

Novel optical probes allow quantification of bile salt hydrolase activity in bacteria, clinical fecal samples, mice, and humans.
Abstract
The microbiome-produced enzyme bile salt hydrolase (BSH) plays a central role in human health, but its function remains unclear due to the lack of suitable methods for measuring its activity. Here, we have developed a novel optical tool based on ultrasensitive bioluminescent imaging and demonstrated that this assay can be used for quick and cost-effective quantification of BSH activity across a broad range of biological settings including pure enzymes and bacteria, intact fecal slurries, and noninvasive imaging in live animals, as well as for the assessment of BSH activity in the entire gastrointestinal tract of mice and humans. Using this assay, we showed that certain types of prebiotics are capable of increasing BSH activity of the gut microbiota in vivo and successfully demonstrated potential application of this assay as a noninvasive diagnostic test to predict the clinical status of inflammatory bowel disease (IBD) patients.
INTRODUCTION
The gut microbiota plays a major role in human health and has been characterized as an extremely dynamic and chemically diverse community. Gut microbes greatly affect host physiology by influencing host homeostasis including metabolism, neurobiology, and immune function (1, 2). Bile salt hydrolase (BSH; EC 3.5.1.24) is an enzyme that is produced by gut bacteria and plays a central role in the gastrointestinal (GI) survival of these bacteria, bile homeostasis, and modulation of multiple host signaling pathways (3–7). BSH enzyme cleaves the amide bond in conjugated bile acids (BAs) from glycine/taurine residues to open up the BA pool to 7α-dehydroxylation that subsequently results in the formation of secondary BAs (4). Secondary BAs are known to be critical mediators of intestinal bacterial composition and many aspects of host metabolism, such as fat digestion and utilization (3–5, 8). BAs allow differential effects on nuclear receptors such as farnesoid X receptor (FXR) and membrane receptors including the G protein–coupled receptor (TGR5). For example, enterohepatic circulation of secondary BAs allows differential effects on nuclear receptors such as FXR and G protein–coupled receptor (e.g., TGR5) in various tissues that directly affect lipid and glucose metabolism, making the rates of secondary BA formation central to the maintenance of metabolic homeostasis (3–5). Secondary BAs such as deoxycholic acid also contribute to the development of colon cancer, and elevated fecal deoxycholic acid concentration is found in populations with high risk of colon cancer (8, 9). Recently, restoration of BA metabolism leading to the enrichment of the secondary BA pool has been linked to increased life span in mice with potential to provide protection against age-related diseases (10). Despite the important role of BAs in human health, the underlying mechanisms by which the gut microbiota regulates BSH enzymatic function is largely unknown. For example, very little is understood about complex interplay of indigestible fibers such as a prebiotics and BA metabolism (11, 12).
Increased BSH activity in the gut via treatment with BSH-positive probiotics or fecal transplants has been shown to confer multiple health benefits to the host (3, 4, 6, 13, 14). Some examples include reduction in inflammation and blood cholesterol levels, protection against colon cancer and urinary tract infections, and amelioration of the symptoms of Crohn’s disease and atopic dermatitis (3, 4, 6, 13, 14). High BSH activity in probiotic strains is also important because this activity maximizes their survival in the hostile environment of the GI tract, resulting in enhanced beneficial effects. In addition, increased BSH activity in fecal transplants has been linked to improved digestive functions, hypercholesterolemia, and Clostridium difficile infection (3, 4, 6, 13, 14). However, the precise mechanism of action of BSH remains elusive due to a lack of noninvasive tools. For example, very little is understood about interplay of indigestible fibers such as prebiotics and BSH activity of gut microbiota.
Estimation of BSH activity in the GI tract is very challenging given the unique chemical environment, variable distribution, and highly dynamic nature of the microbiota (1, 2). Currently, no methods exist for noninvasive evaluation of the BSH activity of the gut microbiota in its intact environment. The existing methods involve ex vivo evaluation of fecal samples, in vitro studies of isolated cultured bacteria (in vitro tests), and in silico methods (7, 15–20). However, all of these approaches have considerable limitations for assessment of the composition and function of the microbiota. For example, results based on the fecal sample analysis do not represent a whole chemical and microbial complexity of the gut microbiota, which can be found along the length of the GI tract, therefore limiting our knowledge to a very small part of the microbiome. In addition, none of the common methods are practical for the analysis of intact fecal samples because these methods are associated with assay interference (17).
Analysis of microbes independent of the host has multiple important drawbacks, including disruption of organismal interactions that may be essential for regulation of various enzymatic functions (19). While in vitro testing of BSH activity in cultured bacteria has been widely used to examine the putative functions of some isolated microbes, current chromatographic methods [e.g., high-performance liquid chromatography (HPLC) and thin-layer chromatography] and spectrophotometric detection methods are generally low throughput and require extensive sample manipulation (15–17, 20). Another important shortcoming of the in vitro methods is that the microbes are placed in a relatively low-complexity environment that differs in both aqueous and atmospheric chemical composition compared to the gut. In addition, bsh genes are widely distributed in multiple bacterial taxa, and many of these bacteria cannot be cultured by the currently available methods. Advances in sequencing technology have allowed functional prediction from large numbers of samples in silico with genes identified in metagenomic surveys (7). However, in addition to being very costly, in silico methods have the potential to underestimate BSH activity because orthologous functions are hidden in the so-called unannotated “microbial dark matter” (7). While there is no doubt that culture-based methods and sequencing are required to identify specific microbe-BA interactions, the above-mentioned issues limit the application of these methods for examination of large cohorts in vitro, and these methods cannot be extended in vivo to account for the dynamic and highly complex environmental conditions in the gut. These shortcomings have created a knowledge gap in our effort to understand the important role of BSH in gut microbial communities and the metabolism of BAs, including the potential impact of BAs on health and diseases. Thus, both basic and translational research would benefit from a low-cost, extensible functional assay that can be adapted for studies of BSH activity in many biological settings, including noninvasive quantification of BSH activity in vivo.
To address this unmet need, we developed a novel quantitative optical readout-based method for noninvasive imaging of BSH activity entitled “BSH-activatable luciferin” (BAL), which produces bioluminescent light proportional to the level of the BSH activity. We applied this assay to demonstrate several exciting biological applications such as in vivo screening of probiotics and the use of the assay as potential noninvasive diagnostic test to predict the clinical status of patients with inflammatory bowel disease (IBD). We also demonstrated that certain types of prebiotics like fructo-oligosacharides (FOSs) could significantly increase BSH activity of intact gut microbiome using noninvasive in vivo imaging. Since the BSH activity of probiotic bacteria and fecal transplants has been linked to bacterial persistence and to multiple health benefits for the host, we believe that the BAL method could be used as a powerful screening tool for the identification of novel efficient strategies that provide long-term host benefits and further understanding of the role of BSH in human health.
RESULTS
Design and synthesis of BAL probes
The design of the method is based on bioluminescence imaging, which is a highly sensitive preclinical imaging modality, due to the extremely low background associated with this method (21–24). The design of the probes is based on a caged-luciferin approach that relies on “caging” luciferin with a small chemical group. The addition of this group to the luciferin scaffold results in a caged molecule that can no longer be recognized as a substrate by firefly luciferase (22–24). Subsequent photon emission is then dependent on the cleavage of the “cage” by a specific biological process to liberate free luciferin scaffolds; this strategy has been widely adopted in studies of many important biological processes (22–24).
One of the main roles of the BSH enzyme in gut microbial metabolism is the deconjugation of glycine or taurine from bile salts (Fig. 1A) (3, 4). Therefore, the design strategy of the BAL assay is based on the conjugation of the d-aminoluciferin scaffold (luciferin) to a specific BA via an amide bond that is specifically cleaved by the BSH enzyme. This cleavage results in uncaging of free luciferin, which can subsequently react with luciferase for light production proportional to the BSH activity (Fig. 1B). We optimized the use of the BAL assay for imaging and quantification of BSH activity in different settings, including pure enzymes, cultured bacteria, and mouse and human fecal samples, as well as for noninvasive imaging and quantification directly in vivo in both normal and altered physiological conditions (e.g., antibiotic/probiotic treatment) and the assessment of BSH activity in the entire GI tract of healthy human volunteer (Fig. 1C). We chose the four most abundant BAs, namely, cholic acid (CA), deoxycholic acid (DCA), chenodeoxycholic acid (CDCA), and lithocholic acid (LCA), as luciferin “cages,” with the goal of investigating the interplay between different steroid scaffolds and BSH activity in various biological settings (Fig. 1D). This approach can be easily extended to studies of many other BA sterol cores to expand our understanding of the roles of these cores in the gut microbiota with respect to BSH activity. A full description of all the synthetic procedures and characterization of the four BAL probes is described in scheme S1.
Fig. 1. Design and applications of BAL probes.
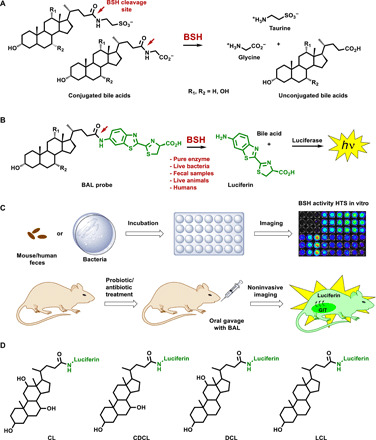
(A) Enzymatic deconjugation mechanism of naturally occurring conjugated BAs. (B) Schematic representation of the BAL assay. The technology is based on BSH-specific uncaging of free luciferin, which is subsequently processed by the luciferase enzyme to produce bioluminescent light. Hence, the signal output is proportional to the level of BSH activity. (C) The BAL method is suitable for imaging and quantification of BSH activity in different settings, including pure in vitro enzymes, cultured bacteria, and mouse and human fecal samples, as well as noninvasive real-time imaging and quantification directly in vivo in both normal and altered physiological conditions (e.g., antibiotic/probiotic treatment). The BAL probes administered to the animals expressing luciferase (FBV + luc) via oral gavage. Immediately after gavage, the animals are imaged using a CCD camera to quantify the resulting signal as a function of time. GIT, gastrointestinal tract. (D) Structures of the BAL probes used here. The design is based on four principal BAs: two primary BAs, namely, choloyl-luciferin (CL) and chenodeoxycholoyl-luciferin (CDCL); and two secondary BAs, namely, deoxycholoyl-luciferin (DCL) and lithocholoyl-luciferin (LCL).
Investigation of the selectivity of the BAL assay using enzymatic assays
We first tested the BAL probes under several in vitro conditions to determine whether we could detect the hydrolysis and subsequent release of free luciferin by bacterial amidases and whether this process is specific to the BSH family of enzymes. As a positive control, we chose choloylglycine hydrolase (CH; EC 3.5.1.24) from Clostridium perfringens, which belongs to the BSH family of hydrolases. As a negative control, we selected penicillin amidase (PA; EC 3.5.1.11) from Escherichia coli because this enzyme hydrolyzes similar amide bonds (CO─N) as CH but differs considerably in specificity (25). We first evaluated the CH and PA activities by incubation with the natural substrates sodium taurodeoxycholate (NaTDC) and penicillin G sodium salt (26), respectively. Using HPLC, we observed complete hydrolysis of both substrates within less than 10 min of incubation with the corresponding enzymes. Then, we monitored the release of luciferin and the corresponding unconjugated BA from the BAL probes as a result of enzymatic hydrolysis using HPLC. We observed complete conversion (>95%) for the choloyl-luciferin (CL), chenodeoxycholoyl-luciferin (CDCL), and deoxycholoyl-luciferin (DCL) probes and low hydrolysis (<2%) for the lithocholoyl-luciferin (LCL) probe upon incubation with CH (fig. S1A). Each probe exhibited a unique hydrolysis rate (LCL << DCL < CL < CDCL), suggesting that the selectivity in the substrate-enzyme interactions could be due to differences in the structures of the sterol cores. As expected, no cleavage of the BAL probes was observed when CH was not added to the samples (fig. S1B). There was no detectable hydrolysis of the BAL probes upon incubation with PA, demonstrating the specificity of the BAL probes toward BSH and not toward a closely related bacterial hydrolase (fig. S1C).
