

Through integration of multiple large database, two oncogenic signatures (YAP conserved and TGFβ‐up signatures) were commonly upregulated in the cancer cell lines with the mesenchymal properties. AXL, the expression of which is highly induced in EMT, contributes the doxorubicin resistance in EMT. Doxorubicin treatment induced YAP‐dependent gene response and promoted AXL expression coordinated with TGFβ‐SMAD4.

Keywords: AXL, doxorubicin, EMT, resistance, TGFβ‐SMAD4, YAP
Abstract
The acquisition of chemoresistance remains a major cause of cancer mortality due to the limited accessibility of targeted or immune therapies. However, given that severe alterations of molecular features during epithelial‐to‐mesenchymal transition (EMT) lead to acquired chemoresistance, emerging studies have focused on identifying targetable drivers associated with acquired chemoresistance. Particularly, AXL, a key receptor tyrosine kinase that confers resistance against targets and chemotherapeutics, is highly expressed in mesenchymal cancer cells. However, the underlying mechanism of AXL induction in mesenchymal cancer cells is poorly understood. Our study revealed that the YAP signature, which was highly enriched in mesenchymal‐type lung cancer, was closely correlated to AXL expression in 181 lung cancer cell lines. Moreover, using isogenic lung cancer cell pairs, we also found that doxorubicin treatment induced YAP nuclear translocation in mesenchymal‐type lung cancer cells to induce AXL expression. Additionally, the concurrent activation of TGFβ signaling coordinated YAP‐dependent AXL expression through SMAD4. These data suggest that crosstalk between YAP and the TGFβ/SMAD axis upon treatment with chemotherapeutics might be a promising target to improve chemosensitivity in mesenchymal‐type lung cancer.
Abbreviations
- AUC
area under the curve
- AXL
AXL receptor tyrosine kinase
- BCL2
B‐cell lymphoma 2
- CTD2
cancer target discovery and development
- CTGF
connective tissue growth factor
- DEG
differentially expressed genes
- DOXO
doxorubicin
- EMT
epithelial–mesenchymal transition
- Eto
etoposide
- FDA
Food and Drug Administration
- ITGB3
integrin beta‐3
- MAPK
mitogen‐activated protein kinase
- MMP2
matrix metalloproteinase‐2
- MMP9
matrix metalloproteinase‐9
- mRNA
messenger RNA
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- SBE
SMAD binding element
- SERPINE1
serpin family E member 1
- siRNA
small interfering RNA
- ssGSEA
single‐sample gene set enrichment analysis
- TCGA
The Cancer Genome Atlas
- TGFβ
transforming growth factor beta
- YAP
Yes‐associated protein
- YAP8SA
mutants of inhibitory phosphorylation site at eight serine to Alanine of YAP
- ZEB1
zinc finger E‐box binding homeobox 1
- ZEB2
zinc finger E‐box‐binding homeobox 2
1. Introduction
Despite recent remarkable advances in cancer therapeutics, conventional chemotherapy remains the standard anti‐cancer therapy for lung cancer patients who are not eligible for the most current target therapy or immunotherapies. Therapy resistance acquired during conventional chemotherapy is a major cause of cancer‐related death, and therefore understanding the molecular mechanisms underlying acquired chemoresistance is critical to identify a novel molecular target for chemosensitization [1]. Emerging evidence suggests that molecular alterations during epithelial–mesenchymal transition (EMT) may confer therapy resistance, which results in tumor relapse and mortality [2]. This is consistent with reports that cancer patients with mesenchymal gene signatures exhibit poor prognoses [3].
Therefore, considerable efforts have been made to identify molecular targets associated with EMT‐mediated chemoresistance. Moreover, instead of targeting key transcription factors that govern the EMT process such as ZEB1, SNAIL, SLUG or TWIST, which are poorly druggable, there is a growing interest in identifying druggable targets or developing strategies to ‘drug undruggable targets’ [4, 5, 6] to inhibit EMT‐mediated therapy resistance. For instance, a pharmacological inhibitor of BCL2, a well‐characterized pro‐survival protein whose expression is positively correlated to mesenchymal gene signatures, has been found to inhibit chemoresistance in EMT [7]. Alternatively, ITGB3 induction driven by the activation of the MAPK pathway leads not only to metastatic potential [8] but also to chemoradioresistance via the NF‐κB survival pathway [9]. Moreover, atorvastatin, an Federal Drug Administration (FDA)‐approved drug, was identified in silico to reverse ITGB3‐dependent NF‐κB survival gene response, as similar as ITGB3 depletion to abrogate the ITGB3‐dependent chemoresistance [10].
The Hippo pathway, which plays pivotal roles in organ development, cell proliferation, survival and homeostasis through the control of YAP/TAZ phosphorylation and nuclear translocation [11], is often deregulated in many types of cancers [12]. The subsequent increase in the transactivation activity of YAP/TAZ through binding to TEA domain transcription factors (TEAD, i.e, highly conserved transcriptional factors in the Hippo pathway, is thought to be a key mechanism for resistance of target therapy [13], as well as conventional chemotherapeutics [14, 15]. Therefore, pharmacological approaches to reactivate the Hippo signaling pathway or interfere with the transactivation activity of YAP/TAZ have been suggested as promising therapeutic alternatives [16, 17]. Moreover, instead of searching FDA‐approved drugs to inhibit YAP‐TEAD indirectly [18], chemical inhibitors to inhibit protein interactions directly between YAP and TEAD have been developed [19, 20].
On the other hand, identifying druggable targets in the downstream effectors of YAP‐TEAD responsible for chemoresistance is another feasible alternative. AXL receptor tyrosine kinase (AXL), one of the downstream targets of YAP‐TEAD [21], is readily druggable with tyrosine kinase inhibitors [22] and triggers diverse mitogenic and survival signals. Moreover, AXL also plays key roles in metastasis, invasion and cancer proliferation [22, 23], as well as EMT [24]. Additionally, AXL bypasses the anti‐mitogenic effect of several tyrosine kinase inhibitors (TKI), leading to resistance to target therapeutics [25, 26, 27], and also confers resistance against conventional chemotherapies by promoting survival signals. Thus, AXL inhibition can improve the chemotherapy response [28, 29, 30]. Despite recent efforts to demonstrate the roles of AXL in typical EMT features (e.g. promoted metastasis and therapy resistance) [24], the molecular mechanisms of AXL induction during EMT have not been fully characterized.
In this study, we took advantage of the lung cancer cell model with clear mesenchymal‐like phenotype acquired via EMT (mesenchymal‐type lung cancer cells) [7, 10] and demonstrated that high‐AXL expression in mesenchymal‐type lung cancers, which contributes to doxorubicin resistance, was induced by high YAP/TAZ activity upon doxorubicin treatment. Particularly, doxorubicin treatment in mesenchymal‐type lung cancer cells also activated TGFβ‐SMAD4 signaling, which in turn primed YAP/TAZ activity toward AXL expression. This priming effect of the TGFβ‐SMAD4 axis toward YAP/TAZ‐dependent AXL expression was also validated via a meta‐analysis of lung cancer patient clinical data.
