fig. 4. IS enhances psDC migratory capacity in response to Ccr7.
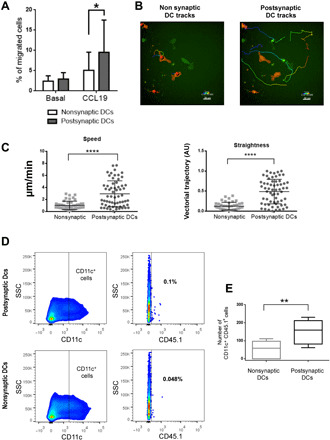
(A) Transwell assay of psDCs and nsDCs in response to CCL19 (100 ng/ml) for 3 hours. Medium without cytokines was set as a control (basal). Cell numbers were normalized to the initial input of cells (n = 6; Wilcoxon matched-paired signed ranks test; *P < 0.05). (B) Image showing the tracks and speed (color coded) of psDCs (green) versus nsDCs (red) in an under-agarose assay in response to a CCL19 gradient (focus on the right side of the image). (C) Quantification of the speed and straightness of the chemotactic movement of DCs in the under-agarose assay (n ≥ 44, Student’s t test; ****P < 0.0001). (D) Dot plot describing the gating strategy for the analysis of the number of CD11c+ (gated in the CD3− population) and CD45.1+ cells in psDCs and nsDCs. (E) Quantification of the number of migrated psDCs and nsDCs in the popliteal lymph node is shown (n = 5, paired t test; **P < 0.01).
In summary, our data support that during antigen-driven interactions, T cells educate DCs, endowing them with enhanced capability to fight additional threats. This opens the door to targeting psDC to design novel therapeutic approaches aimed at improving vaccination or adoptive transfer in cancer or immune deficiencies.
MATERIALS AND METHODS
Reagents
Antibodies and reagents used for flow cytometry were as follows: CD11c-BV421 (585452), MHCII-fluorescein isothiocyanate (FITC) (553623), 7-aminoactinomycin D (7AAD) (51-68981E), CD69-FITC (553236), CD11c-phycoerythrin (PE) (557401), and anti-mouse CD16/CD32 (Fc block) (553142) provided by Becton Dickinson (BD); CD40-allophycocyanin (APC) (20-8050-U025), CD44-V450 (75-0441-U025), CD4-PECy7 (60-0041-U100), CD45.1-PerCPCy5 (65-0453-U500), CD3-FITC (35-0031-U500), CD8-PECy7 (60-0081), and Ghost Red 780 Viability Dye (13-0865-T100) from Tonbo Biosciences; CD4-FITC (130-109-498), CD62L-PerCP-Vio700 (130-107-072), CD25-APC (130-109-052), CD4-PerCPCy5.5 (130-109-497), and CD11c-FITC (130-102-466) provided by Miltenyi; CCR7-PE (120105) and TCRβ-APCCy7 (109220) from BioLegend; and AbC RH capture beads (A10389) and LIVE/DEAD Fixable Yellow Dead Cell Stain (L34968) from Invitrogen. In addition, cell dyes 5-(and-6)-(((4-chloromethyl)benzoyl)amino)tetramethylrhodamine (CMTMR) (C2927) and carboxyfluorescein diacetate succinimidyl ester (CFSE) (C34554) were obtained from Thermo Fisher Scientific, and recombinant murine chemokine MIP-3β (CCL19) (250-27B) was provided by Peprotech.
Mice
Male C57/BL6 wild-type mice, TCR (Vα2, Vβ5) transgenic mice (OT-II), and TCR (Vα2, Vβ5) transgenic mice (OT-I), from 8 to 12 weeks, were housed in the pathogen-free animal facility of the Centro Nacional de Investigaciones Cardiovasculares Carlos III (Madrid) in accordance with the animal care standards of the institution. Animal experiments were approved by the local ethics committee and conformed to EU Directive 86/609/EEC and Recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes, enforced in Spanish law under Real Decreto 53/2013.