Optimization of the sensitivity and high-throughput screening capacity of the BAL assay
We next tested the potential of a commonly used cell-based bioluminescence detection system to quantify the uncaging of the BAL probe under increasingly complex reaction conditions, such as in live bacteria and fecal samples. Our goal was to attain unprecedented sensitivity (i.e., decrease the amount of substrate needed to obtain a quantifiable signal) while increasing the throughput capacity of the assay compared to the HPLC-based method. In addition, we aimed to reduce the number of sample manipulation steps and ideally facilitate the use of the assay with intact aqueous fecal slurries. To perform this optimization, we decided to test two commonly used methods for quantification of luciferin concentration. These methods include direct addition of pure recombinant luciferase enzyme [firefly luciferase (FLuc)] to the experimental samples and the use of whole-cell–based readouts when experimental solutions are added to mammalian cells stably transfected with luciferase followed by bioluminescence signal acquisition.
Quantification of BSH activity using pure recombinant luciferase enzyme (FLuc)
We first investigated the light readout-based method by direct addition of FLuc to the biological samples. As a control experiment, we first tested whether the addition of CH interferes with the luciferase-luciferin reaction, and we quantified the efficiency of light production from the FLuc enzyme and luciferin in the presence and absence of pure CH enzyme. We found no remarkable influence of CH on the activity of FLuc, even at high CH concentrations (fig. S2A). Since many in vitro “caged” luciferin assays rely on FLuc enzyme readout (24, 27), we decided to test whether the efficiency of BSH-specific deconjugation of BAL probes can be measured by the simple addition of FLuc enzyme directly to a bacterial culture that is known to express high levels of BSH. The CL probe was incubated with various concentrations of Lactobacillus plantarum for 1 hour at 37°C, followed by the addition of FLuc enzyme in the corresponding buffer and signal acquisition. The resulting signal was normalized to the signal from luciferin alone under the same experimental conditions. A linear dependence between the normalized bioluminescence and concentration of the BSH-expressing bacterial culture indicates that the assay is suitable for measurement of BSH activity in pure bacterial cultures (fig. S2B).
Quantification of BSH activity using a whole-cell–based bioluminescent readout
Next, we tested whether the FLuc assay can be used for measurement of BSH activity in fecal samples. For this purpose, we quantified the efficiency of light production from the FLuc enzyme and luciferin in the presence and absence of intact aqueous fecal slurries. We found that the light output decreased markedly when FLuc enzyme was exposed to diluted fecal material, rendering pure FLuc measurement unsuitable for use with fecal samples (fig. S2C).
To address this issue, we investigated the possibility of using a cell-based readout in which solutions of intact aqueous fecal slurries were incubated with mammalian cells stably transfected with luciferase, followed by bioluminescence signal acquisition. In this case, the luciferase enzyme is protected by the cell membrane and is not directly exposed to chemically complex fecal content. We decided to use 4T1-luc and Caco2-luc cell lines stably transfected with luciferase for these experiments, because 4T1-luc breast cancer cells are fast growing and exhibit strong luciferase expression, while Caco2-luc cells are known to be highly resistant to external factors because they originate from the intestinal lining of the GI tract (19). As a control experiment, we first tested whether the addition of intact aqueous fecal slurries would alter luciferase expression in these cells. We incubated 4T1-luc and Caco2-luc cells with equimolar amounts of luciferin and increasing concentrations of fecal homogenates. Unlike the results obtained with the pure recombinant FLuc system, we observed robust and stable light production in the cells with different amounts of prepared fecal samples, indicating that the presence of fecal homogenates has no effect on luciferase expression in cells (fig. S2C).
In the next step, we investigated whether the whole-cell–based readout is suitable for quantification of BSH activity in pure bacterial cultures. The CL probe was incubated with various concentrations of L. plantarum for 1 hour at 37°C, followed by centrifugation and addition of supernatants to the 4T1-luc cells in a 96-well plate and then by signal acquisition. The resulting signal was normalized to the signal from luciferin under the same experimental conditions. The linear dependence between the normalized bioluminescence and concentration of the BSH-expressing bacterial culture indicates that the assay is suitable for measurement of BSH activity in pure bacterial cultures (fig. S2B).
Next, we investigated whether the cell-based assay is quantitative and yields a dose-dependent signal from BSH-specific deconjugation of the four BAL probes. For this purpose, we plated 4T1-luc cells in 96-well plates and incubated the cells with different concentrations of all four BAL probes [0 to 50 μM in Hanks’ balanced salt solution (HBSS) buffer] in the presence or absence of CH (5 U/ml). Robust dose-dependent bioluminescent signals were detected from all the BAL probes as the result of BSH-specific deconjugation and subsequent luciferin formation (fig. S3). Only a small amount of background signal was observed from the samples incubated with buffer alone, with signal-to-background ratios ranging from 8- to 74-fold for different BAL probes at 10 μM concentration, suggesting that the selectivity in the substrate-enzyme interactions could be due to differences in the structures of the sterol cores (fig. S3, A to D). Notably, the cell-based assay was able to successfully detect robust BSH-specific deconjugation of the LCL probe (fig. S3D), which was not possible with the HPLC-based assay, even at high enzyme/probe concentrations (fig. S1A).
We then tested whether very low concentrations of BAL probes could still provide reliable signals above the background. Notably, we were able to detect a significant signal above the background with the BAL probes at concentrations as low as 2 μM (fig. S3E). While these data were consistent with our observations in the HPLC-based assay (except for the LCL probe), the use of the cell-based assay requires 250-fold less substrate and twofold less enzyme than the HPLC-based assay.
To further confirm that the amide bond in the BAL probes was specifically hydrolyzed by the BSH family of enzymes and was resistant to hydrolysis by enzymes with similar function, such as PA, we repeated the HPLC-based assay using the whole-cell–based readout method. We also tested deconjugation level of BAL probes in the presence of pancreatic carboxypeptidases A (CPA; EC 3.4.17.1) and B (CPB; EC 3.4.17.2) because they were previously reported to hydrolyze BA–neutral amino acid conjugates (28). BAL probes incubated with PA, CPA, or CPB did not produce any detectable signal over the control samples in which the BAL probes were incubated in the buffer alone, indicating complete resistance of the BAL probes to hydrolysis by these enzymes and overall stability of the BAL probes for the duration of the assay (2 hours) in HBSS buffer at 37°C (Fig. 2A). The only exception was the LCL probe, which produced weak increase (20%) in signal upon incubation with high amounts of PA.
Fig. 2. Investigation of the selectivity of the BAL assay in various biological settings using bioluminescent whole-cell readout.
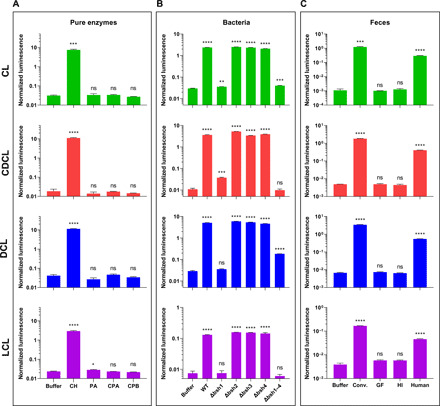
(A) Normalized bioluminescent signal resulting from deconjugation of BAL probes (10 μM) incubated in the presence or absence of the choloylglycine hydrolase (CH), penicillin amidase (PA), pancreatic carboxypeptidase A (CPA) and B (CPB) enzymes (5 U/ml). (B) Normalized bioluminescent signal obtained from BAL probes incubated with L. plantarum WCFS1 and the corresponding bsh deletion mutants at 108 CFU/ml. (C) Comparison of light production from BAL probes incubated with intact conventional mouse (conv.), healthy human donor (human), germ-free (GF) mouse, and heat-inactivated (HI) conventional mouse fecal slurries (5 g/liter). In all cases, luciferase-transfected mammalian cells were used as a readout of BSH-specific deconjugation of the BAL probes. To assure that the experimental conditions did not alter the luciferase-luciferin interaction, all the data obtained for the BAL probes were normalized to the signal of the control experiment with luciferin alone (1 μM) in the same experimental conditions. Data are presented as means ± SEM. Statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) was calculated using a two-tailed unpaired t test. ns, not significant.
In conclusion, these data suggest that the BAL probes are selective for the BSH family of enzymes and can be used for sensitive measurement of BSH activity using HPLC, pure recombinant FLuc enzyme, and whole-cell–based bioluminescence assays. Among all, the cell-based assay provides the highest sensitivity, the capacity for high-throughput screening (HTS), and the ability to measure BSH activity directly in intact supernatants of intact aqueous fecal slurries. In addition, each of the BAL probes exhibited unique rates of BSH deconjugation by CH, suggesting that the selectivity in the substrate-enzyme interactions could be due to differences in the structures of the four most abundant BAs (CA, DCA, CDCA, and LCA).
Investigation of the BSH selectivity of the BAL assay in live bacterial cultures
To further investigate the specificity of the BAL assay, we incubated the probes with the wild-type (WT) or bsh deletion mutants of L. plantarum WCFS1. We selected this bacterial strain for validation purposes because it has been previously reported that only the bsh1 gene is mainly responsible for the BSH activity of L. plantarum WCFS1 (29). Using BAL assay, we observed that deletion of bsh1 resulted in complete suppression of BSH activity, as indicated by the lack of signal from all four BAL probes (Fig. 2B). Mutant strains harboring bsh1 but lacking bsh2, bsh3, or bsh4 (∆bsh2, ∆bsh3, and ∆bsh4) demonstrated efficient uncaging of all the four BAL probes. Deletion of all four bsh genes (∆bsh1–4) resulted in only a background signal for all four BAL probes. These results are consistent with previously reported studies and further demonstrate that live cultures of bacteria expressing active BSH can efficiently uncage the BAL probes and that the signal from the probes is BSH specific (29).
Investigation of the BSH specificity of the BAL assay in mouse and human fecal samples
Monitoring of BSH activity in the intact fecal samples is very challenging given the complex environments of mouse and human feces. Therefore, most of the current assays require extensive sample processing before the BSH activity measurements (30). To test the performance and specificity of the BAL probes in mouse and human feces, we first compared the signals from the BAL probes resulting from incubation with the feces of conventional and germ-free mice that completely lack gut microbial communities (Fig. 2C). We also measured light production from conventional mouse feces in which the enzyme was deactivated by heating to 121°C for 30 min (“heat-inactivated” control, Fig. 2C).
Our data indicate that incubation of all four of the BAL probes with intact aqueous fecal slurries of conventional mice resulted in a significant signal increase compared to the background hydrolysis of the probes in phosphate-buffered saline (PBS). As expected, no signal above background was detected from the BAL probes incubated with germ-free or heat-inactivated mouse feces (Fig. 2C). These results further demonstrate a direct connection between BSH activity and the commensal gut bacteria and are consistent with previous studies on BSH activity in feces from mice with modified gut microbial communities (3, 6).
Next, we investigated whether the BAL assay can be used to quantify the deconjugation potential of BSH in human fecal samples. We analyzed BSH activity in a commercially available healthy human fecal sample collected for fecal transplant (OpenBiome). Similar to the procedure performed with the mouse feces, intact aqueous fecal slurries were incubated with all four BAL probes, followed by the measurement of light production using the whole-cell–based assay. The data demonstrated successful deconjugation of all four BAL probes as a result of BSH activity (Fig. 2C).
