

Summary
The altered molecular proteins and pathways in response to COVID-19 infection are still unclear. Here, we performed a comprehensive proteomics-based investigation of nasopharyngeal swab samples from patients with COVID-19 to study the host response by employing simple extraction strategies. Few of the host proteins such as interleukin-6, L-lactate dehydrogenase, C-reactive protein, Ferritin, and aspartate aminotransferase were found to be upregulated only in COVID-19-positive patients using targeted multiple reaction monitoring studies. The most important pathways identified by enrichment analysis were neutrophil degranulation, interleukin-12 signaling pathways, and mRNA translation of proteins thus providing the detailed investigation of host response in COVID-19 infection. Thus, we conclude that mass spectrometry-detected host proteins have a potential for disease severity progression; however, suitable validation strategies should be deployed for the clinical translation. Furthermore, the in silico docking of potential drugs with host proteins involved in the interleukin-12 signaling pathway might aid in COVID-19 therapeutic interventions.
Subject areas: specimen, molecular biology, proteomics
Graphical Abstract
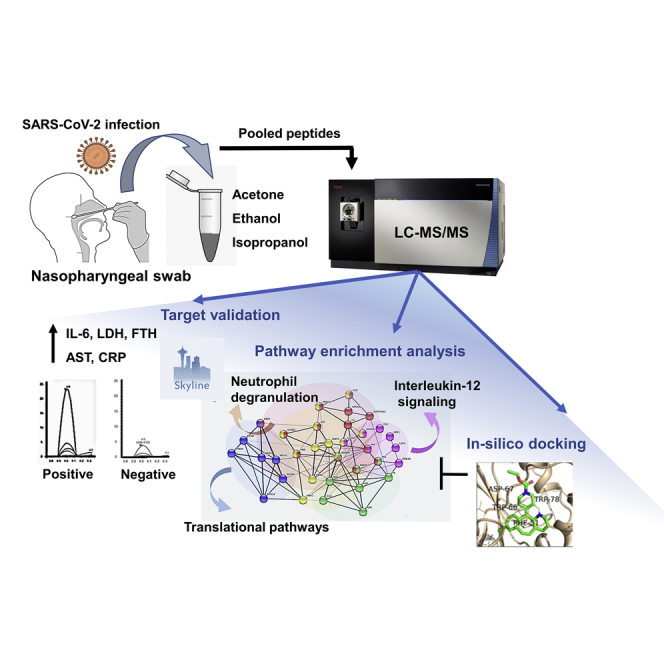
Highlights
-
•
High-resolution mass spectrometry of swab identified 164 significant host proteins
-
•
Upregulation of LDH, Ferritin, and AST was validated by MRM assays
-
•
Significant alteration of immune response and translational pathways was observed
-
•
In silico docking identified Loratadine binding to interleukin signaling proteins
Specimen; molecular biology; proteomics
Introduction
The COVID-19 outbreak first identified in China in December 2019 has been officially declared a global pandemic by the World Health Organization (WHO). Last year, pneumonia-like symptoms caused by a new flu-like virus seemed to wipe out a part of the population in Wuhan, China, which was later named as 2019-novel coronavirus or severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) by WHO. This viral disease manifests with mild fever, throat pain, dry cough, and severe respiratory difficulties to acute respiratory distress leading to breathlessness, pneumonia, and later multiple organ failure, which is the major cause of fatality rates (Chen et al., 2020). A positive viral infection is often diagnosed by either of the two ways; detection of viral load by real-time polymerase chain reaction (RT-PCR) and antibody-based tests (Chan et al., 2020). However, these techniques experience several drawbacks, such as RT-PCR faces several technical artifacts, including a high risk of cross-reaction with other potential coronaviruses and the amplicons to be contaminated; whereas, serological tests have very low-sensitivity in the early infection phase (Tang et al., 2020). Although it is important to screen individuals, mostly asymptomatic individuals, for a country with a large population such as India, it is crucial to use all its available infrastructure based on RT-PCR, next-generation sequencing (NGS), and mass spectrometry platforms, as well as antibody-based assays to expedite the testing. Further, for patients with COVID-19 with mild symptoms, identification of biological markers that define COVID-19 disease severity could help clinicians to provide better health-care facilities to patients in need. Therefore, exploring alternate and effective testing strategies, as well as identification of predictive biomarkers are very crucial to combat this pandemic.
One of the major concerns is the lack of any established therapy or treatment after COVID-19 infection and health-care systems around the world are relying on symptomatic care and supportive medicine. Currently, the disease is being treated by a combination of available antiviral drugs such as remdesivir, lopinavir, and ritonavir (Kim, 2020), and so on. Here, the scenario becomes burdensome when it comes to treating post-infection lung injury followed by multi-organ failure (Matthay et al., 2020). To target such key issues, proteomics-based investigations for the identification of biomarkers for COVID-19 has already begun among the scientific community all over the globe. Proteomics study on SARS-CoV-2-infected Caco-2 cells presented evidence of remodeling of cellular translation to overcome the changes occurring in host translation capacity as a response to the viral attack (Bojkova et al., 2020). Mass spectrometry-based proteomics from diluted gargle samples was able to identify the presence of a unique SARS-CoV-2 peptide, which belongs to viral nucleoprotein (Ihling et al., 2020).
Nasopharyngeal swab has a high viral titer, and it is one of the easiest, cheapest, and non-invasive ways of sample acquisition, currently being used for the RT-PCR-based detection of SARS-CoV-2 virus. Study conducted by Nikolaev et al. resulted in identification of SARS-CoV-2 nucleocapsid (N) protein, which pioneered the idea of developing COVID-19 biomarkers using nasopharyngeal swab samples (Nikolaev et al., 2020). It is evident that nasopharyngeal swab could be used to identify viral peptides for COVID-19 detection as well as for studying the host protein alteration using high-resolution mass spectrometry.