2. Materials and methods
2.1. Reagents
The primary antibodies against α‐tubulin (#G1417), β‐actin (#sc‐47778), AXL (#sc‐166269), PARP (#J2215), YAP (#sc‐101199) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against cleaved Caspase 3 (#9661), E‐cadherin (#3195), Flag (#14793), phospho‐YAP (#4911s) and Smad4 (#46535) were purchased from Cell Signaling Technology (Danvers, MA, USA). TGFβ (#cyt‐716) was purchased from PROSPEC (Ness‐Ziona, Israel). SB‐431542 (#HYY‐10431) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). Doxorubicin hydrochloride (#44583) was purchased from MERCK (Burlington, MA, USA). Y‐27632 (#1293823) was purchased from Biogems (Westlake Village, CA, USA). Small interfering (si)RNA targeting Negative Control (#SN‐1003) and the others (listed at Tables S1 and S2) were obtained from Bioneer (Daejeon, Korea).
2.2. Cell culture
A549, TD and H1299 cell lines were maintained in Dulbecco’s modified Eagle Medium (DMEM) from Hyclone™ (#SH30243.01; Logan, UT, USA), and H358 cell lines were maintained RPMI‐1640 medium from Sigma‐Aldrich (#R8578; St. Louis, MO, USA). DMEM and RPMI‐1640 media were supplemented with 10% (v/v) fetal bovine serum, gentamicin (50 µg·mL−1). All cell lines were cultured at 37 °C in a humidified atmosphere of 5% CO2 in the air.
2.3. Cancer cell‐line transcriptome data analysis
RNA sequencing (RNA‐seq) data and drug‐response profiles of 181 lung cancer cell lines were obtained from the Cancer Cell Line Encyclopedia (CCLE) database (https://portals.broadinstitute.org/ccle) and the Cancer Target Discovery and Development (CTD2) data portal (https://ocg.cancer.gov/programs/ctd2/data‐portal/), respectively. In the RNA‐seq data, 19 082 protein‐coding genes were used for subsequent analysis. Transcripts per million (TPM) and expected counts were taken as gene expression levels for single‐sample GSEA (ssGSEA) and differential gene expression analyses, respectively. The cell‐line enrichment scores for 189 known oncogenic signatures were measured by conducting ssGSEA on 181 lung cancer cell lines. The oncogenic signatures were obtained from MSigDB C6 (https://www.gsea‐msigdb.org/gsea/msigdb). For GSEA, differential gene expression analysis between groups (e.g. mesenchymal vs epithelial, YAP‐high vs YAP‐low) was performed using the R package ‘DESeq2’, yielding a ranked list of genes. Using the gene list as the input, GSEA was run via the R package ‘fgsea’ for public annotated gene sets of interest (e.g. KEGG pathways, oncogenic signatures).
2.4. The Cancer Genome Atlas (TCGA) transcriptome data analysis
RNA‐seq and clinical data of lung adenocarcinoma patients in the TCGA LUAD cohort were obtained from the Broad GDAC firehose (https://gdac.broadinstitute.org/). Overall, 515 patients with clinical information tracked over at least 1 month were used. For survival analysis, patients were divided into three groups according to tertiles of the expression level of the gene of interest (e.g. AXL or YAP1) or mesenchymal score. TPM was taken as gene expression levels. Upper and lower groups excluding the intermediate were defined as ‘high’ and ‘low’ groups. Recurrence‐free survival analysis was performed to test the difference in the recurrence‐free survival rate between the groups by using the R package ‘survival’. The hazard ratio (HR) and P‐value (P) were computed using Cox proportional hazards regression analysis and the log‐rank test, respectively. Differential gene expression analysis between AXL‐high and AXL‐low groups was performed and genes that met the criteria of |log2 fold change | ≥ log23 and FDR ≤ 10−3 were selected as differentially expressed genes (DEG).
2.5. Transfection (plasmid DNA and siRNA)
Transient transfection of plasmid DNA or siRNA was performed with Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) following the manufacturer’s protocol. The DNA construct used is described as follows: 8X GTIIC‐luciferase, Myc‐TEAD4, Flag‐YAP wild type (WT) and Flag‐YAP 8SA were kindly gifted by M. Jung‐Soon (Ajou University, Suwon, Republic of Korea).
2.6. RNA extraction and quantitative real‐time PCR
Total RNA was extracted using easy‐BLUE™ Total RNA Extraction Kit (#17061; iNtRON, Seongnam, Korea) followed by RT‐PCR to generate the cDNA (cat#RR036A; Takara, Shiga, Japan), and the cDNA was applied to real‐time PCR with TB‐Green (cat#RR420; Takara) following the manufacturer’s protocol. Sequence information for RT‐PCR primer and siRNA information is as follows.
| Gene symbol | Primer sequence (5′ to 3′) |
| GAPDH |
F: GCA TCC TGC ACC ACC AAC TG R: GCC TGC TTC ACC ACC TTC TT |
| AXL |
F: GTG GGC AAC CCA GGG AAT ATC R: GTA CTG TCC CGT GTC GGA AAG |
| ANKRD1 |
F: AGT AGA GGA ACT GGT CAC TGG R: TGG GCT AGA AGT GTC TTC AGA T |
| AREG |
F: GCC GCT GCG AAG GAC CAA TG R: CCA GCA GCA TAA TGG CCT GAG CC |
| BIRC5 |
F: GGA CCA CCG CAT CTC TAC R: GCA CTT TCT TCG CAG TTT |
| CDH1 |
F: TGC CCA GAA AAT GAA AAA CG R: GTG TAT GTG GCA ATG CGT TC |
| CDH2 |
F: GAC AAT GCC CCT CAA GTG TT R: CCA TTA AGC CGA GTG ATG GT |
| MMP2 |
F: TAC AGG ATC ATT GGC TAC ACA CC R: GGT CAC AT GCT CCA GAC T |
| MMP9 |
F: TGT ACC GCT ATG GTT ACA CTC G R: GGC AGG GAC AGT TGC TTC T |
| SERPINE1 |
F: TTG AAT CCC ATA GCT GCT TGA AT R: ACC GCA ACG TGG TTT TCT CA |
| SMAD4 |
F: GTC TGG CTT AAG GAG AGC CAT ACT R: GATACCTGCAACTCACCTTCCTAC |
| TGFB1 |
F: CAA GTG GAC ATC AAC GGG TTC AC R: GTC CTT GCG GAA GTC AAT GTA CAG |
| YAP |
F: GTG AGC CTG TTT GGA TGA TG R: CAC TGG ACA AAG GAA GCT GA |
| ZEB1 |
F: AAG AAT TCA CAG TGG AGA GAA GCC A R: CGT TTC TTG CAG TTT GGG CAT T |
| ZEB2 |
F: GAA GAC AGA CAG TGG CAT GTA TGC R: GAG TGC TCG ATA AGG TGG TGC TTG |
| Gene Symbol | siRNA sequence (5′ to 3′) |
| AXL |
S: GAC UGU CUG GAU GGA CUG U AS: ACA GUC CAU CCA GAC AGU C |
| SMAD4 |
S: GAG ACA UUU AAG GUU CCU U AS: AAG GAA CCU UAA AUG UCU C |
| TEAD4 |
S: CCG CCA AAU CUA UGA CAA ATT AS: UUU GUC AUA GAU UUG GCG GTT |
| YAP |
S: CAG AAG AUC AAA GCU ACU U AS: AAG UAG CUU UGA UCU UCU G |
2.7. Immunoblotting and immunofluorescence
Cell lysates were extracted by RIPA buffer supplemented with 1% protease inhibitor cocktail and 0.1% sodium orthovanadate. After 1 h incubation on ice, total protein was extracted by centrifugation. The concentration of total protein was quantified by BCA protein assay kit (#23225; Thermo Scientific™; Waltham, MA, USA). Approximately 25 µg of total protein was separated on various concentrations of SDS/PAGE (7.5%, 10% or 15%). Separated protein in the gel was transferred to PVDF membrane. Membrane with protein was blocked with 5% skim milk in Tris‐buffered saline with 0.1% Tween‐20 (TBS‐T) for 1 h and then washed three times by TBS‐T for 5 min each. The membrane was incubated with primary antibody in TBS‐T (1 : 1000) with 0.1% sodium azide at 4 °C overnight. Incubated membrane was washed three times with TBS‐T for 5 min each. The membrane was incubated at room temperature with HRP‐conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) in TBS‐T (1 : 10 000) for 1 h. Incubated membrane was washed three times with TBS‐T for 15 min each. Immunoreactivity was detected by Chemi‐Doc using WEST‐Queen™ (#16026; iNtRON Biotechnology) kit. For immunofluorescence, cells were fixed 4% paraformaldehyde or cold methanol for 10 min, and permeabilized with 0.1% Triton X‐100 in PBS for 3 min (in the case of methanol, the permeabilization step is not necessary). The fixed cells were then incubated overnight at 4 °C in TBS‐T with 3% BSA for blocking. Next, the cells were washed three times in TBS‐T for 15 min each and incubated with primary antibody (1 : 200) in TBS‐ T in a humidity chamber for 1 h. The cells were then washed three times in TBS‐T for 5 min each and incubated with Alexa 594 conjugated secondary antibody or Cy2‐conjugated secondary antibody (1 : 200) and 0.5 mg·mL−1 40, 6‐diamidino‐2‐phenylindole (DAPI) in a dark humidity chamber for 1 h. Finally, the cells were washed in three times TBS‐T for 15 min each in the dark and attached to slide glass using Mowiol solution.
