

Abstract
A randomized controlled trial,the Proactive IV Iron Therapy in Haemodialysis Patients (PIVOTAL), has recently shown that a high-dose (‘proactive’) intravenous iron regimen was superior to a low-dose (‘reactive’) regimen for hemodialysis patient outcomes and overall safety. However, even in the low-dose group, a substantial amount of iron was administered to maintain serum ferritin >200 ng/mL. This type of comparison may have strongly affected the safety results. Iron has two opposite effects on erythropoiesis: it activates erythroid differentiation directly by supplying iron but inhibits it indirectly by stimulating hepcidin and enhancing oxidative stress. Hepcidin plays an essential role not only in iron homeostasis and the anemia of chronic kidney disease, but also in its complications such as atherosclerosis and infection. Its main stimulation by iron—and to a lesser degree by inflammation—should urge clinicians to avoid prescribing excessive amounts of iron. Furthermore, as serum ferritin is closely correlated with serum hepcidin and iron storage, it would seem preferable to rely mainly on serum ferritin to adjust iron administration, defining an upper limit for risk reduction. Based on our estimations, the optimal range of serum ferritin is ∼50–150 ng/mL, which is precisely within the boundaries of iron management in Japan. Considering the contrasting ranges of target ferritin levels between end-stage renal disease patients in Japan and the rest of the world, the optimal range proposed by us will probably be considered as unacceptable by nephrologists abroad. Only well-balanced, randomized controlled trials with both erythropoiesis-stimulating agents and iron will allow us to settle this controversy.
Keywords: anemia, bone marrow, cardiovascular, erythropoiesis-stimulating agents, hepcidin, infection, iron, oxidative stress
INTRODUCTION
Recently, the randomized controlled trial Proactive IV Iron Therapy in Haemodialysis Patients (PIVOTAL) examined the safetyand efficacy and the administered dose of erythropoiesis-stimulating agents (ESAs)in patients on long-term hemodialysis (HD) treated with two different dynamic intravenous (IV) iron-dosing regimens. The trial provided the first conclusive evidence of the safety of high-dose IV iron therapy including hard outcomes. It must be noted that the iron was given in a proactive fashion, and the results were compared with those of a low-dose regimen of IV iron administered reactively, with a ferritin concentration of <200 ng/mL or a transferrin saturation (TSAT) of <20% being a trigger for iron administration. The results of this trial probably have led the nephrology community at large to alleviate their concerns regarding high-dose IV iron supplementation [1]. Unquestionably, this trial supports the results of a number of reports that IV iron therapy over a wide dose range poses no discernible hazard in incident dialysis patients [2–4]. Most of these studies, however, had not been properly adjusted using inflammation markers, e.g. C-reactive protein (CRP) or interleukin (IL)-6.
However, the results of PIVOTAL are inconsistent with numerous observational studies that identified higher risks of mortality, cardiovascular disease (CVD) and infection in patients with chronic kidney disease (CKD) exposed to long-term high-dose IV iron therapy [5–10]. In addition to the risk of inducing hemosiderosis, excess iron has been shown to cause oxidative stress and cytotoxic effects, favor the growth of micro-organisms and promote atherosclerosis [11, 12]. Therefore, when examining such discrepant results, we decided to have a closer look at the characteristics of the patients in PIVOTAL who served as control or reference group for the purpose of comparison, since this might strongly affect the reported safety results [13].
In the Kidney Disease: Improving Global Outcome Clinical Practice Guideline for Anemia in Chronic Kidney Disease, iron administration is suggested for anemic CKD patients with TSAT <30% and serum ferritin <500 ng/mL if an increase in hemoglobin level is desired, and an upper limit of 500–800 ng/mL for the ferritin level is considered to be acceptable [14]. Some researchers working in the nonrenal hematology field regard anemia treatment for patients on maintenance HD (MHD) as a very special clinical condition, outside the prevailing view of normal iron metabolism and optimal iron handling [15, 16].
The Japanese Society for Dialysis Therapy (JSDT) Guidelines recommend that a minimal amount of iron be given to CKD patients with anemia, and this only in case of evident iron deficiency [17] (Table 1). In the general policy of the guideline, the risk/benefit ratio for iron supplementation is set at a higher level than that accepted in Western countries [14, 17, 18]. From the JSDT registry, patients with a serum ferritin level <200 ng/mL accounted for ∼80% of all dialysis patients at the end of 2012 [19]. In the reply to our comment on the PIVOTAL trial, our opinion has been dismissed on the basis that Japanese standards are certainly not the case for the rest of the world [13, 20].
Table 1.
Excerpt of a passage from the 2015 JSDT Guidelines for renal anemia in chronic kidney disease (evidence level in parentheses) [17]
| Chapter 4: Evaluation of iron status and iron therapy |
| CQ3: What are the criteria for the initiation and discontinuation of iron therapy? |
| Statement 3-1 |
| (1) For patients who are not treated with ESAs or iron and cannot maintain target hemoglobin levels, we suggest iron therapy prior to ESA therapy if the serum ferritin level is <50 ng/mL. (2D) |
| (2) For patients who are treated with ESAs and cannot maintain the target hemoglobin level, we recommend iron therapy if the serum ferritin level is <100 ng/mL and TSAT is <20%. (1B) |
| Statement 3-2 |
| (3) For patients who are treated with ESAs and cannot maintain target hemoglobin levels, we suggest iron therapy if both the following conditions are satisfied: (2C) |
| Absence of disease that decreases iron utilization rate |
| Serum ferritin level <100 ng/mL or TSAT <20% |
| (4) We do not recommend iron administration that targets the serum ferritin levels to rise to ≥300 ng/mL. (2D) |
The international Dialysis Outcomes and Practice Patterns Study (DOPPS) performed a prospective cohort study to better understand the association between ferritin and mortality. Since serum ferritin levels markedly differ by region, the analyses were conducted separately for the USA, Europe and Japan. To compare the differences among the three regions, a summary table was made from three separate tables for these regions in this report [9]. The table shows the remarkable finding that the ESA doses used in Japan are much smaller than the doses used in the other regions, together with smaller iron dose and lower serum ferritin (Table 2). In addition, recent report shows that in Japan the mean hemoglobin increased to the levels of the other regions (http://www.jinzouzaidan.or.jp/j-dopps/data/).
Table 2.
Characteristics of renal anemia treatment from the DOPPS among the USA, Europe and Japan (adapted from Karaboyas et al.) [9]
| US DOPPS | Europe DOPPS | Japan DOPPS | |
|---|---|---|---|
| Patients (n) | 8510 | 6757 | 2994 |
| Age (years) (mean ± SD) | 62.4 ± 15.1 | 65.9 ± 14.9 | 64.9 ± 12.0 |
| BMI (kg/m2) (mean ± SD) | 28.7 ± 7.0 | 26.1 ± 5.5 | 21.5 ± 3.5 |
| Hemoglobin (g/dL) (mean ± SD) | 11.2 ± 1.2 | 11.5 ± 1.4 | 10.7 ± 1.2 |
| ESA (×1000 U/week) (mean ± SD) | 15 ± 15 | 10 ± 8 | 6 ± 5 |
| IV iron (%)/dose (mg/week) (mean ± SD) | 77%/75 ± 52 | 79%/74 ± 47 | 33%/32 ± 21 |
| Bolus >500 mg/month in 3–4 months | Bolus 38% | Bolus 22% | Bolus 17% |
| Ferritin (ng/mL) (mean ± SD) | 774 ± 467 | 486 ± 380 | 145 ± 205 |
| CRP (mg/L) | NA | 6 (3–14) | 1 (1–3) |
| Crude all-cause mortality (number of deaths) | 0.146/year (962) | 0.139/year (757) | 0.051/year (137) |
Adapted with permission from NDT. BMI, body mass index; NA, not available.
Therefore, the Japanese approach to renal anemia treatment could be considered to be consistent with that practiced for anemia in clinical conditions other than nephrology. Rostoker et al. have recently supported the Japanese approach, as the lower use of IV iron products might be related to the better overall survival of Japanese HD patients, as we presumed that they also raised the concerns about the effect of cumulative doses of IV iron over many years [21, 22].
In the present review, going back to grassroots, we focus on contentious subjects related to the treatment of CKD patients, particularly dealing with differences in iron metabolism between individuals in Japan and those in the rest of the world, asking the following questions: (i) Is there any difference in iron balance or metabolism between the CKD and non-CKD status? (ii) Is there any rationale for iron administration in a patient with already plentiful iron stores or functional iron deficiency? (iii) What range of ferritin is optimal for anemia treatment?