Host pathobiology plays a significant role in RNA virus infection. Pathomechanism of SARS-CoV-2 shows virus hijacks and modulates host cell machinery in multiple ways upon its entry. Till now, it is unclear how SARS-CoV-2 alters the host molecular pathways, although recent research has made some progress identifying and characterizing the biomolecular findings for disease onset and progression (Bock and Ortea, 2020). Previously, Bock et al. have re-analyzed the publicly available proteomic data generated by infecting SARS-CoV-2 in a cell line model and found several host proteins alteration, which were linked to various intracellular pathways (Bock and Ortea, 2020; Bojkova et al., 2020). In a study when nasopharyngeal swab samples from COVID-19-infected patients were analyzed, proteins belonging to the innate immune system were found to be significantly altered. Recent study on plasma proteomics revealed the potential clinical classifiers of COVID-19 infection (Messner et al., 2020). Such observations made at the macromolecular level may explain why a large part of innate immune machinery experiences the quintessential change. Till date, very few studies are available, which focuses on the unique biomolecular signature, that can play a crucial role in understanding the disease mechanism and clinical translation aiming at prognosis and cure.
In this study, we have used high-resolution mass spectrometry-based proteomics to identify viral peptides for detection and host proteins for the prognosis of COVID-19 disease severity. We have validated differentially expressed host proteins using multiple reaction monitoring (MRM)-based mass spectrometry assay. This is the first comprehensive proteomics study from an Indian hot spot COVID region, Mumbai, displaying the potential of proteins identified from mass spectrometry-based approaches for understanding the COVID-19 pathogenesis.
Results
Detection of viral peptides from COVID-19 nasopharyngeal swab samples
The mass spectrometry-based label-free quantification is an efficient technique for a simple, rapid, and high-throughput analysis of the peptides from the clinical samples. One of the major challenges associated with the successful mass spectrometry-based detection of the unique peptides and proteins depends on the initial steps of protein precipitation and peptide enrichment. Here, we investigated the use of three different organic solvents (ethanol, acetone, and isopropanol) for the enrichment of peptides from a total of 101 nasopharyngeal swab samples collected in viral transport media (VTM). The swab samples in VTM tubes were heat-inactivated before addition of solvents. The detailed workflow for the mass spectrometry-based analysis of respiratory specimen is shown in Figure 1A. To study the disease progression, only those patients with COVID-19 who were confirmed by RT-PCR and presented non-severe-to-severe symptoms were selected. The clinical details of all the patient samples are shown in Table S1.
Figure 1.

The workflow of sample preparation for detection of viral load and mass spectrometry analysis
Swab samples were collected in viral transfer media and RNA was extracted followed by cDNA preparation which was analyzed by RT-PCR for determining the viral load. For MS analysis, proteins in swab samples were precipitated using three organic solvents namely acetone, ethanol and isopropanol. The proteins were digested, desalted and subjected to mass spectrometry analysis. Raw files from Mass Spectrometry were analyzed using Maxquant and significant proteins were selected for MRM analysis.
(A) Schematic representation for RT-PCR and mass spectrometry-based detection of swab proteins.
(B) Comparison of various extraction methods. The sample pool used in this study was prepared from all three solvents, which yielded the maximum average number of unique viral peptides as compared to the other extraction methods.
(C) Representative MRM spectra for three viral peptides as observed in COVID-19 positive swab samples. The peptides belong to Replicase 1ab, Nucleocapsid and Spike proteins respectively.
At the initial phase of the study, we performed shotgun mass spectrometry analysis for swab samples in three different solvent conditions. Comparison of the three solvents in detecting unique viral peptides showed that for spike glycoprotein and nucleoprotein similar number of peptides were obtained using each of the three solvents, whereas, in the case of replicase polyprotein 1ab protein, acetone exclusively yielded a higher number of enriched peptides (Figure 1B).
To further validate the viral peptides detected from the discovery study comprising a set of 13 patients, 12 peptides were selected, and MRM assays were performed on a set of 5 COVID-19-positive, 5 COVID-19-negative, and 3 non-COVID/healthy controls (samples collected before COVID pandemic). The workflow and quality control checks for TSQ Altis mass spectrometer used for MRM assay is shown in Figures S2 and S3. The representative MRM spectra for three viral peptides (Replicase 1ab, Nucleocapsid and Spike proteins) as observed in COVID-19-positive swab samples are shown in Figure 1C. The final MRM list had 4 viral peptides (IQDSLSSTASALGK, NTQEVFAQVK, FLPFQQFGR, and QIAPGQTGK) belonging to SARS-CoV-2 Spike glycoprotein (P0DTC2); 4 peptides belonging to SARS-CoV-2 nucleoprotein (P0DTC9)(NSTPGSSR, GGSQASSR, DGIIWVATEGALNTPK, and RPQGLPNNTASWFTALTQHGK; 4 more peptides (NGSIHLYFDK, ELLQNGMNGR, DGHVETFYPK, DFMSLSEQLR) (P0DTD1) belonging to Replicase polyprotein 1ab (Table S5) and a SpikeTide (Schnatbaum et al., 2011) having sequence FEDGVLDPDYPR that was spiked into all these samples as an internal standard. Distinct MRM spectra were seen for all the selected viral peptides in the COVID samples but not in healthy controls (refer to the Skyline files in the data availability section). This study should be further validated on the larger cohort of COVID-19-positive and COVID-19-negative samples.
