

In vitro conjugate vaccine expression technology enables rapid and portable biosynthesis of protective antibacterial vaccines.
Abstract
Conjugate vaccines are among the most effective methods for preventing bacterial infections. However, existing manufacturing approaches limit access to conjugate vaccines due to centralized production and cold chain distribution requirements. To address these limitations, we developed a modular technology for in vitro conjugate vaccine expression (iVAX) in portable, freeze-dried lysates from detoxified, nonpathogenic Escherichia coli. Upon rehydration, iVAX reactions synthesize clinically relevant doses of conjugate vaccines against diverse bacterial pathogens in 1 hour. We show that iVAX-synthesized vaccines against Francisella tularensis subsp. tularensis (type A) strain Schu S4 protected mice from lethal intranasal F. tularensis challenge. The iVAX platform promises to accelerate development of new conjugate vaccines with increased access through refrigeration-independent distribution and portable production.
INTRODUCTION
Drug-resistant bacteria are predicted to threaten up to 10 million lives per year by 2050 (1), necessitating new strategies to develop and distribute antibiotics and vaccines. Conjugate vaccines, typically composed of a pathogen-specific capsular (CPS) or O-antigen polysaccharide (O-PS) linked to an immunostimulatory protein carrier, are among the safest and most effective methods for preventing life-threatening bacterial infections (2–4). In particular, implementation of meningococcal and pneumococcal conjugate vaccines has significantly reduced the occurrence of bacterial meningitis and pneumonia worldwide (5, 6), in addition to reducing the prevalence of antibiotic resistant infections (7). However, despite their proven safety and efficacy, global childhood vaccination rates for conjugate vaccines remain as low as ~30%, with lack of access or low immunization coverage accounting for the vast majority of the remaining disease burden (8). In addition, the 2018 World Health Organization prequalification of Typhbar-TCV to prevent typhoid fever represents the first conjugate vaccine approval in nearly a decade. To address emerging drug-resistant pathogens, new platform technologies to accelerate the development and global distribution of conjugate vaccines are urgently needed.
Contributing to the slow pace of conjugate vaccine development and distribution is the fact that these molecules are particularly challenging and costly to manufacture. The conventional process to produce conjugate vaccines involves chemical conjugation of carrier proteins with polysaccharide antigens purified from large-scale cultures of pathogenic bacteria. Large-scale fermentation of pathogens results in high manufacturing costs due to associated biosafety hazards and process development challenges. In addition, chemical conjugation can alter the structure of the polysaccharide, resulting in loss of the protective epitope (9). To address these challenges, it was recently demonstrated that polysaccharide-protein conjugates can be made in Escherichia coli using protein glycan coupling technology (PGCT) (10). In this approach, engineered E. coli cells covalently attach heterologously expressed CPS or O-PS antigens to carrier proteins via an asparagine-linked glycosylation reaction catalyzed by the Campylobacter jejuni oligosaccharyltransferase enzyme PglB (CjPglB) (11–17). Despite this advance, both chemical conjugation and PGCT approaches rely on living bacterial cells, requiring centralized production facilities from which vaccines are distributed via a refrigerated supply chain.
Refrigeration of conjugate vaccines is critical to avoid spoilage due to aggregate formation and loss of the pathogen-specific polysaccharide upon heating and freezing (18–23). Because of complexities and costs associated with cold chain refrigeration, vaccines with even short-term thermostability offer substantial advantages. The availability of effective and thermostable freeze-dried vaccines is cited as a key technological innovation that enabled the global eradication of smallpox (24). Development of MenAfriVac, a meningococcal conjugate vaccine shown to remain active outside of the cold chain for up to 4 days, enabled increased vaccine coverage and an estimated 50% reduction in costs during vaccination in the meningitis belt of sub-Saharan Africa (25). However, this required a sizable ($70 million) investment in the development and validation of a thermostable vaccine. Furthermore, conjugate vaccine thermostability varies for different O-PS antigens both across pathogens and between serotypes of the same pathogen, even in lyophilized formulations (20–23). Thus, generalizable strategies to achieve thermostability in the context of current manufacturing and distribution strategies may prove elusive. Broadly, the need for cold chain refrigeration creates economic and logistical challenges that limit the reach of vaccination campaigns and present barriers to the eradication of disease, especially in low- and middle-income countries (8, 26).
Cell-free protein synthesis (CFPS) offers opportunities to both accelerate vaccine development and enable decentralized, cold chain–independent biomanufacturing by using cell lysates, rather than living cells, to synthesize proteins in vitro (27). CFPS platforms (i) enable point-of-care protein production, as relevant amounts of protein can be synthesized in vitro within a few hours, (ii) can be freeze-dried for distribution at ambient temperature and reconstituted by just adding water (28), and (iii) circumvent biosafety concerns associated with the use of living cells outside of a controlled laboratory setting. CFPS has recently been used to enable on-demand and portable production of aglycosylated protein subunit vaccines (28, 29). However, the production of efficacious glycoprotein products, which represent 70% of approved therapeutics (30), from decentralized biomanufacturing platforms has not yet been demonstrated. As a result, there remains a need for additional technologies that enable decentralized production of glycosylated protein products, including conjugate vaccines.
To address this technological gap, here, we describe the iVAX (in vitro conjugate vaccine expression) platform that enables rapid development and cold chain–independent biosynthesis of conjugate vaccines in cell-free reactions (Fig. 1). iVAX was designed to have the following features. First, iVAX is fast, with the ability to produce multiple conjugate vaccine doses in 1 hour. Second, iVAX is robust, yielding equivalent amounts of conjugate over a range of operating temperatures. Third, iVAX is modular, offering the ability to rapidly interchange carrier proteins, including those used in licensed conjugate vaccines, as well as conjugated polysaccharide antigens. We leverage this modularity to create an array of vaccine candidates targeted against diverse bacterial pathogens, including the highly virulent Francisella tularensis subsp. tularensis (type A) strain Schu S4, enterotoxigenic (ETEC) E. coli O78, and uropathogenic (UPEC) E. coli O7. Fourth, iVAX is shelf-stable, derived from freeze-dried cell-free reactions that operate in a just-add-water strategy. Fifth, iVAX is safe, leveraging lipid A engineering that effectively avoids the high levels of endotoxin present in wild-type (WT) E. coli. Our results demonstrate that an F. tularensis O-PS conjugate derived from freeze-dried, low-endotoxin iVAX reactions outperformed a conjugate produced using the established cell-based PGCT approach in its ability to elicit pathogen-specific antibodies. Moreover, the iVAX-derived conjugate afforded complete protection in a mouse model of intranasal F. tularensis infection. Overall, the iVAX platform offers a new way to deliver the protective benefits of an important class of antibacterial vaccines to both the developed and developing world.
Fig. 1. iVAX platform enables on-demand and portable production of antibacterial vaccines.
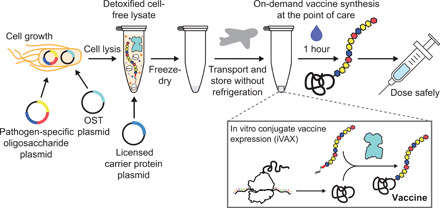
The iVAX platform provides a rapid means to develop and distribute conjugate vaccines against bacterial pathogens. Expression of pathogen-specific polysaccharide antigens (e.g., CPS and O-PS) and a bacterial oligosaccharyltransferase enzyme in nonpathogenic E. coli with detoxified lipid A yields low-endotoxin lysates containing all of the machinery required for synthesis of conjugate vaccines. Reactions catalyzed by iVAX lysates can be used to produce conjugates containing licensed carrier proteins and can be freeze-dried without loss of activity for refrigeration-free transportation and storage. Freeze-dried reactions can be activated at the point of care via simple rehydration and used to reproducibly synthesize immunologically active conjugate vaccines in ~1 hour.
RESULTS
In vitro synthesis of licensed vaccine carrier proteins
To demonstrate proof of principle for cell-free conjugate vaccine production, we first set out to express a set of carrier proteins that are currently used in approved conjugate vaccines. Producing these carrier proteins in soluble conformations in vitro represented an important benchmark because their expression in living E. coli has proven challenging, often requiring multistep purification and refolding of insoluble product from inclusion bodies (31, 32), fusion of expression partners such as maltose-binding protein (MBP) to increase soluble expression (32, 33), or expression of truncated protein variants in favor of the full-length proteins (33). In contrast, CFPS approaches have recently shown promise for difficult-to-express proteins (34). Carrier proteins of interest included nonacylated Haemophilus influenzae protein D (PD), the Neisseria meningitidis porin protein (PorA), and genetically detoxified variants of the Corynebacterium diphtheriae toxin (CRM197) and the Clostridium tetani toxin (TT). We also tested expression of the fragment C (TTc) and light chain (TTlight) domains of TT as well as E. coli MBP. While MBP is not a licensed carrier, it has demonstrated immunostimulatory properties (35) and, when linked to O-PS, was found to elicit polysaccharide-specific humoral and cellular immune responses in mice (13). Similarly, the TT domains, TTlight and TTc, have not been used in approved vaccines but are immunostimulatory and individually sufficient for protection against C. tetani challenge in mice (33). To enable conjugation with O-PS antigens, all carriers were modified at their C termini with four tandem repeats of an optimized bacterial glycosylation motif, DQNAT (36). A C-terminal 6×His-tag was also included to enable purification and detection via Western blot analysis. A variant of superfolder green fluorescent protein that contained an internal DQNAT glycosylation site (sfGFP217-DQNAT) (37) was used as a model protein to facilitate system development.
All eight carriers were synthesized in vitro with soluble yields of ~50 to 650 μg ml−1 as determined by 14C-leucine incorporation (Fig. 2A). In particular, the MBP4xDQNAT and PD4xDQNAT variants were nearly 100% soluble, with yields of 500 μg ml−1 and 200 μg ml−1, respectively, and expressed as exclusively full-length products according to Western blot and autoradiogram analysis (Fig. 2C and fig. S1A). Notably, similar soluble yields were observed for all carriers at 25°, 30°, and 37°C, with the exception of CRM1974xDQNAT (fig. S1B), which is known to be heat sensitive (19). These results suggest that our method of cell-free carrier biosynthesis is robust over a 13°C range in temperature and could be used in settings where precise temperature control is not feasible.
Fig. 2. In vitro synthesis of licensed conjugate vaccine carrier proteins.
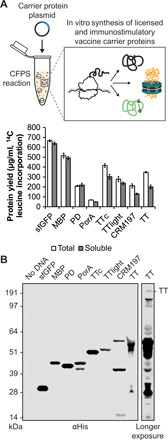
(A) All four carrier proteins used in approved conjugate vaccines were synthesized solubly in vitro, as measured via 14C-leucine incorporation. These include H. influenzae PD, the N. meningitidis porin protein (PorA), and genetically detoxified variants of the C. diphtheriae toxin (CRM197) and the C. tetani toxin (TT). Additional immunostimulatory carriers, including E. coli MBP and the fragment C (TTc) and light chain (TTlight) domains of TT, were also synthesized solubly. Values represent means and error bars represent SDs of biological replicates (n = 3). (B) Full-length product was observed for all proteins tested via Western blot. Different exposures are indicated with solid lines. Molecular weight ladder is shown at left. αHis, anti–hexa-histidine antibody.