IRON METABOLISM IN CKD
Iron is a vital element for almost all cells because it is required for many biological processes, including erythropoiesis [23]. The amount of iron stored in the body is ∼3–4 g, and excess iron can be very harmful for cells since it can lead to the generation of free radicals by the Fenton reaction [23, 24]. Importantly, there is no physiological mechanism of iron excretion, even when iron intake and stores are excessive, as iron losses occur predominantly through desquamation of epithelial cells in the intestine and the skin and through minor red blood cell (RBC) loss into the gut. To minimize the risk of oxidative stress, body iron homeostasis is tightly regulated by hepcidin. This hormone has emerged as the central regulator of systemic iron homeostasis [24, 25]. It is mainly produced by hepatocytes when iron is abundant, as evidenced by plasma diferric transferrin and stored iron in hepatocytes. Hepcidin synthesis responds to multiple stimuli, including inflammation, erythropoiesis and hypoxia, yet the major regulator is iron. Circulating hepcidin represses the exit of iron from cells by binding to ferroportin (the only known iron export protein) and inducing its degradation, which causes iron sequestration in cells, including macrophages, enterocytes and vascular cells [26, 27]. Thereby, hepcidin restricts iron availability for erythropoiesis, by inhibiting both intestinal iron absorption and the release of stored iron originating from senescent RBCs.
The role of free (ferrous) iron in promoting the synthesis of highly toxic free hydroxyl radicals through the Fenton reaction is well documented. Free iron could exist not only in plasma but also intracellularly [28]. Major technical difficulties hampered the quantification of intracellular iron [28]. Iron sequestration by hepcidin could increase free iron intracellularly, which accelerates the synthesis of cytosolic ferritin. Intracellular iron could be coupled with ferritin, but unbound iron should be increased with the rise of intracellular iron, which may be related to the growth of intracellular pathogens, the activation of macrophages and organ disorders [27].
In iron overload, excess iron may be deposited in almost all tissues, but the bulk of iron is found in association with two cell types: reticuloendothelial (RE) cells and parenchymal tissues [28].
There had been a belief that parenchymal, especially hepatic, iron accumulation is toxic, while iron deposition in the RE system (RES) is considerably less toxic [29]. In the aggressive use of IV iron for anemia treatment in HD patients, it frequently caused hepatic iron accumulation including hepatocytes, but did not imply iron accumulation in other organs, such as the heart [30, 31]. From these data, the authors suggested that current dosing of iron in HD is safe from a cardiac perspective, with no sign of cardiac iron excess. Macrophage intracellular iron level, however, has been a matter of concern to the cardiovascular events, as it affected their polarization and the expression of pro-inflammatory cytokines. A recent review demonstrated that iron affects all among the cell types that participate in the process of atherosclerosis (monocytes/macrophages, endothelial cells, vascular smooth muscle cells and platelets) [12]. Another review strongly endorsed the deleterious role of macrophage iron in the pathogenesis of atherosclerosis and coronary artery disease, where high hepcidin levels and resulting intracellular iron sequestration may be the principal culprits [32]. However, there are still controversial debates regarding the role of macrophage iron and the relationship between iron and atherosclerosis [12, 32–36]. In addition, in the PIVOTAL trial patients assigned to the proactive approach had lower risk of acute myocardial infarction without a higher risk of infection, and hence we need a further investigation into the role of iron in the prevention from atherosclerosis.
When reviewing the role of iron in the pathogenesis of infection, we mentioned that some organisms, categorized as intracellular pathogens, are capable of growing inside macrophages, avoiding their destruction and even increasing their viability in the intracellular iron-rich environment induced by hepcidin [11, 37].
In CKD, elevated serum levels of hepcidin and ferritin were explained by inflammation, the malnutrition–inflammation complex syndrome or diminished renal clearance, although it was not known through which mechanisms and to which extent the uremic state enhanced inflammation precisely [38–40]. This assumption ignored the fact that the main mechanism of hepcidin stimulation is iron.
Research on the relation between hepcidin and the estimated glomerular filtration rate (eGFR) has shown conflicting results [41]. In the research analyzing the factors affecting hepcidin in non-HD CKD patients, serum hepcidin levels were not associated with eGFR or CRP but with ferritin in multivariate analysis, although univariate analysis showed that hepcidin levels negatively correlated with eGFR [42]. Thus, adjustment for confounders dissipates the correlation between eGFR and hepcidin. Similar observation was also reported [43].
The determinants of serum hepcidin have been explored in numerous studies, including patients on HD therapy [44–49] (Table 3). We demonstrated that in such patients serum hepcidin levels were correlated with serum ferritin and transferrin, but not IL-6 and tumor necrosis factor-α [46]. If the high-sensitive C-reactive protein level was >0.3 mg/dL, both IL-6 and ferritin were selected as significant predictors of hepcidin. However, even in this latter patient subgroup, the association with IL-6 was much less tight than with ferritin or iron storage. These observations have been confirmed in animal studies with inflammation combined with iron deficiency [50–53]. Therefore, we presume that, in most cases, the major dependable determinant of hepcidin is serum ferritin or iron storage, and that inflammation, which may be caused by uremic milieu, affects its expression much less. Then, we may not need to distinguish between CKD and non-CKD in the supplementation of iron.
Table 3.
Correlation of serum hepcidin with ferritin and/or IL-6
| Reference | Patient characteristics | Correlated with |
||
|---|---|---|---|---|
| Ferritin | IL-6 | |||
| Tomosugi et al. | [44] | HD patients (n = 40) | (+) | (−) |
| Acute pyelonephritis (n = 2), pneumonia (n = 6), sepsis (n = 8) | NA | (+) | ||
| Healthy volunteers (n = 16) | (+) | (−) | ||
| Weiss et al. | [45] | HD patients (n = 20) | (+) | (−) |
| Kuragano et al. | [46] | HD patients (n = 198) | (+) | (−) |
| HD patients CRP >0.3 mg/dL (n = 36) | (+) | (+) | ||
| van der Putten et al. | [47] | CKD + heart failure (n = 33) | (+) | (−) |
| Stoffel et al. | [48] | Young women with iron deficiency + vaccination (n = 21) | NA | (−) |
| Young women without iron deficiency + vaccination (n = 25) | NA | (+) | ||
NA, not available.
In addition, several recent reports demonstrated that high hepcidin levels and resulting intracellular iron sequestration might be the principal pathogenesis of comorbidities in CKD, infection and CVD [11, 12, 34, 35]. Therefore, limiting hepcidin levels could be critical for the prognosis of CKD patients.
IRON PARAMETERS FOR DIAGNOSING BODY IRON STATUS
Serum ferritin concentration and TSAT are widely used in clinical practice to monitor IV iron therapy in patients treated with ESAs.
TSAT [serum iron × 100 divided by total iron-binding capacity (TIBC)] is the most commonly used measure of iron availability to support erythropoiesis but is not a reliable parameter in the detection of depleted iron stores or a predictor of responsiveness to IV iron therapy in patients with CKD [14, 54, 55]. TSAT is highly variable as it has a circadian and day-to-day variability, and can also fluctuate during inflammation and iron administration [56]. Inflammation decreases transferrin synthesis and plasma iron level, which could be mainly caused by high hepcidin. Iron supplementation increases plasma iron and decreases plasma transferrin level or TIBC, despite no change in some iron preparations [57, 58]. From the clinical studies showing the order of the increases in iron parameters after iron administration, they changed in the order of TSAT, hepcidin and ferritin, irrespective of the routes of iron administration, oral or IV [59, 60]. IV iron can definitely elevate TSAT at least transiently, and this may be effective in increasing iron availability for erythroblasts and accelerating erythropoiesis, which links to a decline in the dose of ESA (ESA sparing effect) [61, 62]. There is no physiological mechanism of iron excretion, even when iron stores are excessive. Thus, these increases in TSAT and iron storage cause an additional increment in hepcidin synthesis. Consequently, elevated hepcidin should decrease TSAT gradually due to the inhibition of iron recycling from old RBCs (intrinsic iron supply), although a small amount of iron can be supplied to plasma from overabundant iron stores. Erythropoiesis then inevitably depends on the extrinsic iron supply, and regular iron administration is necessary to maintain TSAT at sufficiently high levels to guarantee continued erythropoiesis.