Deconjugation levels of the BAL probes in various biological settings with different levels of BSH activity
To develop an analytical tool for accurate quantification of BSH activity with the BAL assay, we measured the deconjugation level of the BAL probes in various biological settings (pure enzyme, bacterial culture, and fecal samples) with different levels of BSH activity. We first started with dilutions of purified CH enzyme (EC 3.5.1.24) from C. perfringens and pure bacterial culture of L. plantarum WCFS1 and lastly moved to mouse and human feces (Fig. 3, A to D, respectively). In all cases, increasing levels of BSH resulted in increased levels of bioluminescent signal from all four BAL probes. We also observed a linear dependency of the bioluminescent signal on the concentration of BSH at low concentrations of the enzyme, which eventually developed into a saturation plateau because of the limited amount of the probe in the reaction mixture (all probes were used at 10 μM). Incubation with DCL normally induced the brightest bioluminescent signal, followed by CDCL, CL, and LCL; a very similar pattern was observed in all four biological settings, with minor variation between the uncaging potentials of CL and CDCL.
Fig. 3. Deconjugation level of the BAL probes in various biological settings with different levels of BSH activity.
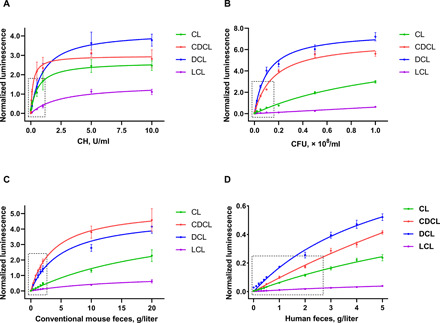
(A) Normalized bioluminescent signal resulting from incubation of BAL probes with different dilutions of pure CH enzyme at a concentration range of 0 to 10 U/ml. (B) Normalized bioluminescent signal resulting from incubation of BAL probes with different dilutions of L. plantarum WCFS1 slurries (0 to 1 × 108 CFU/ml). Normalized bioluminescent signal resulting from incubation of BAL probes with different dilutions of intact aqueous fecal slurries: (C) from conventional mice (0 to 20 g/liter) and (D) human (0 to 5 g/liter). In all cases, luciferase-transfected mammalian cells were used as a readout of BSH-specific deconjugation of the BAL probes. To verify that the experimental conditions did not alter the luciferase-luciferin interaction, all the data obtained for the BAL probes were normalized to the signal from luciferin alone in the same experimental conditions. Statistical significances in all data sets (P < 0.0001) were calculated using an ordinary one-way analysis of variance (ANOVA) test.
Together, these data represent the first example of robust measurement of BSH activity in intact aqueous human and mouse fecal slurries without any need for extensive sample manipulation. In addition, these results further confirm the BSH specificity of the novel BAL probes, making these probes valuable as tools for understanding the complex role of BSH in various pathologies and for the identification of disease biomarkers.
Real-time noninvasive quantification of BSH activity in vivo using the BAL assay
Because no tools currently exist for in vivo real-time noninvasive imaging and quantification of BSH activity directly in living animals, we decided to investigate whether the BAL assay can be used to address this unmet need. We used commercially available transgenic luciferase reporter mice that constitutively express luciferase under a β-actin promoter in every cell of the body (FVB + luc, the Jackson laboratory, USA). First, we decided to investigate whether the whole-body bioluminescent signal resulting from FVB + luc mice that received oral gavage of increasing doses of luciferin would be proportional to different luciferin concentrations. As shown in fig. S4A, the total body signal from the mice linearly correlated with the amount of administered luciferin within a large dynamic range corresponding to working concentrations of BAL probes used for our in vivo experiments. Similar results were obtained from the analysis of the mouse feces where the animals were gavaged with increasing concentrations of luciferin, suggesting strong linear dependence between luciferin concentrations in the feces and the resulting bioluminescent signal (fig. S4B). These results indicate that the detection of free luciferin resulting from the BSH-specific uncaging of BAL probes could be accurately quantified using both whole-body signal quantification in FVB + luc mice or subsequent fecal analysis.
We then measured bioluminescent light production in mice that received the CL and DCL probes via oral gavage because these probes exhibited the highest signal to background (S/B) ratio in vitro. We found that administration of as little as 50 nmol of the probe per mouse resulted in an approximately 100-fold increase in the signal over the instrumental background. In the next step, we decided to test whether the BAL probes could noninvasively detect changes in the overall level of BSH activity in vivo. For this purpose, we performed real-time imaging experiments before and after treatment of mice with a broad-spectrum antibiotic cocktail [ampicillin, vancomycin, neomycin, and metronidazole (AVNM)] that is known to drastically reduce the bacterial load in the GI tract (31), thus suppressing overall BSH activity in the gut microbiota. The luminescence resulting from AVNM-treated mice was 4.7- and 2.8-fold lower compared to untreated animals for both CL and DCL probes, respectively (Fig. 4A). These data indicate that the antibiotic cocktail was effective at reducing, but not completely abolishing, the overall BSH activity of gut bacteria against two abundant BA sterols, which is consistent with previous observations (6). These in vivo data demonstrate that BSH-mediated deconjugation of BAL probes requires functional microbial activity and that the remaining bacterial communities after antibiotic treatment retain the ability to uncage BAL probes, albeit at much lower rates than intact microbial communities.
Fig. 4. Real-time noninvasive quantification of BSH activity in vivo using the BAL assay.
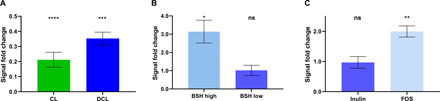
(A) The ratio of bioluminescent signal resulting from FVB + luc mice treated with the antibiotic cocktail (AVNM) for 7 days over untreated animals. The animals received oral gavage of CL (green bar) or DCL (blue bar) probes before and after AVNM treatment (n = 5 per group). (B) The ratio of bioluminescent signal resulting from FVB + luc mice that received oral gavage of DCL probe before and after administration L. plantarum WT (BSH-high) or L. plantarum Δbsh1–4 (BSH-low) bacteria (n = 5 per group). (C) The ratio of bioluminescent signal resulting from the DCL probe administered to FVB + luc mice treated with prebiotics added in drinking water (Inulin or FOS mixture) for 20 days over untreated animals (n = 5 per group). Bar graphs represent the mean ratio of signals after and before the corresponding treatment for each animal with error bars representing ± SEM. Statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001) calculated using a one-sample t test with a theoretical mean of 1.
Next, we investigated whether BAL probes can be used to quantify the increase in BSH activity upon administration of BSH-positive strains of bacteria. As discussed above, increased BSH activity is a highly desirable property of probiotic bacteria, but no assays currently exist for noninvasive assessment of this activity in the complex whole-gut microbiota (in vivo). In this study, we examined the impact of L. plantarum WCFS1 supplementation on BSH-specific deconjugation of the DCL probe in FVB + luc mice. As a negative control, we used the L. plantarum WCFS1 Δbsh1–4 mutant lacking BSH activity (29). The results revealed a 3.1-fold increase in light production from the basal state in mice supplemented with WT L. plantarum WSCF1 (Fig. 4B). In contrast, the mice supplemented with the WSCF1 bsh1–4 deletion mutants showed no difference in luminescence in comparison to the basal state (Fig. 4B). These data are consistent with the results of in vitro testing (Fig. 2B) and represent the first example of noninvasive real-time in vivo imaging of BSH activity resulting from individual probiotic bacterial strains. In conclusion, the results of antibiotic cocktail treatment and probiotic bacterial supplementation clearly demonstrate that the BAL probes represent a powerful tool for noninvasive quantification of BSH activity in the intestinal microbiomes of live animals.
Furthermore, we investigated whether BAL probes allow to detect potential changes in BSH activity caused by shift in abundance of specific bacterial taxa upon supplementation with dietary fibers, known as prebiotics. Since DCL probe resulted in highest light production in previous experiments (e.g., Fig. 3), we used this probe to monitor change in BSH activity in the intact guts of mice treated with inulin and FOS mixture added to drinking water for the duration of 3 weeks. While treatment with inulin alone caused no change in signal from DCL probe (P = 0.88 compared to the untreated control), treatment with FOS resulted in twofold increase of light output (P = 0.0059) (Fig. 4C), suggesting significant increase of abundance of BSH-positive microbes as the result of prebiotic treatment. To the best of our knowledge, this is the first demonstration of prebiotic treatment alone resulting in increase of BSH activity in the intact GI tract.
To exclude the potential contribution of host’s enzymes to deconjugation of BAL probes, we decided to investigate whether the signal production also depends on the activity of ileal BA transporters (IBATs) that potentially may expose conjugates to liver enzymes via the systemic circulation. Therefore, we compared the resulting bioluminescent signals from the DCL probe (100 nmol) administered orally to mice before and after treatment with a potent IBAT inhibitor GSK2330672 or linerixibat (10 mg/kg) (32). The results demonstrate no difference in light production from DCL probe before and after treatment (P = 0.96, n = 4), indicating no significant contribution of to probe unconjugation by host enzymes (fig. S6), further proving specificity of BAL probes toward BSH-producing gut bacteria.
Correlation of the BAL assay results with the conjugation status of BAs in human fecal samples
Deconjugation of BAs by BSH is a microbial function and is critical for the biotransformation of primary to secondary BAs. Therefore, a microbiome with high BSH activity should be associated with a BA profile that is low in conjugated BAs and high in secondary BAs. We decided to examine the relationship between the BAL assay results and fecal BA profile and to investigate whether the BAL assay can be used to predict the BA conjugation status in healthy human volunteers. To examine this relationship in humans, we obtained 25 human fecal samples from healthy donors and determined the BSH deconjugation potential using our whole-cell–based bioluminescent BAL assay. We decided to focus on the CL probe because this probe previously exhibited the highest dynamic range for quantification of BSH activity in vivo when used in healthy versus antibiotic-treated mice (5.6-fold; Fig. 4A). The results of the human fecal assay were correlated with the concentrations of several different BA species quantified using a targeted quantitative HPLC–mass spectrometry (MS) method (30), as summarized in table S1.
Our results showed that the BSH activity estimated by the CL probe negatively correlated with the ratio of conjugated to total BA (Spearman rank correlation r = −0.53, P = 0.013). Fecal BSH activity also broadly correlated with the ratios of the levels of major BA classes to total BA levels. For example, the primary to total BA proportion was negatively correlated (Spearman rank correlation r = −0.53, P = 0.014), whereas the secondary BA proportion was positively correlated with the CL deconjugation potential (Spearman rank correlation r = 0.56, P = 0.008; table S1). These results demonstrated the expected consequences of BA deconjugation by BSH and further confirmed that the high BSH activity quantified by the BAL assay indeed favored secondary BA formation.
Comparison of the BAL assay results with common DNA-based methods to predict BSH activity in human fecal samples
Next, we decided to investigate whether the BSH-specific deconjugation of the BAL probes correlates with the bsh gene abundance measured by quantitative polymerase chain reaction (qPCR) and with the predicted metagenomic results based on 16S analysis of human fecal samples. One of the most common ways to infer BSH activity, as reported in the literature, is measurement of bsh gene copy number using real-time qPCR. We tested primer sets representing bsh genes from three bacteria (Eubacterium ventriosum, Blautia obeum, and Bacteroides ovatus) that are known to inhabit the human GI tract and express bsh enzymes (33). The primer pair for E. ventriosum consistently amplified detectable gene fragments in all samples, and we used these data for comparison with BA abundances and the BSH deconjugation potential determined by the BAL assay. The results obtained by the bsh probe did not correlate with the bsh gene copy number (table S1). No correlation was observed with the conjugation status of BAs in fecal samples or with the ratio of primary or secondary BA to total BA (table S1).