The open nature of our cell-free system enabled facile manipulation of the reaction environment to improve production of more complex carriers. For example, in the case of the membrane protein PorA4xDQNAT, lipid nanodiscs were added to increase soluble expression (fig. S1C). Nanodiscs provide a cellular membrane mimic to stabilize hydrophobic regions of membrane proteins (38). For expression of TT, which contains an intermolecular disulfide bond, expression was carried out for 2 hours in oxidizing conditions (39), which improved assembly of the heavy and light chains into full-length product (fig. S1D). Western blot analysis of in vitro synthesized CRM1974xDQNAT and TT4xDQNAT revealed species that were comparable in size to commercially available purified diphtheria toxin (DT) and TT protein standards (fig. S1E). Lower–molecular weight species were also observed, which we hypothesize result from protease degradation of full-length CRM1974xDQNAT and TT4xDQNAT following translation. These species could be mitigated by incorporating a size-based separation step into decentralized production, as previously described (29). In addition, full-length CRM1974xDQNAT and TT4xDQNAT were reactive with αDT and αTT antibodies, respectively (fig. S1F), indicating that immunogenic epitopes were faithfully synthesized. This is notable as CRM197 and TT are U.S. Food and Drug Administration (FDA)–approved vaccine antigens for diphtheria and tetanus, respectively, when they are administered without conjugated polysaccharides. Together, our results highlight the ability of CFPS to express licensed conjugate vaccine carrier proteins in soluble conformations over a range of temperatures.
On-demand biosynthesis of conjugate vaccines
We next sought to synthesize polysaccharide-conjugated versions of these carrier proteins by coactivating their in vitro expression with cell-free glycosylation in a one-pot reaction. As a model vaccine target, we focused on the highly virulent F. tularensis subsp. tularensis (type A) strain Schu S4, a gram-negative, facultative coccobacillus and the causative agent of tularemia. This bacterium is categorized as a class A bioterrorism agent due to its high fatality rate, low dose of infection, and ability to be aerosolized (40, 41). Vaccines targeting F. tularensis will likely need to be deployed rapidly using ring vaccination strategies in response to an outbreak, similar to those planned in the event of a smallpox attack (42) and used to eradicate smallpox in the 1960s and 1970s (24). We thus sought to develop rapidly deployable, thermostable conjugate vaccines against F. tularensis as an initial demonstration of the iVAX technology.
Although there are currently no approved vaccines against F. tularensis, several studies have independently confirmed the important role of antibodies directed against F. tularensis lipopolysaccharide (LPS), specifically the O-PS repeat unit, in providing protection against the Schu S4 strain (43, 44). More recently, a conjugate vaccine comprising the F. tularensis Schu S4 O-PS (FtO-PS) conjugated to the Pseudomonas aeruginosa exotoxin A (EPADNNNS-DQNRT) carrier protein produced using PGCT (14, 15) was shown to be protective against challenge with the Schu S4 strain in a rat inhalation model of tularemia (15). In light of these earlier findings, we investigated the ability of the iVAX platform to produce anti–F. tularensis conjugate vaccine candidates on-demand by conjugating the FtO-PS structure to diverse carrier proteins in vitro.
The FtO-PS is composed of the 826-Da repeating tetrasaccharide unit Qui4NFm-(GalNAcAN)2-QuiNAc (Qui4NFm: 4,6-dideoxy-4-formamido-d-glucose; GalNAcAN: 2-acetamido-2-deoxy-d-galacturonamide; QuiNAc: 2-acetamido-2,6-dideoxy-d-glucose) (45). We and other groups have previously shown that the authentic FtO-PS structure can be synthesized in K-12 strains of E. coli via recombinant expression of the FtO-PS biosynthetic pathway (14, 45, 46). To glycosylate proteins with FtO-PS, we produced an iVAX lysate from E. coli cells expressing the FtO-PS biosynthetic pathway and the oligosaccharyltransferase enzyme CjPglB (Fig. 3A). This lysate, which contained lipid-linked FtO-PS and active CjPglB, was used to catalyze iVAX reactions primed with plasmid DNA encoding sfGFP217-DQNAT. Control reactions, in which attachment of the FtO-PS was not expected, were performed with lysates from cells that lacked either the FtO-PS pathway or the CjPglB enzyme. We also tested reactions that lacked plasmid encoding the target protein sfGFP217-DQNAT or were primed with plasmid encoding sfGFP217-AQNAT, which contained a mutated glycosylation site (AQNAT) that is not modified by CjPglB (47). In reactions containing the iVAX lysate and primed with plasmid encoding sfGFP217-DQNAT, immunoblotting with anti-His antibody or a commercial monoclonal antibody specific to FtO-PS revealed a ladder-like banding pattern (Fig. 3B). This ladder is characteristic of FtO-PS attachment, resulting from O-PS chain length variability through the action of the Wzy polymerase (10, 14, 45). Glycosylation of sfGFP217-DQNAT was observed only in reactions containing a complete glycosylation pathway and the preferred DQNAT glycosylation sequence (Fig. 3B). This glycosylation profile was further reproducible across biological replicates from the same lot of lysate (Fig. 3C, left) and using different lots of lysate (Fig. 3C, right), with an average efficiency of conjugation with FtO-PS of 69 ± 5% by densitometry analysis. In vitro protein synthesis and glycosylation was observed after 1 hour, with the amount of conjugated polysaccharide reaching a maximum between 45 and 75 min (fig. S2). Similar glycosylation reaction kinetics were observed at 37°C, 30°C, 25°C, and room temperature (~21°C), indicating that iVAX reactions are robust over a range of temperatures (fig. S2).
Fig. 3. Reproducible glycosylation of proteins with FtO-PS in iVAX.
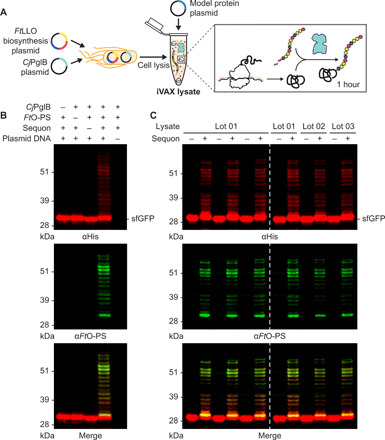
(A) iVAX lysates were prepared from cells expressing CjPglB and the biosynthetic pathway encoding FtO-PS. (B) Glycosylation of sfGFP217-DQNAT with FtO-PS was only observed when CjPglB, FtO-PS, and the preferred DQNAT glycosylation sequence (sequon) were present in the reaction (lane 3). When plasmid DNA was omitted, sfGFP217-DQNAT synthesis was not observed. (C) Biological replicates of iVAX reactions producing sfGFP217-DQNAT using the same lot (left) or different lots (right) of iVAX lysates demonstrated reproducibility of reactions and lysate preparation. Top panels show signal from probing with αHis to detect the carrier protein, middle panels show signal from probing with commercial anti–FtO-PS antibody (αFtO-PS), and bottom panels show αHis and αFtO-PS signals merged. Images are representative of at least three biological replicates. Dashed lines indicate that samples are from nonadjacent lanes of the same blot with the same exposure. Molecular weight ladders are shown at the left of each image.
Next, we investigated whether immunologically relevant carriers could be similarly conjugated with FtO-PS in iVAX reactions. Following addition of plasmid DNA encoding MBP4xDQNAT, PD4xDQNAT, PorA4xDQNAT, TTc4xDQNAT, TTlight4xDQNAT, CRM1974xDQNAT, or the most common PGCT carrier protein, EPADNNNS-DQNRT (12, 14–17), glycosylation of each with FtO-PS was observed for iVAX reactions enriched with lipid-linked FtO-PS and CjPglB but not control reactions lacking CjPglB (Fig. 4). Notably, our attempts to synthesize the same panel of conjugates using the established PGCT approach in living E. coli yielded less promising results. Specifically, only limited expression of conjugates composed of PorA and CRM197, two of the carriers used in licensed conjugate vaccines, was achieved in vivo (fig. S3). Collectively, these data indicate that iVAX may provide advantages over the established PGCT approach for production of conjugate vaccine candidates composed of diverse and potentially membrane-bound carrier proteins.
Fig. 4. On-demand production of conjugate vaccines against F. tularensis using iVAX.
(A) iVAX reactions were prepared from lysates containing CjPglB and FtO-PS and primed with plasmid encoding immunostimulatory carriers, including those used in licensed vaccines. (B) We observed on-demand synthesis of anti-F. tularensis conjugate vaccines for all carrier proteins tested. Conjugates were purified using Ni-NTA agarose from 1-ml iVAX reactions lasting ~1 hour. Top panels show signal from probing with αHis to detect the carrier protein, middle panels show signal from probing with αFtO-PS, and bottom panels show αHis and αFtO-PS signals merged. Images are representative of at least three biological replicates. Dashed lines indicate that samples are from nonadjacent lanes of the same blot with the same exposure. Molecular weight ladders are shown at the left of each image.
We next asked whether the yields of conjugates produced using iVAX were sufficient to enable production of relevant vaccine doses. To assess expression titers, we focused on MBP4xDQNAT and PD4xDQNAT because these carriers expressed in vitro with high soluble titers and as exclusively full-length protein (Fig. 2 and fig. S1A). In addition, PD has been shown to be a safe and effective conjugate vaccine carrier protein (48, 49) and may have advantages over DT and TT in generating robust immune responses to polysaccharide antigens (50–53). Recent clinical data show that 1- to 10-μg doses of conjugate vaccine candidates are well tolerated and effective in stimulating the production of antibacterial antibodies (54–56). We found that reactions lasting ~1 hour produced ~20 μg ml−1, or two 10-μg doses ml−1, of FtO-PS–conjugated MBP4xDQNAT and PD4xDQNAT as determined by 14C-leucine incorporation and densitometry analysis (fig. S4A). It should be noted that vaccines are currently distributed in vials containing 1 to 20 doses of vaccine to minimize wastage (57). Our yields indicate that multiple doses per milliliter can be synthesized in 1 hour using the iVAX platform.