The serum ferritin assay has become the standard test for the assessment of iron stores [14, 55, 63]. Ferritin is a cytosolic protein that stores iron. Small amounts are secreted into the plasma where it functions as an iron carrier. It is a marker of total body iron. Confounding variables may alter the regulation of ferritin synthesis and reduce its direct association with body iron stores. Most clinicians are aware of the ‘acute phase’ nature of serum ferritin, but the degree to which this variable deflects from the true measure of iron stores is less often taken into consideration. In the nephrology community, the questionable opinion that high ferritin levels generally do not indicate iron overload is commonly accepted, although serum ferritin is widely used for estimating body iron stores in the clinic. Of particular interest is the role of inflammation/infection in shifting serum ferritin upwards.
In a World Health Organization (WHO) report, it has been mentioned that correction factors can be used to modify raw ferritin data to remove the effects of inflammation when inflammation is detected in a blood sample. After categorizing data according to the inflammatory status of subjects in the incubation, early- and late-convalescence groups, these values can be individually corrected using multipliers 0.77, 0.53 and 0.75, respectively [64, 65].
Cook verified this concept using data previously published by Lipschitz et al. [66, 67]. In this study, bone marrow aspiration was performed, and iron stores were graded into the following four categories: absent, decreased, moderate and increased. ‘Decreased’ corresponded to the average stores of anemic adult females, whereas ‘moderate’ corresponded to the average stores found in healthy males. Marrow iron in patients with severe infection or inflammation whose serum ferritin ranged from 10 to 1650 ng/mL was compared with controls without these conditions at each level of marrow iron, and the serum ferritin was ∼3 times higher in patients with infection or inflammation than in controls.
Theurl et al. made a similar observation [68]. In their study, the log ferritin/soluble transferrin receptor ratio was used to categorize patients as having either anemia of chronic disease (ACD) or ACD with iron deficiency anemia (ACD/IDA), as the ratio has been demonstrated to discriminate ACD from ACD/IDA appropriately [69]. ACD patients experienced relatively severe infections, including pneumonia. As expected, the IL-6 and CRP levels were significantly higher in ACD and ACD/IDA patients than in healthy controls, whereas no difference was found between ACD and ACD/IDA patients. Serum ferritin and hepcidin levels were significantly elevated in ACD patients compared with controls, but significantly lower in ACD/IDA patients than in ACD patients. Based on these observations, ferritin and hepcidin levels did not increase in the condition of iron deficiency even with inflammation and/or infection.
Thus, serum ferritin might roughly but reliably enable the diagnosis of depleted iron storage. This concept is also supported by the Guidelines for the Management of Iron Deficiency Anaemia of the British Society of Gastroenterology [70].
IS THERE ANY RATIONALE FOR IRON ADMINISTRATION IN A PATIENT WITH ALREADY PLENTIFUL IRON STORES OR FUNCTIONAL IRON DEFICIENCY?
It is widely believed that patients with CKD are prone to iron deficiency, and its etiology is multifactorial including blood loss during HD session [71]. The symptoms and signs of iron deficiency are partially explained by the presence of anemia. In CKD patients treated with ESA, iron deficiency necessitates an increase in the dose of ESA for maintaining hemoglobin levels. In addition, iron deficiency-induced thrombocytosis may have evolved to maintain or increase the coagulation capacity [72]. Iron deficiency consists of two main categories: absolute and functional iron deficiency. Absolute iron deficiency is relatively easily diagnosed as a decline in serum ferritin levels when there is a deficiency of total body iron stores.
Functional iron deficiency, defined by normal or elevated serum ferritin levels but TSAT <20%, occurs if iron stores are full but iron release from the storage site is impaired, leading to impaired iron supply to erythroid progenitors in the bone marrow [73]. The discovery of hepcidin, the master regulator of iron homeostasis, has clarified the underlying mechanism. High hepcidin levels inhibit iron recycling from senescent RBCs, intestinal iron absorption and iron release from regular storage sites, such as the liver and RES, and consequently, diminish iron availability for erythropoiesis.
As mentioned above, in our opinion, one should pay more attention to iron retention and/or increased iron stores caused by the excessive use of IV iron compounds than to the uremic milieu for other factors stimulating hepcidin production.
It has long been established that even in iron-replete patients, those supplemented with IV iron have an enhanced hemoglobin response to ESAs, with better maintenance of iron stores and lower dosage requirements of ESA compared with those patients receiving oral iron and no iron supplementation [74].
As mentioned above, IV iron inevitably increases hepcidin and ferritin levels, and consequently decreases iron availability for erythropoiesis. Then we question whether it makes sense to administer iron to patients with functional iron deficiency and whether such a practice is safe. It has been established by several reports that hyporesponsiveness to ESAs is associated with a poor prognosis in CKD patients on MHD [75–80]. In the presence of absolute iron deficiency, an insufficient iron supply to the bone marrow depresses erythropoiesis, and iron administration definitely attenuates ESA hyporesponsiveness [81]. However, iron administration to patients with functional iron deficiency will raise hemoglobin more or less depending on each patient’s condition. The rise in hemoglobin alone in response to IV iron is not necessarily associated with an improvement in ESA responsiveness because the effect may not last long due to the increase in serum hepcidin, and in a vicious circle, additional administration is required. In a meta-analysis determining the benefits and harms of IV iron supplementation, there was no significant relationship between total iron dose or iron dose per month and hemoglobin, while the standard mean differences of ferritin increased with increasing total iron dose [82]. Similarly, in another meta-analysis only using data of randomized controlled studies, IV iron therapy for functional IDA in HD patients resulted in a significant increase in TSAT but did not change hemoglobin level significantly [58]. Thus, we cannot be sure that iron administration increases hemoglobin levels in patients with functional iron deficiency on a long-term basis, which might be related to the factors inhibiting erythropoiesis, e.g. inflammation and hepcidin. Even in the PIVOTAL trial, the mean hemoglobin level increased from 10.6 g/dL at baseline to 11.1 g/dL at study end, with only 110 mg of iron being used for erythropoiesis, as the estimated blood volume is 6.3 L and iron content in hemoglobin is 3.4 mg/g hemoglobin, whereas the rest of the administered iron, ∼9000 mg, was stored as ferritin or lost with blood during HD sessions [1, 83].
IRON OVERLOAD AND ESA HYPORESPONSIVENESS
Based on recent reports, we suspect that bone marrow iron overload may also inhibit erythropoiesis, although it is commonly believed that an increase in iron stores or serum ferritin attenuates hyporesponsiveness to ESAs in patients with CKD [84–86].
In the database of the Japanese Society for Dialysis Therapy Renal Data Registry, relationships of the ESA resistance index (ERI) with serum ferritin were examined in patients who underwent HD therapy at various facilities [19], ERI being defined as ‘ESA dose/(hemoglobin level × post-dialysis body weight)’. For patients with TSAT levels ≥20%, there was no clear association between TSAT and ERI, whereas a U-shaped relationship was observed between the ERI and ferritin, with a nadir of ERI at ∼100 ng/mL of ferritin [87]. The ERI also tended to be high for serum ferritin levels ≥300 ng/mL [19]. These tendencies were observed in both patients on darbepoetin and those on epoetin beta pegol therapy. We would like to emphasize that CRP levels were <2.0 mg/dL in most of the patients.
Similar observations have been reported in the USA. Gaweda et al. examined the factors that affected the maximum hemoglobin response to erythropoietin (EPO) in HD patients [88]. Among several parameters, serum ferritin was one of the markers of a nonlinear effect modification of the erythropoietic response in HD patients, and the maximum hemoglobin response to EPO was achieved for serum ferritin levels between 350 and 500 ng/mL. Thus, an impaired erythropoietic response could be present when ferritin was elevated >500 ng/mL. In the later study, the same authors observed increasing hemoglobin response to ESA over the range of high ferritin values. It may be presumed that these analyses were not adjusted using the marker of inflammation, CRP [89]. The difference in the range of ferritin for ESA responsiveness between the USA and Japan might be caused by different iron administration policies, baseline ferritin levels and inflammation. In our cohort study, most of the patients had serum ferritin levels <300 ng/mL and only 20.8% of them were treated with IV iron, and then ESA hyporesponsiveness was not related to iron storage or ferritin level [79]. Similarly, the index of ESA resistance expressed as the ratio of the total weekly ESA dose to hematocrit (EPO/Hct) was found to be correlated with log-transformed ferritin [90]. Considering the important controversy regarding the main culprit responsible for the inhibition of erythropoiesis at high ferritin levels, we would like to emphasize the role of excess iron rather than inflammation or the malnutrition–inflammation complex syndrome. A report from the European Clinical Dialysis Database (EuCliD®), showing a significant association between the 2-year iron cumulative dose and ERI using the analysis adjusted by CRP and other factors provides further support for our interpretation of available data [91].