Alteration of host proteome in response to COVID-19 infection
To investigate the alteration in host proteome, mass spectrometry-based label-free quantification was performed on 68 patient’s swab samples, extracted using three different solvents (ethanol, acetone, and isopropanol). These clinical samples were run in two batches, first batch consisted of 44 samples (26 COVID-19-positive, 11 recovered, and 7 negative) and the second batch consisted of 24 samples (11 severe and 13 non-severe). The mass spectrometry generated raw data sets were processed with MaxQuant software against the Human Swiss-Prot database, which includes a total of 20,353 proteins. The total number of proteins identified in all the groups is shown in Table S6. Around 164 common significant proteins were identified from the comparison between 3 groups (COVID-19 positive, negative, and recovered) (Figure S4). Interestingly, Partial least squares-discriminant analysis (PLSDA) indicated sample-ID 30 belonging to the recovered group, to be a part of cluster consisting of positive samples. This analysis indicates that the patient sample-ID 30 might have not recovered completely and thus was showing clustering with the COVID-19-positive samples (Figure 2A). The heatmap of top 25 proteins from 164 common significant proteins between 3 groups revealed that the protein angiotensinogen, complement component C3, T-complex protein 1 subunit delta and signal transducer and activator of transcription 1 (STAT1) was found to be highly upregulated in COVID-19-positive patients when compared with negative and recovered patients (Figure 2B). The two different comparisons of positive samples with recovered samples and true negative samples identified several significant proteins and they were also able to form a distinct cluster based on the dysregulated proteins (Figure S5). These results show that the panel of significant proteins was able to distinguish the three clusters even on different group comparisons.
Figure 2.

Alteration of host proteome in response to COVID-19 infection
To study the host proteome profile the mass spectrometry generated raw datasets were processed with MaxQuant software against the Human Swiss-Prot database.
(A) Map of the Segregation of Positive, True Negative, and Recovered samples using PLSDA. Analysis of 18 positive, 11 recovered and 7 true negative samples showed segregation into three clusters. True negative clusters distinctly classified from recovered and positive samples. Sample 30, a recovered sample found to be placed within the positive sample cluster.
(B) HeatMap of Positive, True Negative and Recovered samples. A list of 164 significant proteins found to be common between Positive vs Negative, Positive vs Recovered and Positive vs True Negative has been used to draw a hierarchical clustering based heatmap. The figure depicts the top 25 significant proteins found to segregate the groups using the ward clustering algorithm.
(C) The unsupervised heatmap of 25 significant protein shows a perturbation between the non-severe and severe group.
(D) Clustering analysis to segregate the Positive severe and non-severe samples. 12 severe positives and 11 non-severe positive samples were found to be segregated into two clusters in PLSDA.
(E) Violin plot showing the expressional difference of clinical protein markers of COVID diagnosis. Violin plot of LDH-A and LDH-B – the subunits of Lactate dehydrogenase found to be significantly upregulated in COVID-19-positive samples when compared with COVID-19-negative samples. STAT1, a key regulator of interleukin also found to be significantly upregulated in COVID-19-positive, having biological connections with D-dimers and creatine phosphokinase which are clinical markers. HBA and HBB subunits of hemoglobin are also found to be significantly upregulated in the COVID-19-positive samples. (Unpaired Welch's T test; ns: 5.00e-02 < p <= 1.00e+00; ∗: 1.00e-02 < p <= 5.00e-02; ∗∗: 1.00e-03 < p <= 1.00e-02; ∗∗∗: 1.00e-04 <p <= 1.00e-03; ∗∗∗∗: p <= 1.00e-04).
Further, the batch of 24 COVID-19-positive samples which includes 11 non-severe and 13 severe patient samples were separately analyzed in MaxQuant using the same parameters discussed above. One sample of 24 samples was found to be an outlier due to its low correlation coefficient and hence removed. From the heatmap of top 25 proteins, 6 significant proteins were identified to be differently expressed in COVID-19 severe when compared to COVID-19 non-severe patient samples (Figure 2C). A few of the significant proteins identified were lipocalin-1, myeloid-derived growth factor, fibrinogen silencer-binding protein, and Cofilin-1. The partial least square-discriminant analysis of 23 samples was performed which found to show a clear distinction between the severe and non-severe cohorts (Figure 2D). VIP score based on PLSDA has been used to select top 15 features and 7 of these 15 features were found to be significantly differentially expressed between non-severe and severe cohort (Figure S6).
The proteomic analysis of COVID-19-positive and COVID-19-negative patients revealed few links with the clinical parameters. We found the protein lactate dehydrogenase A (LDH-A), lactate dehydrogenase B (LDH-B), STAT1 (a key regulator of IL-6), hemoglobin subunit alpha (HBA) and hemoglobin subunit beta (HBB) protein (hemoglobin subunits) to be upregulated in COVID-19-positive patients when compared to COVID-19-negative patients (Figure 2E). These results correlated with the symptoms of the patients and hence the significant proteins identified from positive and negative comparison were taken forward for the validation assay and pathway analysis.