To demonstrate the modularity of the iVAX approach for conjugate vaccine production, we sought to produce conjugates bearing O-PS antigens from additional pathogens including ETEC E. coli strain O78 and UPEC E. coli strain O7. E. coli O78 is a major cause of diarrheal disease in low- and middle-income countries, especially among children, and a leading cause of traveler’s diarrhea (58), while the O7 strain is a common cause of urinary tract infections (59). Like the FtO-PS, the biosynthetic pathways for EcO78-PS and EcO7-PS have been described previously and confirmed to produce O-PS antigens with the repeating units GlcNAc2Man2 (60) and Qui4NAcMan(Rha)GalGlcNAc (61), respectively (GlcNAc: N-acetylglucosamine; Man: mannose; Qui4NAc: 4-acetamido-4,6-dideoxy-d-glucopyranose; Rha: rhamnose; Gal: galactose). Using iVAX lysates from cells expressing CjPglB and either the EcO78-PS and EcO7-PS pathways in reactions that were primed with PD4xDQNAT or sfGFP217-DQNAT plasmids, we observed O-PS conjugation only when both lipid-linked O-PS and CjPglB were present in the reactions (fig. S4, B and C). These results demonstrate modular production of conjugate vaccines against multiple bacterial pathogens using iVAX, enabled by compatibility of multiple heterologous O-PS pathways with in vitro carrier protein synthesis and glycosylation.
Endotoxin editing and freeze-drying yield iVAX reactions that are safe and portable
A key challenge in using any E. coli–based system for biopharmaceutical production is the presence of lipid A, or endotoxin, which is known to contaminate protein products and can cause lethal septic shock (62). As a result, the amount of endotoxin in formulated biopharmaceuticals is regulated by the U.S. FDA, as well as the European Medicines Agency (63). Because iVAX reactions rely on lipid-associated components, such as CjPglB and FtO-PS, standard detoxification approaches involving the removal of lipid A (64) could compromise the activity or concentration of our glycosylation components, in addition to increasing cost and processing complexities. Thus, there is a need for detoxification of the cell-free reactions from a quality-by-design perspective.
To address this issue, we adapted a previously reported strategy to detoxify the lipid A molecule through strain engineering (46, 65). In particular, the deletion of the acyltransferase gene lpxM and the overexpression of the F. tularensis phosphatase LpxE in E. coli have been shown to result in the production of nearly homogeneous pentaacylated, monophosphorylated lipid A with significantly reduced toxicity but retained adjuvanticity (46). This pentaacylated, monophosphorylated lipid A is structurally identical to the primary component of monophosphoryl lipid A (MPL) from Salmonella minnesota R595, an approved adjuvant composed of a mixture of monophosphorylated lipids (66). To generate detoxified lipid A structures in the context of iVAX, we produced lysates from a ∆lpxM derivative of CLM24 that coexpressed FtLpxE and the FtO-PS glycosylation pathway (Fig. 5A). The growth rate of CLM24 ∆lpxM was indistinguishable from the WT strain, facilitating scale-up of fermentations for production of reduced-endotoxin lysates (Fig. 5B). Lysates derived from this strain exhibited significantly decreased levels of toxicity compared to WT CLM24 lysates expressing CjPglB and FtO-PS (Fig. 5C) as measured by human Toll-like receptor 4 (TLR4) activation in HEK-Blue hTLR4 reporter cells (65). The structural remodeling of lipid A did not affect the activity of the membrane-bound CjPglB and FtO-PS components in iVAX reactions (fig. S5A). By engineering the chassis strain for lysate production, we produced iVAX lysates with <1000 endotoxin units (EU) ml−1. This represents a 12.5-fold average reduction in endotoxin content compared to the WT lysate, which we suspected would ensure acceptable endotoxin levels in purified conjugate vaccines without the need for additional removal steps before administration. Indeed, FtO-PS conjugates synthesized and affinity-purified from detoxified iVAX lysates contained 0.21 ± 0.3 EU per 10-μg dose, which is well below endotoxin levels reported in commercial conjugate vaccines (<12 EU/dose) (Fig. 5D) (63, 67).
Fig. 5. Detoxified, lyophilized iVAX reactions produce conjugate vaccines.
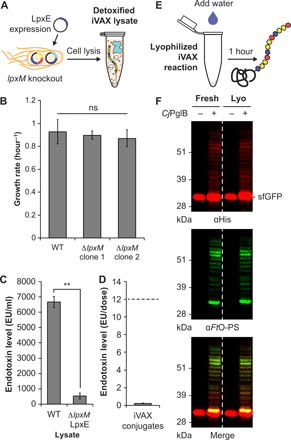
(A) iVAX lysates were detoxified via deletion of lpxM and expression of F. tularensis LpxE in the source strain used for lysate production. (B) Growth rates of CLM24 WT and CLM24 ∆lpxM strains. No growth defects were observed across two clones of the knockout strain. Values represent means and error bars represent SDs of n = 4 replicates. ns, not significant. (C) The resulting lysates exhibited significantly reduced endotoxin activity, as measured by activation of human TLR4 in HEK-Blue hTLR4 reporter cells. **P = 0.003, as determined by two-tailed t test. Values represent means and error bars represent SDs of n = 3 replicates. (D) FtO-PS conjugate vaccines produced and purified from detoxified iVAX reactions contained 0.21 ± 0.3 EU/10-μg dose, as measured by human TLR4 activation. Dashed line represents endotoxin levels reported in commercial conjugate vaccines (<12 EU/dose). Value represents mean and error bars represent SD of n = 6 replicates. (E) iVAX reactions producing sfGFP217-DQNAT were run immediately or following lyophilization and rehydration. (F) Glycosylation activity was preserved following lyophilization, demonstrating the potential of iVAX reactions for portable biosynthesis of conjugate vaccines. Top panel shows signal from probing with αHis to detect the carrier protein, middle panel shows signal from probing with αFtO-PS, and bottom panel shows αHis and αFtO-PS signals merged. Images are representative of at least three biological replicates. Molecular weight ladder is shown at the left of each image.
A major limitation of traditional conjugate vaccines is that they must be refrigerated (19), making it difficult to distribute these vaccines to remote or resource-limited settings. The ability to freeze-dry iVAX reactions for ambient temperature storage and distribution could alleviate the logistical challenges associated with refrigerated supply chains that are required for existing vaccines. To investigate this possibility, detoxified iVAX lysates were used to produce FtO-PS conjugates in two different ways: either by running the reaction immediately after priming with plasmid encoding the sfGFP217-DQNAT target protein or by running after the same reaction mixture was lyophilized and rehydrated (Fig. 5E). In both cases, conjugation of FtO-PS to sfGFP217-DQNAT was observed when CjPglB was present, with similar modification efficiencies (average glycosylation efficiency of sfGFP217-DQNAT in reactions with and without lyophilization was 66 ± 7% and 69 ± 5% by densitometry, respectively) (Figs. 3C and 5F and fig. S6). We also showed that detoxified, freeze-dried iVAX reactions can be scaled to 5 ml for production of FtO-PS–conjugated MBP4xDQNAT and PD4xDQNAT in a manner that was reproducible from lot to lot and indistinguishable from production without freeze-drying (average glycosylation efficiencies of MBP4xDQNAT and PD4xDQNAT were 66 ± 10% and 70 ± 1% by densitometry, respectively) (fig. S5, B and C). In addition, freeze-dried reactions are stable under ambient temperature storage for at least 3 months, with no observable differences in protein synthesis or glycosylation activity (fig. S6). The ability to lyophilize iVAX reactions, store reactions at ambient temperature, and manufacture conjugate vaccines at increasing scales and without specialized equipment highlights the potential for portable, on-demand vaccine production.
In vitro synthesized conjugates elicit pathogen-specific antibodies in mice
We next evaluated the ability of iVAX-derived conjugates to elicit anti-FtLPS antibodies in mice (Fig. 6A). We found that BALB/c mice receiving iVAX-derived FtO-PS–conjugated MBP4xDQNAT or PD4xDQNAT produced high titers of FtLPS-specific IgG antibodies, which were significantly elevated compared to the titers measured in the sera of control mice receiving phosphate-buffered saline (PBS) or unmodified versions of each carrier protein (Fig. 6B and fig. S7). The IgG titers measured in sera from mice receiving PGCT-derived MBP4xDQNAT conjugates were similar to the titers observed in the control groups (Fig. 6B and fig. S7). Notably, both MBP4xDQNAT and PD4xDQNAT conjugates produced using iVAX elicited similar levels of IgG production and neither resulted in any observable adverse events in mice, confirming the modularity and safety of the technology for production of conjugate vaccine candidates.
Fig. 6. iVAX-derived conjugates elicit FtLPS-specific antibodies and protect mice from lethal pathogen challenge.
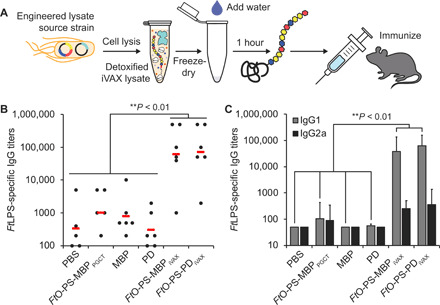
(A) Freeze-dried iVAX reactions assembled using detoxified lysates were used to synthesize anti–F. tularensis conjugate vaccines for immunization studies. (B) Groups of BALB/c mice were immunized subcutaneously with PBS or 7.5 μg of purified, cell-free synthesized unmodified or FtO-PS–conjugated carrier proteins. FtO-PS–conjugated MBP4xDQNAT prepared in living E. coli cells using PCGT was used as a positive control. Each group was composed of six mice except for the PBS control group, which was composed of five mice. Mice were boosted on days 21 and 42 with identical doses of antigen. FtLPS-specific IgG titers were measured by enzyme-linked immunosorbent assay (ELISA) in endpoint (day 70) serum of individual mice (black dots) with F. tularensis LPS immobilized as antigen. Mean titers of each group are also shown (red lines). iVAX-derived conjugates elicited significantly higher levels of FtLPS-specific IgG compared to all other groups (**P < 0.01, Tukey-Kramer post hoc test). (C) IgG1 and IgG2a subtype titers measured by ELISA from endpoint serum revealed that iVAX-derived conjugates boosted production of FtO-PS–specific IgG1 compared to all other groups tested (**P < 0.01, Tukey-Kramer post hoc test). These results indicate that iVAX conjugates elicited a TH2-biased immune response typical of most conjugate vaccines. Values represent means and error bars represent SEs of FtLPS-specific IgGs detected by ELISA.