Two possible mechanisms could be proposed regarding the relationship between high iron stores and ESA hyporesponsiveness. First, iron administration further increases hepcidin levels, which in turn hampers iron recycling from senescent RBCs and decreases the iron supply to erythroblasts. This mechanism is supported by recent animal studies, where hepcidin has emerged as a major factor involved in the development of anemia in CKD, as hepcidin knockout mice with adenine-induced CKD did not exhibit anemia and iron deficiency [81, 92]. Thus, recent hepcidin research suggests to us that the concept of a beneficial effect of pathologically increased iron stores for erythropoiesis may be completely erroneous. Second, a large body of in vitro and in vivo evidence shows that bone marrow iron overload inhibits erythroid differentiation. The increase in reactive oxygen species caused by bone marrow iron overload could impair the proliferation potential of erythroid precursors and disturb the bone marrow-derived mesenchymal stem cells [93], as well as damage bone marrow stromal cells with reduced expression of C-X-C motif chemokine 12 (CXCL12), vascular cell adhesion molecule-1 (VCAM-1), Kit-ligand and insulin-like growth factor-1 (IGF-1) [94] (Figure 1). Thus to improve ESA responsiveness for an efficient anemia therapy in patients with CKD one should consider these two opposite effects on the bone marrow, namely (i) activating the differentiation of erythroid cells by supplying iron and (ii) inhibiting such differentiation by iron-mediated hepcidin and oxidative stress-rich actions, as if stepping on an engine’s brake and accelerator at the same time (Figure 2). We presumed that the evaluation of these opposite effects counterbalanced might be too difficult to perform renal anemia therapy with appropriate iron dose.
FIGURE 1.
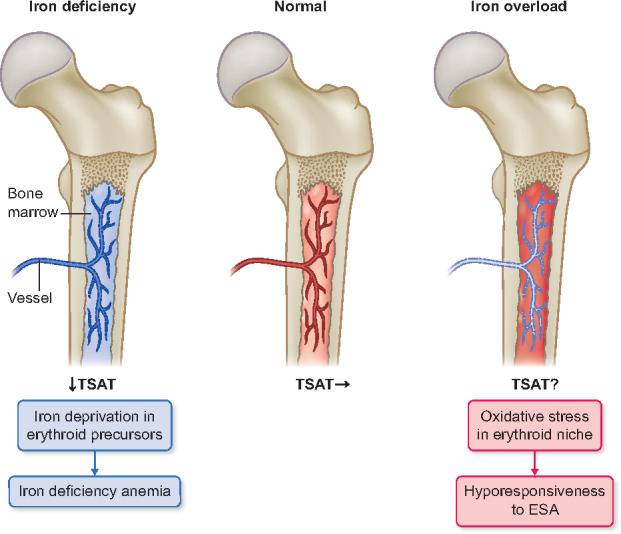
Effect of iron status on the erythropoiesis in bone marrow. In the condition of iron deficiency, TSAT and serum ferritin level are decreased, which diminishes iron supplementation for erythroid precursors. In the condition of iron overload, serum ferritin level is high but TSAT may be variable by the timing of iron administration. Bone marrow iron overload attenuates erythroid differentiation by oxidative stress in erythroid niche, which causes hyporesponsiveness to ESAs.
FIGURE 2.
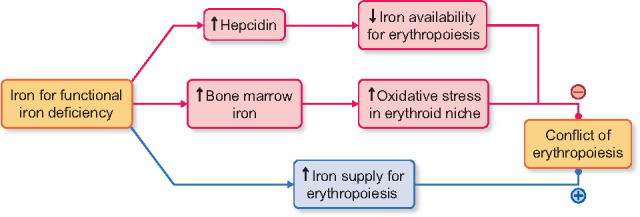
Hypothesized conflict of erythropoiesis, activation versus inhibition, in IV iron administration for functional iron deficiency. (Pink) Activation of the differentiation of erythroid cells by supplying iron for erythropoiesis. (Blue) Inhibition of erythroid differentiation by iron-mediated hepcidin and oxidative stress-rich actions.
Therefore, we do not agree with the opinion that iron supplementation is necessary or essential, as long as iron administration could increase hemoglobin or reduce the required dose of ESAs. Undoubtedly, expanded iron storage could increase the risk of infection and cardiovascular events, even if ESA responsiveness would be improved. We would like to emphasize again that regular high-dose iron administration does not necessarily ameliorate ESA responsiveness. Optimum iron storage and administration should be reconsidered for an efficient and safe management of renal anemia.
WHAT IS THE OPTIMAL RANGE OF SERUM FERRITIN IN CKD?
We would like to take issue with the large recommended target range of serum ferritin in CKD, assuming that iron metabolism does not fundamentally differ between CKD and non-CKD conditions.
Given the pivotal role of hepcidin in maintaining iron homeostasis as well as in the pathogenesis of comorbidities in CKD, one needs to know its optimal range. Although the serum hepcidin level has never been measured in clinical fields treating renal anemia, we might be able to estimate its level from the serum ferritin level as serum ferritin has been demonstrated to be the major predictor of hormone level in nearly all the studies published so far, which also included CRP as covariate [49]. In this study, all previous studies concordantly suggested that the regulation of hepcidin by iron stores was preserved in HD patients, as illustrated by the fact that serum ferritin levels were invariably among the strongest predictors of hepcidin levels.
Serum ferritin could, therefore, be a valuable index of optimal iron storage for erythropoiesis. Patient outcomes could be improved by determining more adequately the lower ferritin limit, reflecting the minimum requirement for optimal erythropoiesis and the upper limit reflecting increased comorbidity risk.
Lower limit of serum ferritin for minimal iron stores required for erythropoiesis
As regards the minimum range of ferritin, an association between hemoglobin concentration and serum ferritin concentration was analyzed in a Norwegian general patient population without inflammation and normal kidney function [95]. The entire hemoglobin distribution was shifted downwards in patients with serum ferritin levels <20 ng/mL in women, while the turning point toward lower hemoglobin was at a ferritin level of 30 ng/mL in men. Similarly, we found that in chronic HD patients the turning point toward lower hemoglobin was a serum ferritin <50 ng/mL [79]. When the patients were stratified into four groups according to their serum ferritin levels (<50, 50–100, 101–300 and >300 ng/mL) a significant correlation (P < 0.001, R = 0.89) between ferritin and hemoglobin levels was observed only in those with levels <50 ng/mL, while no significant relationship was observed in the three other groups. Based on this observation, we concluded that in HD patients the minimal ferritin concentration, corresponding to the minimal amount of stored iron required for erythropoiesis, could be in the range of 50 ng/mL. The treatment policy could avoid absolute iron deficiency, which is supported by guidelines of the British Society of Gastroenterology and the JSDT [17, 70].
Upper limit of serum ferritin to minimize the risks of comorbidities due to iron sequestration and high hepcidin levels
As to the ferritin concentration reflecting iron overload, a guideline from the WHO and Centers for Disease Control states that serum ferritin levels >200 ng/mL in men and >150 ng/mL in women indicate a risk of iron overload [96].
Kalantar-Zadeh et al. [2] analyzed data from a cohort of 58 058 HD patients and found, after time-dependent and multivariate adjustments without CRP, that patients with serum ferritin levels between 200 and 1200 ng/mL were at the lowest risk of cardiovascular and all-cause mortality compared with patients in whom the same iron parameter was outside these ranges. This observation was supportive of the safety of high ferritin levels. However, only 9% among these patients had ferritin levels <100 ng/mL, who had undoubtedly low iron storage even with inflammation, and they had even better outcomes than those with a reference ferritin range between 100 and 200 ng/mL. This range of iron management is currently being applied in about 60% of Japanese dialysis patients [19].
We prospectively monitored 1086 Japanese HD patients for 2 years and evaluated the association between serum ferritin levels and adverse outcomes including mortality. In this study [the Prospective Study of Treatment for Renal Anemia on Prognosis in hemodialysis patients (TRAP) study], covariates showing time-dependent variation were adjusted for confounding using measured values including CRP and albumin at the same time points every 3 months [6]. We found that patients with high serum ferritin levels (>100 ng/mL) or large ferritin level fluctuations above this range were at a higher risk of all adverse events combined, those with cardio-cerebrovascular disease, infections and death as compared with patients with persistently lower serum ferritin levels (<100 ng/mL) [6].