Cell isolation and coculture
All cells were cultured in RPMI 1640, GlutaMAX-I, 25 mM Hepes (Gibco-Invitrogen) supplemented with 10% fetal bovine serum (FBS), penicillin (50 IU/ml), streptomycin (50 μg/ml) (Gibco), and β-mercaptoethanol (50 μM; Sigma-Aldrich). CD4+ or CD8+ T cells for coculture were obtained from spleens and lymph nodes of OT-II or OT-I mice, respectively, after purification using the CD4+ or CD8+ mouse T cell isolation kit from StemCell (19852 and 19853, respectively) and biotinylated anti-CD25 antibody (7D4, BD Biosciences). BMDCs were generated from C57/BL6 wild-type mice and cultured in the presence of mouse GM-CSF (20 ng/ml). After 3 days, cells in suspension were collected and plated with fresh GM-CSF. On days 6 and 9, cells were detached with phosphate-buffered saline (PBS), EDTA (5 mM), and bovine serum albumin (BSA) (0.5%) and plated with fresh medium. Cells were used at days 9 to 10 (80% CD11c+) and 24 hours before coculture DCs were matured with LPS (250 ng/ml).
Next day, LPS was withdrawn and DCs were pulsed with the corresponding peptide (table S1) and cocultured for 24 hours with the OT-II purified CD4+ or OT-I purified CD8+ T cells (ratio, 1:2; DCs:T cells). For OVA protein processing and antigen presentation, DCs were pulsed with Ovalbumin (100 μg/ml) (InvivoGen) simultaneously with LPS and withdrawn the day after. Alive DCs were then sorted either in a Sony Synergy four-laser (4 L) sorter or with an ultrapure CD11c+ MicroBeads positive selection kit (Miltenyi).
qPCR and RNA-seq
For qPCR and RNA-seq, RNA from purified nonsynaptic and postsynaptic DCs was extracted using the RNeasy Micro Kit (Qiagen). RNA was quantified using a fluorimeter (Promega), and RNA integrity was measured with both an Agilent 6000 Pico Kit and Bioanalyzer. For qPCR, 0.5 to 1 μg of RNA was retrotrascribed with a Promega kit. Then, qPCR was performed with a GoTaq Promega kit and SYBR Green using the primers from table S2. For RNA-seq, 200 ng of total RNA was used to generate barcoded RNA-seq libraries using the NEBNext Ultra RNA Library preparation kit (New England Biolabs). Library size was checked using the Agilent 2100 Bioanalyzer DNA 1000 chip, and their concentration was determined using the Qubit fluorometer (Life Technologies).
Libraries were sequenced on a HiSeq 2500 (Illumina) to generate 60 bases of single reads and processed with real time analysis (RTA) v1.18.66.3. FastQ files for each sample were obtained using bcl2fastq v2.20.0.422 software (Illumina).
ATAC-seq
A number of 5 × 104 synaptic DCs were sorted and lysed [10 mM tris-HCl (pH 7.4), 10 mM MgCl2, and 0.1% IGEPAL CA-630] for 15 min at 4°C while centrifuging at 500g. The supernatant was discard, and nuclei were resuspended in transposase reaction buffer with Tn5 and incubated for 1 hour at 37°C and 7 min at 4°C. Later, 0.1% SDS was added to stop the reaction (5 min, room temperature), and DNA was isolated with Agencourt AMPure XP [Beckman Coulter; solid phase reversible immobilization (SPRI) beads, 2×] and measured with Qubit Kit. For DNA library generation, qPCR with i5 and i7 primers (barcoding) was performed (98°C 2′, 98°C 20ʺ, 63°C 30ʺ, 72°C 1′, and 4°C ∞; 9 cycles). A size cut-off was conducted (0.65× SPRI beads), and DNA was isolated with 1.8× SPRI beads. Last, another qPCR was performed with i5 and i7 primers with 6 to 7 cycles for 0.9 ng/μl or with 5 cycles for more than 0.9 ng/μl (98°C 2′, 98°C 20ʺ, 63°C 30ʺ, 72°C 1′, and 4°C ∞), and amplified DNA was isolated with SPRI beads (2×), and its concentration was measured by Qubit. As control, we used qPCR for Actb promoter (open) and Cryaa promoter (closed). An Agilent Pico 6000 bioanalyzer was used to assess peak sizes and percentages.