Intrigued by these results, we also performed an additional microbiota analysis by sequencing the V4 region of the 16S ribosomal DNA (rDNA) genes followed by estimation of bsh gene copy number by analysis of the predicted metagenome (PICRUSt) (34). The predicted bsh gene abundance also did not correlate with most BA classifications (table S1). The only significant correlation was observed between the ratio of primary BA levels to total BA levels and the predicted bsh gene abundance (Spearman rank correlation r = 0.45, P = 0.038; table S1). Notably, these results demonstrate that the prediction of BSH functionality based on gene abundance has limitations, while direct measurement of enzymatic activity appears to provide a relatively accurate estimate of the BA conjugation status of fecal BA pool.
Quantification of BSH activity in clinical samples of gut microbiota of patients with IBD
IBD is characterized by profound changes in bacterial community composition, and it was previously suggested that BSH function may also be compromised in patients with IBD (35, 36). Therefore, we decided to investigate the level of BSH activity in intact fecal slurries of IBD patients with “active” versus “remission” clinical status of disease. The results obtained from incubation of intact fecal slurries of IBD patients with both CL and DCL probes clearly indicate significant decrease in BSH activity in patients with an active clinical state of IBD in comparison to those in the remission state (Fig. 5). These data were in agreement with primary/secondary BA abundance in feces of patients with IBD measured by standard HPLC-MS method on the same set of samples (fig. S5) (37), further proving the efficiency of the BAL tool in accurately analyzing the BA conjugation status of clinical fecal samples. Given the urgent need for IBD biomarkers and simple-to-perform diagnostic tests, we envision broad application of BAL assay in the field of IBD diagnostics.
Fig. 5. Quantification of BSH activity in clinical stool samples of patients with IBD.

Normalized bioluminescent signal resulting from deconjugation of (A) CL probe and (B) DCL probe in feces of patients with active (n = 7) versus remission (n = 18) IBD clinical status. Bar graphs represent the mean total photon flux resulting from the corresponding probe normalized to the total photon flux from luciferin under the same conditions with error bars representing ±SEM. Statistical significance (*P < 0.05) calculated using unpaired t test.
Comparison of BSH activity measured in the entire GI tract versus end-point fecal samples
Since fecal samples represent only a small portion of the gut microbiota environment, we decided to compare the BSH deconjugation potential of the entire GI tract to the end-point fecal analysis. Two independent experiments were conducted to address this question. In the first experiment, Swiss nude mice were divided in two groups (n = 3) and placed in individually vented cages supplied with sterile bedding, standard mouse chow diet, and water. The drinking water of the first group of mice was supplemented with a combination of four antibiotics referred to as “AVNM cocktail” (31) for 7 days, while the control group received no antibiotics. To assess the BSH activity of the entire GI tract, both groups of mice received oral gavage of CL probe (200 nmol of probe in 200 μl of sterile PBS) on the 8th day of the experiment. The feces were collected 6 hours after gavage and analyzed using whole-cell–based bioluminescence assay. The total photon flux resulting from the feces of control group of mice was about 300 times higher than that of the antibiotic-treated group (Fig. 6A).
Fig. 6. Noninvasive quantification of BSH activity in human and mouse GI tract.

(A) Total photon flux resulting from the feces of mice that received AVNM antibiotic cocktail for 7 days (n = 3) versus control untreated group (n = 3). Both groups of mice received the oral gavage of 200 nM of CL probe in 200 μl of PBS probe 6 hours before feces collection and analysis. P = 0.0002, n = 9 (three samples × three replicates). (B) Total photon flux from incubation of CL probe with the feces of mice treated with AVNM antibiotic cocktail for 7 days versus control untreated group (n = 5). P < 0.0001, n = 15 (five samples × three replicates). (C) Total photon flux resulting from feces of a healthy 42-year-old female collected 12 hours after oral administration of two gelatin-based capsules containing a total of 6.8 mg of CL probe as one dose. BG, background. P = 0.0021, n = 3 (one sample × three replicates). Data are presented as means ± SEM. P values were calculated using a two-tailed unpaired t test.
In the second in vivo experiment, two groups of Swiss nude mice (n = 5) were subjected to the same conditions (AVNM cocktail for 7 days versus no antibiotics). On the 8th day of the experiment, the feces were collected, and the BSH activity was quantified by incubation with CL probe using whole-cell–based bioluminescence assay. Notably, the total photon flux in antibiotic-treated group was only four times lower than control (Fig. 6B). This difference is 72-fold lower in comparison to the first experiment where the CL probe was given orally and allowed to pass through the entire GI tract. These data indicate that the end-point fecal analysis has considerable limitations for the assessment of the composition and the function of the entire gut microbiota because it represents only a small portion of the gut environment.
Quantification of BSH activity in intact GI tract of healthy human adult
To demonstrate the potential of BAL method for clinical translation, we conducted a human experiment where a healthy 42-year-old female self-administered two gelatin-based capsules containing a total of 6.8 mg of CL probe orally. In preparation for this experiment, we first assessed an acute hepatotoxicity of the CL probe in mice by measuring aspartate aminotransferase (AST) blood levels using a commercially available kit (Sigma-Aldrich, MAK055) before and 24 hours after the administration of the probe. No significant differences in AST levels were detected in blood plasma samples before and after treatment with the CL probe, indicating the absence of potential hepatotoxicity (fig. S7).
The final human dose of the compound was 0.12 mg/kg, which is about 16 times lower that the dose used in previous AST mouse experiment. A stool sample was acquired 12 hours after the probe consumption, and the amount of free luciferin was quantified using whole-cell–based bioluminescence assay as described above. The data shown on Fig. 6C demonstrate a strong bioluminescent signal as the result of BSH deconjugation of CL probe by the entire intact human gut microbiota, which was 1300-fold higher than the instrumental background. These data are the first example of the use of bioluminescent probes for a functional readout of a biological process in humans.
DISCUSSION
Despite the important role of BSH in human health and metabolism, no efficient methods currently exist for measurement of BSH activity in vitro and in vivo, creating a large gap in our understanding of this basic function of the gut microbiota. Here, we developed a novel optical tool based on sensitive bioluminescent imaging and a caged-luciferin approach (BAL) in which light production is proportional to BSH activity. The four BAL probes described herein are based on the four most abundant BAs, allowing differentiation of BSH activity between different sterol components, namely, CA, DCA, CDCA, and LCA. The metabolic roles of various BA cores with respect to BSH activity and their interplay in human pathologies remain largely unknown. In the course of this work, we optimized the use of this novel BAL assay for imaging and quantification of BSH activity in settings ranging from pure enzyme and bacterial culture to mouse and human fecal samples. Unlike other methods, the BAL assay requires minimal sample manipulation and is suitable for the HTS format. In addition, various tests with purified CH, PA, CPA, CPB hydrolases, and WT/bsh gene knockout bacterial strains demonstrated high selectivity of the BAL assay for the BSH family of enzymes.
We also demonstrated that BSH activity can be imaged and quantified directly in vivo using real-time noninvasive bioluminescent imaging. To further investigate the specificity of the BAL probes deconjugation by microbial enzymes in vivo, we decided to analyze whether the signal production also depends on the activity of IBATs that potentially could expose BAL probes to the liver enzymes via the systemic circulation. Therefore, we compared signals from the mice treated with the DCL probe before and after administration of a potent IBAT inhibitor (GSK2330672, also known as linerixibat) (32). Administration of this inhibitor at a dose of 10 mg/kg significantly reduces reabsorption of BAs in rats from 95 to 50% (32). In our experiment, we applied the same dose of IBAT inhibitor and were expecting to get 50% reduction in light output if the signal was resulting from deconjugation of BAL probes by the hepatic enzymes rather than microbial enzymes. However, no changes in bioluminescence signal was observed in IBAT inhibitor–treated animals, supporting the argument that most of the BAL probe deconjugation is a result of microbial BSH activity and is not because of the nonspecific enzymatic reactions in the liver. The ability to noninvasively image BSH activity of gut microbiota directly in living animals provides an opportunity for investigation of many important biological functions of the BSH enzyme directly in the intact gut microbial environment in both healthy and pathological conditions.
The capacity of exogenously cultured bacteria to persist in the gut ecosystem is currently an area of intense research, as probiotics and fecal microbiota transplants are proposed as powerful remedies for a number of ailments (3, 5, 6, 13). While high BSH activity is one of the main selection criteria for probiotics potentially leading to multiple health benefits (3, 5, 6, 13), no tools currently exist that would allow its assessment in intact GI tract in noninvasive, real-time fashion. In this work, we demonstrated the application of the BAL method for evaluation of the activity of BSH-positive probiotic strains directly in vivo, making this tool highly valuable for probiotic and fecal microbiota transplantation research.
Prebiotics are classified as indigestible fibers and have been previously shown to increase survival of probiotics when used in a “synbiotic” fashion (11–14), reduce levels of genotoxicity of known mutagens in the gut, and inhibit growth of human colon adenocarcinoma (38). In addition, previous studies reported that increase in metabolic production of short-chain fatty acid profile might be the most beneficial effects of prebiotics on the gut microbiota (11). Although only a very few studies have been reported on the symbiotic assessment of probiotics and prebiotics (11–14), very little is known about the impact of prebiotics alone on BA and cholesterol metabolism. Using the novel DCL probe, we decided to investigate potential influence of two commonly used prebiotics, inulin and FOS, on BSH activity directly in vivo. FOS and inulin are indigestible polysaccharides known to differ in the degree of polymerization (39). Recent investigations have reported a notable influence of dietary inulin on intestinal microbiota based on fecal bacterial 16S ribosomal RNA (rRNA) sequence analysis. While many bacterial groups increased the abundance after prebiotic feeding, the extent of increase in Bifidobacterium family was stronger with FOS than with inulin (40). Our data demonstrate that the addition of FOS prebiotic to the drinking water for 3 weeks results in approximately twofold increase in BSH activity, while no change was detected for inulin-treated mice. Since bsh gene is commonly found in probiotics including Bifidobacteirum species, selective increase of BSH activity after FOS but not after inulin might be due to the preferential growth of Bifidobacterium under the FOS feeding. These results represent the first example that prebiotics alone are capable of increasing BSH activity of healthy intact gut microbiome directly in vivo and further confirm complex interplay between dietary fibers and important functions of gut microbiome. They also indicate that previously reported BSH increase of probiotic/prebiotic synbiotic applications in some cases could be explained by direct effect of prebiotics alone. Because manufacturing and storage of prebiotics is simpler in comparison to probiotics, this discovery could become a foundation for identification of novel set of powerful prebiotics resulting in significant increase of abundance of BSH-positive gut microbes in the gut leading to multiple health benefits of the host.
In addition to a wide range of in vivo imaging applications, our results also demonstrate that the new BAL assay can accurately reflect the conjugation status of BAs in complex microbial environments, such as human fecal samples from healthy donors. The data obtained with the BAL assay show the expected outcome, with high BSH activity being correlated with secondary BA formation, as determined by the classic HPLC-MS technique. Note that the main goal of BAL assay is to quantify and monitor the actual deconjugation process of BAs by BSH, a gatekeeper of BA metabolism (4). However, traditional liquid chromatography–MS (LC-MS) techniques are still required to characterize the full BA metabolome and all the follow-up transformations of BAs such as oxidation and 7α/7β dehydroxylation. Unlike other methods for measuring BSH activity, the BAL assay requires minimal sample manipulation and is suitable for the HTS format. Inspired by these results, we also investigated whether the bsh gene copy number determined by classic real-time PCR is correlated with the actual BSH activity and conjugation status of BAs. We observed no correlation between the results obtained by the novel BAL assay and the bsh gene copy number determined by qPCR. No correlation was observed between the qPCR data and the conjugation status of BAs in human fecal samples or the ratio of primary or secondary BAs to total BAs obtained using HPLC-MS. In addition, we also performed additional microbiota analysis by sequencing the V4 region of 16S rDNA genes followed by estimation of bsh gene abundance with analysis of the predicted metagenome (PICRUSt) (34). Consistent with qPCR data, the predicted bsh gene abundance was also not correlated with the classification of most BAs. These results clearly demonstrate that prediction of BSH functionality based on gene abundance has considerable limitations, while direct measurement of enzymatic activity appears to allow relatively accurate quantification of BSH activity, thereby facilitating elucidation of the BA metabolic process of fecal communities.