Finally, we focused on those ferritin levels corresponding to the range of hepcidin levels that are known to affect cellular iron exit, based on the close relationship between serum ferritin and hepcidin. The representative model of cellular iron exit, intestinal absorption of nonheme iron has been well explored with serum hepcidin or ferritin levels. As mentioned above, the iron absorption rate is ≥10% in a subject with a ferritin level <25 ng/mL, while it decreases to <2% if the ferritin level is >100 ng/mL. Although these values varied slightly between studies they were mostly consistent in non-CKD and CKD patients [97–100] (Figure 3). Regarding the relationship between early iron exits from the RES and serum ferritin levels, radiolabeled iron release from heat-damaged RBCs by the RES was examined in patients with iron deficiency, idiopathic hemochromatosis, inflammation, marrow aplasia or hyperplastic erythropoiesis [101]. In this study, the percentage of early release clearly correlated with the plasma ferritin level, and it appeared to reach the bottom line if the ferritin level was >100 ng/mL. Based on these observations, we presume that in the range of hepcidin levels, which correspond to serum ferritin levels >100 ng/mL, there is a substantial decrease in cellular iron exit via the hepcidin induced decrease in ferroportin-dependent iron export [102].
FIGURE 3.
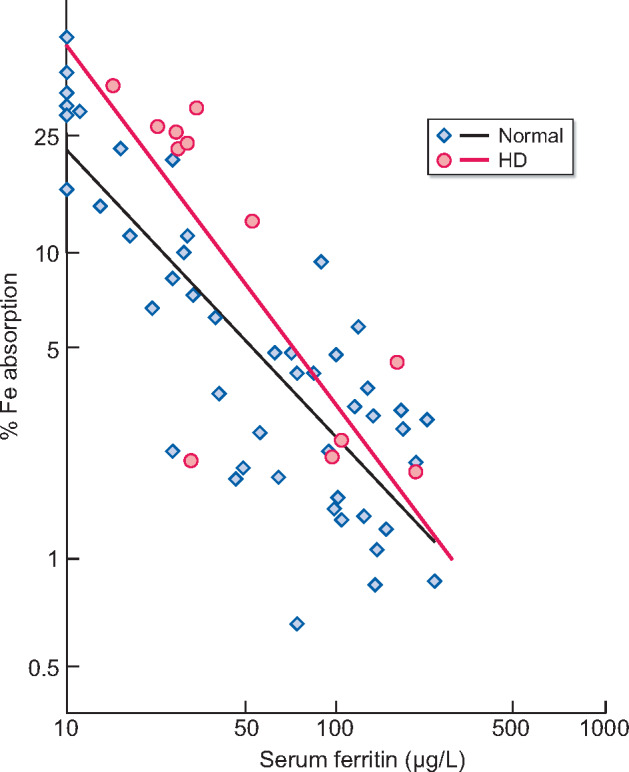
Food iron absorption in chronic HD patients in comparison with control. The black line and blue square plots show the relationship between serum ferritin and food iron absorption in 47 normal volunteers [97]. Increasing log-transformed percent iron absorption is associated with a linear decrease in log-transformed serum ferritin. The red line and red circle plots show the same relationship in 13 HD patients, whose ferritin levels are within the range from 10 to 200 µg/L from the previous reports, on the same graph [100]. Interestingly, the line linking iron absorption to serum ferritin in HD patients overlaps that of the healthy controls for ferritin levels. At that time, water purification for HD was far from endotoxin free. Thus even in a condition of poor dialysate purity, i.e. a highly probable cause of inflammation, iron absorption in HD patients was mainly dependent on ferritin.
Given the presumed hepcidin concentration threshold for risks such as CVD and infection and the strong correlation between hepcidin and ferritin, one could use ferritin instead of hepcidin as a valid marker of iron stores. From the observations above, we could propose the acceptable ferritin upper limit as 150 ng/mL with its clinically fluctuating range [6, 103, 104]. Finally, we estimate that the optimal ferritin range might be about 50–150 ng/mL when considering both safety margin and ESA responsiveness. These values will probably provoke reactions of skepticism by nephrologists in Western countries, but we would like to emphasize that they are within the accepted ferritin range in disciplines other than nephrology [105–111]. As a matter of course, in considering the beneficial effect of IV iron in the setting of congestive heart failure, further investigation should be essential for ascertaining whether our optimal ferritin range is appropriate in CKD patients [112, 113].
CONCLUSION
The recently published, randomized controlled trial, PIVOTAL demonstrated the safety of high-dose IV iron therapy, including hard outcomes. However, we would like to take issue with PIVOTAL’s evidence of safety, since the comparator group of patients also received a substantial amount of IV iron, and this low-dose iron group had relatively high serum ferritin levels as well, at least from a Japanese perspective. It is noteworthy that iron exerts two opposite actions in erythropoiesis. On one hand, it activates erythroid differentiation by supplying iron to red cell precursors, but on the other hand, it inhibits this differentiation by an iron-mediated increase in serum hepcidin and induction of oxidative stress. Moreover, recent remarkable advances in the understanding of iron metabolism have taught us that hepcidin plays an essential role in the pathogenesis of renal anemia as well as comorbidities in CKD patients, including atherosclerosis and infection. As this risk is predominantly increased by iron—and to a smaller degree by inflammation—the dose of iron administered to patients with CKD should be more appropriately controlled. In this review, we propose the optimal ferritin range as 50–150 ng/mL from the recent observations, which is precisely the range for iron management in HD patients in Japan. The PIVOTAL trial, however, goes in the opposite direction of the Japanese approach and has demonstrated better outcomes with a proactive approach. Considering the opposite extremes of ferritin status in Japan and the rest of the world in patients with CKD, our proposition will probably encounter profound skepticism in the nephrology community outside of Japan, although it is within the range accepted by medical disciplines other than nephrology [114]. More importantly, we have not yet determined whether the Japanese approach can be extrapolated to Western end-stage renal disease populations, as it has been demonstrated that there is variation in the prevalence of inflammation, and in practice and nonpractice factors between countries [115, 116]. To solve this controversy, it would seem essential to explore in future randomized controlled trials the efficacy and safety of a well-balanced therapy with ESAs and iron in anemic patients with CKD. Whether the introduction of hypoxia-inducible factor prolyl hydroxylase inhibitors into the treatment of renal anemia will lead to different iron parameter targets remains to be seen.
ACKNOWLEDGEMENTS
We gratefully acknowledge the advice and editorial assistance of Tilman B. Drueke, MD, Villejuif, France.
CONFLICT OF INTEREST STATEMENT
T.N. reports personal fees from Kyowa-Kirin, personal fees from Chugai , personal fees from Torii Pharmaceuticals, personal fees from Bayer Yakuhin and personal fees from Tanabe-Mitsubishi, outside the submitted work. T.K. reports personal fees from Chugai, personal fees from Kyowa-Kirin, personal fees from Astellas, personal fees from Bayer Yakuhin, grants and personal fees from Ono, personal fees from Fuso, grants from kissei and personal fees from Tanabe Mitsubishi, outside the submitted work. The results presented in this article have not been published previously in whole or part, except in abstract format.