Libraries were sequenced on a HiSeq 2500 (Illumina) to generate 2 × 100–base pair (bp) reads and processed with RTA v1.18.66.3. FastQ files for each sample were obtained using bcl2fastq v2.20.0.422 software (Illumina). Sequencing reads were trimmed off Illumina adapters and transposase sequence using cutadapt 1.16. Then, they were aligned to the mouse reference genome (GRCh38 v91) using bowtie 1.2.2 with a limit of 2000-bp paired distance. The aligned bam files were converted to bw for visualization purposes using bamCoverage from deepTools.
Bioinformatics analysis
RNA-seq and ATAC-seq data were analyzed by the Bioinformatics Unit of Centro Nacional de Investigaciones Cardiovasculares (CNIC). Sequencing reads were preprocessed with a pipeline that used FastQC to assess read quality and Cutadapt to trim sequencing reads, eliminating Illumina adaptor remains, and to discard reads that were shorter than 30 bp. In the case of ATAC-seq, Nextera transposase adapter contaminations were also removed with Cutadapt.
Preprocessed RNA-seq reads were mapped against reference transcriptome NCBIM37.65 and quantified using RNA-Seq by expectation maximization (RSEM). Percentages of reads participating in at least one reported alignment were around 85%. Expected expression counts were then processed with an analysis pipeline using Bioconductor package Limma for normalization [trimmed mean of M-values (TMM) method] and differential expression testing. Only those genes expressed with at least one count per million in a number of samples equal to the number of replicate samples of the condition with less replicates were taken into account. Gene expression changes were considered significant if associated to Benjamini and Hochberg adjusted P value < 0.05. Given that the number of genes detected as differentially expressed was very high (5211 genes), abs(logFC) > 1 was used as an additional filter to obtain a smaller collection of genes for further analyses. The resulting collection of 1108 genes was then used for functional enrichment analyses with Ingenuity Pathway Analysis (IPA) and the R package clusterProfiler.
Preprocessed ATAC-seq reads were mapped against reference genome mm9 (same assembly than NCBIM37) with a pipeline that used the Burrows-Wheeler Aligner-Maximal Exact Match (BWA-MEM) algorithm as aligner, Piccard to mark duplicate alignments, and samtools to eliminate duplicates, chimerics, and suboptimally multimapped alignments. Only properly paired and mapped reads were kept. Alignments against the mitochondrial genome or chromosomes X and Y were also removed. The final number of read pairs was 10 to 20 × 106 for any of the samples. Transcription start site (TSS) enrichment values, calculated with HOMER’s annotate Peaks function, had values between 4 and 11 for any of the samples. Once filtered alignments had been obtained, peaks (accessible DNA regions) were called with MACS2, using parameters “--nomodel --shift -100 --extsize 200”, and “-q 0.05” as the FDR cut-off. Around 100,000 peaks were detected for any of the samples. Next, filtered alignments and peaks, in bam and bed formats, respectively, were processed with the R package DiffBind to define a consensus set of 98,731 peaks, to calculate and normalize their coverage in various samples, and to identify differentially accessible regions, using edgeR as analysis method. The fraction of reads in peaks (FRiP) score, as calculated by DiffBind, was around 0.5 for any of the samples. Functional enrichment analyses for the set of differentially accessible regions were performed with the genomic regions enrichment of annotations tool (GREAT) (29), which also provided information about the genes located at the shortest distance from any of the regions. Other data manipulations and graphical representations (heatmaps, bars, and scatterplots) were produced with R.
ChIP-qPCR
ChIP was performed with the iDeal ChIP-seq Kit for Histones (Diagenode) following the provider’s instructions. Briefly, isolated DCs were fixed with formaldehyde (1%) for 10 min at 37°C, and reaction was quenched with cold glycine. Cells were lysed with radioimmunoprecipitation assay buffer 20 min at 4°C, and nuclei were pelleted at 2300g for 5 min. Later, nuclear lysis buffer was applied (130 μl), and chromatin was sheared by using a Covaris sonicator and the program “Duty 10% PIP” 175 cycles 200 for 15 min. Sheared chromatin was incubated with the corresponding anti-histone antibody or immunoglobulin G control (1.5 μg/ml) conjugated with magnetic beads overnight at 4°C. After incubation, beads were washed and DNA fragments were eluted and decross-linked for 8 hours at 65°C. Last, after fixation reaction was reversed (8 hours, 65°C), DNA was isolated with the iPure beads v2 as indicated by the manufacturer protocol. qPCR was performed using primers against already described promoter regions for the indicated genes (ChIP-seq) as well as against the open regions detected for those genes in the ATAC-seq (table S2). Control primers for glyceraldehyde-3-phosphate dehydrogenase promoter region were used as a positive control.