The lack of correlation between qPCR/16S data with the BA pool and BSH-specific deconjugation of the BAL probes may be a result of several factors. First, qPCR of the bsh genes in a single organism represents only a fraction of the genetic potential directed toward BA deconjugation. Since it is generally not possible to design a “universal” qPCR primer, this feature is typically considered as one of the common limitations of qPCR method (41). Recent evidence suggests that metagenomic sequencing can reveal a wide array of potential bsh and bsh-like genes that likely contribute to the BA deconjugation potential and, thus, may be relatively well correlated with the BA pool (7, 18, 36). Second, the sterols in our novel probes are linked to luciferin rather than glycine or taurine, which are the typical substrates used in current methods to estimate BA deconjugation. Thus, the hydrolysis of the amide bond linking the BA to luciferin in our probes represents specificity toward the sterol core rather than the combined effect of the recognition of the sterol and amino acid side chain on deconjugation potential. Preferential deconjugation of sterols has been reported for individual bacteria such as Lactobacillus buchneri JCM1069, which can deconjugate taurodeoxycholic but not taurocholic acid (42), although future research on substrate specificity is needed to better understand how our probes could be used to reveal the dynamics of BA transformation in mixed microbial communities. Together, these results show that our novel BAL probes better capture associations between BA deconjugation activity and BA abundance than qPCR and 16S sequencing because these probes reflect the overall BSH deconjugation potential of fecal microbial communities.
To investigate the potential application of BAL assays as a diagnostic tool, we quantified BSH activities of intact fecal slurries of IBD patients in active versus remission clinical status of disease. We choose to look at IBD because it is a very common human pathology for which no simple noninvasive diagnostic tools currently exists. In addition, it has been previously shown that IBD patients with an active clinical status of disease have increased concentration of conjugated BAs in feces, while the concentration of total secondary BAs is reduced (35, 36). In addition, metagenomics analysis of gut microbiota of patients with IBD demonstrates loss in the abundance of BSH gene (36). Both of these findings imply impairments in BSH activity in active versus remission clinical states. However, BSH activity of gut microbiota has never been directly quantified. The results obtained from incubation of intact fecal slurries of IBD patients with both CL and DCL probes clearly indicate significant reduction in BSH activity in patients with active clinical states of IBD. These findings position BSH as an important potential biomarker of gut inflammation in IBD and open an opportunity for simple noninvasive assessment of the response to therapy by monitoring restoration of fecal microbiota function while in remission.
Inspired by the exciting results in clinical fecal samples, we decided to further investigate the potential of the BAL method for clinical translation. To do it, we first decided to compare BSH activity measured in the entire GI tract versus end-point fecal samples in mice. Fecal samples are known to represent only a small portion of the gut microbiome environmental, chemical, and microbial complexity that can be found along the length of the GI tract. However, no methods currently exist for the assessment of enzymatic activity of the entire intact GI tract, limiting our knowledge to a very small part of the microbiome contributing to host health. The results obtained using antibiotic-treated mice revealed 72-fold difference between the signals obtained with CL probe given orally versus direct incubation of the probe with the feces. These data indicate that end-point fecal analysis has limitations for assessment of the function of the entire gut microbiota and provides only limited information.
To demonstrate whether the BSH activity measurements in the entire GI tract can be clinically translated, we conducted a human experiment where a healthy 42-year-old female self-administered CL probe orally. While the final dose of the compound was about 35 times lower than the dose used in the mouse in vivo experiment, a strong signal was acquired that was 1300-fold higher than the instrumental background. These data are the first example of a functional assessment of the entire human microbiome demonstrating the potential of clinical translation of this assay as a simple noninvasive diagnostics for IBD and, potentially, many other human pathologies. In addition, this is the first example of the use of bioluminescent probes for a functional readout of a biological process in humans.
In conclusion, the BAL assay is the first example of quantification of BSH activity in multiple formats including pure enzyme and pure bacteria, intact fecal slurries, noninvasive imaging in live animals, and assessment of BSH activity in the entire GI tract of mice and humans. Using this novel tool, we showed that accurate measurements of BSH activity can be an important potential biomarker of gut inflammation. Because the BSH activity of probiotic bacteria and fecal transplants has been linked to bacterial persistence and to multiple health benefits for the host, we believe that our probes are powerful screening tools for the identification of potent bacterial strains that can inhabit the gut and provide long-term benefits. In addition, this tool can be used for screening of existing and novel prebiotics that could significantly increase BSH activity of GI tract by causing the shift of specific bacterial taxa. Given the importance of BSH in human health, this novel method could also become a powerful tool for elucidation of the role of BSH-mediated BA deconjugation and the relationship of this process to long-term metabolic health in response to diet, drug therapies, or bacterial supplementation. In addition, the current applications of the novel BAL assay could be expanded further when used in combinations with novel bioluminescent imaging platforms.
MATERIALS AND METHODS
Chemical and biochemical reagents
The procedures for chemical synthesis and characterization of novel compounds are summarized in the Supplementary Materials. All chemical reagents obtained from commercial suppliers were used without further purification unless noted. d-Luciferin was acquired from Promega, USA. 6-aminobenzo[d]thiazole-2-carbonitrile was obtained from Fluorochem Ltd., UK. Magnesium sulfate heptahydrate was obtained from Alfa Aesar GmbH, Germany. Adenosine 5′-triphosphate (ATP) disodium salt was obtained from AppliChem GmbH, Germany. All other reagents were purchased from Sigma-Aldrich Chemie GmbH, Switzerland, including d-cysteine, CA, DCA, CDCA, LCA, 6′-amino-d-luciferin (d-aminoluciferin), 2-mercaptoethanol (ME), dimethyl sulfoxide (DMSO) BioUltra for molecular biology, luciferase from Photinus pyralis (FLuc), CH from C. perfringens (Clostridium welchii), PA from E. coli, NaTDC, and penicillin G sodium salt. Linerixibat (MedChem Express) was obtained from Lucerna-Chem AG (Switzerland).
Chemical synthesis
The BAL probes were synthesized according to the synthetic scheme depicted in scheme S1. The detailed synthetic procedures, purification, and characterization [1H and 13C nuclear magnetic resonance, high-resolution mass spectrometry (HRMS)] of all the BAL probes and corresponding intermediates are summarized in the “Chemical Synthesis and Characterization” section in the Supplementary Materials . Briefly, 6-amino-benzo[d]thiazole-2-carbonitrile was acylated by a corresponding derivative of BA ethyl carbonic anhydride, followed by a reaction with d-cysteine to yield the final BAL probe (CL, CDCL, DCL, and LCL). Stock solutions of the BAL probes in DMSO at a concentration of 10 or 25 mM were prepared and stored at −20°C. Immediately before each experiment, the stock solutions of the BAL probes were diluted with appropriate buffers to achieve necessary concentrations.
Investigation of the selectivity of the BAL assay using HPLC-based enzymatic assays
Each of the BAL probes at a concentration of 500 μM was first incubated with a BSH family hydrolase, namely, CH from C. perfringens, at 10 U/ml at 37°C in a mixture of 10% DMSO in PBS supplemented with 10 mM ME for 0 to 3 hours. The same experiment was repeated with another bacterial amidase, namely, PA, which is known to cleave similar amide bonds, at 1000 U/ml under the same experimental conditions. As a negative control, all the BAL probes were incubated with the buffer alone to account for nonspecific hydrolysis. Aliquots of the reaction mixtures were taken every 10 min after reaction onset, and the conversion of the BAL probes to free luciferin was monitored by HPLC. Analytical ultraperformance liquid chromatography (UPLC) was performed on a Waters ACQUITY UPLC Class H instrument using a BEH C18 1.7 μm 2.1 × 50 mm column with a 4-min gradient from 5 to 100% acetonitrile in water containing 0.1% formic acid. The conversion of the BAL probes was calculated as the percentage of free luciferin measured, as described in a previous report (27). To ensure that both the CH and PA enzymes were functional, NaTDC and penicillin G sodium salt were used as positive controls to confirm enzymatic activity according to the manufacturer’s instructions (26).
Bioluminescence readout by direct addition of recombinant FLuc
A freshly prepared solution of recombinant FLuc (20 μg/ml) in PBS supplemented with the necessary cofactors, namely, Mg2SO4 (2 mM) and ATP (2 mM), was used to quantify the amount of free luciferin released as the result of BSH-specific deconjugation of the BAL probes. In each experiment, the analyzed samples were mixed with luciferase solution, and the resulting mixture was transferred immediately to a black flat-bottomed 96-well assay plate (reference no. 3915, Corning, Germany), followed by quantification of the bioluminescent signal using an IVIS 100 imaging system (PerkinElmer, USA). Total photon flux was quantified using the region of interest (ROI) analysis of each individual well, and the average signal expressed as the total number of photons emitted per second per square centimeter per steradian was quantified using Living Image software (PerkinElmer, USA). A detailed description of each experiment is provided below.
Cell cultures
RPMI 1640 cell culture medium (reference no. A10491-01), fetal bovine serum (FBS) (reference no. 10270-106), PBS (pH 7.2; reference no. 20012-068), and HBSS (reference no. 14025-050) were obtained from Gibco, USA. Murine breast cancer 4T1-luc cells were obtained from PerkinElmer (USA). Human colon cancer Caco-2 cells that were stably transduced with a red-luciferase–expressing gene construct (referred to here as Caco2-luc cells; CLS960002, PerkinElmer, USA) were a gift from SwissLumix, Switzerland. The 4T1-luc and Caco2-luc cells were maintained in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin-streptomycin (P/S) at 37°C in a 5% CO2 atmosphere. The cells were seeded at a density of 2 to 3 × 103 cells/cm2 and split at 60% density to avoid differentiation.
Whole-cell–based bioluminescence readout
In this assay, we used two mammalian cell lines stably transfected with luciferase, namely, the 4T1-luc and Caco2-luc cells described above. The 4T1-luc cells were used for the experiments with pure enzymes, such as CH and PA, and Caco2-luc were used for all the other experiments because these cells produced robust and dose-dependent signals in complex biological environments, such as intact aqueous fecal slurries. In a typical experiment, the cells were plated at a density of 3 × 104 cells per well on clear-bottomed black-walled 96-well tissue-culture–treated plates (reference no. 353219, Corning, Germany) with RPMI 1640 medium supplemented with 10% FBS and incubated at 37°C for 24 to 40 hours in a 5% CO2 atmosphere to achieve 80 to 100% confluence. Immediately before the experiment, the growth medium of the cells was replaced with corresponding solutions of biological samples, and the resulting bioluminescent signal was quantified using an IVIS 100 charge-coupled device (CCD) camera as photon emission over time. As a positive control, we incubated cells with either 5 μM d-luciferin or 1 μM d-aminoluciferin to account for the potential influence of experimental conditions on luciferase-luciferin light emission efficacy. Total photon flux was quantified using ROI analysis of each individual well, and the average signal expressed as the total number of photons emitted per second per square centimeter per steradian was quantified using Living Image software (PerkinElmer, USA). The experiments were repeated three times (three wells per experiment). Total luminescence was calculated by integrating the area under the corresponding kinetic curve for the duration of the experiment. Normalized bioluminescence values were calculated as a ratio of total photon flux resulting from BSH-specific deconjugation of BAL probes and the total photon flux resulting from positive-control experiments (luciferin alone) in the same experimental conditions.