We further characterized IgG titers by analysis of IgG1 and IgG2a subtypes and found that both iVAX-derived FtO-PS–conjugated MBP4xDQNAT and PD4xDQNAT boosted production of IgG1 antibodies by more than two orders of magnitude relative to all control groups as well as to PGCT-derived MBP4xDQNAT conjugates (Fig. 6C). Observed IgG subclass titers elicited by iVAX-derived conjugates (IgG1 > IgG2a) are further consistent with a T helper 2 (TH2)–biased response, which is characteristic of most conjugate vaccines (68), although additional studies are needed to confirm this immunological phenotype. Together, these results provide evidence that the iVAX platform supplies vaccine candidates that are capable of eliciting strong, pathogen- and polysaccharide-specific humoral immune responses.
iVAX-derived vaccines protect mice from lethal intranasal F. tularensis challenge
Lastly, we tested the ability of iVAX-derived vaccines to protect mice in an intranasal model of F. tularensis infection (Fig. 7A). We selected an intranasal infection model because it represents the most challenging and relevant route of infection in potential bioterrorism attacks (69, 70). Mice were immunized with either iVAX- or PGCT-derived MBP4xDQNAT, PD4xDQNAT, or EPADNNNS-DQNRT conjugates, as well as unmodified controls. Notably, across all three carrier proteins, only iVAX-derived vaccines elicited FtO-PS–specific antibody titers that were significantly higher than those measured in the PBS immunized control group (Fig. 7B). All immunized mice were challenged intranasally with 6000 colony-forming units (CFU) [60 times the intranasal median lethal dose (LD50)] of the virulent F. tularensis subsp. holarctica LVS Rocky Mountain Laboratories. All iVAX-derived vaccine candidates provided complete protection against intranasal challenge, which was indistinguishable from the protection conferred by PGCT-derived versions of the same vaccines (Fig. 7, C to E). These results demonstrate that the iVAX platform produces protective vaccine candidates that are at least as effective as those produced using a state-of-the-art biomanufacturing approach.
Fig. 7. iVAX-derived conjugates protect mice from lethal F. tularensis challenge.
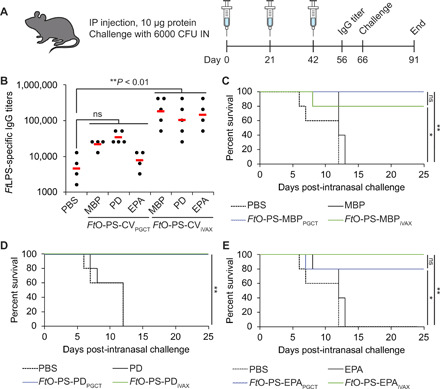
(A) Groups of five BALB/c mice were immunized intraperitoneally (IP) with PBS or 10 μg of purified, cell-free synthesized unmodified or FtO-PS–conjugated carrier proteins. FtO-PS–conjugated carriers prepared in living E. coli cells using PCGT were used as positive controls. Mice were boosted on days 21 and 42 with identical doses of antigen. IN, intranasally. (B) On day 56, FtLPS-specific IgG titers were measured by ELISA in serum of individual mice immunized with PBS or anti–F. tularensis conjugate vaccines (FtO-PS-CV) (black dots) with F. tularensis LPS immobilized as antigen. Mean titers of each group are also shown (red lines). Only iVAX-derived conjugates elicited significantly higher levels of FtLPS-specific IgG compared to PBS immunized controls across all carrier proteins tested (**P < 0.01, Tukey-Kramer post hoc test; CV, conjugate vaccine). On day 66, mice were challenged intranasally with 6000 CFU (60 times the intranasal LD50) F. tularensis subsp. holarctica LVS Rocky Mountain Laboratories and monitored for survival for an additional 25 days. Kaplan-Meier curves for immunizations with (C) MBP4xDQNAT, (D) PD4xDQNAT, and (E) EPADNNNS-DQNRT as the carrier protein are shown. iVAX-derived vaccines protected mice from lethal pathogen challenge as effectively as vaccines synthesized using the state-of-the-art PGCT approach. *P < 0.05; **P < 0.01, Fisher’s exact test.
DISCUSSION
In this work, we have established iVAX, a cell-free platform for portable, on-demand, and scalable production of protective conjugate vaccines. We show that iVAX reactions can be detoxified to ensure the safety of conjugate vaccine products, freeze-dried for cold chain–independent distribution, and reactivated for high-yielding conjugate production by simply adding water. As a model vaccine candidate, we show that anti–F. tularensis conjugates produced in iVAX elicited pathogen-specific IgG antibodies and protected mice from lethal intranasal challenge with F. tularensis. Broadly, iVAX has the potential to facilitate conjugate vaccine distribution and increase vaccination rates by reducing reliance on refrigerated supply chains.
The iVAX platform has several important features. First, iVAX is modular, which we have demonstrated through the interchangeability of (i) carrier proteins, including those used in licensed conjugate vaccines, and (ii) bacterial O-PS antigens from F. tularensis subsp. tularensis (type A) Schu S4, ETEC E. coli O78, and UPEC E. coli O7. iVAX is the first example of enrichment of large and polymeric O-antigen carbohydrates as substrates for cell-free protein glycosylation. Moreover, to our knowledge, this work represents the first demonstration of oligosaccharyltransferase-mediated O-PS conjugation to the carrier proteins used in licensed conjugate vaccine formulations, likely due to historical challenges associated with the expression of approved carriers in living E. coli (31–33). The modularity of iVAX has the potential to enable rapid development of conjugate vaccines against diverse bacteria as well as multiple serotypes of a single bacterial pathogen that can be co-formulated to yield multivalent conjugate vaccines. Furthermore, iVAX could accelerate efforts to develop alternative carrier proteins that address issues related to carrier-induced epitopic suppression. Specifically, immunity to DT and TT generated via existing vaccines is known to diminish immune responses to bacterial polysaccharides conjugated to these carriers (50–53). The modular iVAX platform promises to facilitate production and evaluation of additional carrier proteins in order to expand the repertoire of safe and effective conjugate vaccines that are compatible with preexisting immunity within the population.
Second, iVAX reactions are inexpensive, costing ~$12 ml−1 (table S1) with the ability to synthesize ~20-μg conjugate ml−1 in 1 hour (fig. S4A). Assuming a dose size of 10 μg, our iVAX reactions can produce a vaccine dose for ~$6. For comparison, the Centers for Disease Control and Prevention (CDC) cost per dose for conjugate vaccines ranges from ~$9.50 for the H. influenzae vaccine ActHIB to ~$75 and ~$118 for the meningococcal vaccine Menactra and pneumococcal vaccine Prevnar 13, respectively (71).
Third, while conjugates derived from both PGCT and iVAX protected mice from lethal challenge with F. tularensis LVS, iVAX-derived conjugates were significantly more effective at eliciting FtLPS-specific IgGs than those derived from PGCT in the context of multiple carrier proteins (Fig. 6, B and E). Achieving high titers of polysaccharide-specific antibodies is broadly recognized as a correlate of conjugate vaccine efficacy (20–23, 50–56). Given the importance of antibodies in protection against infection with the highly virulent Schu S4 strain (43, 44), and the fact that antibody titers wane over time, achieving higher initial titers of FtLPS-specific IgGs could provide protection against higher doses of pathogen and/or extend the duration of protection afforded by vaccination. Future comparative studies of iVAX- and PGCT-derived vaccines could identify immunogen features responsible for the enhanced FtLPS-specific IgGs elicited by iVAX-derived vaccines, revealing design rules for the production of more effective conjugate vaccines.
Fourth, iVAX addresses a key gap in both cell-free and decentralized biomanufacturing technologies. Production of glycosylated products has not yet been demonstrated in cell-based decentralized biomanufacturing platforms (72, 73), and existing cell-free platforms using E. coli lysates lack the ability to synthesize glycoproteins (28, 74–76). While glycosylated human erythropoietin has been produced in a cell-free biomanufacturing platform based on freeze-dried Chinese hamster ovary cell lysates, its in vivo efficacy was not evaluated and challenges achieving efficient glycosylation were noted (29). Furthermore, this and the vast majority of other eukaryotic cell-free and cell-based systems rely on endogenous protein glycosylation machinery, and so are not compatible with conjugation of bacterial O-PS antigens. In contrast, the iVAX platform is enabled by our previous work to activate glycosylation in E. coli lysates that lack endogenous protein glycosylation pathways, which allows bottom-up engineering of desired glycosylation activities (37). Here, we show that further development of this approach allows rapid and portable production of protective conjugate vaccines. This required (i) demonstration of modular cell-free synthesis and glycosylation of approved carrier proteins with O-PS antigens over a range of temperatures; (ii) rigorous assessment of reproducibility, scalability, and thermostability of reactions; (iii) detoxification of cell-free lysates; and (iv) evaluation of in vivo conjugate vaccine efficacy.
In summary, iVAX represents a platform technology for rapid development and on-demand, cold chain–independent biomanufacturing of conjugate vaccines. Conjugate vaccines are one of the safest and most effective methods for preventing bacterial infections (2–4), but their development and distribution is limited by current manufacturing approaches. The iVAX platform addresses these limitations and further provides the first example, to our knowledge, of efficacious glycoprotein product synthesis in a decentralized manufacturing platform. iVAX joins an emerging set of technologies (28, 29, 72–76) that have the potential to promote increased access to complex, life-saving drugs through decentralized production.
MATERIALS AND METHODS
Bacterial strains and plasmids
E. coli NEB 5-alpha (NEB) was used for plasmid cloning and purification. E. coli CLM24 or CLM24 ∆lpxM strains were used for preparing cell-free lysates. E. coli CLM24 was used as the chassis for expressing conjugates in vivo using PGCT. As a derivative of the W3110 E. coli K strain, CLM24 does not synthesize an endogenous O-PS antigen due to an inactivating mutation in the gene encoding the WbbL O-PS glycosyltransferase, enabling unimpeded biosynthesis of heterologous O-PS antigens on undecaprenyl diphosphate (UndPP). CLM24 further has a deletion in the gene encoding the WaaL ligase that transfers O-PS antigens from UndPP to lipid A, facilitating the accumulation of preassembled glycans on UndPP as substrates for CjPglB-mediated protein glycosylation (10). CLM24 ∆lpxM has an endogenous acyltransferase deletion and serves as the chassis strain for production of detoxified cell-free lysates.
All plasmids used in the study are listed in table S2. Plasmids pJL1-MBP4xDQNAT, pJL1-PD4xDQNAT, pJL1-PorA4xDQNAT, pJL1-TTc4xDQNAT, pJL1-TTlight4xDQNAT, pJL1-CRM1974xDQNAT, and pJL1-TT4xDQNAT were generated via polymerase chain reaction (PCR) amplification and subsequent Gibson assembly of a codon-optimized gene construct purchased from IDT with a C-terminal 4×DQNAT–6×His-tag (77) between the Nde I and Sal I restriction sites in the pJL1 vector. Plasmid pJL1-EPADNNNS-DQNRT was constructed using the same approach, but without the addition of a C-terminal 4×DQNAT–6×His-tag. Plasmids pTrc99s-ssDsbA-MBP4xDQNAT, pTrc99s-ssDsbA-PD4xDQNAT, pTrc99s-ssDsbA-PorA4xDQNAT, pTrc99s-ssDsbA-TTc4xDQNAT, pTrc99s-ssDsbA-TTlight4xDQNAT, and pTrc99s-ssDsbA-EPADNNNS-DQNRT were created via PCR amplification of each carrier protein gene and insertion into the pTrc99s vector between the Nco I and Hind III restriction sites via Gibson assembly. Plasmid pSF-CjPglB-LpxE was constructed using a similar approach, but via insertion of the lpxE gene from pE (65) between the Nde I and Nsi I restriction sites in the pSF vector. Inserts were amplified via PCR using Phusion High-Fidelity DNA polymerase (NEB) with forward and reverse primers designed using the NEBuilder Assembly Tool (www.nebuilder.neb.com) and purchased from IDT. The pJL1 vector (Addgene 69496) was digested using restriction enzymes Nde I and SalI-HF (NEB). The pSF vector was digested using restriction enzymes Nde I and Not I (NEB). PCR products were gel-extracted using the EZNA Gel Extraction Kit (Omega Bio-Tek), mixed with Gibson assembly reagents, and incubated at 50°C for 1 hour. Plasmid DNA from the Gibson assembly reactions was transformed into E. coli NEB 5-alpha cells, and circularized constructs were selected using kanamycin at 50 μg ml−1 (Sigma-Aldrich). Sequence-verified clones were purified using the EZNA Plasmid Midi Kit (Omega Bio-Tek) for use in CFPS and iVAX reactions.