Targeted MRM assay and clinical validation of the host proteins for COVID-19 prognosis
A few proteins such as L-lactate dehydrogenase A chain (P00338), L-lactate dehydrogenase B chain (P07195), Ferritin heavy chain (P02794), Ferritin light chain (P02792), aspartate aminotransferase, mitochondrial (P00505), aspartate aminotransferase, cytoplasmic (P17174) and albumin (P02768) which were found to be upregulated in COVID-19-positive patients when compared to COVID-19-negative patients in the LFQ data were selected and used for a targeted MRM study. Also, we checked for the clinical markers such as interleukin – 6 (IL-6), C-reactive protein, as well as proinflammatory cytokines such as tumor necrosis factor (TNF-α) which we could not identify in LFQ data but is clinically significant using targeted MRM assay. The following analysis was performed on the swab samples of 6 COVID-19-negative and 16 COVID-19-positive patients (consisting of 8 severe and 8 mild samples) were run on a triple quadrupole mass spectrometer coupled to an HPLC. The peak shapes of representative peptides for clinically significant proteins validated using MRM, as seen in Skyline are shown in Figures 3A and S7. Table S7 shows the list of MRM identified peptides between 16 COVID-19-positive and 6 COVID-19-negative samples, used for validation. Intensities of the peptide for proteins interleukin-6 (IL-6), L-lactate dehydrogenase A chain (LDH-A) and aspartate aminotransferase, and cytoplasmic (GOT1) were observed to be statistically significant between COVID-19-positive and COVID-19-negative patient samples (p < 0.05; Fold change > 1.5 at a confidence interval of 99%). A refined list of transitions that provided validated clinical marker proteins using MRM and gave statistically significant peptides (adjusted p value < 0.05; fold change > 1.5) between COVID-19-positive and COVID-19-negative swab samples are shown in Figure 3B and Table S8.
Figure 3.

MRM validation of the host proteins upregulated in COVID-19-positive samples
The MRM analysis was performed on 6 COVID-negative and 16 COVID-positive patient swab samples. 1 μg peptide from each sample was injected into a Vanquish HPLC coupled to a TSQ Altis™ triple quadrupole and run against a transition list of peptides belonging to important clinical markers. The list of transitions was prepared for unique peptides of these selected proteins using Skyline (Version 20.2.1.286). This list included a spiked-in synthetic heavy peptide (THCLYTHVCDAIK) used for monitoring the consistency of the mass spectrometry runs. For identification of the sensitivity of the peptide detection, we performed serial dilution of two crude synthetic peptides (heavy and light). The concentration of peptide was calculated using the Scopes method from its O.D. value at 205 nm and 280 nm. Different concentrations of the peptides starting from 25 to 125 ng were run in TSQ Altis™ Triple Quadrupole Mass Spectrometer.
(A) Peak shapes of representative peptides for clinical marker proteins validated using MRM. The statistically significant change in the expression of the proteins was observed between COVID-19-positive and COVID-19-negative patient samples (T-test, ∗∗p < 0.05; Fold change > 1.5 at a confidence interval of 99% - determined by Skyline).
(B) Box plots of representative peptides for clinical marker proteins validated using MRM.
(C) Standard curve of heavy synthetic peptide HSGFEDELSEVLENQSSQAELK. The crude heavy synthetic peptide was diluted in the range of 25 to 125 ng concentration. The standard curve for this peptide was plotted using the peak area against the concentration of the peptide. The intensity of the peak area was proportional to the amount of the synthetic peptide. The lowest amount of synthetic peptide detected was at 30.9 ng.
(D) Standard curve of light synthetic peptide HSGFEDELSEVLENQSSQAELK. The crude light synthetic peptide was diluted in the range of 25 to 125 ng concentration. The standard curve for this peptide was plotted using the peak area against the concentration of the peptide. The intensity of the peak area was proportional to the amount of the synthetic peptide. The lowest amount of synthetic peptide detected was at 19.8 ng.
Molecular pathway perturbations with the progression of COVID-19 infection
A list of 452 significant proteins from COVID-19-positive samples, when compared to COVID-19-negative samples, were taken forward for Gene Ontology (GO) enrichment analysis using Metascape. The most prominent pathways identified were neutrophil degranulation, platelet degranulation, interleukin-12 signaling pathways, mRNA translation of proteins, and co-factor metabolomic process (Figures 4 and S8). The abundance of few proteins mapping into the prominent pathways such as peroxiredoxin-6 (PRDX6), macrophage migration inhibitory factor (MIF), glutathione S-transferase omega-1, 40S ribosomal protein S28 (RPS28), eukaryotic translation initiation factor 4H (EIF4H) and elongation factor 2 (EEF2) for COVID-19-positive and COVID-19-negative controls are shown in the violin plot (Figure 4).
Figure 4.

Molecular pathway perturbations with the progression of COVID-19 infection
The figure represents the enriched GO biological processes with their co-expressed proteins in the form a bipartite network where few proteins has been shown in the form of violin plot for COVID-19-positive (high and low viral load patients) and COVID-19-negative controls. (Unpaired Welch’s T-test, p-value annotation legends: ns: 5.00e-02 < p <= 1.00e+00; : 1.00e-02 < p <= 5.00e-02; ∗: 1.00e-03 < p <= 1.00e-02; ∗: 1.00e-04 < p <= 1.00e-03; ∗∗: p <= 1.00e-04). The representative images from the Reactome depicting the key pathways perturbed in the host are also shown.
Although we could not identify some of the proinflammatory cytokines in LFQ analysis, the GO enrichment analysis did reveal several proteins that might be upregulated in response to the stimulus from proinflammatory cytokines or might be inducing its release. For example, proteins such as RPS3 with the role in biological process response to TNF-α agonist, MIF protein involved in the interleukin-12 family signaling capable of activating T cells to produce proinflammatory cytokines such as TNF-α, interleukin (IL) 1β, IL-2, IL-6, IL-8, and interferon-γ, leading to inflammatory responses. Thus, the GO enrichment analysis showed several proteins belonging to the IL-6, IL-1, IL-23, IL-12, and IL-7 signaling pathway which plays an important role in the COVID-19 progression to severity (Table S9). We also identified ILK pathways such as interleukin-12 family and interleukin-1 signaling which belongs from cytokine signaling in the immune system pathway. We also found STAT1, a key regulator of IL-6, significantly upregulated in most of the COVID-19-positive when compared with the negative sample group (Figure S8).