Comparison of recombinant FLuc-based and whole-cell–based readout methods in various biological settings
To test the influence of pure CH enzyme on luciferase activity, a solution of d-luciferin (10 μM, in 300 μl of HBSS) was mixed with a solution of CH (60 U/ml, in 300 μl of HBSS supplemented with 20 mM of ME). A solution of d-luciferin (5 μM, 600 μl in HBSS supplemented with 10 mM ME) was used as a positive control. After incubation at 37°C for 1 hour, the reaction mixtures were analyzed by direct addition of recombinant FLuc enzyme. The resulting bioluminescence intensities expressed as total photon flux over 1 hour.
To compare the FLuc-based and whole-cell–based readout methods for analysis of BSH activity in pure bacterial cultures, solutions of different concentrations of L. plantarum WCFS1 [0, 105, 106, 107, and 108 CFU (colony-forming units)/ml] in 300 μl of PBS supplemented with 20 mM ME were mixed with a solution of the CL probe (20 μM in 300 μl of PBS). After incubation at 37°C for 1 hour, the samples were centrifuged at 13,000 rpm and analyzed by the recombinant FLuc and whole-cell–based (4T1-luc cells) readouts as described above. As a positive control, we measured light production in the d-aminoluciferin solution (2 μM in PBS) under the same experimental conditions. The bioluminescence intensities from CL probe hydrolysis expressed as total photon flux were normalized to that of the positive control (luciferin alone) in the corresponding medium to calculate normalized bioluminescence.
To test the influence of fecal aqueous slurries on luciferase activity, a solution of d-aminoluciferin (1 μM, in 300 μl of PBS) was mixed with different dilutions of conventional mouse fecal slurries (1.0 and 2.0 g/liter, in 300 μl of PBS supplemented with 20 mM ME). A solution of d-aminoluciferin (1 μM, in 600 μl of PBS supplemented with 10 mM ME) was used as a positive control. After incubation of samples at 37°C for 1 hour, the solutions were centrifuged at 13,000 rpm and analyzed by both bioluminescence assays with recombinant FLuc, 4T1-luc, and Caco2-luc cells as described above. All bioluminescence intensities expressed as total photon flux were divided by that of the positive control (luciferin alone) to calculate normalized bioluminescence.
BSH-specific deconjugation of BAL probes by the CH, PA, CPA, and CPB enzyme using cell-based readout
Each of the BAL probes at concentrations of 10 μM was incubated with CH, PA, CPA, or CPB enzymes (5 U/ml) in HBSS supplemented with 10 mM ME at 37°C for 1 hour. As a negative control, all of the BAL probes were incubated with the buffer alone. The reaction mixtures were transferred to plates (three wells × 100 μl) containing 4T1-luc reporter cells for signal quantification in an IVIS 100 system for 1 hour as described above. d-Luciferin (1 μM) in the same experimental conditions was used as a positive control to assess luciferase activity.
BSH-specific deconjugation of the BAL probes by a pure bacterial culture of L. plantarum WSCF1 and its bsh deletion mutants
WT and bsh deletion mutants of L. plantarum WSCF1 were grown in de Man, Rogosa and Sharpe (MRS) broth (Sigma-Aldrich) at 37°C for 48 hours in an anaerobic jar (Oxoid, 2.5 liters) using AnaeroGen 2.5-liter anaerobic atmosphere generation pads. Aliquots (15 ml) of each culture were centrifuged at 1500 rpm to pellet cells, and the cell pellets were washed three times with 10 ml of PBS, followed by an additional centrifugation at 1500 rpm and resuspension in PBS (10 ml) supplemented with ME (20 mM). Aliquots of the resulting suspension (200 μl) were mixed with the corresponding BAL probe (20 μM, in 200 μl of PBS) or d-aminoluciferin (2 μM, in 200 μl of PBS) to form 400-μl reaction mixtures at final concentrations of 10 μM, 1 μM, and 10 mM for the BAL probes, d-aminoluciferin, and ME, respectively. After incubation at 37°C for 1 hour, the reaction mixtures were centrifuged at 13,000 rpm, and the amounts of the resulting deconjugation product (luciferin) in the supernatants were quantified in triplicate using Caco2-luc luciferase-expressing cells as described above (see cell-based bioluminescence assay). CFU concentrations were assessed by inoculating serial dilutions on MRS agar (Roth, Germany). WT and bsh deletion mutants of L. plantarum WCFS1 were a gift of W. M. de Vos from Wageningen University, Finland.
BAL probe deconjugation in mouse and human fecal samples
Fresh fecal pellets were collected from conventional mice, frozen, and stored at −20°C. For germ-free and conventional mice, fecal samples (ca. 100 mg) were soaked in PBS supplemented with 20 mM ME (2 ml) for 30 min and then mechanically disrupted by vortex. After precipitation of coarse particles, supernatant was taken, and the volume was adjusted by the corresponding buffer to the desired concentration (g/liter) immediately before use in the BAL assay. To generate heat-inactivated mouse feces, a freshly prepared sample was heated in a screw-capped glass vial at 121°C for 30 min, followed by the same analysis.
Similar to the procedure with mouse feces, frozen human fecal samples (ca. 1 to 2 mg) were suspended in PBS supplemented with 20 mM ME at a concentration of 1 g/liter. In each experiment, aliquots of 200 μl were taken from individual samples, mixed, and incubated with the corresponding solutions of BAL probes (20 μM in 200 μl of PBS) or d-aminoluciferin control (2 μM in 200 μl of PBS) for 1 hour at 37°C followed by centrifugation at 13,000 rpm and transfer of the resulting supernatants to plates (three wells × 100 μl) containing Caco2-luc reporter cells for signal quantification in an IVIS 100 system for 1 hour as described above. Feces collection and analyses of healthy human fecal samples were approved by the Lausanne ethical committee under the license of CER-VD 489/14 and by the University of North Carolina Chapel Hill Institutional Review Board approval no. 14-2049. Feces collection and analysis of fecal samples of patients with IBD were approved by the Health Ethics Committee and Clinical Research Committee of the Beatriz Ângelo Hospital, Loures, Portugal, reference no: 0028/2014_RMEB.
Deconjugation levels of the BAL probes in various biological settings with different levels of BSH activity
In the experiments with pure CH enzyme, solutions of each of the BAL probes (20 μM in 200 μl of HBSS) or firefly d-luciferin control (10 μM in 200 μl of HBSS) were mixed with solutions of CH (200 μl of HBSS supplemented with 20 mM ME at 0, 0.01, 0.02, 0.1, 0.2, 1, 2, 10, and 20 U/ml). After incubation at 37°C for 1 hour, the solutions were transferred to a 96-well plate (three wells × 100 μl) containing 4T1-luc reporter cells, and the resulting bioluminescent signal was quantified for 1 hour as described above (see whole-cell–based bioluminescence assay).
In the experiments with pure bacterial cultures, solutions of each of the BAL probes (20 μM in 200 μl of PBS) or d-aminoluciferin control (2 μM in 200 μl of PBS) were mixed with serial dilutions of WT L. plantarum WCFS1 (200 μl of PBS supplemented with 20 mM ME at 0, 0.002, 0.004, 0.01, 0.02, 0.04, 0.1, 0.2, 0.4, 1, and 2 × 108 CFU/ml). After incubation at 37°C for 1 hour, the reaction mixtures were centrifuged at 13,000 rpm and transferred to a 96-well plate (three wells × 100 μl) containing Caco2-luc reporter cells for signal quantification for 1 hour as described above.
In the experiments with mouse fecal samples, solutions of each of the BAL probes (20 μM in 200 μl of PBS) or d-aminoluciferin control (2 μM in 200 μl of PBS) were mixed with dilutions of vortexed mouse fecal slurries (200 μl in PBS supplemented with 20 mM ME at 0, 0.8, 1.6, 2.4, 3.2, 4.0, 20.0, and 40.0 g/liter). After incubation at 37°C for 1 hour, the solutions were transferred to a 96-well plate (three wells × 100 μl) containing Caco2-luc reporter cells for signal quantification for 1 hour as described above.
In the experiments with human fecal samples, solutions of each of the BAL probes (20 μM in 200 μl of PBS) or d-aminoluciferin control (2 μM in 200 μl of PBS) were mixed with dilutions of human fecal transplant from OpenBiome, USA (200 μl in PBS supplemented with 20 mM ME at 0, 0.2, 0.4, 0.6, 0.8, 1.0, 2.0, 4.0, 6.0, 8.0, and 10.0 g/liter). After incubation at 37°C for 1 hour, the reaction mixtures were transferred to a 96-well plate (three wells × 100 μl) containing Caco2-luc reporter cells for signal quantification using an IVIS 100 system for 1 hour as described above.
Fecal DNA extraction and sequencing
Microbial and BA analyses of healthy human fecal samples were approved by the Lausanne ethical committee under the license of CER-VD 489/14 and by the University of North Carolina Chapel Hill Institutional Review Board approval no. 14-2049. Frozen aliquots (250 mg) of fecal samples were prepared with a coring technique using a CryoXtract CXT350 (Woburn MA, USA). Approximately 50 mg of each sample was processed using the MO BIO PowerMag Soil DNA Isolation Kit (MO BIO Laboratories Inc., CA, USA) according to the manufacturer’s specifications. An amplicon library of partial 16S rRNA genes was produced according to the methods outlined by Caporaso et al. (43). Briefly, we used primers targeting the V4 region of the 16S ribosomal genes at positions 515 to 806 (E. coli numbering), composed of the Illumina adapters, pad, barcode (forward primer only), and gene-specific nucleotides. Details on the primer design can be found at http://earthmicrobiome.org/emp-standard-protocols/16s/. The resulting PCR products from three reactions per sample were combined and cleaned using the Agencourt AMPure XP system (Beckman Coulter Life Sciences, IN, USA). The cleaned amplicons were then quantified using PicoGreen double-stranded DNA reagent, and an equal amount of amplicon from each sample was pooled into a single tube and sequenced on a MiSeq instrument using v2 chemistry.
Data processing
Sequences were demultiplexed from fastq files according to the specific barcode introduced at the PCR step. Quality filtering and chimera removal were performed with MOTHUR (44) and UCHIME (45). The resulting sequences were then clustered into operational taxonomic units (OTUs) at 97% sequence identity in QIIME (46) using the closed reference picking method against the Greengenes gg_13_5 dataset (https://greengenes.secondgenome.com/?prefix=downloads/greengenes_database/gg_13_5/), followed by rarefaction to 25,000 sequences per sample from which Bray-Curtis distances (47), and taxon abundances were determined (34).
BA analysis
BA quantification was performed using a UPLC-HRMS system. Materials and detailed methods are listed in the Supplementary Materials. Briefly, 1500 μl of MeOH:H2O (2:1) with 0.1% (v/v) formic acid was added to 100 mg of lyophilized feces. The samples were homogenized with ceramic beads, and the extracted supernatant was removed and stored at −80°C. The fecal extracts (50 μl) were mixed with internal standards before solid-phase extraction. The dry extracts were reconstituted in 100 μl of 30% acetonitrile in water (v/v). To quantify BAs in the samples, 20 μl of each sample was injected for UPLC-HRMS. BA quantification was performed on a QExactive Hybrid Quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, MA, USA). The instrument was equipped with a Thermo Accela 1250 UPLC pump and CTC PAL Analytics autosampler (Zwingen, Switzerland) for UPLC. The reversed-phase chromatographic method consisted of the following mobile phase system: (i) 5 mM ammonium acetate + 0.1% (v/v) formic acid in water and (ii) 0.1% (v/v) formic acid in acetonitrile. An ACQUITY UPLC HSS T3 1.8 μm 2.1 × 100 mm column was used for BA separation at an operating temperature of 40°C. The MS parameters were as follows: full MS = 370 to 522 (centroid acquisition), resolution = 70,000, negative polarity, and automatic gain control target = 5 × 105. The LC-MS HR system was operated using XCalibur 2.2 (Thermo Fisher Scientific). The data files were processed into result files using TraceFinder 3.0 (Thermo Fisher Scientific).