2.8. Dual‐luciferase assay
Cells were transfected with specific promoter‐luciferase vector and pRL vector following the above procedures. Cells lysates were extracted with 1× passive lysis buffer. After 1 h incubation on ice, total lysate was extracted by centrifugation. The supernatant was reacted using LARII and Stop & Glo reagent. The reporter assay was performed according to the Dual‐Luciferase Reporter Assay System (#E1980; Promega).
2.9. Transwell invasion assay
Transwell (6.5 mm) with an 8‐µm pore polycarbonate membrane insert (from Corning, Corning, NY, USA) was embedded with 120 µg Matrigel (from Biosesang, Seongnam, Gyeonggi, Korea) and 100 µg gelatin (from Sigma‐Aldrich) coats at the bottom of the membrane. Cells were added into the Matrigel‐embedded insert with serum‐free RPMI media, and the inserts were placed into the bottom chambers containing 10% FBS media. Cells were incubated for 24–48 h. The images were taken with a light microscope.
2.10. Zymography
Cells were starved for 24 h with DMEM containing 2% FBS. Supernatants were concentrated using a 30‐kDa cut centricon. After adding non‐reducing sample buffer, concentrated proteins were separated on 8% gel containing gelatin by electrophoresis. The gel was washed three times by secondary washing buffer for 30 min each and incubated with reaction buffer at 37 °C overnight. After reaction, the gel was stained with 0.25% Coomassie brilliant blue solution (#C1031; Biosesang) and destained with destaining buffer.
2.11. Cell proliferation
JuLI stage (NanoEntek, Seoul, Korea) was used to measure the cell proliferation rate. After cell seeding, plates were loaded on the JuLI stage for 2 days. With the JuLI stage, time‐dependent live images were obtained and then analyzed using JuLI‐Stat software (NanoEntek, Seoul, Korea) to determine time‐dependent growth potential, according to the manufacturer’s manual.
2.12. Statistical analysis
Graphical data are presented as mean ± standard deviation (SD). Statistical significance among three groups and between groups was determined using Student’s t‐test. Significance was assumed for P < 0.05(*), P < 0.01(**) and P < 0.001(***).
3. Results
3.1. High YAP activity in mesenchymal‐type lung cancer cells
To identify dysregulated oncogenic pathways driving lung cancer EMT and therapy resistance, we evaluated the gene expression data of 181 lung cancer cell lines with cell‐line mesenchymal scores calculated as described in a previous study [31]. Using 189 publicly available oncogenic gene sets from MSigDB [32], enrichment scores were computed for 181 lung cancer cell lines via ssGSEA. Correlation analyses between cell‐line oncogenic enrichment scores and mesenchymal scores identified YAP as a key oncogene activated in mesenchymal‐type lung cancer cells, in addition to TGFβ, a well‐characterized EMT inducer (Fig. 1A, Table S1). YAP/TAZ target genes associated with the Hippo signaling pathway [33] were also significantly enriched in mesenchymal state lung cancer cells obtained from the CCLE (Fig. 1B and Fig. S1) or GSE4824 (Fig. 1C and Fig. S1B) transcriptome datasets, which is consistent with previous studies demonstrating the EMT‐promoting activity of YAP [34, 35].
Fig. 1.
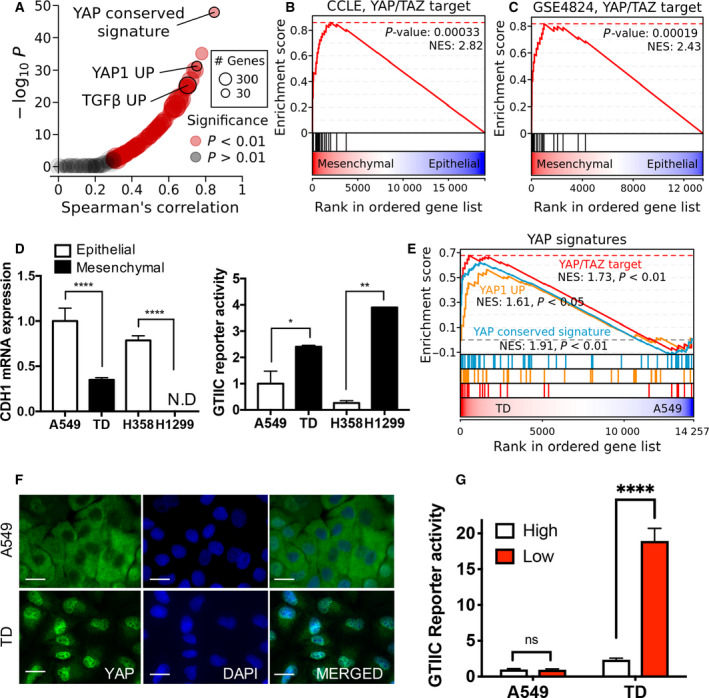
High YAP activity in chemoresistant mesenchymal cancer cell. (A) Correlation between cell‐line mesenchymal score and enrichment score across 189 oncogenic signatures. (B,C) Gene set enrichment analysis (GSEA) plots showing the enrichment of YAP/TAZ target genes in a ranked list of genes differentially expressed between mesenchymal and epithelial cell groups in (B) CCLE and (C) GEO (GSE4824) datasets. (D) CDH1 mRNA expression level in H358, H1299, A549 and TD cells (left) and reporter activity of GTIIC promoter (right), N.D., not determined. (E) GSEA plot showing the enrichment of three YAP signatures in a ranked list of genes differentially expressed between TD and A549 cells. (F) Immunofluorescence microscopic image for YAP (green) of A549 and TD cells, DAPI (blue) for nuclear staining (600×, scale bar: 50 μm). (G) Reporter assay of GTIIC promoter activity in A549 and TD cells (High: High cell density, Low: Low cell density), Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**) and P < 0.0001 (****), n = 3.