Molecular docking of drugs with host proteins
We used the in silico approach of molecular docking to analyze the inhibition mechanism of few drugs on pathways related to immune response. In our study, we have docked the identified proteins from neutrophil degranulation, translation and interleukin pathway against 29 FDA-approved, 9 clinical, and 20 pre-clinical trial drugs (Table S10). The inhibitors well established in the literature (Gordon et al., 2020) were used as a positive control against these target proteins. The control inhibitor gives us a possible cutoff for the docking score. The selection of drugs for the protein was done by considering a couple of criteria. First, the binding energy of the drug was expected to be equal or higher than that of the control inhibitor. Second, the binding pocket of the drug was expected to be similar to the control drug. The proteins involved in the neutrophil degranulation, translation, and interleukin pathways are the mediators to distinguish between two cohorts (COVID-19-positive and COVID-19-negative) and hence were taken forward as potential drug targets to perform further computational studies. Based on our docking analysis, we observed that 1 pre-clinical trial drug binds to protein involved in the translation pathway (Figure S9) and 7 FDA-approved drugs to target neutrophil degranulation pathway (Figure S10); and 2 FDA-approved and 2 pre-clinical drugs for interleukin pathway (Figure 5). One FDA-approved drug loratadine (ZINC537931) and another, currently in pre-clinical studies, UCPH-101 (ZINC000040914195) was found to bind three major proteins (ADP-ribosylation factor 1, carbonic anhydrase 1 and MIF) from the interleukin pathway. Both, loratadine and UCPH-101 bind to the control ligand-binding pocket with a binding affinity more negative than control (<-8 Kcal/mol) (Figure 5). Similarly, few more drugs were found to bind proteins belonging to eukaryotic translation, as well as neutrophil degranulation pathway.
Figure 5.

Molecular docking studies of drugs binding to the host proteins involved in interleukin-12 signaling pathways
Autodock Vina (1.1.2) was used to identify the drugs binding to the host proteins involved in the neutrophil degranulation, translation, and interleukin pathway. A screening of 29 FDA-approved, 9 clinical, and 20 pre-clinical trial drugs against the host protein identified several potential drug candidates targeting the interleukin-12 signaling pathway.
(A) shows the drug Loratadine (green) docked with three proteins from the interleukin pathway; ADP-ribosylation factor 1 (binding energy or BE -9.8 kcal/mol), carbonic anhydrase 1 (BE -8.3 kcal/mol) and macrophage migration inhibitory factor (MIF) (BE -8 kcal/mol).
(B) shows the same proteins docked with the drug UCPH-101 (cyan); ADP-ribosylation factor 1 (BE -9.9 Kcal/mol), carbonic anhydrase 1 (BE -8.5 kcal/mol) and MIF (BE -8.8 kcal/mol).
Both of the drugs bind to all three proteins with negative binding energy greater than their respective control inhibitor. The interacting amino acid residues, which are present on the ligand-binding pocket are labeled. Almost all of the interacting residues belong to hydrophobic amino acids.
Discussion
We have performed a comprehensive proteomics study from an Indian hot spot COVID region, Mumbai, displaying the potential of mass spectrometry-based approaches for severity progression of COVID-19 and clinical translation. To obtain highly efficient peptide enrichment from COVID-19-infected swab samples, we precipitated protein from swab using three different organic solvents namely, ethanol, isopropanol, and acetone, as well as prepared a pool of peptides from all three conditions. A few MS-based studies have relied on a complex sample preparation protocol, using TCA-acetone and in-gel digestion or a single organic solvent for the extraction of proteins identifying only a few unique peptides (Gouveia et al., 2020; Nikolaev et al., 2020). However, we observed the pool of peptides from three methods identified the maximum number of viral peptides using Orbitrap Fusion mass spectrometer. The MRM-based assay could specifically detect SARS-CoV-2 peptides from spike glycoprotein, nucleoprotein, and replicase polyprotein 1ab protein. These peptides showed a distinct elution profile and were eluted between 0.5 to 8 minutes. Thus, MRM-based assay confirmed the presence of SARS-CoV-2 peptides in COVID-19-positive samples as compared to healthy controls.
Currently, several researcher groups have studied the host proteome alteration in response to COVID-19 infection (Shen et al., 2020; Overmyer et al., 2020). However, still panel of host proteins to be routinely monitored in the patients or the pathways to be targeted are not yet identified and validated. Thus, to identify the potential prognostic markers and therapeutic targets, we performed a deep proteome profiling of COVID-19 respiratory samples based on label-free quantification using a high-resolution mass spectrometer. The nasopharynx being the primary site of the viral entry, the nasopharyngeal swab might be the better sample to study the complex molecular and immune response events in the infected host. From over 3749 host proteins, we could identify around 164 significant proteins in COVID-19-positive when compared to COVID-19-negative and recovered patients. Using targeted MRM assay, we could detect a statistically significant change in the intensity of the peptide of proteins such as interleukin-6, L-lactate dehydrogenase A chain, L-lactate dehydrogenase B chain, Ferritin heavy chain, Ferritin light chain, aspartate aminotransferase (mitochondrial), aspartate aminotransferase (cytoplasmic), C-reactive protein, and proinflammatory cytokines, TNF-α in COVID-19-positive patients when compared to COVID-19-negative patients. However, targeted assays based on these host proteins on a large cohort of patients using longitudinal samples are required for the successful clinical translation.