Imaging and quantification of BSH activity in transgenic reporter mice (FVB + luc) using the CL and DCL probes
Transgenic luciferase reporter mice [referred to as FVB + luc, an abbreviation of FVBTg (CAG-luc,-GFP)L2G85Chco/J] were purchased from the Jackson laboratory (USA) and maintained in the Center of PhenoGenomics (CPG) at the Swiss Federal Institute of Technology (EPFL) animal facility under a standard 12-hour light/12-hour dark cycle with food and water provided ad libitum. Animal experiments were approved by the Veterinary Authority of the Canton Vaud, Switzerland (license no. 2849c).
Demonstration of linear correlation between the whole-body bioluminescent signal and oral doses of luciferin
Nine female FVB + luc mice (46 to 50 weeks old) were divided into three groups (three mice per group). Each group of mice received oral gavage of aminoluciferin solution in 100 μl of sterile PBS in the doses of 10, 50, or 100 nmol per mouse. Five minutes later, the mice were anesthetized using isoflurane, placed in the IVIS Spectrum (PerkinElmer, USA), and imaged for 1 hour. Images were acquired every minute using the following settings: auto exposure, field of view D, aperture F1, and binning medium. Total photon flux was calculated by integration of the corresponding kinetic curves. Mean value of total photon flux per each group was plotted against the aminoluciferin concentration. Line fitting was performed using least-squares method.
Demonstration of linear correlation between the bioluminescent signal from the fecal samples and oral doses of luciferin
Eight Swiss nude mice (42 to 44 weeks old) divided in four groups received an oral gavage of different doses of luciferin (0.02, 0.1, 0.2, or 0.5 mg dissolved in 100 μl of sterile PBS). Animals were placed separately in clean cages (one mouse per cage), and feces were collected within a 24-hour period. Feces were then dried and weighed. For the detection of luciferin, dry fecal material was homogenized in water (2 × 5 ml) and spun down by centrifugation (3500 rpm). Resulting supernatants were diluted with Eagle’s Minimum Essential Medium (EMEM) medium (1:10). Amounts of luciferin were quantified using whole-cell–based bioluminescent readout in 4T1-luc cell as described above.
Antibiotic treatment experiment
The level of the BSH activity was measured using BAL probes before and after intervention in vivo using FVB + luc mice (e.g., antibiotic or probiotic treatments). To measure the uncaging potential in the basal state, the mice (n = 5 per group) were maintained for 7 days in individually vented cages and supplied with sterile bedding, food (standard mouse chow), and water. CL and DCL probes were administered to mice via oral gavage (50 nmol of probe in 100 μl of sterile PBS). The mice were allowed to rest for 30 min before being anesthetized with isofluran and imaged in an IVIS Spectrum system (PerkinElmer, USA). The whole-body bioluminescence signal was quantified using automatic acquisition settings over the course of 3 hours (baseline control). For the antibiotic treatment, the mice were transferred to new cages with fresh bedding, and their drinking water was supplemented with a combination of four antibiotics referred as AVNM cocktail [ampicillin (1.0 g/liter), vancomycin (0.5 g/liter), neomycin (1.0 g/liter), and metronidazole (1.0 g/liter)] supplemented with glucose (50 g/liter) as a sweetener for 7 days as described previously (31). On day 8, each animal was administered 50 nmol of either CL or DCL of BAL probes via oral gavage in 100 μl of sterile PBS and allowed to rest for 30 min before being anesthetized and imaged using an IVIS Spectrum system. The whole-body bioluminescence signal was quantified using automatic acquisition settings over the course of 3 hours (after AVNM). The ratio of the total photon flux for each animal after antibiotic treatment (after AVNM) over untreated state (baseline control) gavaged with either CL or DCL probe was calculated.
Probiotic treatment experiment
In the probiotic experiment, the signal resulting from DCL probe was measured from each mouse (baseline control) as described above for two study groups (n = 5) after administration of 50 nmol of the DCL probe in 100 μl of sterile PBS. After 3 days, the first group of mice was treated with type of L. plantarum WCFS1 (L. plantarum W), while the second group received bsh1–4 deletion mutant (L. plantarum Δbsh1–4) by oral gavage of ca. 1 × 109 CFU/ml in 100 μl of sterile PBS, followed by oral gavage of 50 nmol of the DCL probe in 100 μl of sterile PBS. The mice were allowed to rest for 30 min before being anesthetized and imaged in an IVIS Spectrum system as described above. The whole-body bioluminescence signal was quantified using automatic acquisition settings over the course of 3 hours in untreated state (baseline control) and after treatment with either L. plantarum WT or L. plantarum Δbsh1–4. The ratio of the total photon flux for each animal treated with either L. plantarum WT or L. plantarum Δbsh1–4 and its corresponding untreated state (baseline control) was calculated.
Prebiotic treatment experiment
In the prebiotic experiment, the signal resulting from DCL probe was measured from each mouse (baseline control) for three study groups (n = 5) after oral administration of 100 nmol of the DCL probe in 100 μl of sterile PBS and 30 min of resting after probe administration. Then, the first group of mice received inulin (Alfa Aesar) added to drinking water (10%), the second group received an inulin-FOS 1:1 mixture (PiLeJe Laboratiore, France) suspended in drinking water (10%), and the control group received normal drinking water. After treatment for 20 days, the signal from DCL probe was measured again from each animal as described above. The whole-body bioluminescence signal was quantified using automatic acquisition settings 3 hours after administration of the probe in untreated state (baseline control) and after treatment with prebiotics (inulin or FOS). The ratio of the total photon flux for each animal treated with either inulin or FOS to corresponding untreated state (baseline control) was calculated and normalized to an average signal of the control group.
In vivo IBAT inhibition
To exclude contribution of IBATs to the unspecific cleavage of the BAL probes, we tested the effect of the IBAT inhibitor GSK2330672 or linerixibat (10 mg/kg) (32) on the bioluminescent signal from the DCL probe in mice. A group of FVB + luc (n = 4) were imaged with the DCL probe (100 nmol per animal orally, at 3-hour time point) to estimate a basal level of bioluminescent signal (control). Two days later, the mice orally received a dose of linerixibat (10 mg/kg in 100 μl of sterile PBS). After 2 hours, the animals were imaged with the DCL probe (100 nmol per animal orally, at 3-hour time point) to assess the level of bioluminescent signal under inhibition conditions (linerixibat). The whole-body bioluminescence signal was quantified using automatic acquisition settings. The data represent the total photon flux normalized to control experiment.
Hepatotoxicity assay
Toxicity of the CL probe in mice was assessed by an AST activity assay kit (Sigma-Aldrich). The AST assay was performed in blood samples taken before and 24 hours after oral gavage of the probe (100 nmol per animal) in a group of FVB + luc mice (n = 3) according to the supplier’s instruction. The AST levels were within a normal range for mice (50 to 100 U/ml), and no statistically significant increase in signal was observed after the treatment.
Comparison of BSH activity measured in the entire GI tract versus end-point fecal samples
Swiss nude mice were purchased from Charles River Laboratories (France) and maintained in the CPG at the EPFL animal facility under a standard 12-hour light/12-hour dark cycle with food and water provided ad libitum. In the in vivo experiments when CL probes were orally gavaged, the mice were divided in two groups (n = 3 per group), placed in individually vented cages, and supplied with sterile bedding, food (standard mouse chow), and water. The drinking water of the first group was supplemented with a combination of four antibiotics referred as AVNM cocktail for 7 days as described previously (31), while the control group received no antibiotic. On the 8th day of the experiment, the mice received oral gavage of CL probe (200 nmol of probe in 200 μl of sterile PBS). The feces were collected 6 hours after gavage and analyzed using whole-cell–based bioluminescence assay as described above (see whole-cell–based bioluminescence assay).
In a second in vivo experiment, two groups of Swiss nude mice (n = 5) were subjected to the same conditions, where one of the groups received AVNM cocktail for 7 days as described previously (31), while the control group received no antibiotic. On the 8th day of the experiment, the feces of the mice were collected, and the BSH activity was quantified by direct incubation with CL probe using whole-cell–based bioluminescence assay.
Quantification of BSH activity in intact GI tract of healthy human adult
A healthy 42-year-old female self-administered two gelatin-based capsules containing a total of 6.8 mg of CL probe orally at one dose. A sample of gut microbiota was acquired 12 hours later and analyzed for the presence of free luciferin using whole-cell–based bioluminescence assay as described above (see whole-cell–based bioluminescence assay). According to the internationally accepted practice and previously reported precedents (48, 49) as well as the conclusion of “Commission cantonale (VD) d’éthique de la recherche sur l’être humain” as of 23 July 2019, this study was not a subject to “human subjects research” on the basis of informed self-administration.
Data analysis
Integrated photon emission and subject measurements were analyzed in R version 3.4.3 with “Kite-Eating Tree” (https://cran.r-project.org/) using the base stats, tidyverse (v1.2.1), and broom (v0.4.4) packages. We used Spearman rank correlations calculated with the Hmisc package (v4.1-1) to qualitatively explore the relationship between BAs, probe uncaging, and qPCR values. One-sample t tests (in vivo tests only) were performed using GraphPad Prism v8.1.2 for Windows GraphPad Software, San Diego, CA, USA (www.graphpad.com). All means and SEs for the in vitro tests are from three replicate reactions. Means and SEs for the in vitro tests are from five mice per group.