Our previous studies [7, 8, 10, 36] and other authors [37] have previously reported that EMT characteristics such as high invasiveness and chemoresistance in an isogenic lung cancer cell model (A549 transdifferentiated cells: A549TD, hereafter referred to as TD) were established by long‐term exposure to TGFβ. Along with this isogenic pair, we characterized the YAP reporter activity in four in‐house non‐small cell lung carcinoma (NSCLC) lines with epithelial or mesenchymal characteristics (epithelial: H359 and A549, mesenchymal: H1299 and TD). As predicted, H1299 and TD cells with a low expression of CDH1 (encoding E‐cadherin), a typical epithelial marker (Fig. 1D, left panel), and a high expression of ZEB2 (Fig. S1C) exhibited relatively high GTIIC reporter activity, a reporter system used as an indicator of YAP/TAZ transcriptional activity [38] (Fig. 1D, right panel). To examine further the positive correlation between YAP activity and EMT properties, we took advantage of the A549 and TD isogenic pair to minimize any cell‐type‐specific bias. Consistent with the upregulation of YAP signatures in TD compared with A549 (Fig. 1E), the nuclear localization of YAP (Fig. 1F and Fig. S1D), a robust indicator of YAP activation, and high reporter activity at low cell density, which inhibits Hippo signaling activity (Fig. 1G), were observed in TD cells. These results demonstrated that YAP activity was elevated in mesenchymal‐type lung cancer cell lines including TD cells.
3.2. Identification of AXL as a factor in doxorubicin chemoresistance
We previously demonstrated that TD cells were highly resistant to doxorubicin [10] and etoposide [7]. To examine whether YAP activity also drives doxorubicin‐specific chemoresistance in lung cancer cells, we compared measurements of cell‐line sensitivity to doxorubicin in the CTD2 dataset with the cell‐line oncogenic enrichment scores. Notably, the YAP conserved signature was markedly enriched in the doxorubicin‐resistant cell lines along with the TGFβ signature, which was shown to be highly correlated with the EMT signature via a comprehensive meta‐analysis [39] (Fig. 2A, Table S2). Similar to doxorubicin, other chemotherapeutics such as topotecan and gemcitabine also showed a strong correlation to these signatures across all lung cancer cell lines (Fig. S2A).
Fig. 2.
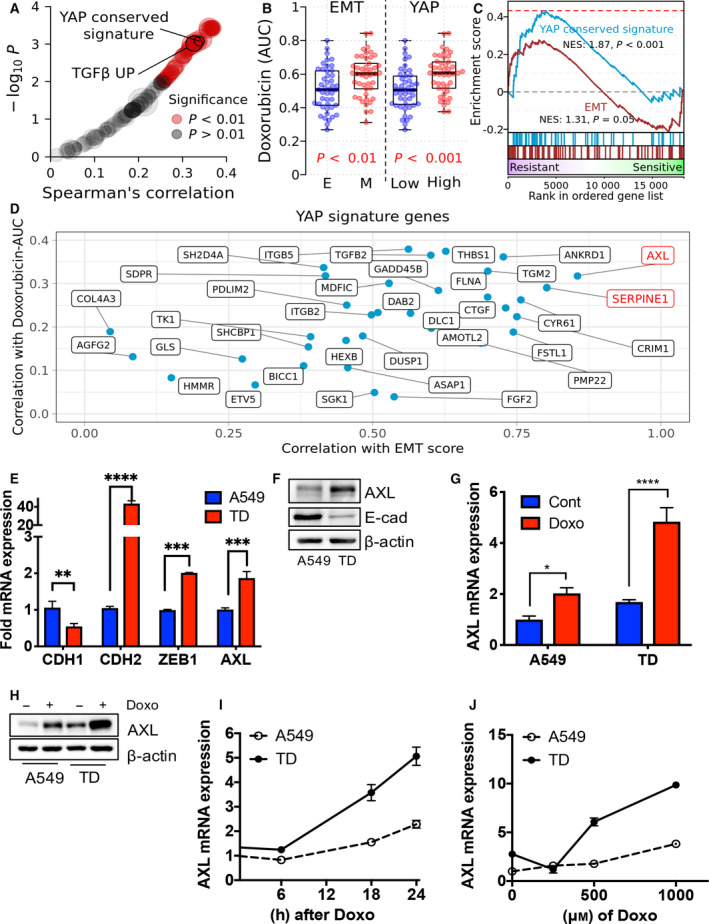
Identification of AXL as a player for doxorubicin chemoresistance. (A) Correlation between cell‐line drug sensitivity measurements (AUC) and enrichment score across oncogenic signatures. (B) Distribution of doxorubicin sensitivity by the cell groups of EMT (epithelial: E and mesenchymal: M) and YAP activity (YAP‐low: Low and YAP‐high: High) in lung cancer cell line. P‐values were calculated with two‐tailed t‐tests. (C) GSEA plot of EMT and YAP signatures in a ranked list of genes differentially expressed between doxorubicin‐resistant and doxorubicin‐sensitive cell groups. (D) YAP signature genes scattered by their expression correlation with mesenchymal score and correlation with sensitivity to doxorubicin. (E) Real‐time PCR analysis of each indicative gene in A549 and TD cells. (F) Immunoblotting analysis for indicative proteins in A549 and TD cells, β‐actin for equal protein loading. (G) AXL mRNA expression in A549 and TD cells with or without doxorubicin (Doxo, 1 μm) treatment. (H) Immunoblotting analysis for AXL protein in A549 and TD cells with or without doxorubicin (1 μm) treatment. (I) AXL mRNA expression in A549 and TD cells at the indicative time after doxorubicin (1 μm) treatment. (J) AXL mRNA expression in A549 and TD cells for each concentration of doxorubicin treatment, Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**), P < 0.001(***) and P < 0.0001(****), n = 3.
According to CTD2 data, mesenchymal cell lines and cell lines with high YAP activity had an increased resistance to doxorubicin (Fig. 2B and Fig. S2B). In particular, high YAP activity appeared to be more closely related to lung cancer resistance to doxorubicin compared with the mesenchymal state (Fig. 2C and Fig. S2C). Therefore, we aimed to identify key genes among the YAP signature genes associated with mesenchymal status and lung cancer chemoresistance. AXL and SERPINE1 were selected as candidate genes because their expression was most positively correlated with the cell‐line mesenchymal score and drug resistance to doxorubicin (Fig. 2D). Consistent with this, the role of AXL and PAI‐1 (encoded by SERPINE1) in therapy resistance has been previously reported [26].
TD cells that exhibited resistance to doxorubicin [10] and etoposide [7] had a high‐AXL expression compared with their parent A549 epithelial cells (Fig. 2E,F). AXL was markedly induced upon doxorubicin treatment (Fig. 2G,H) in a dose‐ and time‐dependent manner (Fig. 2I,J). Furthermore, SERPINE1 was markedly induced in TD cells upon doxorubicin treatment (Fig. S2D). Etoposide, another genotoxic chemotherapeutic, induced SERPINE1 and AXL in TD cells (Fig. S2E). These data suggest that YAP activation (Fig. 1) may induce AXL or SERPINE1 in mesenchymal‐type lung cancer cells upon treatment with genotoxic chemotherapeutic compounds.