REFERENCES
- 1. Macdougall IC, White C, Anker SD. et al. ; PIVOTAL Investigators and Committees. Intravenous iron in patients undergoing maintenance hemodialysis. N Engl J Med 2019; 380: 447–458 [DOI] [PubMed] [Google Scholar]
- 2. Kalantar-Zadeh K, Regidor DL, McAllister CJ. et al. T ime-dependent associations between iron and mortality in hemodialysis patients. J Am Soc Nephrol 2005; 16: 3070–3080 [DOI] [PubMed] [Google Scholar]
- 3. Kshirsagar AV, Freburger JK, Ellis AR. et al. Intravenous iron supplementation practices and short-term risk of cardiovascular events in hemodialysis patients. PLoS One 2013; 8: e78930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hougen I, Collister D, Bourrier M. et al. Safety of intravenous iron in dialysis: a systematic review and meta-analysis. Clin J Am Soc Nephrol 2018; 13: 457–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kletzmayr J, Horl WH.. Iron overload and cardiovascular complications in dialysis patients. Nephrol Dial Transplant 2002; 17: 25–29 [DOI] [PubMed] [Google Scholar]
- 6. Kuragano T, Matsumura O, Matsuda A. et al. Association between hemoglobin variability, serum ferritin levels, and adverse events/mortality in maintenance hemodialysis patients. Kidney Int 2014; 86: 845–854 [DOI] [PubMed] [Google Scholar]
- 7. Miskulin DC, Tangri N, Bandeen-Roche K. et al. Developing Evidence to Inform Decisions about Effectiveness (DEcIDE) Network Patient Outcomes in End Stage Renal Disease Study Investigators: intravenous iron exposure and mortality in patients on hemodialysis. Clin J Am Soc Nephrol 2014; 9: 1930–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bailie GR, Larkina M, Goodkin DA. et al. Data from the Dialysis Outcomes and Practice Patterns Study validate an association between high intravenous iron doses and mortality. Kidney Int 2015; 87: 162–168 [DOI] [PubMed] [Google Scholar]
- 9. Karaboyas A, Morgenstern H, Pisoni RL. et al. Association between serum ferritin and mortality: findings from the USA, Japan and European Dialysis Outcomes and Practice Patterns Study. Nephrol Dial Transplant 2018; 33: 2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li X, Cole SR, Kshirsagar AV. et al. Safety of dynamic intravenous iron administration strategies in hemodialysis patients. Clin J Am Soc Nephrol 2019; 14: 728–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakanishi T, Kuragano T, Nanami M. et al. Iron localization and infectious disease in chronic kidney disease patients. Am J Nephrol 2016; 43: 237–244 [DOI] [PubMed] [Google Scholar]
- 12. Kraml P. The role of iron in the pathogenesis of atherosclerosis. Physiol Res 2017; 66: S55–S67 [DOI] [PubMed] [Google Scholar]
- 13. Nakanishi T, Kuragano T.. Intravenous iron and maintenance hemodialysis. N Engl J Med 2019; 380: e46. [DOI] [PubMed] [Google Scholar]
- 14. KDIGO: KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int 2012; 2 (Suppl 2): 279–335 [Google Scholar]
- 15. Peyrin-Biroulet L, Williet N, Cacoub P.. Guidelines on the diagnosis and treatment of iron deficiency across indications: a systematic review. Am J Clin Nutr 2015; 102: 1585–1594 [DOI] [PubMed] [Google Scholar]
- 16. Garcia-Casal MN, Pasricha SR, Martinez RX. et al. Are current serum and plasma ferritin cut-offs for iron deficiency and overload accurate and reflecting iron status? A systematic review. Arch Med Res 2018; 49: 405–417 [DOI] [PubMed] [Google Scholar]
- 17. Yamamoto H, Nishi S, Tomo T. et al. 2015 Japanese Society for Dialysis Therapy: guidelines for renal anemia in chronic kidney disease. Ren Replace Ther 2017; 3: 36 [Google Scholar]
- 18. Yamamoto H, Tsubakihara Y.. Limiting iron supplementation for anemia in dialysis patients-the basis for Japan’s conservative guidelines. Semin Dial 2011; 24: 269–271 [DOI] [PubMed] [Google Scholar]
- 19. Nakai S, Hanafusa N, Masakane I. et al. An overview of regular dialysis treatment in Japan (as of 31 December 2012). Ther Apher Dial 2012; 18: 535–602 [DOI] [PubMed] [Google Scholar]
- 20. Macdougall IC, White C, Anker SD. et al. Intravenous iron and maintenance hemodialysis. N Engl J Med 2019; 380: e46–e458 [DOI] [PubMed] [Google Scholar]
- 21. Rostoker G, Vaziri ND.. Iatrogenic iron overload and its potential consequences in patients on hemodialysis. Presse Med 2017; 46: e312–e328 [DOI] [PubMed] [Google Scholar]
- 22. Rostoker G, Griuncelli M, Loridon C. et al. Maximal standard dose of parenteral iron for hemodialysis patients: an MRI-based decision tree learning analysis. PLoS One 2014; 9: e115096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Daher R, Karim Z.. Iron metabolism: state of the art. Transfus Clin Biol 2017; 24: 115–119 [DOI] [PubMed] [Google Scholar]
- 24. Ganz T. Systemic iron homeostasis. Physiol Rev 2013; 93: 1721–1741 [DOI] [PubMed] [Google Scholar]
- 25. Ganz T, Nemeth E.. Hepcidin and disorders of iron metabolism. Annu Rev Med 2011; 62: 347–360 [DOI] [PubMed] [Google Scholar]
- 26. Nemeth E, Tuttle MS, Powelson J. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004; 306: 2090–2093 [DOI] [PubMed] [Google Scholar]
- 27. Nakanishi T, Hasuike Y, Otaki Y. et al. Hepcidin: another culprit for complications in patients with chronic kidney disease? Nephrol Dial Transplant 2011; 26: 3092–3100 [DOI] [PubMed] [Google Scholar]
- 28. Hershko C, Link G, Cabantchik I.. Pathophysiology of iron overload. Ann NY Acad Sci 1998; 850: 191–201 [DOI] [PubMed] [Google Scholar]
- 29. Kshirsagar AV, Li X.. Long-term risks of intravenous iron in end-stage renal disease patients. Adv Chronic Kidney Dis 2019; 26: 292–297 [DOI] [PubMed] [Google Scholar]
- 30. Tolouian R, Mulla ZD, Diaz J. et al. Liver and cardiac iron deposition in patients on maintenance hemodialysis by magnetic resonance imaging T2. Iran J Kidney Dis 2016; 10: 68–74 [PubMed] [Google Scholar]
- 31. Holman R, Olynyk JK, Kulkarni H. et al. Characterisation of hepatic and cardiac iron deposition during standard treatment of anaemia in haemodialysis. Nephrology (Carlton )2017; 22: 114–117 [DOI] [PubMed] [Google Scholar]
- 32. Cornelissen A, Guo L, Sakamoto A. et al. New insights into the role of iron in inflammation and atherosclerosis. EBioMedicine 2019; 47: 598–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saeed O, Otsuka F, Polavarapu R. et al. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol 2012; 32: 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li JJ, Meng X, Si HP. et al. Hepcidin destabilizes atherosclerotic plaque via overactivating macrophages after erythrophagocytosis. Arterioscler Thromb Vasc Biol 2012; 32: 1158–1166 [DOI] [PubMed] [Google Scholar]
- 35. Habib A, Polavarapu R, Karmali V. et al. Hepcidin-ferroportin axis controls toll-like receptor 4 dependent macrophage inflammatory responses in human atherosclerotic plaques. Atherosclerosis 2015; 241: 692–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gill D, Del Greco MF, Walker AP. et al. The effect of iron status on risk of coronary artery disease: a Mendelian randomization study. Arterioscler Thromb Vasc Biol 2017; 37: 1788–1792 [DOI] [PubMed] [Google Scholar]
- 37. Liu D, Gan ZS, Ma W. et al. Synthetic porcine hepcidin exerts opposite roles in Escherichia coli and Salmonella infections. Antimicrob Agents Chemother 2017; 61: e02638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kalantar-Zadeh K, Rodriguez RA, Humphreys MH.. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol Dial Transplant 2004; 19: 141–149 [DOI] [PubMed] [Google Scholar]
- 39. Nemeth E, Rivera S, Gabayan V. et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sedlackova T, Racek J, Rajdl D. et al. Relationship between hepcidin and ferritin in haemodialysed patients. Wien Klin Wochenschr 2013; 125: 448–452 [DOI] [PubMed] [Google Scholar]
- 41. van der Weerd NC, Grooteman MP, Nubé MJ. et al. Hepcidin in chronic kidney disease: not an anaemia management tool, but promising as a cardiovascular biomarker. Neth J Med 2015; 73: 108–118 [PubMed] [Google Scholar]