Construction of CLM24 ∆lpxM strain
E. coli CLM24 ∆lpxM was generated using the Datsenko-Wanner gene knockout method (78). Briefly, CLM24 cells were transformed with the pKD46 plasmid encoding the λ red system. Transformants were grown to an OD600 (optical density at 600 nm) of 0.5 to 0.7 in 25 ml of LB-Lennox medium [tryptone (10 g liter−1), yeast extract (5 g liter−1), and NaCl (5 g liter−1)] with carbenicillin (50 μg ml−1) at 30°C, harvested and washed three times with 25 ml of ice-cold 10% glycerol to make them electrocompetent, and resuspended in a final volume of 100 μl of 10% glycerol. In parallel, an lpxM knockout cassette was generated by PCR amplifying the kanamycin resistance cassette from pKD4 with forward and reverse primers with homology to lpxM. Electrocompetent cells were transformed with 400 ng of the lpxM knockout cassette and plated on LB agar with kanamycin (30 μg ml−1) for selection of resistant colonies. Plates were grown at 37°C to cure cells of the pKD46 plasmid. Colonies that grew on kanamycin were confirmed to have acquired the knockout cassette via colony PCR and DNA sequencing. These confirmed colonies were then transformed with pCP20 to remove the kanamycin resistance gene via Flp-FRT recombination. Transformants were plated on LB agar with carbenicillin (50 μg ml−1). Following selection, colonies were grown in liquid culture at 42°C to cure cells of the pCP20 plasmid. Colonies were confirmed to have lost both lpxM and the knockout cassette via colony PCR and DNA sequencing and confirmed to have lost both kanamycin and carbenicillin resistance via replica plating on LB agar plates with carbenicillin (50 μg ml−1) or kanamycin. All primers used for construction and validation of this strain are listed in table S3.
Cell-free lysate preparation
E. coli CLM24 source strains were grown in 2xYTP medium [yeast extract (10 g/liter), tryptone (16 g/liter), NaCl (5 g/liter), K2HPO4 (7 g/liter), KH2PO4 (3 g/liter) (pH 7.2)] in shake flasks (1-liter scale) or a Sartorius Stedim BIOSTAT Cplus bioreactor (10-liter scale) at 37°C. To generate CjPglB-enriched lysate, CLM24 cells carrying plasmid pSF-CjPglB (79) were used as the source strain. To generate FtO-PS–enriched lysates, CLM24 carrying plasmid pGAB2 (14) was used as the source strain. To generate one-pot lysates containing both CjPglB and FtO-PS, EcO78-PS, or EcO7-PS, CLM24 carrying pSF-CjPglB and one of the following bacterial O-PS biosynthetic pathway plasmids was used as the source strain: pGAB2 (FtO-PS), pMW07-O78 (EcO78-PS), and pJHCV32 (EcO7-PS). CjPglB expression was induced at an OD600 of 0.8 to 1.0 with 0.02% (w/v) l-arabinose, and cultures were moved to 30°C. Cells were grown to a final OD600 of ~3.0, at which point cells were pelleted by centrifugation at 5000g for 15 min at 4°C. Cell pellets were then washed three times with cold S30 buffer [10 mM tris-acetate (pH 8.2), 14 mM magnesium acetate, 60 mM potassium acetate] and pelleted at 5000g for 10 min at 4°C. After the final wash, cells were pelleted at 7000g for 10 min at 4°C, weighed, flash-frozen in liquid nitrogen, and stored at −80°C. To make cell lysate, cell pellets were resuspended to homogeneity in 1 ml of S30 buffer per 1 g of wet cell mass. Cells were disrupted via a single passage through an Avestin EmulsiFlex-B15 (1-liter scale) or EmulsiFlex-C3 (10-liter scale) high-pressure homogenizer at 20,000 to 25,000 psi. The lysate was then centrifuged twice at 30,000g for 30 min to remove cell debris. Supernatant was transferred to clean microcentrifuge tubes and incubated at 37°C with shaking at 250 rpm for 60 min. Following centrifugation (15,000g) for 15 min at 4°C, supernatant was collected, aliquoted, flash-frozen in liquid nitrogen, and stored at −80°C. S30 lysate was active for about three freeze-thaw cycles and contained total protein (~40 g/liter) as measured by Bradford assay.
Cell-free protein synthesis
CFPS reactions were carried out in 1.5-ml microcentrifuge tubes (15-μl scale), 15-ml conical tubes (1-ml scale), or 50-ml conical tubes (5-ml scale) with a modified PANOx-SP system (80). The CFPS reaction mixture consists of the following components: 1.2 mM adenosine triphosphate (ATP); 0.85 mM each of guanosine triphosphate (GTP), uridine triphosphate (UTP), and cytidine triphosphate (CTP); L-5-formyl-5, 6, 7, 8-tetrahydrofolic acid (folinic acid) (34.0 μg ml−1); E. coli tRNA mixture (170.0 μg ml−1); 130 mM potassium glutamate; 10 mM ammonium glutamate; 12 mM magnesium glutamate; 2 mM each of 20 amino acids; 0.4 mM nicotinamide adenine dinucleotide (NAD); 0.27 mM coenzyme A (CoA); 1.5 mM spermidine; 1 mM putrescine; 4 mM sodium oxalate; 33 mM phosphoenolpyruvate (PEP); 57 mM Hepes; plasmid (13.3 μg ml−1); and 27% (v/v) cell lysate. For reaction volumes ≥1 ml, plasmid was added at 6.67 μg ml−1, as this lower plasmid concentration conserved reagents with no effect on protein synthesis yields or kinetics. For expression of PorA, reactions were supplemented with nanodiscs at 1 μg ml−1, which were prepared as previously described (81) or purchased (Cube Biotech). For expression of CRM1974xDQNAT, CFPS was carried out at 25°C for 20 hours, unless otherwise noted. For all other carrier proteins, CFPS was run at 30°C for 20 hours, unless otherwise noted.
For expression of TT4xDQNAT, which contains intermolecular disulfide bonds, CFPS was carried out under oxidizing conditions, as previously reported (39). For oxidizing conditions, lysate was preconditioned with 750 μM iodoacetamide at room temperature for 30 min to covalently bind free sulfhydryls (-SH), including the active site cysteines of the thioredoxin reductase (trxB) and glutathione reductase (gor) enzymes that represent the primary disulfide bond reducing enzymes in the E. coli cytoplasm. The CFPS reaction mix was then supplemented with 200 mM glutathione at a 4:1 ratio of oxidized and reduced forms and 10 μM recombinant E. coli DsbC (39).
In vitro conjugate vaccine expression
For in vitro expression and glycosylation of carrier proteins in crude lysates, a two-phase scheme was implemented. In the first phase, CFPS was carried out for 15 min at 25° to 30°C as described above. In the second phase, protein glycosylation was initiated by the addition of MnCl2 and DDM (n-Dodecyl β-D-maltoside) at a final concentration of 25 mM and 0.1% (w/v), respectively, and allowed to proceed at 30°C for a total reaction time of 1 hour. Reactions were then centrifuged at 20,000g for 10 min to remove insoluble or aggregated protein products, and the supernatant was analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Purification of unmodified and O-PS–conjugated carriers from iVAX reactions was carried out using Ni-NTA agarose (Qiagen) according to the manufacturer’s protocols. Briefly, 0.5-ml Ni-NTA agarose per 1-ml cell-free reaction mixture was equilibrated in buffer 1 (300 mM NaCl, 50 mM NaH2PO4) with 10 mM imidazole. Soluble fractions from iVAX reactions were loaded on Ni-NTA agarose and incubated at 4°C for 2 to 4 hours to bind 6×His-tagged protein. Following incubation, the cell-free reaction/agarose mixture was loaded onto a polypropylene column (Bio-Rad) and washed twice with six-column volumes of buffer 1 with 20 mM imidazole. Protein was eluted in four fractions, each with 0.3-ml buffer 1 with 300 mM imidazole per milliliter of cell-free reaction mixture. All buffers were used and stored at 4°C. Protein was stored at a final concentration of 1 to 2 mg ml−1 in sterile 1× PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 (pH 7.4)] at 4°C.
Lyophilization of iVAX reactions
iVAX reactions were prepared according to the recipe above and lyophilized using a VirTis BenchTop Pro lyophilizer (SP Scientific) at 100 mtorr and −80°C overnight or until fully freeze-dried. Following lyophilization, freeze-dried pellets were rehydrated with nuclease-free water (Ambion) and run as described above. If reactions were stored at ambient temperature after lyophilization, they were stored under vacuum using economic packaging materials. Specifically, we vacuum-sealed reactions using a commercial FoodSaver appliance with Dri-Card desiccant cards enclosed to prevent rehydration of the iVAX pellets. Our results show that freeze-dried iVAX pellets can be stored at ambient temperature for up to 3 months with no loss of activity (fig. S7).
Expression of conjugates in vivo using PGCT
Plasmids encoding conjugate carrier protein genes preceded by the DsbA leader sequence for translocation to the periplasm were transformed into CLM24 cells carrying pGAB2 and pSF-CjPglB. CLM24 carrying only pGAB2 was used as a negative control. Transformed cells were grown in 5-ml LB medium [yeast extract (10 g liter−1), tryptone (5 g liter−1), NaCl (5 g liter−1)] overnight at 37°C. The next day, cells were subcultured into 100-ml LB and allowed to grow at 37°C for 6 hours, after which the culture was supplemented with 0.2% arabinose and 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to induce expression of CjPglB and the conjugate carrier protein, respectively. Protein expression was then carried out for 16 hours at 30°C, at which point cells were harvested. Cell pellets were resuspended in 1-ml sterile PBS [137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 (pH 7.4)] and lysed using a Q125 sonicator (Qsonica, Newtown, CT) at 40% amplitude in cycles of 10 s on/10 s off for a total of 5 min. Soluble fractions were isolated following centrifugation at 15,000 rpm for 30 min at 4°C. Protein was purified from soluble fractions using Ni-NTA spin columns (Qiagen), following the manufacturer’s protocol.