Currently, we have limited understanding of molecular pathways altered during the SARS-CoV-2 infection (Shen et al., 2020; Overmyer et al., 2020). Using enrichment analysis, we could identify proteins involved in neutrophil degranulation, platelet degranulation, interleukin-12 signaling related pathway, translational mechanism, and co-factor metabolomic process to be key GO enriched pathways altered in the COVID-19-positive patients. Recent studies state that there is an evidence of increased peripheral neutrophil-to-lymphocyte-ratio in the case of severe COVID-19 patients (Zheng et al., 2020), suggesting the fact that neutrophil degranulation is one of the major events that modulate the immune system post-infection. We identified several host proteins such as complement C3, glucose-6-phosphate isomerase, SERPINB3, peroxiredoxin-6, and transitional endoplasmic reticulum ATPase (VCP) mapping to neutrophil degranulation pathway. In recent studies, it has also been argued that neutrophils play a dual role during pathogen attack. They help our immune system to get rid of viruses or bacteria by releasing granule-derived mediators and activating complement, but accumulation or overreaction due to these mediators may lead to hyperinfection, tissue injury, or septic shock (Lacy, 2006). However, it is quite unclear whether SARS-CoV-2 directly targets the neutrophil degranulation pathway or is just a consequence of severe immune complications of SARS-CoV-2 infection. The interleukin IL-6 is a hallmark of the cytokine storm observed in the COVID-19 severe patients (Sun, 2020). We also identified several proteins such as STAT1 (a key regulator of IL-6) belonging to the interleukin signaling pathway upregulated in COVID-19-positive patients as compared to the COVID-19-negative patients (Matsuyama, 2020).
We also identified several host proteins belonging to platelet activation, aggregation, and degranulation pathways such as fibrinogen gamma chain, profilin-1, albumin, serotransferrin, and alpha-2-macroglobulin. Platelets function as exocytotic cells, secreting a plethora of effector molecules at sites of vascular injury. The increased fibrin formation and breakdown correlated with the high level of D-dimers observed in the COVID-19 patients with the worst outcomes (Tang, 2020). The increasing levels of FGG in severe cases might be due to liver injury, impairing hepatic fibrinogen secretion with acquired fibrinogen storage disease (Fraga, 2020).
Translation event inside a eukaryote system requires several hundred proteins to act in sync. It becomes impossible for a virus to carry that much information in its genome thereby increasing its dependence on the host to make viral proteins. To achieve that, viral mRNAs compete with the host mRNAs to access limited cellular translational resources (Katze et al., 1985). Almost all coronavirus families initiate their translation in a ‘cap-dependent pathway’ with the help of SARS-CoV-2 N protein (Nakagawa et al., 2016). We identified several proteins such as EEF2 (P13639), ATP-dependent RNA helicase DDX18 (Q9NVP1), 60S acidic RP P1 (P05386), 40S RP S4, X isoform (P62701), 40S RP S12 (P25398), elongation factor 1-gamma (Q15056), EIF4H (Q15056) belonging to the translational pathway. One of the proteins EIF4H was known to stimulate the RNA helicase activity of EIF4A in the translation initiation complex (Cencic et al., 2011). Krogan et al. reported that eIF4H, which interacts with NSP9 SARS-CoV-2 protein, is a partner of eIF4A, and observed a strong antiviral effect after treatment with the eIF4A inhibitor Zotatifin (Gordon et al., 2020). After infection by some viruses, the translation of the host mRNA is often suppressed, whereas translation initiation of RPs mRNA might increase and persist late (Shuo, 2019). We observed significant upregulation of 40S RP and 60S RP which might be a result of the virus-host interaction for enhancing the production of few RPs to maintain viral propagation. These results reveal that the composition of ribosome proteins may be different between COVID-19-infected hosts and in uninfected populations.
Interleukin-12 signaling pathway plays an important role in the coordination of the innate and adaptive immune response (Liu, 2005). We identified several host proteins such as plastin-2, MIF, glutathione S-transferase A2, carbonic anhydrase 1 (CA1), and ADP-ribosylation factor 1 (ARF1) involved in the interleukin-12 signaling pathways. The MIF secreted in response to viral infection can activate the T cells and macrophages to produce inflammatory cytokines, including TNF-α, IL-6 and others, leading to elevated inflammatory response also called “cytokine storm” (Vandenbark, 2020). In our study, we have detected high levels of MIF, TNF-α and IL-6 in COVID-19 patients which correlates with the symptoms of acute respiratory distress syndrome (ARDS) in severe patients. Thus, targeting these host proteins involved in the interleukin-12 signaling or inflammatory response might protect the patients from developing ARDS.
Further, we investigated the binding of the drug to the host proteins involved in the molecular pathways altered in the infected host. An extensive docking study of 29 FDA-approved, 9 clinical, and 20 pre-clinical trial drugs was performed to target neutrophil degranulation, translation, and interleukin-12 signaling pathways. We identified Loratadine and UCPH-101 as a potential drug to target the host proteins such as MIF, CA1, and ARF1 involved in the interleukin-12 signaling. Loratadine is an anti-histamine used for the treatment of allergies and is known for reducing the excessive cytokine proinflammatory storm (Canonica, 2011). UCPH-101 an inhibitor of excitatory amino acid transporter subtype 1 (EAAT1), is also shown to bind to these host proteins and thus might play an important role in blocking inflammatory response (Abrahamsen, 2013). We also identified several drugs and small molecules inhibiting the proteins involved in the translational and neutrophil degranulation pathways. These small molecules and drug candidates should further be validated using in vitro human cell line model.
In conclusion, our data emphasize that nasopharyngeal swab respiratory samples, which are routinely collected for the COVID-19 RT-PCR testing; could also be used for mass spectrometry-based detection of host proteins. Further, we have validated these proteins using MRM-based mass spectrometry assay. The significant proteins involved in the translational and neutrophil degranulation pathways might be potential targets for the COVID-19 therapeutics. This study opens up new opportunity for the researchers to understand the alteration of the host response to SARS-CoV-2 in the Indian population.