3.3. YAP activity induces AXL upon doxorubicin treatment
To examine whether YAP activity promotes AXL expression, we depleted YAP/TAZ in TD cells and determined AXL expression thereafter. As expected, the significant repression of AXL resulted from depletion of YAP or TAZ along with a marked suppression of connective tissue growth factor (CTGF), a well‐established direct downstream target of YAP/TAZ [40] (Fig. 3A). The dependency of AXL expression on YAP/TAZ upon doxorubicin treatment, was confirmed by lower AXL protein levels following the depletion of YAP by siRNA (Fig. 3B). In contrast, the ectopic expression of constitutively active mutant of YAP (YAP8SA: inhibitory phosphorylation dead mutant) [41] was sufficient to induce AXL and SERPINE1 (Fig. 3C), which was further validated by immunoblotting analysis (Fig. 3D). Therefore, reactivating the Hippo signaling pathway to inhibit YAP/TAZ transcriptional activation with two independent chemical inhibitors (dasatinib, an SRC inhibitor [42] or Y27632, a ROCK inhibitor [43]) markedly suppressed AXL expression in A549 and TD cells (Fig. 3E). Suppression of YAP/TAZ action by knockdown or inhibitor treatment clearly repressed AXL expression upon doxorubicin treatment (Fig. 3F). Therefore, the upregulation of AXL induced by doxorubicin treatment or present in doxorubicin‐resistant cancer cells (Fig. 2) and YAP‐dependent AXL expression (Fig. 3) suggests that YAP/TAZ activation may occur by doxorubicin treatment alone. Moreover, as expected, the GTIIC reporter was significantly upregulated in TD cells (Fig. 3G), which was coupled with a clear nuclear translocation of YAP in TD cells after doxorubicin treatment (Fig. 3H,I).
Fig. 3.
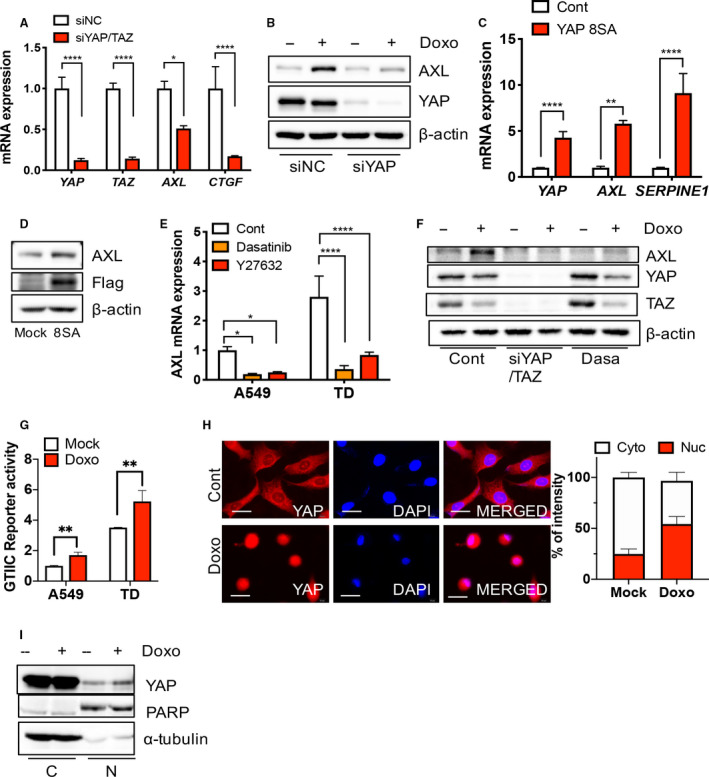
YAP activity for AXL induction upon doxorubicin. (A) Real‐time PCR analysis of each indicative gene after introduction of control (siNC) or YAP/TAZ (siYAP/TAZ) siRNA. (B) Immunoblotting analysis for indicative proteins in TD cells after introduction of control (siNC) or YAP (siYAP) siRNA, upon doxorubicin (Doxo, 1 μm) treatment. (C,D) Real‐time PCR analysis (C) and immunoblotting analysis of the indicative gene or protein after introduction of control or YAP8SA in A549 cells. (E) Real‐time PCR analysis of AXL expression after Dasatinib (100 μm) and Y27632 (10 ng·mL−1) in A549 and TD cells (F) immunoblotting analysis of the indicative proteins with siRNA for YAP/TAZ (siYAP/TAZ) or 100 μm Dasatinib (Dasa), 24 h after 1 μm doxorubicin treatment (Doxo) in TD cells. (G) Reporter assay of GTIIC promoter activity in A549 and TD cells after doxorubicin (Doxo, 1 μm) treatment. (H) Immunofluorescence microscopic images for YAP (red) in TD cells, DAPI (blue) for nuclear staining (left). Graphical presentation of cytoplasmic (Cyto) and nuclear intensity (Nuc) of YAP in A549 cells after doxorubicin (Doxo, 1 μm) (right) (600×, scale bar: 50 μm). (I) Immunoblotting analysis of cytoplasmic (C) and nuclear (N) fraction in TD cells after 1 μm of doxorubicin treatment (Doxo), PARP and α‐tubulin for equal protein loading in nuclear fraction and cytoplasmic fraction respectively. Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**), P < 0.001(***) and P < 0.0001(****), n = 3.
3.4. AXL expression induces doxorubicin resistance in TD cells
Consistent with the data presented in Fig. 1, high YAP/TAZ activity derived from the inactivation of the Hippo signaling pathway is known to be linked to EMT [44, 45]. Based on the high YAP activity in TD cells (Fig. 1D,G), we examined whether two clear mesenchymal characteristics of TD cells – high invasiveness [8, 36] and chemoresistance [7, 10] – were affected by high YAP activity.
Chemoresistance to doxorubicin was first assessed based on cell growth potential. Although this potential remained unaffected even under doxorubicin treatment (siNC, Fig. 4A), significant attenuation of cell growth after depletion of YAP (siYAP) was observed after doxorubicin treatment (siYAP, Fig. 4A), which corresponded with high active caspase 3 activity due to YAP knockdown via doxorubicin treatment (Fig. 4B). Moreover, the depletion of YAP also inhibited cell invasiveness (Fig. 4C), suggesting that YAP‐dependent gene responses promoted chemoresistance and high invasiveness. AXL and SERPINE1 were associated with downstream YAP signals via transcriptome analysis of doxorubicin‐resistant and mesenchymal‐type lung cancer cells (Fig. 2A–D); therefore, we investigated the effect of the knockdown of each gene on invasiveness or chemoresistance. Consistent with recent studies reporting that AXL upregulation contributed to target and conventional chemotherapy resistance [28, 29, 30], TD cells exhibited attenuated apoptotic responses coupled with AXL induction upon doxorubicin treatment compared with A549 cells and were sensitized to doxorubicin treatment by AXL depletion (Fig. 4D,E). In contrast, AXL knockdown had negligible effects on cell invasion (data not shown) and MMP2 and MMP9 activity (Fig. S3A,B). Additionally, knockdown of SERPINE1 had an insignificant effect on chemoresistance (Fig. S3C,D) but did affect the invasiveness of TD cells (Fig. S3E). These results suggest that YAP‐dependent AXL expression contributes to doxorubicin resistance in doxorubicin‐treated TD cells.
Fig. 4.