Western blot analysis
Samples were run on 4-12% bis-tris SDS-PAGE gels (Invitrogen). Following electrophoretic separation, proteins were transferred from gels onto immobilon-P polyvinylidene difluoride membranes (0.45 μm) according to the manufacturer’s protocol. Membranes were washed with PBS [NaCl (80 g liter−1), KCl (0.2 g liter−1), Na2HPO4 (1.44 g liter−1), KH2PO4 (0.24 g liter−1) (pH 7.4)] followed by incubation for 1 hour in Odyssey Blocking Buffer (LI-COR). After blocking, membranes were washed six times with PBST [NaCl (80 g liter−1), KCl (0.2 g liter−1), Na2HPO4 (1.44 g liter−1), KH2PO4 (0.24 g liter−1), Tween 20 (1 ml liter−1) (pH 7.4)] with a 5-min incubation between each wash. For iVAX samples, membranes were probed with both an anti–6×His-tag antibody and an anti–O-PS antibody or antisera specific to the O antigen of interest, if commercially available. Probing of membranes was performed for at least 1 hour with shaking at room temperature, after which membranes were washed with PBST in the same manner as described above and probed with fluorescently labeled secondary antibodies. Membranes were imaged using an Odyssey Fc imaging system (LI-COR). CRM197 and TT production were compared to commercial DT and TT standards (Sigma-Aldrich) and orthogonally detected by an identical SDS-PAGE procedure followed by Western blot analysis with a polyclonal antibody that recognizes diphtheria or tetanus toxin, respectively. All antibodies and dilutions used are listed in table S4.
TLR4 activation assay
HEK-Blue hTLR4 cells (InvivoGen) were cultured in Dulbecco’s modified Eagle medium, high glucose/l-glutamine supplemented with 10% fetal bovine serum, penicillin (50 U ml−1), streptomycin (50 mg ml−1), and Normacin (100 μg ml−1) at 37°C in a humidified incubator containing 5% CO2. After reaching ~50 to 80% confluency, cells were plated into 96-well plates at a density of 1.4 × 105 cells/ml in HEK-Blue detection medium (InvivoGen). Antigens were added at the following concentrations: purified protein (100 ng μl−1) and total protein in lysate (100 ng μl−1). Purified E. coli O55:B5 LPS (Sigma-Aldrich) and detoxified E. coli O55:B5 (Sigma-Aldrich) were added at 1.0 ng ml−1 and served as positive and negative controls, respectively. Plates were incubated at 37°C, 5% CO2 for 10 to 16 hours before measuring absorbance at 620 nm. Statistical significance was determined using paired t tests.
Mouse immunization and F. tularensis challenge
Groups of five to six 6-week-old female BALB/c mice (Harlan Sprague Dawley) were immunized with 100 μl of PBS (pH 7.4) alone or containing purified MBP, FtO-PS–conjugated MBP, PD, FtO-PS–conjugated PD, EPA, or FtO-PS–conjugated EPA, as previously described (82). The amount of antigen in each preparation was normalized to ensure that an equivalent amount of unmodified protein or conjugate was administered in each case. Purified protein groups formulated in PBS were mixed with an equal volume of incomplete Freund’s adjuvant (Sigma-Aldrich) before injection. Before immunization, material for each group (5 μl) was streaked on LB agar plates and grown overnight at 37°C to confirm sterility and endotoxin activity was measured by TLR4 activation assay. Each group of mice was boosted with an identical dosage of antigen 21 and 42 days after the initial immunization. Mice were observed 24 and 48 hours after each injection for changes in behavior and physical health, and no abnormal responses were reported.
For initial antibody titering studies, mice were immunized subcutaneously with 7.5 μg of vaccine or controls according to the protocol described above. Blood was obtained on days −1, 21, 35, 49, and 63 via submandibular collection and at study termination on day 70 via cardiac puncture.
For pathogen challenge studies, mice were immunized intraperitoneally with 10 μg of vaccine or controls according to the protocol described above. Blood for antibody titering was obtained on day 56 via submandibular collection. Mice were challenged intranasally on day 66 with 6000 CFU (60 times the intranasal LD50) F. tularensis subsp. holarctica LVS Rocky Mountain Laboratories. Survival was monitored for an additional 25 days following pathogen challenge, during which mice were examined daily for signs of disease and sacrificed according to the approved protocol when moribund. Statistical significance was determined via endpoint comparison using Fisher’s exact test.
All procedures were carried out in accordance with Protocol 2012-0132 approved by the Cornell University Institutional Animal Care and Use Committee and/or Protocol 1305086 approved by the University of Iowa Animal Care and Use Committee.
Enzyme-linked immunosorbent assay
F. tularensis LPS-specific antibodies elicited by immunized mice were measured via indirect enzyme-linked immunosorbent assay (ELISA) using a modification of a previously described protocol (82). Briefly, sera were isolated from the collected blood draws after centrifugation at 5000g for 10 min and stored at −20°C; 96-well plates (MaxiSorp; Nunc Nalgene) were coated with F. tularensis LPS (BEI Resources) at a concentration of 5 μg ml−1 in PBS and incubated overnight at 4°C. The next day, plates were washed three times with PBST (PBS, 0.05% Tween 20, 0.3% bovine serum albumin) and blocked overnight at 4°C with 5% nonfat dry milk (Carnation) in PBS. Samples were serially diluted by a factor of 2 in triplicate between 1:100 and 1:12,800,000 in blocking buffer and added to the plate for 2 hours at 37°C. Plates were washed three times with PBST and incubated for 1 hour at 37°C in the presence of one of the following horseradish peroxidase–conjugated antibodies (all from Abcam and used at 1:25,000 dilution): goat anti-mouse IgG, anti-mouse IgG1, and anti-mouse IgG2a. After three additional washes with PBST, 3,3′-5,5′-tetramethylbenzidine substrate (1-Step Ultra TMB-ELISA; Thermo Fisher Scientific) was added, and the plate was incubated at room temperature in the dark for 30 min. The reaction was halted with 2 M H2SO4, and absorbance was quantified with a microplate spectrophotometer (Tecan) at a wavelength of 450 nm. Serum antibody titers were determined by measuring the lowest dilution that resulted in signal 3 SDs above no serum background controls. Statistical significance was determined in RStudio 1.1.463 using one-way analysis of variance (ANOVA) and the Tukey-Kramer post hoc honest significant difference test.
Quantification and statistical analysis
Quantification of CFPS yields and autoradiography
To quantify the amount of protein synthesized in iVAX reactions, two approaches were used. Fluorescence units of sfGFP were converted to concentrations using a previously reported standard curve (83). Yields of all other proteins were assessed via the addition of 10 μM l-14C-leucine (11.1 GBq mmol−1, PerkinElmer) to the CFPS mixture to yield trichloroacetic acid–precipitable radioactivity that was measured using a liquid scintillation counter as described previously (84). Soluble fractions were also run on an SDS-PAGE gel and exposed by autoradiography. Autoradiographs were imaged with Typhoon 7000 (GE Healthcare Life Sciences).
Statistical analysis
Statistical parameters including the definitions and values of n, P values, SDs, and SEs are reported in the figures and corresponding figure legends. Analytical techniques are described in Materials and Methods.
Acknowledgments
We thank Yung-Fu Chang for help with the mouse immunization at Cornell Univeristy. Funding: This work was supported by Defense Threat Reduction Agency (HDTRA1-15-10052/P00001 and HDTRA1-20-1-0004 to M.P.D. and M.C.J.), National Science Foundation (grant nos. CBET 1159581 and CBET 1264701 both to M.P.D. and MCB 1413563 to M.P.D. and M.C.J.), the Bill and Melinda Gates Foundation (OPP1217652 to M.P.D. and M.C.J.), the David and Lucile Packard Foundation, and the Dreyfus Teacher-Scholar program. J.C.S. and T.C.S. were supported by National Science Foundation Graduate Research Fellowships. T.J. was supported by a Royal Thai Government Fellowship. T.D.M. was supported by a National Institutes of Health T32 Training Grant Fellowship and a Cornell Fleming Graduate Scholarship. J.M.H. and K.F.W. were supported by National Defense Science and Engineering Graduate Fellowships. R.S.D. was funded, in part, by the Northwestern University Chemistry of Life Processes Summer Scholars program. Author contributions: J.C.S. and T.J. designed research, performed research, analyzed data, and wrote the paper. T.D.M. designed research, performed research, and analyzed data. J.M.H., K.F.W., B.S.M., A.M.M., R.S.D., and K.J.H. performed research. T.C.S. aided in research design. B.D.J. directed research. M.C.J. and M.P.D. directed research, analyzed data, and wrote the paper. Competing interests: M.P.D. has interests in Glycobia Inc. and Versatope Inc. M.P.D. and M.C.J. have an interest in SwiftScale Biologics. M.P.D.’s and M.C.J.’s interests are reviewed and managed by Cornell University and Northwestern University, respectively, in accordance with their conflict of interest policies. All other authors declare no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All plasmid constructs used in this study including complete DNA sequences are deposited on Addgene (constructs 128389 to 128404).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe9444/DC1
REFERENCES AND NOTES
- 1.Review on Antimicrobial Resistance, Antimicrobial resistance: Tackling a crisis for the health and wealth of nations (2014); https://wellcomecollection.org/works/rdpck35v.