Limitations of the study
Considering the high viral count, swab samples are clinically used for the detection of COVID-19 by RT-PCR, however, using these samples for routine monitoring of biomarkers is a challenge. Swab samples being highly infectious, in the clinical settings most of the biochemical and immunoassay are designed for application using blood-based sample. The VTM components in the swab samples will also interfere with the immunoassay, thus might not be suitable for the clinical application. However, suitable validation strategies could be developed directly from swab samples and should be validated on large cohort of patients before the identified biomarkers could be taken forward for clinical translation.
Resource availability
Lead contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sanjeeva Srivastava (sanjeeva@iitb.ac.in).
Materials availability
This study did not generate any new unique reagents and/or materials.
Data and code availability
All proteomics data associated with this study are present in the manuscript or the Supplementary information. The accession number for the raw MS data and search output files for proteomics data sets depositeded to the Proteome Xchange Consortium via the PRIDE partner repository is “PRIDE: PXD020580” and “PRIDE: PXD023016” (Link: https://www.ebi.ac.uk/pride/archive/loginreviewer20505@ebi.ac.uk. https://www.ebi.ac.uk/pride/archive/loginreviewer_pxd023016@ebi.ac.uk) The targeted proteomics data is deposited in the Panorama Public and can be accessed through this link: “Panorama Public: https://panoramaweb.org/COVID_Swab_MRM.url” panorama+srivastava@proteinms.net. The additional supplemental items are available from “Mendeley Data: https://doi.org/10.17632/rnfn3vhg63.1”. The present research did not use any new codes.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
The active support from Prof. Ambarish Kunwar from the Department of Biosciences & Bioengineering to fabricate UV transport device for sample transport and Prof. Anirban Banerjee for the BSL-2 biosafety aspects is gratefully acknowledged. We are also grateful to Dr. Jaishree Garhyan, JNU, New Delhi for discussion on biosafety guidelines and Dr. Arup Acharjee, BHU, Varanasi for the critical discussion on the COVID project. The authors thank Kasturba Hospital for Infectious Diseases for sample collection and sharing sample related information. The study was supported through Science and Engineering Research Board (SERB), Department of Science & Technology, Ministry of Science and Technology, MinisGovernment of India (SB/S1/Covid-2/2020), and a special COVID seed grant (RD/0520-IRCCHC0-006) from IRCC, IIT Bombay to SS. MASSFIIT (Mass Spectrometry Facility, IIT Bombay) from the Department of Biotechnology (BT/PR13114/INF/22/206/2015) is gratefully acknowledged for MS-based proteomics work. The authors also acknowledge the Thermo Fisher Scientific engineers and application scientists for their support to our MASSFIIT facility during the extreme lock-down time. The authors thank the PRIDE team for helping with the mass spectrometric data downloading and deposit in ProteomeXChange/PRIDE and Reactome.org for pathway related figures. A.B acknowledge CSIR-SRF funding, M.C and K.S thank IIT Bombay-IPDF funding.
Author contributions
Concept and design of the study were carried out by S.S., J.S., and O.S. Sample collection was done by S.A., R.B, V.P, and A.S. and sample preparation was done by R.B., S.G., A.B., K.S., and M.C. Mass spectrometry analysis, and method optimization was done by A.B, S.G, S.S. MRM (multiple reaction monitoring) were done by S.G, S.S, M.G.J.P, Al. S, and J.R. Statistical Data Analysis, and Data Visualization were done by D.B., A.S., A.V., M.C., K.S., and S.S. Docking study was performed by A.B. Data submission was done by A.S. Manuscript writing and Review by S.S., K.S., M.C., A.B., S.G., R.B., D.B., A.V., A.S., and A.M.
Declaration of interests
The authors have filed an Indian patent related to this work “Protein markers and method for prognosis of COVID-19 in individuals”. (Application number: 202021034688). The authors declare no competing interests. “Proteomics-based method for detection of Coronavirus in a sample” (Application number 202021034687).
Published: March 19, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2021.102135.
Supplemental information
References
- Abrahamsen B., Schneider N, Erichsen MN Allosteric modulation of an excitatory amino acid transporter: the subtype-selective inhibitor UCPH-101 exerts sustained inhibition of EAAT1 through an intramonomeric site in the trimerization domain. J Neurosci. 2013;33(3):1068–1087. doi: 10.1523/JNEUROSCI.3396-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock J.O., Ortea I. Re-analysis of SARS-CoV-2-infected host cell proteomics time-course data by impact pathway analysis and network analysis: a potential link with inflammatory response. Aging (Albany. NY) 2020;12:11277–11286. doi: 10.18632/aging.103524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. SARS-CoV-2 Infected Host Cell Proteomics Reveal Potential Therapy Targets. Nature. 2020;583(7816):469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PubMed] [Google Scholar]