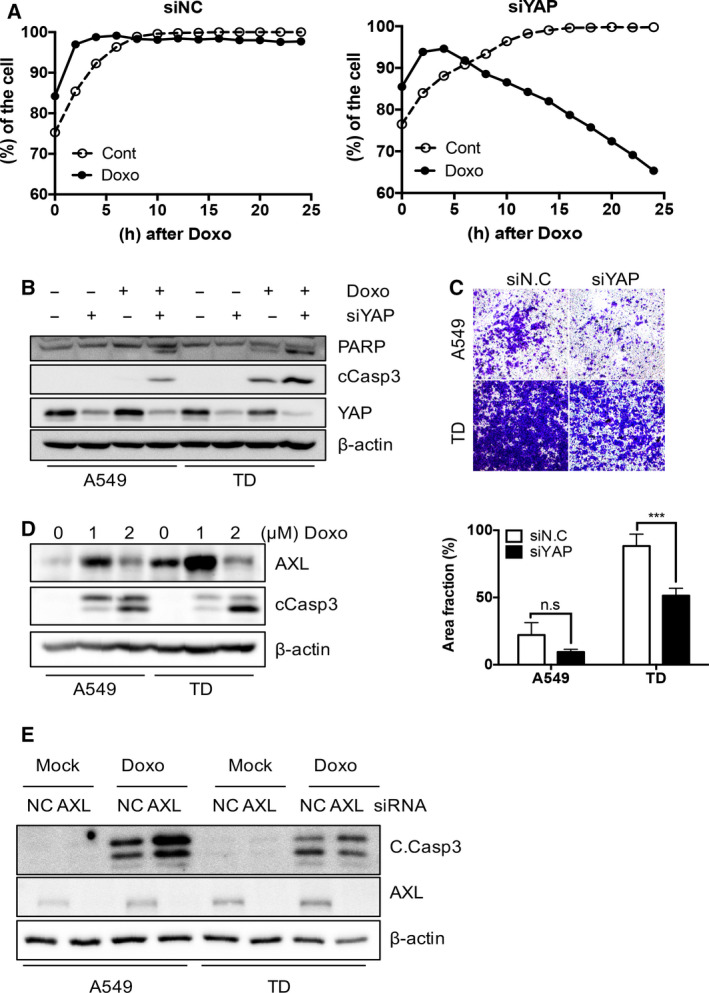
AXL expression for doxorubicin resistance in TD cells. (A) Cell growth of TD cells determined by JuLI stage, after introducing control (siNC: left) or YAP (siYAP: right) siRNA at indicative times after 2 μm doxorubicin treatment (Doxo). (B) Immunoblotting analysis for indicative proteins in A549 and TD cells after introduction control (–) or YAP (siYAP) siRNA, 24 h after 2 μm of doxorubicin (Doxo) treatment (C) Representative images of two chamber invasion assay of A549 and TD cells after introducing control (siNC) or YAP (siYAP) siRNA treatment (top) and graphical presentation of invaded area (bottom). (D) Immunoblotting of A549 and TD cells after doxorubicin (Doxo, 1 or 2 μm) treatment, β‐actin for equal protein loading, (E) Immunoblotting of the indicative proteins in A549 and TD cells of introducing control (siNC) or YAP (siYAP) siRNA treatment at 24 h after doxorubicin (Doxo, 2 μm) treatment, Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**), P < 0.001(***) and P < 0.0001(****), n.s.: not significant, n = 3.
3.5. TGFβ‐SMAD4 axis primes AXL expression upon doxorubicin treatment
Upon doxorubicin treatment, AXL expression (Fig. 2G) and nuclear YAP levels (Fig. 3H) were significantly elevated in TD cells relative to A549 cells. This suggested that the transcriptional dysregulation of upstream signaling, which was accompanied by EMT, contributed to YAP/TAZ activity and subsequent AXL induction. Therefore, we searched for other factors associated with the Hippo signaling pathway that could affect YAP‐dependent gene responses in lung cancer cells with high AXL expression. Lung cancer cell lines were divided into high‐, intermediate‐ and low‐AXL groups based on the tertiles of the basal AXL expression level (Fig. S4A) and we visualized the differential expression between the high‐ and low‐AXL groups in the KEGG Hippo signaling pathway (Fig. 5A). Interestingly, TGFβ and SMAD2/3 were significantly upregulated in the high‐AXL group (Fig. 5A), suggesting that the activation of the TGFβ‐SMAD signaling pathway might contribute to YAP‐dependent AXL induction. To test the relevance of TGFβ‐SMAD signaling on AXL expression, we depleted SMAD4 to abolish TGFβ‐dependent gene expression and monitored AXL expression in our cell model. As shown in Fig. 5B, AXL expression was moderately modulated by SMAD4 knockdown in A549 and TD cells (Fig. 5B and Fig. S4B). Surprisingly, TGFβ treatment markedly induced AXL expression in TD cells, whereas the effect of TGFβ on AXL expression was negligible in A549 cells (Fig. 5C,D). This pattern was similar in ZEB1, a typical TGFβ‐mediated gene (Fig. S4C). The effect of TGFβ signaling on AXL expression was fully abrogated by SMAD4 knockdown, suggesting that the TGFβ‐SMAD axis contributes to AXL expression (Fig. 5E). Based on these results, we hypothesized that increased TGFβ‐SMAD signaling in mesenchymal‐type lung cancer cells after doxorubicin treatment potentiates YAP‐dependent AXL expression. As predicted, doxorubicin treatment increased SMAD binding element (SBE) reporter activity in TD cells in a time‐dependent manner (Fig. 5F). Moreover, SMAD4 depletion partially attenuated AXL or SERPINE1 induction upon doxorubicin treatment (Fig. 5G and Fig. S4D).
Fig. 5.
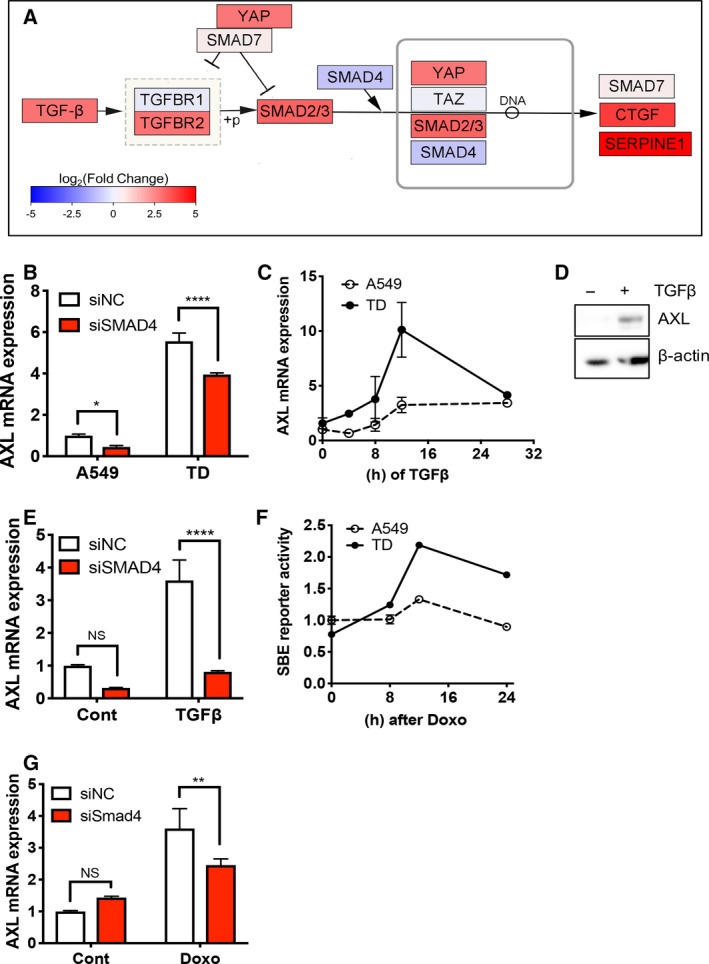
TGFβ‐SMAD4 axis to prime AXL expression upon doxorubicin. (A) Simplified TGFβ‐SMAD axis in Hippo signaling pathway. Each gene is colored according to log2 fold change values between high AXL and low AXL patient groups. (B,C) Real‐time PCR analysis of AXL in A549 and TD cells after introducing control (siNC) or SMAD4 (siSMAD4) siRNA (B) or at indicative time after 5 ng·mL−1 of TGFβ treatment (D) Immunoblotting analysis for AXL protein in TD cell upon TGFβ treatment (5 ng·mL−1). (E) Real‐time PCR analysis of AXL in A549 and TD cells, introducing control (siNC) or SMAD4 24 h after 5 ng·mL−1 of TGFβ treatment. (F) Reporter assay of SBE promoter activity at the indicative time after 1 μm of doxorubicin treatment (Doxo). (G) Real‐time PCR analysis of AXL in TD cells, introducing control (siNC) AXL or SMAD4 (siSMAD4) siRNA after doxorubicin treatment (Doxo, 1 μm). Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**), P < 0.001(***) and P < 0.0001(****), NS: not significant, n = 3.