In vitro migration assay (Transwell)
Postsynaptic DCs were purified after 48 hours of IS formation and resuspended in RPMI medium with 0.1% BSA. Cells were settled in 100 μl in a Transwell permeable support (5-μm pore size, polycarbonate membrane, Costar) over 600 μl of RPMI medium with 0.1% BSA alone (basal) or containing mouse CCL19 (100 ng/ml). A well was filled with the same amount of cells without the membrane as input. Cells were let to migrate for 3 hours at 37°C. Afterward, membrane was removed and migrated cells were recovered. Then, cells were stained with a viability dye (7AAD) and CD11c+ antibodies and analyzed by flow cytometry. The number of migrated cells was counted and normalized using Counting Beads (BD).
Under-agarose assay
For this assay, a manufactured microscopy chamber was customed from a modified protocol previously described (30). Briefly, a 17-mm-diameter circle was cut into the center of 60-mm dishes. The hole was sealed from the bottom part of the dish using aquarium silicone (Marina) and a 24-mm glass coverslip. After the silicone dried, we overlaid a 5-mm-high ring cut from a 15-ml Falcon tube and sealed the borders with low melting point paraffin. Later, the glass was coated for 1 hour at 37°C with 10% FBS in PBS, and a 1.2% agarose gel (in Hanks’ balanced salt solution/RPMI medium supplemented with 10% FBS) was let to polymerize over the glass. Nonsynaptic and postsynaptic DCs were stained with CMTMR and CFSE (respectively), mixed at a 1:1 ratio (5 × 105 cells globally), and plated in a 2-mm-diameter hole performed in the agarose gel. In parallel, CCL19 solution (100 ng/ml) was added to another 2-mm hole. The migration from the original place of the cells toward the chemokine was measured with a GE DeltaVision Elite wide-field fluorescence microscope, taking pictures with a 20× dry objective (U plan, S Apo, numerical aperture 0.75) every minute for 2 hours. Images were analyzed with Imaris software (Bitplane).
Homing experiment
After coculture, DCs were purified by CD4+ T cell depletion with a CD90.2-positive selection depletion kit (StemCell). Between 5 and 1 × 106 cells, in RPMI supplemented with 10% FBS were injected in the footpad of C57B/L6 wild-type mice (CD45.2). To reduce animal variability, nonsynaptic or postsynaptic DCs were injected in different limbs of the same mice. After 24 hours, popliteal, inguinal, and axillary lymph nodes were removed and processed with Liberase and deoxyribonuclease. Cells were then stained using CD45.1, CD11c, CD3, and Ghost Red 780 Viability Dye antibodies and analyzed in a BD FACS Canto II cytometer. Total number of cells was normalized using Counting Beads (BD).
Statistical analyses
Samples were tested for normality through a Kolmogorov-Smirnov normality test. Accordingly, parametric or nonparametric test (Student’s t test or Mann-Whitney U test) was applied. Samples with more than two groups were analyzed with one-way analysis of variance (ANOVA) test with Tukey’s posttest for parametric data or Kruskal-Wallis with Dunn’s posttest for nonparametric. When working with dependent samples, a paired t test was conducted. Every experiment was analyzed through at least three biological replicates. Statistical analysis was performed using GraphPad Prism 6.