- 2.Weintraub A., Immunology of bacterial polysaccharide antigens. Carbohydr. Res. 338, 2539–2547 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Trotter C. L., Vernon J. M., Ramsay M. E., Whitney C. G., Mulholland E. K., Goldblatt D., Hombach J., Kieny M.-P.; SAGE subgroup , Optimising the use of conjugate vaccines to prevent disease caused by Haemophilus influenzae type b, Neisseria meningitidis and Streptococcus pneumoniae. Vaccine 26, 4434–4445 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Jin C., Gibani M. M., Moore M., Juel H. B., Jones E., Meiring J., Harris V., Gardner J., Nebykova A., Kerridge S. A., Hill J., Thomaides-Brears H., Blohmke C. J., Yu L.-M., Angus B., Pollard A. J., Efficacy and immunogenicity of a Vi-tetanus toxoid conjugate vaccine in the prevention of typhoid fever using a controlled human infection model of Salmonella Typhi: A randomised controlled, phase 2b trial. Lancet 390, 2472–2480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novak R. T., Kambou J. L., Diomandé F. V., Tarbangdo T. F., Ouédraogo-Traoré R., Sangaré L., Lingani C., Martin S. W., Hatcher C., Mayer L. W., Laforce F. M., Avokey F., Djingarey M. H., Messonnier N. E., Tiendrébéogo S. R., Clark T. A., Serogroup A meningococcal conjugate vaccination in Burkina Faso: Analysis of national surveillance data. Lancet Infect. Dis. 12, 757–764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poehling K. A., Talbot T. R., Griffin M. R., Craig A. S., Whitney C. G., Zell E., Lexau C. A., Thomas A. R., Harrison L. H., Reingold A. L., Hadler J. L., Farley M. M., Anderson B. J., Schaffner W., Invasive pneumococcal disease among infants before and after introduction of pneumococcal conjugate vaccine. JAMA 295, 1668–1674 (2006). [DOI] [PubMed] [Google Scholar]
- 7.A. von Gottberg, L. de Gouveia, S. Tempia, V. Quan, S. Meiring, C. von Mollendorf, S. A. Madhi, E. R. Zell, J. R. Verani, K. L. O’Brien, C. G. Whitney, K. P. Klugman, C. Cohen; GERMS-SA Investigators, Effects of vaccination on invasive pneumococcal disease in South Africa. N. Eng. J. Med. 371, 1889-1899 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Wahl B., O’Brien K. L., Greenbaum A., Majumder A., Liu L., Chu Y., Lukšić I., Nair H., McAllister D. A., Campbell H., Rudan I., Black R., Knoll M. D., Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: Global, regional, and national estimates for 2000-15. Lancet Glob. Health 6, e744–e757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhushan R., Anthony B. F., Frasch C. E., Estimation of group B streptococcus type III polysaccharide-specific antibody concentrations in human sera is antigen dependent. Infect. Immun. 66, 5848–5853 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman M. F., Wacker M., Hernandez M., Hitchen P. G., Marolda C. L., Kowarik M., Morris H. R., Dell A., Valvano M. A., Aebi M., Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 102, 3016–3021 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Quintanilla F., Iwashkiw J. A., Price N. L., Stratilo C., Feldman M. F., Production of a recombinant vaccine candidate against Burkholderia pseudomallei exploiting the bacterial N-glycosylation machinery. Front. Microbiol. 5, 381 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wetter M., Kowarik M., Steffen M., Carranza P., Corradin G., Wacker M., Engineering, conjugation, and immunogenicity assessment of Escherichia coli O121 O antigen for its potential use as a typhoid vaccine component. Glycoconj. J. 30, 511–522 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Ma Z., Zhang H., Shang W., Zhu F., Han W., Zhao X., Han D., Wang P. G., Chen M., Glycoconjugate vaccine containing Escherichia coli O157:H7 O-antigen linked with maltose-binding protein elicits humoral and cellular responses. PLOS ONE 9, e105215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cuccui J., Thomas R. M., Moule M. G., D’Elia R. V., Laws T. R., Mills D. C., Williamson D., Atkins T. P., Prior J. L., Wren B. W., Exploitation of bacterial N-linked glycosylation to develop a novel recombinant glycoconjugate vaccine against Francisella tularensis. Open Biol. 3, 130002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marshall L. E., Nelson M., Davies C. H., Whelan A. O., Jenner D. C., Moule M. G., Denman C., Cuccui J., Atkins T. P., Wren B. W., Prior J. L., An O-antigen glycoconjugate vaccine produced using protein glycan coupling technology is protective in an inhalational rat model of tularemia. J. Immunol. Res. 2018, 8087916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wacker M., Wang L., Kowarik M., Dowd M., Lipowsky G., Faridmoayer A., Shields K., Park S., Alaimo C., Kelley K. A., Braun M., Quebatte J., Gambillara V., Carranza P., Steffen M., Lee J. C., Prevention of Staphylococcus aureus infections by glycoprotein vaccines synthesized in Escherichia coli. J. Infect. Dis. 209, 1551–1561 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ihssen J., Kowarik M., Dilettoso S., Tanner C., Wacker M., Thöny-Meyer L., Production of glycoprotein vaccines in Escherichia coli. Microb. Cell Fact. 9, 61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frasch C. E., Preparation of bacterial polysaccharide–protein conjugates: Analytical and manufacturing challenges. Vaccine 27, 6468–6470 (2009). [DOI] [PubMed] [Google Scholar]
- 19.World Health Organization, Temperature Sensitivity of Vaccines (World Health Organization, 2014). [Google Scholar]
- 20.Gao F., Lockyer K., Burkin K., Crane D. T., Bolgiano B., A physico-chemical assessment of the thermal stability of pneumococcal conjugate vaccine components. Hum. Vaccin. Immunother. 10, 2744–2753 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beresford N. J., Martino A., Feavers I. M., Corbel M. J., Bai X., Borrow R., Bolgiano B., Quality, immunogenicity and stability of meningococcal serogroup ACWY-CRM197, DT and TT glycoconjugate vaccines. Vaccine 35, 3598–3606 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Ho M. M., Mawas F., Bolgiano B., Lemercinier X., Crane D. T., Huskisson R., Corbel M. J., Physico-chemical and immunological examination of the thermal stability of tetanus toxoid conjugate vaccines. Vaccine 20, 3509–3522 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Ho M. M., Bolgiano B., Corbel M. J., Assessment of the stability and immunogenicity of meningococcal oligosaccharide C-CRM197 conjugate vaccines. Vaccine 19, 716–725 (2000). [DOI] [PubMed] [Google Scholar]
- 24.Hopkins J. W., The eradication of smallpox: Organizational learning and innovation in international health administration. J. Dev. Areas 22, 321–332 (1988). [PubMed] [Google Scholar]
- 25.Lydon P., Zipursky S., Tevi-Benissan C., Djingarey M. H., Gbedonou P., Youssoufe B. O., Zaffran M., Economic benefits of keeping vaccines at ambient temperature during mass vaccination: The case of meningitis A vaccine in Chad. Bull. World Health Organ. 92, 86–92 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashok A., Brison M., LeTallec Y., Improving cold chain systems: Challenges and solutions. Vaccine 35, 2217–2223 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Silverman A. D., Karim A. S., Jewett M. C., Cell-free gene expression: An expanded repertoire of applications. Nat. Rev. Genet. 21, 151–170 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Pardee K., Slomovic S., Nguyen P. Q., Lee J. W., Donghia N., Burrill D., Ferrante T., Mc Sorley F. R., Furuta Y., Vernet A., Lewandowski M., Boddy C. N., Joshi N. S., Collins J. J., Portable, on-demand biomolecular manufacturing. Cell 167, 248–259.e12 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Adiga R., Al-adhami M., Andar A., Borhani S., Brown S., Burgenson D., Cooper M. A., Deldari S., Frey D. D., Ge X., Guo H., Gurramkonda C., Jensen P., Kostov Y., Course W. L., Liu Y., Moreira A., Mupparapu K. S., Peñalber-Johnstone C., Pilli M., Punshon-Smith B., Rao A., Rao G., Rauniyar P., Snovida S., Taurani K., Tilahun D., Tolosa L., Tolosa M., Tran K., Vattem K., Veeraraghavan S., Wagner B., Wilhide J., Wood D. W., Zuber A., Point-of-care production of therapeutic proteins of good-manufacturing-practice quality. Nat. Biomed. Eng. 2, 675–686 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Sethuraman N., Stadheim T. A., Challenges in therapeutic glycoprotein production. Curr. Opin. Biotechnol. 17, 341–346 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Haghi F., Peerayeh S. N., Siadat S. D., Montajabiniat M., Cloning, expression and purification of outer membrane protein PorA of Neisseria meningitidis serogroup B. J. Infect. Dev. Ctries. 5, 856–862 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Stefan A., Conti M., Rubboli D., Ravagli L., Presta E., Hochkoeppler A., Overexpression and purification of the recombinant diphtheria toxin variant CRM197 in Escherichia coli. J. Biotechnol. 156, 245–252 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Figueiredo D., Turcotte C., Frankel G., Li Y., Dolly O., Wilkin G., Marriott D., Fairweather N., Dougan G., Characterization of recombinant tetanus toxin derivatives suitable for vaccine development. Infect. Immun. 63, 3218–3221 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perez J. G., Stark J. C., Jewett M. C., Cell-free synthetic biology: Engineering beyond the cell. Cold Spring Harb. Perspect. Biol. 8, a023853 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez S., Palmer D. R., Simmons M., Sun P., Bisbing J., Clain S. M., Mani S., Burgess T., Gunther V., Sun W., Potential role for Toll-like receptor 4 in mediating Escherichia coli maltose-binding protein activation of dendritic cells. Infect. Immun. 75, 1359–1363 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen M. M., Glover K. J., Imperiali B., From peptide to protein: Comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46, 5579–5585 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Jaroentomeechai T., Stark J. C., Natarajan A., Glasscock C. J., Yates L. E., Hsu K. J., Mrksich M., Jewett M. C., De Lisa M. P., Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun. 9, 2686 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bayburt T. H., Sligar S. G., Membrane protein assembly into Nanodiscs. FEBS Lett. 584, 1721–1727 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D. M., Swartz J. R., Efficient production of a bioactive, multiple disulfide-bonded protein using modified extracts of Escherichia coli. Biotechnol. Bioeng. 85, 122–129 (2004). [DOI] [PubMed] [Google Scholar]
- 40.Oyston P. C. F., Sjostedt A., Titball R. W., Tularaemia: Bioterrorism defence renews interest in Francisella tularensis. Nat. Rev. Microbiol. 2, 967–978 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Maurin M., Francisella tularensis as a potential agent of bioterrorism? Expert Rev. Anti Infect. Ther. 13, 141–144 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Centers for Disease Control and Prevention, Ring Vaccination (Centers for Disease Control and Prevention, 2019). [Google Scholar]
- 43.Fulop M., Mastroeni P., Green M., Titball R. W., Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis. Vaccine 19, 4465–4472 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Lu Z., Madico G., Roche M. I., Wang Q., Hui J. H., Perkins H. M., Zaia J., Costello C. E., Sharon J., Protective B-cell epitopes of Francisella tularensis O-polysaccharide in a mouse model of respiratory tularaemia. Immunology 136, 352–360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prior J. L., Prior R. G., Hitchen P. G., Diaper H., Griffin K. F., Morris H. R., Dell A., Titball R. W., Characterization of the O antigen gene cluster and structural analysis of the O antigen of Francisella tularensis subsp. tularensis. J. Med. Microbiol. 52, 845–851 (2003). [DOI] [PubMed] [Google Scholar]