3.6. TGFβ‐SMAD4 primes YAP‐dependent AXL expression
TGFβ is involved in the induction of EMT and stromal environment modulation, both of which are essential for tumor progression [46]. Moreover, cancer cells may be chronically exposed to TGFβ in the tumor microenvironment [37, 47]. Under these conditions, additional TGFβ stimulation further activated the TGFβ‐SMAD axis in mesenchymal cancer cells, resulting in increased YAP/TAZ activity. This was evidenced by the time‐dependent increase in YAP levels in the nucleus coupled with SMAD4 (Fig. 6A) and GTIIC reporter activity (Fig. 6B) upon TGFβ treatment. Even basal levels of YAP reporter activity were significantly modulated by SMAD4 knockdown in TD cells (Fig. 6C), suggesting that constant SMAD4‐dependent gene transcription contributed to maintaining high YAP activity in mesenchymal‐type lung cancer cells. Given that YAP/TAZ forms a protein complex with SMAD2/3 and SMAD4 and promotes nuclear translocation [48], the interaction between TGFβ‐SMAD4 and YAP/TAZ has been extensively characterized [49, 50]. Interestingly, a wild type (YAPWT) and a constitutively active YAP mutant (YAP8SA) failed to affect SBE reporter activity (Fig. S5), suggesting that the TGFβ‐SMAD axis might uni‐directionally prime YAP/TAZ‐dependent gene expression.
Fig. 6.
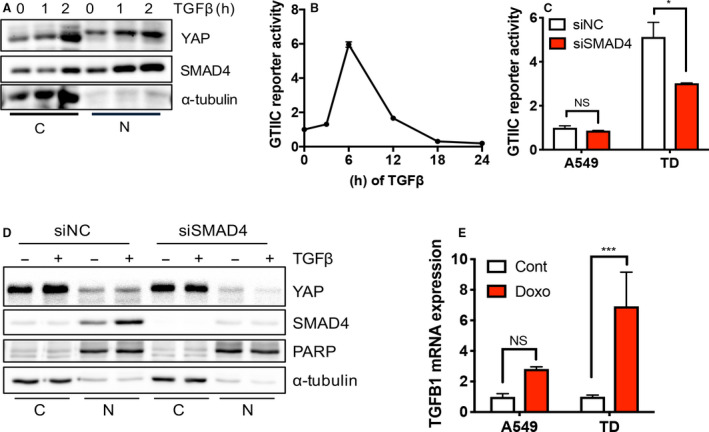
TGFβ‐SMAD4 primes YAP‐dependent AXL expression. (A) Immunoblotting analysis of cytoplasmic (C) and nuclear (N) fraction in TD cells at indicative time after 5 nm of TGFβ treatment, α‐tubulin for equal protein loading. (B,C) Reporter assay of GTIIC promoter activity in TD cell at indicative time after 5 ng·mL−1 TGFβ treatment (B) and in A549 and TD cells after introducing control (siNC) or SMAD4 (siSMAD4) siRNA. (D) Immunoblotting analysis of cytoplasmic (C) and nuclear (N) fraction in TD cells with siRNA of control (siNC) or SMAD4 (siSMAD4) 24 h after 5 nm TGFβ treatment, PARP and α‐tubulin for equal protein loading in nuclear fraction and cytoplasmic fraction respectively. (E) Real‐time PCR analysis of TGFB1 in A549 and TD cells after doxorubicin treatment (Doxo, 1 μm), Mean ± SD, Student’s t‐test, P < 0.05(*), P < 0.01(**), P < 0.001(***) and P < 0.0001(****), NS, not significant, n = 3.
3.7. Clinical significance of AXL in mesenchymal‐type lung cancer patients
As determined by our isogenic cell‐line model, AXL expression in mesenchymal‐type lung cancer cells, which confers doxorubicin resistance, was upregulated by YAP/TAZ activity primed by the TGFβ‐SMAD axis. These results were evaluated in primary tumors using the transcriptome data of 515 lung cancer patients from TCGA database. Similar to the high expression of AXL in TD cells and its close correlation to YAP/TAZ activity (Figs 2 and 3), AXL expression was closely correlated to typical EMT genes including VIM (encoding vimentin), SNAI2 (encoding SLUG), ZEB1, ZEB2, SERPINE1 and CTGF (Fig. 7A and Fig. S6A). Given that our results suggested that the upregulation of AXL conferred therapy‐resistant mesenchymal properties, we investigated the association between AXL expression and recurrence‐free patient survival. We found that a higher expression of AXL and higher mesenchymal scores were strongly associated with a poor prognosis, similar to other typical EMT genes and YAP1 (Fig. 7B and Fig. S6B). To determine further whether AXL expression was indicative of mesenchymal properties in primary tumors, the patients were divided into high‐, intermediate‐ and low‐AXL groups based on the tertiles of AXL expression (Fig. S6C), after which the differentially expressed genes (DEG) between groups were examined (Fig. 5A). Functional enrichment analysis indicated that the organizing functions of cell structure, which are mostly affected during EMT [51], were significantly upregulated in high‐AXL patients (Fig. 7C). As expected, EMT and YAP signatures were strongly enriched in high‐AXL patients, thereby confirming the YAP‐dependent expression of AXL in mesenchymal‐type lung cancer cells (Fig. 7D). Consistent with the priming effect of TGFβ‐SMAD for YAP‐dependent AXL expression (Figs 5 and 6), the TGFβ signaling and Hippo signaling were also highly enriched in high‐AXL patients (Fig. 7D).
Fig. 7.