Acknowledgments
We thank Microscopy, Cellomic, Genomic, and Bioinformatic Units from CNIC. Funding: This study was supported by grant SAF2017-82886-R from the Spanish Ministry of Economy and Competitiveness (MINECO), grant S2017/BMD-3671-INFLAMUNE-CM from the Comunidad de Madrid, a grant from the Ramón Areces Foundation “Ciencias de la Vida y la Salud” (XIX Concurso-2018), a grant from Ayudas Fundación BBVA a Equipos de Investigación Científica (BIOMEDICINA-2018), the Fundació Marató TV3 (grant 122/C/2015), “la Caixa” Banking Foundation (HR17-00016), BIOIMID (PIE13/041) from Instituto de Salud Carlos III, CIBER Cardiovascular (CB16/11/00272), and Fondo de Investigación Sanitaria del Instituto de Salud Carlos III and co-funding by Fondo Europeo de Desarrollo Regional FEDER). D.C.-F. is supported by a Fellowship from “la Caixa” Foundation (LCF/BQ/DR19/11740010). I.F.-D. is supported by a Fellowship from the Spanish Ministry of Science, Innovation, and Universities (FPU15/02539). The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Spanish Ministry of Economy and Competitiveness (MINECO) and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505). Funding agencies did not intervene in the design of the studies, with no copyright over the study. Author contributions: A.A.-S., E.B.-M., I.F.-D., D.C.-F., and F.S.-M. designed most experimentation and analyzed results; D.T. helped with the collection of some data and with project design. E.M.-Z. and A.R. collaborated in the ChIP-qPCR approach. E.L.-V., E.V., and A.D. participated in the collection of the genomic and epigenomic data. M.J.G. and F.S.-C. participated in the bioinformatics analysis of the genomic and epigenomic data. J.B.A., N.R., and J.V.S. participated in the under-agarose assay. A.A.-S., E.B.-M., I.F.-D., and D.C.-F. made the figures and wrote the manuscript with input from the rest of the authors. F.S.-M. supervised and revised all the work. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-seq and ATAC-seq presented in this study were deposited in Gene Expression Omnibus (GEO) under accession code GSE162361 and are available at www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162361. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabb9965/DC1
REFERENCES AND NOTES
- 1.Dustin M. L., Choudhuri K., Signaling and polarized communication across the T cell immunological synapse. Annu. Rev. Cell Dev. Biol. 32, 303–325 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Alvarez D., Vollmann E. H., von Andrian U. H., Mechanisms and consequences of dendritic cell migration. Immunity 29, 325–342 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Worbs T., Hammerschmidt S. I., Forster R., Dendritic cell migration in health and disease. Nat. Rev. Immunol. 17, 30–48 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Riol-Blanco L., Delgado-Martín C., Sánchez-Sánchez N., Alonso-C L. M., Gutiérrez-López M. D., del Hoyo G. M., Navarro J., Sánchez-Madrid F., Cabañas C., Sánchez-Mateos P., Rodríguez-Fernández J. L., Immunological synapse formation inhibits, via NF-κB and FOXO1, the apoptosis of dendritic cells. Nat. Immunol. 10, 753–760 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Benvenuti F., The dendritic cell synapse: A life dedicated to T cell activation. Front. Immunol. 7, 70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gómez-Cabañas L., López-Cotarelo P., Criado-García O., Murphy M. P., Boya P., Rodríguez-Fernández J. L., Immunological synapse formation induces mitochondrial clustering and mitophagy in dendritic cells. J. Immunol. 202, 1715–1723 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Boes M., Cerny J., Massol R., op den Brouw M., Kirchhausen T., Chen J., Ploegh H. L., T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature 418, 983–988 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Foster N., Turnbull E. L., Macpherson G., Migrating lymph dendritic cells contain intracellular CD40 that is mobilized to the immunological synapse during interactions with antigen-specific T lymphocytes. J. Immunol. 189, 5632–5637 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Torralba D., Baixauli F., Villarroya-Beltri C., Fernández-Delgado I., Latorre-Pellicer A., Acín-Pérez R., Martín-Cófreces N. B., Jaso-Tamame Á. L., Iborra S., Jorge I., González-Aseguinolaza G., Garaude J., Vicente-Manzanares M., Enríquez J. A., Mittelbrunn M., Sánchez-Madrid F., Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat. Commun. 9, 2658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mittelbrunn M., Gutiérrez-Vázquez C., Villarroya-Beltri C., González S., Sánchez-Cabo F., González M. Á., Bernad A., Sánchez-Madrid F., Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2, 282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurts C., Robinson B. W., Knolle P. A., Cross-priming in health and disease. Nat. Rev. Immunol. 