- 46.Chen L., Valentine J. L., Huang C.-J., Endicott C. E., Moeller T. D., Rasmussen J. A., Fletcher J. R., Boll J. M., Rosenthal J. A., Dobruchowska J., Wang Z., Heiss C., Azadi P., Putnam D., Trent M. S., Jones B. D., De Lisa M. P., Outer membrane vesicles displaying engineered glycotopes elicit protective antibodies. Proc. Natl. Acad. Sci. U.S.A. 113, E3609–E3618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowarik M., Young N. M., Numao S., Schulz B. L., Hug I., Callewaert N., Mills D. C., Watson D. C., Hernandez M., Kelly J. F., Wacker M., Aebi M., Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 25, 1957–1966 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palmu A. A., Jokinen J., Borys D., Nieminen H., Ruokokoski E., Siira L., Puumalainen T., Lommel P., Hezareh M., Moreira M., Schuerman L., Kilpi T. M., Effectiveness of the ten-valent pneumococcal Haemophilus influenzae protein D conjugate vaccine (PHiD-CV10) against invasive pneumococcal disease: A cluster randomised trial. Lancet 381, 214–222 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Silfverdal S. A., Hogh B., Bergsaker M. R., Skerlikova H., Lommel P., Borys D., Schuerman L., Immunogenicity of a 2-dose priming and booster vaccination with the 10-valent pneumococcal nontypeable Haemophilus influenzae protein D conjugate vaccine. Pediatr. Infect. Dis. J. 28, e276–e282 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Dagan R., Eskola J., Leclerc C., Leroy O., Reduced response to multiple vaccines sharing common protein epitopes that are administered simultaneously to infants. Infect. Immun. 66, 2093–2098 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burrage M., Robinson A., Borrow R., Andrews N., Southern J., Findlow J., Martin S., Thornton C., Goldblatt D., Corbel M., Sesardic D., Cartwight K., Richmond P., Miller E., Effect of vaccination with carrier protein on response to meningococcal C conjugate vaccines and value of different immunoassays as predictors of protection. Infect. Immun. 70, 4946–4954 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dagan R., Goldblatt D., Maleckar J. R., Yaïch M., Eskola J., Reduction of antibody response to an 11-valent pneumococcal vaccine coadministered with a vaccine containing acellular pertussis components. Infect. Immun. 72, 5383–5391 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knuf M., Szenborn L., Moro M., Petit C., Bermal N., Bernard L., Dieussaert I., Schuerman L., Immunogenicity of routinely used childhood vaccines when coadministered with the 10-valent pneumococcal non-typeable Haemophilus influenzae protein D conjugate vaccine (PHiD-CV). Pediatr. Infect. Dis. J. 28, S97–S108 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Hatz C. F., Bally B., Rohrer S., Steffen R., Kramme S., Siegrist C.-A., Wacker M., Alaimo C., Fonck V. G., Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella dysenteriae type 1 administered to healthy adults: A single blind, partially randomized Phase I study. Vaccine 33, 4594–4601 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Huttner A., Hatz C., van den Dobbelsteen G., Abbanat D., Hornacek A., Frölich R., Dreyer A. M., Martin P., Davies T., Fae K., van den Nieuwenhof I., Thoelen S., de Vallière S., Kuhn A., Bernasconi E., Viereck V., Kavvadias T., Kling K., Ryu G., Hülder T., Gröger S., Scheiner D., Alaimo C., Harbarth S., Poolman J., Fonck V. G., Safety, immunogenicity, and preliminary clinical efficacy of a vaccine against extraintestinal pathogenic Escherichia coli in women with a history of recurrent urinary tract infection: A randomised, single-blind, placebo-controlled phase 1b trial. Lancet Infect. Dis. 17, 528–537 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Riddle M. S., Kaminski R. W., Paolo C. D., Porter C. K., Gutierrez R. L., Clarkson K. A., Weerts H. E., Duplessis C., Castellano A., Alaimo C., Paolino K., Gormley R., Fonck V. G., Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella flexneri 2a administered to healthy adults: A single blind, randomized phase I study. Clin. Vaccine Immunol. 23, 908–917 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.G. Humphreys, Vaccination: Rattling the supply chain. World Health Organization. Bull. World Health Organ. 89, 324 (2011). [DOI] [PMC free article] [PubMed]
- 58.Qadri F., Svennerholm A.-M., Faruque A. S. G., Sack R. B., Enterotoxigenic Escherichia coli in developing countries: Epidemiology, microbiology, clinical features, treatment, and prevention. Clin. Microbiol. Rev. 18, 465–483 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johnson J. R., Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 4, 80–128 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jansson P.-E., Lindberg B., Widmalm G., Leontein K., Structural studies of the Escherichia coli O78 O-antigen polysaccharide. Carbohydr. Res. 165, 87–92 (1987). [DOI] [PubMed] [Google Scholar]
- 61.L’vov V. L., Shashkov A. S., Dmitriev B. A., Kochetkov N. K., Jann B., Jann K., Structural studies of the O-specific side chain of the lipopolysaccharide from Escherichia coli O:7. Carbohydr. Res. 126, 249–259 (1984). [DOI] [PubMed] [Google Scholar]
- 62.Russell J. A., Management of sepsis. N. Engl. J. Med. 355, 1699–1713 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Brito L. A., Singh M., Acceptable levels of endotoxin in vaccine formulations during preclinical research. J. Pharm. Sci. 100, 34–37 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Petsch D., Anspach F. B., Endotoxin removal from protein solutions. J. Biotechnol. 76, 97–119 (2000). [DOI] [PubMed] [Google Scholar]
- 65.Needham B. D., Carroll S. M., Giles D. K., Georgiou G., Whiteley M., Trent M. S., Modulating the innate immune response by combinatorial engineering of endotoxin. Proc. Natl. Acad. Sci. U.S.A. 110, 1464–1469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casella C. R., Mitchell T. C., Putting endotoxin to work for us: Monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell. Mol. Life Sci. 65, 3231–3240 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bolgiano B., Mawas F., Burkin K., Crane D. T., Saydam M., Rigsby P., Corbel M. J., A retrospective study on the quality of Haemophilus influenzae type B vaccines used in the U.K. between 1996 and 2004. Hum. Vaccin. 3, 176–182 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Bogaert D., Hermans P. W. M., Adrian P. V., Rümke H. C., de Groot R., Pneumococcal vaccines: An update on current strategies. Vaccine 22, 2209–2220 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Conlan J. W., Shen H., Webb A., Perry M. B., Mice vaccinated with the O-antigen of Francisella tularensis LVS lipopolysaccharide conjugated to bovine serum albumin develop varying degrees of protective immunity against systemic or aerosol challenge with virulent type A and type B strains of the pathogen. Vaccine 20, 3465–3471 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Sebastian S., Pinkham J. T., Lynch J. G., Ross R. A., Reinap B., Blalock L. T., Conlan J. W., Kasper D. L., Cellular and humoral immunity are synergistic in protection against types A and B Francisella tularensis. Vaccine 27, 597–605 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Centers for Disease Control and Prevention, CDC Vaccine Price List (Centers for Disease Control and Prevention, 2019). [Google Scholar]
- 72.Crowell L. E., Lu A. E., Love K. R., Stockdale A., Timmick S. M., Wu D., Wang Y., Doherty W., Bonnyman A., Vecchiarello N., Goodwine C., Bradbury L., Brady J. R., Clark J. J., Colant N. A., Cvetkovic A., Dalvie N. C., Liu D., Liu Y., Mascarenhas C. A., Matthews C. B., Mozdzierz N. J., Shah K. A., Wu S.-L., Hancock W. S., Braatz R. D., Cramer S. M., Love J. C., On-demand manufacturing of clinical-quality biopharmaceuticals. Nat. Biotechnol. 36, 988–995 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Perez-Pinera P., Han N., Cleto S., Cao J., Purcell O., Shah K. A., Lee K., Ram R., Lu T. K., Synthetic biology and microbioreactor platforms for programmable production of biologics at the point-of-care. Nat. Commun. 7, 12211 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salehi A. S. M., Smith M. T., Bennett A. M., Williams J. B., Pitt W. G., Bundy B. C., Cell-free protein synthesis of a cytotoxic cancer therapeutic: Onconase production and a just-add-water cell-free system. Biotechnol. J. 11, 274–281 (2016). [DOI] [PubMed] [Google Scholar]
- 75.Murphy T. W., Sheng J., Naler L. B., Feng X., Lu C., On-chip manufacturing of synthetic proteins for point-of-care therapeutics. Microsyst. Nanoeng. 5, 13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boles K. S., Kannan K., Gill J., Felderman M., Gouvis H., Hubby B., Kamrud K. I., Venter J. C., Gibson D. G., Digital-to-biological converter for on-demand production of biologics. Nat. Biotechnol. 35, 672–675 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Fisher A. C., Haitjema C. H., Guarino C., Çelik E., Endicott C. E., Reading C. A., Merritt J. H., Ptak A. C., Zhang S., DeLisa M. P., Production of secretory and extracellular N-linked glycoproteins in Escherichia coli. Appl. Environ. Microbiol. 77, 871–881 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Datsenko K. A., Wanner B. L., One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ollis A. A., Chai Y., Natarajan A., Perregaux E., Jaroentomeechai T., Guarino C., Smith J., Zhang S., DeLisa M. P., Substitute sweeteners: Diverse bacterial oligosaccharyltransferases with unique N-glycosylation site preferences. Sci. Rep. 5, 15237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jewett M. C., Swartz J. R., Mimicking the Escherichia coli cytoplasmic environment activates long-lived and efficient cell-free protein synthesis. Biotechnol. Bioeng. 86, 19–26 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Schoborg J. A., Hershewe J. M., Stark J. C., Kightlinger W., Kath J. E., Jaroentomeechai T., Natarajan A., DeLisa M. P., Jewett M. C., A cell-free platform for rapid synthesis and testing of active oligosaccharyltransferases. Biotechnol. Bioeng. 115, 739–750 (2017). [DOI] [PubMed] [Google Scholar]
- 82.Chen D. J., Osterrieder N., Metzger S. M., Buckles E., Doody A. M., DeLisa M. P., Putnam D., Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc. Natl. Acad. Sci. U.S.A. 107, 3099–3104 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hong S. H., Ntai I., Haimovich A. D., Kelleher N. L., Isaacs F. J., Jewett M. C., Cell-free protein synthesis from a release factor 1 deficient Escherichia coli activates efficient and multiple site-specific nonstandard amino acid incorporation. ACS Synth. Biol. 3, 398–409 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim D.-M., Swartz J. R., Regeneration of adenosine triphosphate from glycolytic intermediates for cell-free protein synthesis. Biotechnol. Bioeng. 74, 309–316 (2001). [PubMed] [Google Scholar]
- 85.Ollis A. A., Zhang S., Fisher A. C., DeLisa M. P., Engineered oligosaccharyltransferases with greatly relaxed acceptor-site specificity. Nat. Chem. Biol. 10, 816–822 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Çelik E., Ollis A. A., Lasanajak Y., Fisher A. C., Gür G., Smith D. F., DeLisa M. P., Glycoarrays with engineered phages displaying structurally diverse oligosaccharides enable high-throughput detection of glycan-protein interactions. Biotechnol. J. 10, 199–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valvano M. A., Crosa J. H., Molecular cloning and expression in Escherichia coli K-12 of chromosomal genes determining the O7 lipopolysaccharide antigen of a human invasive strain of E. coli O7:K1. Infect. Immun. 57, 937–943 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe9444/DC1