Acknowledgments
We would like to thank Nestlé S.A. for financial support for BA analysis, qPCR analysis of E. ventriosum bsh, and microbial 16S sequencing analysis. We thank J. M. Lambert and W. M. De Vos from Wageningen University, Finland for the L. plantarum strains. We also thank SwissLumix, Switzerland for the gift of Caco-2-luc cells. We would like to thank D. Rainteau at French Institute of Health and Medical Research for performing BA analysis of subset of human fecal samples reported in the paper. We also thank J. W. Collins for discussion of in vivo experiments on antibiotic and probiotic treatment. We thank C. Fournier and A. Charpagne for performing 16S sequencing. We thank Lumigenics (USA), a CRO company specializing in optical assays, for performing analysis of human fecal samples. Funding: This work was partially funded by the grant from the Integrative Food and Nutrition Centre (IFNC, EPFL). Author contributions: E.A.G. conceptualized the study and acquired the funding. E.A.G, P.V.K, and C.J.C. designed the experiment. P.V.K. synthesized and characterized the probes. P.V.K., A.Y., A.A.B., B.B., and S.B. performed the experiments. P.V.K. and C.L.L. analyzed the data. E.A.G., P.V.K., and C.L.L. wrote the paper. Y.R. and T.R.-K. provided human fecal samples and associated clinical data. Y.R., T.R.-K., and C.J.C. provided critical scientific comments to the manuscript. J.T. and J.H. provided IBD human fecal samples and associated clinical data. Competing interests: C.J.C. and B.B. are current employees of Société des Produits Nestlé S.A. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eaaz9857/DC1
REFERENCES AND NOTES
- 1.B. Tungland, in Human Microbiota in Health and Disease, B. Tungland, Ed. (Academic Press, 2018), pp. 107–134. [Google Scholar]
- 2.Cani P. D., Van Hul M., Lefort C., Depommier C., Rastelli M., Everard A., Microbial regulation of organismal energy homeostasis. Nat. Metab. 1, 34–46 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Joyce S. A., MacSharry J., Casey P. G., Kinsella M., Murphy E. F., Shanahan F., Hill C., Gahan C. G., Regulation of host weight gain and lipid metabolism by bacterial bile acid modification in the gut. Proc. Natl. Acad. Sci. U.S.A. 111, 7421–7426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foley M. H., O’Flaherty S., Barrangou R., Theriot C. M., Bile salt hydrolases: Gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLOS Pathog. 15, e1007581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wahlstrom A., Sayin S. I., Marschall H. U., Backhed F., Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab. 24, 41–50 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Mullish B. H., McDonald J. A. K., Pechlivanis A., Allegretti J. R., Kao D., Barker G. F., Kapila D., Petrof E. O., Joyce S. A., Gahan C. G. M., Glegola-Madejska I., Williams H. R. T., Holmes E., Clarke T. B., Thursz M. R., Marchesi J. R., Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut 68, 1791–1800 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones B. V., Begley M., Hill C., Gahan C. G. M., Marchesi J. R., Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 105, 13580–13585 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ou J., DeLany J. P., Zhang M., Sharma S., O’Keefe S. J. D., Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr. Cancer 64, 34–40 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Keefe S. J. D., Kidd M., Espitalier-Noel G., Owira P., Rarity of colon cancer in Africans is associated with low animal product consumption, not fiber. Am. J. Gastroenterol. 94, 1373–1380 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Barcena C., Valdes-Mas R., Mayoral P., Garabaya C., Durand S., Rodriguez F., Fernandez-Garcia M. T., Salazar N., Nogacka A. M., Garatachea N., Bossut N., Aprahamian F., Lucia A., Kroemer G., Freije J. M. P., Quiros P. M., Lopez-Otin C., Healthspan and lifespan extension by fecal microbiota transplantation into progeroid mice. Nat. Med. 25, 1234–1242 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Poeker S. A., Geirnaert A., Berchtold L., Greppi A., Krych L., Steinert R. E., de Wouters T., Lacroix C., Understanding the prebiotic potential of different dietary fibers using an in vitro continuous adult fermentation model (PolyFermS). Sci. Rep. 8, 4318 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adebola O. O., Corcoran O., Morgan W. A., Prebiotics may alter bile salt hydrolase activity: Possible implications for cholesterol metabolism. PharmaNutrition 12, 100182 (2020). [Google Scholar]
- 13.Kerry R. G., Patra J. K., Gouda S., Park Y., Shin H. S., Das G., Benefaction of probiotics for human health: A review. J. Food Drug Anal. 26, 927–939 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandey K. R., Naik S. R., Vakil B. V., Probiotics, prebiotics and synbiotics—A review. J. Food Sci. Technol. 52, 7577–7587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huijghebaert S. M., Hofman A. F., Influence of the amino acid moiety on deconjugation of bile acid amidates by cholylglycine hydrolase or human fecal cultures. J. Lipid Res. 27, 742–752 (1986). [PubMed] [Google Scholar]
- 16.Mullish B. H., Pechlivanis A., Barker G. F., Thursz M. R., Marchesi J. R., McDonald J. A. K., Functional microbiomics: Evaluation of gut microbiota-bile acid metabolism interactions in health and disease. Methods 149, 49–58 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandvold K. R., Weaver J. M., Whidbey C., Wright A. T., A continuous fluorescence assay for simple quantification of bile salt hydrolase activity in the gut microbiome. Sci. Rep. 9, 1359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heinken A., Ravcheev D. A., Baldini F., Heirendt L., Fleming R. M. T., Thiele I., Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. bioRxiv , 229138 (2017). [Google Scholar]
- 19.Pearce S. C., Coia H. G., Karl J. P., Pantoja-Feliciano I. G., Zachos N. C., Racicot K., Intestinal in vitro and ex vivo models to study host-microbiome interactions and acute stressors. Front. Physiol. 9, 1584 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee Y. P., Takahashi T., An improved colorimetric determination of amino acids with the use of ninhydrin. Anal. Biochem. 14, 71–77 (1966). [Google Scholar]
- 21.Sweeney T. J., Mailänder V., Tucker A. A., Olomu A. B., Zhang W., Cao Y., Negrin R. S., Contag C. H., Visualizing the kinetics of tumor-cell clearance in living animals. Proc. Natl. Acad. Sci. U.S.A. 96, 12044–12049 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mezzanotte L., van’t Root M., Karatas H., Goun E. A., Lowik C., In vivo molecular bioluminescence imaging: New tools and applications. Trends Biotechnol. 35, 640–652 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Rathbun C. M., Prescher J. A., Bioluminescent probes for imaging biology beyond the culture dish. Biochemistry 56, 5178–5184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su T. A., Bruemmer K. J., Chang C. J., Caged luciferins for bioluminescent activity-based sensing. Curr. Opin. Biotechnol. 60, 198–204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar R. S., Brannigan J. A., Prabhune A. A., Pundle A. V., Dodson G. G., Dodson E. J., Suresh C. G., Structural and functional analysis of a conjugated bile salt hydrolase from Bifidobacterium longum teveals an evolutionary relationship with penicillin V acylase. J. Biol. Chem. 281, 32516–32525 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Olsson A., Hagstrom T., Nilsson B., Uhlen M., Gatenbeck S., Molecular cloning of Bacillus sphaericus penicillin V amidase gene and its expression in Escherichia coli and Bacillus subtilis. Appl. Environ. Microbiol. 49, 1084–1089 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones L. R., Goun E. A., Shinde R., Rothbard J. B., Contag C. H., Wender P. A., Releasable luciferin−transporter conjugates: Tools for the real-time analysis of cellular uptake and release. J. Am. Chem. Soc. 128, 6526–6527 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Huijghebaert S. M., Hofman A. F., Pancreatic carboxypeptidase hydrolysis of bile acid-amino acid conjugates: Selective resistance of glycine and taurine amidates. Gastroenterology 90, 306–315 (1986). [DOI] [PubMed] [Google Scholar]
- 29.Lambert J. M., Bongers R. S., de Vos W. M., Kleerebezem M., Functional analysis of four bile salt hydrolase and penicillin acylase family members in Lactobacillus plantarum WCFS1. Appl. Environ. Microbiol. 74, 4719–4726 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakrabarti A., Membrez M., Morin-Rivron D., Siddharth J., Chou C. J., Henry H., Bruce S., Metairon S., Raymond F., Betrisey B., Loyer C., Parkinson S. J., Masoodi M., Transcriptomics-driven lipidomics (TDL) identifies the microbiome-regulated targets of ileal lipid metabolism. NPJ Syst. Biol. Appl. 3, 33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reikvam D. H., Erofeev A., Sandvik A., Grcic V., Jahnsen F. L., Gaustad P., McCoy K. D., Macpherson A. J., Meza-Zepeda L. A., Johansen F. E., Depletion of murine intestinal microbiota: Effects on gut mucosa and epithelial gene expression. PLOS ONE 6, e17996 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y., Aquino C. J., Cowan D. J., Anderson D. L., Ambroso J. L., Bishop M. J., Boros E. E., Chen L., Cunningham A., Dobbins R. L., Feldman P. L., Harston L. T., Kaldor I. W., Klein R., Liang X., McIntyre M. S., Merrill C. L., Patterson K. M., Prescott J. S., Ray J. S., Roller S. G., Yao X., Young A., Yuen J., Collins J. L., Discovery of a highly potent, nonabsorbable apical sodium-dependent bile acid transporter inhibitor (GSK2330672) for treatment of type 2 diabetes. J. Med. Chem. 56, 5094–5114 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Korpela K., Salonen A., Virta L. J., Kekkonen R. A., Forslund K., Bork P., de Vos W. M., Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat. Commun. 7, 10410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langille M. G., Zaneveld J., Caporaso J. G., McDonald D., Knights D., Reyes J. A., Clemente J. C., Burkepile D. E., Vega Thurber R. L., Knight R., Beiko R. G., Huttenhower C., Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duboc H., Rajca S., Rainteau D., Benarous D., Maubert M.-A., Quervain E., Thomas G., Barbu V., Humbert L., Despras G., Bridonneau C., Dumetz F., Grill J.-P., Masliah J., Beaugerie L., Cosnes J., Chazouillères O., Poupon R., Wolf C., Mallet J.-M., Langella P., Trugnan G., Sokol H., Seksik P., Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 62, 531–539 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Labbe A., Ganopolsky J. G., Martoni C. J., Prakash S., Jones M. L., Bacterial bile metabolising gene abundance in Crohn’s, ulcerative colitis and type 2 diabetes metagenomes. PLOS ONE 9, e115175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Torres J., Palmela C., Brito H., Bao X., Ruiqi H., Moura-Santos P., Pereira da Silva J., Oliveira A., Vieira C., Perez K., Itzkowitz S. H., Colombel J. F., Humbert L., Rainteau D., Cravo M., Rodrigues C. M., Hu J., The gut microbiota, bile acids and their correlation in primary sclerosing cholangitis associated with inflammatory bowel disease. United European Gastroenterol J 6, 112–122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adebola O., Corcoran O., Morgan W. A., Protective effects of prebiotics inulin and lactulose from cytotoxicity and genotoxicity in human colon adenocarcinoma cells. Food Res. Int. 52, 269–274 (2013). [Google Scholar]
- 39.Chi Z.-M., Zhang T., Cao T.-S., Liu X.-Y., Cui W., Zhao C.-H., Biotechnological potential of inulin for bioprocesses. Bioresour. Technol. 102, 4295–4303 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Zhu L., Qin S., Zhai S., Gao Y., Li L., Inulin with different degrees of polymerization modulates composition of intestinal microbiota in mice. FEMS Microbiol. Lett. 364, (2017). [DOI] [PubMed] [Google Scholar]
- 41.Kim J., Lim J., Lee C., Quantitative real-time PCR approaches for microbial community studies in wastewater treatment systems: Applications and considerations. Biotechnol. Adv. 31, 1358–1373 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Moser S. A., Savage D. C., Bile salt hydrolase activity and resistance to toxicity of conjugated bile salts are unrelated properties in lactobacilli. Appl. Environ. Microbiol. 67, 3476–3480 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caporaso J. G., Lauber C. L., Walters W. A., Berg-Lyons D., Huntley J., Fierer N., Owens S. M., Betley J., Fraser L., Bauer M., Gormley N., Gilbert J. A., Smith G., Knight R., Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., Lesniewski R. A., Oakley B. B., Parks D. H., Robinson C. J., Sahl J. W., Stres B., Thallinger G. G., Van Horn D. J., Weber C. F., Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R., UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caporaso J. G., Kuczynski J., Stombaugh J., Bittinger K., Bushman F. D., Costello E. K., Fierer N., Pena A. G., Goodrich J. K., Gordon J. I., Huttley G. A., Kelley S. T., Knights D., Koenig J. E., Ley R. E., Lozupone C. A., McDonald D., Muegge B. D., Pirrung M., Reeder J., Sevinsky J. R., Turnbaugh P. J., Walters W. A., Widmann J., Yatsunenko T., Zaneveld J., Knight R., QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roger Bray J., Curtis J. T., An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 27, 325–349 (1957). [Google Scholar]
- 48.L. K. Altman, in Who Goes First? The Story of Self-Experimentation in Medicine (University of California Press, ed. 1, 1998), pp. 454. [Google Scholar]
- 49.Trammell S. A., Schmidt M. S., Weidemann B. J., Redpath P., Jaksch F., Dellinger R. W., Li Z., Abel E. D., Migaud M. E., Brenner C., Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 7, 12948 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eaaz9857/DC1