10, 403–414 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Garg S., Oran A., Wajchman J., Sasaki S., Maris C. H., Kapp J. A., Jacob J., Genetic tagging shows increased frequency and longevity of antigen-presenting, skin-derived dendritic cells in vivo. Nat. Immunol. 4, 907–912 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Schiavoni G., Mattei F., Gabriele L., Type I interferons as stimulators of DC-mediated cross-priming: Impact on anti-tumor response. Front. Immunol. 4, 483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Meer J. W., Joosten L. A., Riksen N., Netea M. G., Trained immunity: A smart way to enhance innate immune defence. Mol. Immunol. 68, 40–44 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Gardiner C. M., Mills K. H., The cells that mediate innate immune memory and their functional significance in inflammatory and infectious diseases. Semin. Immunol. 28, 343–350 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Dominguez-Andres J., Joosten L. A., Netea M. G., Induction of innate immune memory: The role of cellular metabolism. Curr. Opin. Immunol. 56, 10–16 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Ritter U., Wiede F., Mielenz D., Kiafard Z., Zwirner J., Körner H., Analysis of the CCR7 expression on murine bone marrow-derived and spleen dendritic cells. J. Leukoc. Biol. 76, 472–476 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Oxenius A., Bachmann M. F., Ashton-Rickardt P. G., Tonegawa S., Zinkernagel R. M., Hengartner H., Presentation of endogenous viral proteins in association with major histocompatibility complex class II: On the role of intracellular compartmentalization, invariant chain and the TAP transporter system. Eur. J. Immunol. 25, 3402–3411 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Packard R. R., Maganto-García E., Gotsman I., Tabas I., Libby P., Lichtman A. H., CD11c+ dendritic cells maintain antigen processing, presentation capabilities, and CD4+ T-cell priming efficacy under hypercholesterolemic conditions associated with atherosclerosis. Circ. Res. 103, 965–973 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tscharke D. C., Karupiah G., Zhou J., Palmore T., Irvine K. R., Haeryfar S. M. M., Williams S., Sidney J., Sette A., Bennink J. R., Yewdell J. W., Identification of poxvirus CD8+ T cell determinants to enable rational design and characterization of smallpox vaccines. J. Exp. Med. 201, 95–104 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saliba D. G., Céspedes-Donoso P. F., Bálint Š., Compeer E. B., Korobchevskaya K., Valvo S., Mayya V., Kvalvaag A., Peng Y., Dong T., Tognoli M. L., O’Neill E., Bonham S., Fischer R., Kessler B. M., Dustin M. L., Composition and structure of synaptic ectosomes exporting antigen receptor linked to functional CD40 ligand from helper T cells. eLife 8, e47528 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez R. M., Suarez-Alvarez B., Lopez-Larrea C., Therapeutic epigenetic reprogramming of trained immunity in myeloid cells. Trends Immunol. 40, 66–80 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Helft J., Böttcher J., Chakravarty P., Zelenay S., Huotari J., Schraml B. U., Goubau D., Reis e Sousa C., GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c+MHCII+ macrophages and dendritic cells. Immunity 42, 1197–1211 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Vander Lugt B., Khan A. A., Hackney J. A., Agrawal S., Lesch J., Zhou M., Lee W. P., Park S., Xu M., DeVoss J., Spooner C. J., Chalouni C., Delamarre L., Mellman I., Singh H., Transcriptional programming of dendritic cells for enhanced MHC class II antigen presentation. Nat. Immunol. 15, 161–167 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Laufer J. M., Kindinger I., Artinger M., Pauli A., Legler D. F., CCR7 is recruited to the immunological synapse, acts as co-stimulatory molecule and drives LFA-1 clustering for efficient T cell adhesion through ZAP70. Front. Immunol. 9, 3115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Sanchez N., Riol-Blanco L., Rodriguez-Fernandez J. L., The multiple personalities of the chemokine receptor CCR7 in dendritic cells. J. Immunol. 176, 5153–5159 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Grakoui A., Bromley S. K., Sumen C., Davis M. M., Shaw A. S., Allen P. M., Dustin M. L., The immunological synapse: A molecular machine controlling T cell activation. Science 285, 221–227 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Stone J. D., Chervin A. S., Kranz D. M., T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 126, 165–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heit B., Kubes P., Measuring chemotaxis and chemokinesis: The under-agarose cell migration assay. Sci. STKE 2003, PL5 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabb9965/DC1