- 42. Uehata T, Tomosugi N, Shoji T. et al. Serum hepcidin-25 levels and anemia in non-dialysis chronic kidney disease patients: a cross-sectional study. Nephrol Dial Transplant 2012; 27: 1076–1083 [DOI] [PubMed] [Google Scholar]
- 43. Peters HP, Laarakkers CM, Swinkels DW. et al. Serum hepcidin-25 levels in patients with chronic kidney disease are independent of glomerular filtration rate. Nephrol Dial Transplant 2010; 25: 848–853 [DOI] [PubMed] [Google Scholar]
- 44. Tomosugi N, Kawabata H, Wakatabe R. et al. Detection of serum hepcidin in renal failure and inflammation by using protein chip system. Blood 2006; 108: 1381–1387 [DOI] [PubMed] [Google Scholar]
- 45. Weiss G, Theurl I, Eder S. et al. Serum hepcidin concentration in chronic haemodialysis patients: associations and effects of dialysis, iron and erythropoietin therapy. Eur J Clin Invest 2009; 39: 883–890 [DOI] [PubMed] [Google Scholar]
- 46. Kuragano T, Shimonaka Y, Kida A. et al. Determinants of hepcidin in patients on maintenance hemodialysis: role of inflammation. Am J Nephrol 2010; 31: 534–540 [DOI] [PubMed] [Google Scholar]
- 47. van der Putten K, Jie KE, van den Broek D. et al. Hepcidin-25 is a marker of the response rather than resistance to exogenous erythropoietin in chronic kidney disease/chronic heart failure patients. Eur J Heart Fail 2010; 12: 943–950 [DOI] [PubMed] [Google Scholar]
- 48. Stoffel NU, Lazrak M, Bellitir S. et al. The opposing effects of acute inflammation and iron deficiency anemia on serum hepcidin and iron absorption in young women. Haematologica 2019; 104: 1143–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Valenti L, Messa P, Pelusi S. et al. Hepcidin levels in chronic hemodialysis patients: a critical evaluation. Clin Chem Lab Med 2014; 52: 613–619 [DOI] [PubMed] [Google Scholar]
- 50. Krijt J, Vokurka M, Chang K-T. et al. Expression of Rgmc, the murine ortholog of hemojuvelin gene, is modulated by development and inflammation, but not by iron status or erythropoietin. Blood 2004; 104: 4308–4310 [DOI] [PubMed] [Google Scholar]
- 51. Darshan D, Frazer DM, Wilkins SJ. et al. Severe iron deficiency blunts the response of the iron regulatory gene Hamp and proinflammatory cytokines to lipopolysaccharide. Haematologica 2010; 95: 1660–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Theurl I, Schroll A, Nairz M. et al. Pathways for the regulation of hepcidin expression in anemia of chronic disease and iron deficiency anemia in vivo. Haematologica 2011; 96: 1761–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lofruthe N, Gallitz I, Traeger L. et al. Intravenous iron carboxymaltose as a potential therapeutic in anemia of inflammation. PLoS One 2016; 11: e0158599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Recommendations to prevent and control iron deficiency in the United States. Centers for Disease Control and Prevention. MMWR Recomm Rep 1998; 47: 1–29 [PubMed] [Google Scholar]
- 55. Thomas DW, Hinchliffe RF, Briggs C. et al. ; British Committee for Standards in Haematology. Guideline for the laboratory diagnosis of functional iron deficiency. Br J Haematol 2013; 161: 639–648 [DOI] [PubMed] [Google Scholar]
- 56. Goodnough LT, Skikne B, Brugnara C.. Erythropoietin, iron, and erythropoiesis. Blood 2000; 96: 823–833 [PubMed] [Google Scholar]
- 57. Canavese C, Bergamo D, Ciccone G. et al. Low-dose continuous iron therapy leads to a positive iron balance and decreased serum transferrin levels in chronic haemodialysis patients. Nephrol Dial Transplant 2004; 19: 1564–1570 [DOI] [PubMed] [Google Scholar]
- 58. Susantitaphong P, Alqahtani F, Jaber BL.. Efficacy and safety of intravenous iron therapy for functional iron deficiency anemia in hemodialysis patients: a meta-analysis. Am J Nephrol 2014; 39: 130–141 [DOI] [PubMed] [Google Scholar]
- 59. Moretti D, Goede JS, Zeder C. et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood 2015; 126: 1981–1989 [DOI] [PubMed] [Google Scholar]
- 60. Talbot NP, Lakhal S, Smith TG. et al. Regulation of hepcidin expression at high altitude. Blood 2012; 119: 857–860 [DOI] [PubMed] [Google Scholar]
- 61. Richardson D, Bartlett C, Will EJ.. Optimizing erythropoietin therapy in hemodialysis patients. Am J Kidney Dis 2001; 38: 109–117 [DOI] [PubMed] [Google Scholar]
- 62. Aronoff GR. Safety of intravenous iron in clinical practice: implications for anemia management protocols. J Am Soc Nephrol 2004; 2: 99–106 [DOI] [PubMed] [Google Scholar]
- 63. Cavill I. Iron status as measured by serum ferritin: the marker and its limitations. Am J Kidney Dis 1999; 34: 12–17 [DOI] [PubMed] [Google Scholar]
- 64. Thurnham DI, McCabe LD, Haldar S. et al. Adjusting plasma ferritin concentrations to remove the effects of subclinical inflammation in the assessment of iron deficiency: a meta-analysis. Am J Clin Nutr 2010; 92: 546–555 [DOI] [PubMed] [Google Scholar]
- 65. Thurnham DI, McCabe GP.. Influence of infection and inflammation on biomarkers of nutritional status with an emphasis on vitamin A and iron. 2012; A2.4: 63–80. https://www.who.int/nutrition/publications/micronutrients/background_paper4_report_assessment_vitAandIron_status.pdf (29 October 2019, date last accessed) [Google Scholar]
- 66. Lipschitz DA, Cook JD, Finch CA.. A clinical evaluation of serum ferritin as an index of iron stores. N Engl J Med 1974; 290: 1213–1216 [DOI] [PubMed] [Google Scholar]
- 67. Cook JD. Clinical evaluation of iron deficiency. Semin Hematol 1982; 19: 6–18 [PubMed] [Google Scholar]
- 68. Theurl I, Aigner E, Theurl M. et al. Regulation of iron homeostasis in anemia of chronic disease and iron deficiency anemia: diagnostic and therapeutic implications. Blood 2009; 113: 5277–5286 [DOI] [PubMed] [Google Scholar]
- 69. Punnonen K, Irjala K, RajamäKi A.. Serum transferrin receptor and its ratio to serum ferritin in the diagnosis of iron deficiency. Blood 1997; 89: 1052–1057 [PubMed] [Google Scholar]
- 70. Goddard AF, James MW, McIntyre AS. et al. ; British Society of Gastroenterology. Guidelines for the management of iron deficiency anaemia. Gut 2011; 60: 1309–1316 [DOI] [PubMed] [Google Scholar]
- 71. Macdougall IC, Bircher AJ, Eckardt KU. et al. ; Conference Participants. Iron management in chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int 2016; 89: 28–39 [DOI] [PubMed] [Google Scholar]
- 72. Evstatiev R, Bukaty A, Jimenez K. et al. Iron deficiency alters megakaryopoiesis and platelet phenotype independent of thrombopoietin. Am J Hematol 2014; 89: 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sunder-Plassmann G, Horl WH.. Erythropoietin and iron. Clin Nephrol 1997; 47: 141–157 [PubMed] [Google Scholar]
- 74. Macdougall IC, Tucker B, Thompson J. et al. A randomized controlled study of iron supplementation in patients treated with erythropoietin. Kidney Int 1996; 50: 1694–1699 [DOI] [PubMed] [Google Scholar]
- 75. Kilpatrick RD, Critchlow CW, Fishbane S. et al. Greater epoetin alfa responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol 2008; 3: 1077–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bradbury BD, Wang O, Critchlow CW. et al. Exploring relative mortality and epoetin alfa dose among hemodialysis patients. Am J Kidney Dis 2008; 51: 62–70 [DOI] [PubMed] [Google Scholar]
- 77. Fukuma S, Yamaguchi T, Hashimoto S. et al. Erythropoiesis-stimulating agent responsiveness and mortality in hemodialysis patients: results from a cohort study from the dialysis registry in Japan. Am J Kidney Dis 2012; 59: 108–116 [DOI] [PubMed] [Google Scholar]
- 78. McCullough PA, Barnhart HX, Inrig JK. et al. Cardiovascular toxicity of epoetin-alfa in patients with chronic kidney disease. Am J Nephrol 2013; 37: 549–558 [DOI] [PubMed] [Google Scholar]