- Canonica G.W., Blaiss M Antihistaminic, Anti-Inflammatory, and Antiallergic Properties of the Nonsedating Second-Generation Antihistamine Desloratadine: A Review of the Evidence. World Allergy Organ J. 2011;4(2):47–53. doi: 10.1097/WOX.0b013e3182093e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R., Desforges M., Hall D.R., Kozakov D., Du Y., Min J., Dingledine R., Fu H., Vajda S., Talbot P.J. Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J. Virol. 2011;85:6381–6389. doi: 10.1128/JVI.00078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.F.W., Yip C.C.Y., To K.K.W., Tang T.H.C., Wong S.C.Y., Leung K.H., Fung A.Y.F., Ng A.C.K., Zou Z., Tsoi H.W. Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-PCR assay validated in vitro and with clinical specimens. J. Clin. Microbiol. 2020;58 doi: 10.1128/JCM.00310-20. e00310-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga M., Moradpour D, Artru F, Romailler E, Tschopp J, Schneider A, Chtioui H, Neerman-Arbez M, Casini A, Alberio L, Sempoux C Hepatocellular type II fibrinogen inclusions in a patient with severe COVID-19 and hepatitis. J Hepatol. 2020;73:967–970. doi: 10.1016/j.jhep.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia D., Miotello G., Gallais F., Gaillard J.-C., Debroas S., Bellanger L., Lavigne J.-P., Sotto A., Grenga L., Pible O. Proteotyping SARS-CoV-2 virus from nasopharyngeal swabs: a proof-of-concept focused on a 3 min mass spectrometry window. J. Proteome Res. 2020;19:4407–4416. doi: 10.1021/acs.jproteome.0c00535. [DOI] [PubMed] [Google Scholar]
- Ihling C., Taenzler D., Hagemann S., Kehlen A., Huettelmaier S., Sinz A. Mass spectrometric identification of SARS-CoV-2 proteins from gargle solution samples of COVID-19 patients. Journal of proteome research. 2020;19(11):4389–4392. doi: 10.1021/acs.jproteome.0c00280. [DOI] [PubMed] [Google Scholar]
- Katze M.G., Detjen B.M., Safer B., Krug R.M. Translational control in influenza virus-infected cells. Virus Res. 1985;3:39. [Google Scholar]
- Kim J.Y. Letter to the editor: case of the index patient who caused tertiary transmission of coronavirus disease 2019 in Korea: the application of lopinavir/ritonavir for the treatment of COVID-19 pneumonia monitored by quantitative RT-PCR. J. Korean Med. Sci. 2020;35:1–4. doi: 10.3346/jkms.2020.35.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006;2:98–108. doi: 10.1186/1710-1492-2-3-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Cao S, Kim S Interleukin-12: an update on its immunological activities, signaling and regulation of gene expression. Curr Immunol Rev. 2005;1(2):119–137. doi: 10.2174/1573395054065115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama Toshifumi, Kubli, S.P., Yoshinaga, S.K An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020;27:3209–3225. doi: 10.1038/s41418-020-00633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay M.A., Aldrich J.M., Gotts J.E. Treatment for severe acute respiratory distress syndrome from COVID-19. Lancet Respir. Med. 2020;8:433–434. doi: 10.1016/S2213-2600(20)30127-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.S., Kreidl M. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020:11–24. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K., Lokugamage K.G., Makino S. Elsevier Inc.; 2016. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev E.N., Indeykina M.I., Brzhozovskiy A.G., Bugrova A.E., Kononikhin A.S., Starodubtseva N.L., Petrotchenko E.V., Kovalev G., Borchers C.H., Sukhikh G.T. Mass Spectrometric detection of SARS-CoV-2 virus in scrapings of the epithelium of the nasopharynx of infected patients via Nucleocapsid N protein. J. Proteome Res. 2020;19:4393–4397. doi: 10.1021/acs.jproteome.0c00412. [DOI] [PubMed] [Google Scholar]
- Overmyer K.A., Shishkova E., Miller I.J., Balnis J., Bernstein M.N., Meyer J.G., Quan Q., Muehlbauer L.K., Trujillo E.A., He Y. Large-scale Multi-Omic Analysis of COVID-19 Severity. Cell Syst. 2020;12:23–40.e7. doi: 10.1016/j.cels.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnatbaum K., Zerweck J., Nehmer J., Wenschuh H., Schutkowski M., Reimer U. SpikeTidesTM—proteotypic peptides for large-scale MS-based proteomics. Nat. Methods. 2011;8 i–ii. [Google Scholar]
- Shen B., Yi X., Sun Y., Bi X., Du J., Zhang C., Quan S., Zhang F., Sun R., Qian L. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020:59–72. doi: 10.1016/j.cell.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuo Li. Regulation of Ribosomal Proteins on Viral Infection. Cells. 2019;8(5):508. doi: 10.3390/cells8050508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Xinjuan, Tianyuan Wang, Dayong Cai, Zhiwei Hu, Jin’an Chen, Hui Liao, Liming Zhi, Hongxia Wei, Zhihong Zhang, Yuying Qiu, Jing Wang, Aiping Wang Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine & Growth Factor Reviews. 2020;53:38–42. doi: 10.1016/j.cytogfr.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y., Schmitz J.E., Persing D.H., Stratton C.W. Laboratory diagnosis of COVID-19: current issues and challenges. J. Clin. Microbiol. 2020;58:1–9. doi: 10.1128/JCM.00512-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark A.A., Meza-Romero R, Offner H Surviving the storm: Dealing with COVID-19. Cellular immunology. 2020;354:104153. doi: 10.1016/j.cellimm.2020.104153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Gao Y., Wang G., Song G., Liu S., Sun D., Xu Y., Tian Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020;17:533–535. doi: 10.1038/s41423-020-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All proteomics data associated with this study are present in the manuscript or the Supplementary information. The accession number for the raw MS data and search output files for proteomics data sets depositeded to the Proteome Xchange Consortium via the PRIDE partner repository is “PRIDE: PXD020580” and “PRIDE: PXD023016” (Link: https://www.ebi.ac.uk/pride/archive/loginreviewer20505@ebi.ac.uk. https://www.ebi.ac.uk/pride/archive/loginreviewer_pxd023016@ebi.ac.uk) The targeted proteomics data is deposited in the Panorama Public and can be accessed through this link: “Panorama Public: https://panoramaweb.org/COVID_Swab_MRM.url” panorama+srivastava@proteinms.net. The additional supplemental items are available from “Mendeley Data: https://doi.org/10.17632/rnfn3vhg63.1”. The present research did not use any new codes.