- 79. Kuragano T, Kitamura K, Matsumura O. et al. ESA hyporesponsiveness is associated with adverse events in maintenance hemodialysis (MHD) patients, but not with iron storage. PLoS One 2016; 11: e0147328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Luo J, Jensen DE, Maroni BJ. et al. Spectrum and burden of erythropoiesis-stimulating agent hyporesponsiveness among contemporary hemodialysis patients. Am J Kidney Dis 2016; 68: 763–771 [DOI] [PubMed] [Google Scholar]
- 81. Nakanishi T, Kimura T, Kuragano T.. The hepcidin-anemia axis: pathogenesis of anemia in chronic kidney disease. Contrib Nephrol 2019; 198: 124–134 [DOI] [PubMed] [Google Scholar]
- 82. Albaramki J, Hodson EM, Craig JC. et al. Parenteral versus oral iron therapy for adults and children with chronic kidney disease. Cochrane Database Syst Rev 2012; 18: 1–97 [DOI] [PubMed] [Google Scholar]
- 83. Olsson KS, Väisänen M, Konar J. et al. The effect of withdrawal of food iron fortification in Sweden as studied with phlebotomy in subjects with genetic hemochromatosis. Eur J Clin Nutr 1997; 51: 782–786 [DOI] [PubMed] [Google Scholar]
- 84. Aucella F, Vigilante M, Scalzulli P. et al. Desferrioxamine improves burst-forming unit-erythroid (BFU-E) proliferation in haemodialysis patients. Nephrol Dial Transplant 1998; 13: 1194–1199 [DOI] [PubMed] [Google Scholar]
- 85. Goch J, Birgegård G, Danielson BG. et al. Treatment of erythropoietin-resistant anaemia with desferrioxamine in patients on haemofiltration. Eur J Haematol 2009; 55: 73–77 [DOI] [PubMed] [Google Scholar]
- 86. Aucella F, Vigilante M, Scalzulli P. et al. Synergistic effect of desferrioxamine and recombinant erythropoietin on erythroid precursor proliferation in chronic renal failure. Nephrol Dial Transplant 1999; 14: 1171–1175 [DOI] [PubMed] [Google Scholar]
- 87. Hamano T, Fujii N, Hayashi T. et al. Thresholds of iron markers for iron deficiency erythropoiesis-finding of the Japanese nationwide dialysis registry. Kidney Int Suppl (2011) 2015; 5: 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Gaweda AE, Goldsmith LJ, Brier ME. et al. Iron, inflammation, dialysis adequacy, nutritional status, and hyperparathyroidism modify erythropoietic response. Clin J Am Soc Nephrol 2010; 5: 576–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gaweda AE, Bhat P, Maglinte GA. et al. TSAT is a better predictor than ferritin of hemoglobin response to epoetin alfa in US dialysis patients. Hemodial Int 2014; 18: 38–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gunnell J, Yeun JY, Depner TA. et al. Acute-phase response predicts erythropoietin resistance in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis 1999; 33: 63–72 [DOI] [PubMed] [Google Scholar]
- 91. Rosati A, Tetta C, Merello JI. et al. Cumulative iron dose and resistance to erythropoietin. J Nephrol 2015; 28: 603–613 [DOI] [PubMed] [Google Scholar]
- 92. Akchurin O, Sureshbabu A, Doty SB. et al. Lack of hepcidin ameliorates anemia and improves growth in an adenine-induced mouse model of chronic kidney disease. Am J Physiol Renal Physiol 2016; 311: F877–F889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang Y, Zhai W, Zhao M. et al. Effects of iron overload on the bone marrow microenvironment in mice. PLoS One 2015; 10: e0120219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Okabe H, Suzuki T, Uehara E. et al. The bone marrow haematopoietic microenvironment is impaired in iron-overloaded mice. Eur J Haematol 2014; 93: 118–128 [DOI] [PubMed] [Google Scholar]
- 95. Asberg A, Mikkelsen G, Thorstensen K. et al. Lower hemoglobin with lower ferritin: it is not just a question of anemia. Scand J Clin Lab Invest 2013; 73: 622–626 [DOI] [PubMed] [Google Scholar]
- 96. World Health Organization (WHO). Iron Deficiency Anaemia: Assessment, Prevention, and Control. A Guide for Programme Managers. Geneva: WHO Magazine 2013: 1–114 [Google Scholar]
- 97. Lynch SR, Skikne BS, Cook JD.. Food iron absorption in idiopathic hemochromatosis. Blood 1989; 74: 2187–2193 [PubMed] [Google Scholar]
- 98. Hallberg L, Hulten L, Gramatkovski E.. Iron absorption from the whole diet in men: how effective is the regulation of iron absorption? Am J Clin Nutr 1997; 66: 347–356 [DOI] [PubMed] [Google Scholar]
- 99. Cook JD, Lipschitz DA, Miles LE. et al. Serum ferritin as a measure of iron stores in normal subjects. Am J Clin Nutr 1974; 27: 681–687 [DOI] [PubMed] [Google Scholar]
- 100. Eschbach JW, Cook JD, Scribner BH. et al. Iron balance in hemodialysis patients. Ann Intern Med 1977; 87: 710–713 [DOI] [PubMed] [Google Scholar]
- 101. Fillet G, Beguin Y, Baldelli L.. Model of reticuloendothelial iron metabolism in humans: abnormal behavior in idiopathic hemochromatosis and in inflammation. Blood 1989; 74: 844–851 [PubMed] [Google Scholar]
- 102. Nakanishi T, Kuragano T, Kaibe S. et al. Should we reconsider iron administration based on prevailing ferritin and hepcidin concentrations? Clin Exp Nephrol 2012; 16: 819–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hasuike Y, Nonoguchi H, Tokuyama M. et al. Serum ferritin predicts prognosis in hemodialysis patients: the Nishinomiya study. Clin Exp Nephrol 2010; 14: 349–355 [DOI] [PubMed] [Google Scholar]
- 104. Ogawa C, Tsuchiya K, Kanda F. et al. Low levels of serum ferritin lead to adequate hemoglobin levels and good survival in hemodialysis patients. Am J Nephrol 2014; 40: 561–570 [DOI] [PubMed] [Google Scholar]
- 105. Kiechl S, Willeit J, Egger G. et al. Body iron stores and the risk of carotid atherosclerosis: prospective results from the Bruneck study. Circulation 1997; 96: 3300–3307 [DOI] [PubMed] [Google Scholar]
- 106. Haidari M, Javadi E, Sanati A. et al. Association of increased ferritin with premature coronary stenosis in men. Clin Chem 2001; 47: 1666–1672 [PubMed] [Google Scholar]
- 107. Mainous AG, Diaz VA.. Relation of serum ferritin level to cardiovascular fitness among young men. Am J Cardiol 2009; 103: 115–118 [DOI] [PubMed] [Google Scholar]
- 108. Friedrich N, Milman N, Volzke H. et al. Is serum ferritin within the reference range a risk predictor of cardiovascular disease? A population-based, long-term study comprising 2874 subjects. Br J Nutr 2009; 102: 594–600 [DOI] [PubMed] [Google Scholar]
- 109. Zacharski LR, Shamayeva G, Chow BK.. Effect of controlled reduction of body iron stores on clinical outcomes in peripheral arterial disease. Am Heart J 2011; 162: 949–957 [DOI] [PubMed] [Google Scholar]
- 110. Zacharski LR, DePalma RG, Shamayeva G. et al. The statin-iron nexus: anti-inflammatory intervention for arterial disease prevention. Am J Public Health 2013; 103: e105–e112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ponikowska B, Suchocki T, Paleczny B. et al. Iron status and survival in diabetic patients with coronary artery disease. Diabetes Care 2013; 36: 4147–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Anker SD, Comin Colet J, Filippatos G. et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N Engl J Med 2009; 361: 2436–2448 [DOI] [PubMed] [Google Scholar]
- 113. Jankowska EA, Tkaczyszyn M, Suchocki T. et al. Effects of intravenous iron therapy in iron-deficient patients with systolic heart failure: a meta-analysis of randomized controlled trials. Eur J Heart Fail 2016; 18: 786–795 [DOI] [PubMed] [Google Scholar]
- 114. Nakanishi T, Hasuike Y, Nagasawa Y. et al. Opposite extremes in hepcidin status between the US and Japan. Am J Med 2013; 126: e11. [DOI] [PubMed] [Google Scholar]
- 115. Goodkin DA, Bragg-Gresham JL, Koenig KG. et al. Association of comorbid conditions and mortality in hemodialysis patients in Europe, Japan, and the United States: the Dialysis Outcomes and Practice Patterns Study (DOPPS). J Am Soc Nephrol 2003; 14: 3270–3277 [DOI] [PubMed] [Google Scholar]
- 116. Stenvinkel P. Inflammation in end-stage renal disease: the hidden enemy. Nephrology (Carlton) 2006; 11: 36–41 [DOI] [PubMed] [Google Scholar]