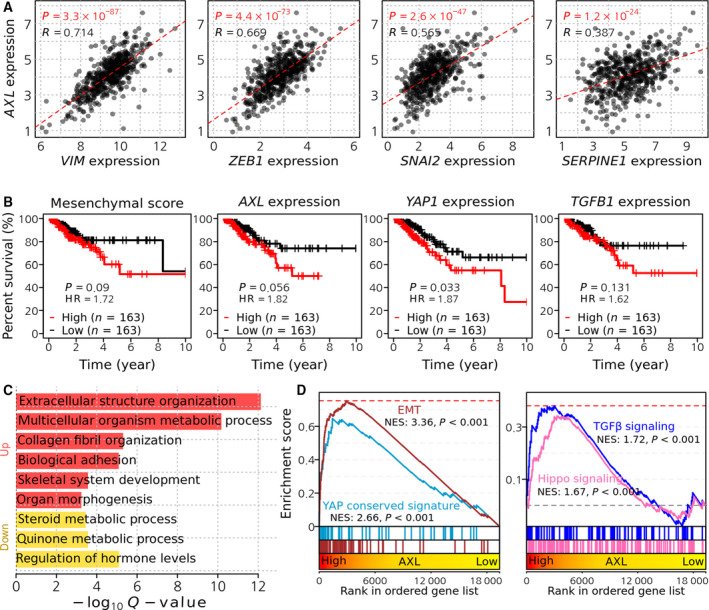
Clinical significance of AXL in the lung cancer patients. (A) Correlation between AXL expression and VIM, ZEB1, SNAI2 and SERPINE1 expression levels in TCGA lung adenocarcinoma patients. (B) Kaplan–Meier curves showing recurrence‐free survival time of lung adenocarcinoma patients. Patients were divided into three groups (high, intermediate, low) by mesenchymal score, expression levels of AXL, YAP1 and TGFB1. High and low groups were used for comparison. (C) Significantly enriched functions in upregulated DEG in high AXL patients. Hypergeometric tests were performed using gene annotation in the biological process of the Gene Ontology database, yielding enrichment scores, defined as −log10 (q‐value). (D) GSEA plots showing the enrichment of EMT and YAP signatures (left) and TGFβ and Hippo signaling pathway (right) in a ranked list of genes differentially expressed between high and low AXL groups in TCGA lung adenocarcinoma patients.
4. Discussion
Most targets responsible for acquired chemoresistance in cancers identified through extensive molecular mechanistic studies are poorly druggable, making it particularly challenging to identify therapeutic agents for chemosensitization [4]. The constant activation of YAP occurs in parallel with EMT to promote survival gene responses and has therefore been suggested to be an important molecular target. However, inhibiting protein binding to TEAD (i.e. its partner transcription factor) using small molecules remains particularly challenging despite many attempts. Therefore, the identification of AXL as a key survival factor in chemoresistant mesenchymal cancer and downstream YAP signaling factor is important because AXL (i.e. a receptor tyrosine kinase) is readily druggable via kinase inhibitors. In fact, many AXL inhibitors are currently being developed or investigated in clinical trials [22]. In this study, we used a doxorubicin‐resistant isogenic pair of lung cancer cell lines to demonstrate that YAP activation in mesenchymal‐type lung cancer cells (Fig. 1) induced AXL expression upon doxorubicin treatment (Fig. 2) and contributed to doxorubicin resistance (Figs 4 and 5). AXL induction in mesenchymal‐type lung cancer cells upon doxorubicin treatment (Fig. 2) was induced by the elevated nuclear translocation of YAP and subsequent increase in YAP transactivation (Fig. 3). Particularly, the activation of TGFβ signaling through SMAD4 after doxorubicin treatment (Fig. 5) contributed to YAP‐dependent AXL expression in mesenchymal‐type lung cancer cells (Fig. 6). These results suggest that the crosstalk between TGFβ (which is highly enriched in cancer microenvironments) and the YAP pathway synergizes with AXL expression during EMT to promote chemoresistance. Furthermore, TGFβ expression was promoted in mesenchymal‐type lung cancer cells (Fig. 6E), further enhancing this synergistic effect.
The crosstalk between TGFβ and YAP/TAZ has been extensively studied. YAP/TAZ regulates TGFβ signaling [52] and promotes SMAD2/3 nuclear translocation [48]. However, the ectopic expression of YAP (WT) and constitutively active YAP (YAP8SA) failed to affect SMAD4‐dependent gene responses (Fig. S6). Furthermore, TGFβ stimuli promoted the nuclear translocation of YAP (Fig. 6A) and YAP‐dependent gene responses, including AXL (Figs 5 and 6), suggesting that a TGFβ‐rich microenvironment favors YAP‐dependent AXL expression, which leads to chemoresistance. These results are consistent with a previous study that reported the positive effects of TGFβ on YAP transactivation [53]. It is also worth noting that the direct interaction of ZEB1 with the YAP/TEAD complex alters the YAP‐dependent gene expression profile, which favors therapy resistance and increases metastatic risk [54]. Additionally, the presence of SMAD2/3 in the YAP–TEAD complex coordinates transcriptional regulation, which promotes an aggressive metastasis response [55]. Similarly, we also observed that the loss of SMAD4 led to alterations in YAP‐dependent gene responses (Fig. S6B). Unfortunately, we did not confirm the presence of SMAD in the AXL promoters (data not shown). Therefore, it is still unclear whether TGFβ‐mediated SMAD elevate YAP‐dependent AXL expression directly or indirectly. Alternatively, microRNA (miR)‐34a, which downregulates AXL [56], was reportedly suppressed by TGFβ [57]. Thus, it would be interesting to quantify miR‐34a levels upon doxorubicin treatment to characterize the role of miR‐34a on AXL expression in mesenchymal‐type lung cancer cells compared with epithelial cancer cells.
5. Conclusions
Our results indicate that a doxorubicin‐induced increase in TGFβ expression contributes to YAP‐dependent AXL induction in mesenchymal‐type lung cancer cells, suggesting that alterations in EMT lead to chemoresistance by enhancing YAP‐dependent survival gene responses. Therefore, the modulation of TGFβ signaling is a promising chemosensitizing alternative strategy. Upregulation of the TGFβ/SMAD pathway primes YAP‐dependent AXL expression upon doxorubicin treatment to promote chemoresistance in mesenchymal‐type lung cancer cells.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
HJ.C conceived the overall study design and led the experiments. JY.C and H.L conducted the experiments and database analyses, and provided critical discussion of the results. OS.K and HJ.K provided the initial preliminary data. All authors contributed to manuscript writing and revision and endorsed the final manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.12857.
Supporting information
Fig. S1. (A) CCLE lung cancer cell lines ranked by mesenchymal score. Distribution of mesenchymal score is shown in the right panel. Cell lines were divided into tertiles by mesenchymal score (mesenchymal, intermediate, epithelial groups). (B) Distribution of mesenchymal score of 78 lung cancer cell lines in GSE4824. Cell lines were divided into three groups according to mesenchymal score (mesenchymal, intermediate, epithelial groups). (C) ZEB2 mRNA expression level in A549, TD, H358 and H1299 cells. (D) Immunostaining of YAP in A549 and TD cells.
Fig. S2. (A) Association between cell‐line enrichment scores of oncogenic signatures and drug sensitivity (AUC) to doxorubicin, topotecan and gemcitabine in 181 lung cancer cell lines in the CTD2 database. (B) Lung cancer cell lines ranked by doxorubicin sensitivity. Distribution of area under the curve (AUC) is shown in the right panel. Cell lines were divided into tertiles of AUC (doxorubicin‐resistant, ‐intermediate and ‐sensitive groups). (C) Correlation between cell‐line sensitivity (AUC) to doxorubicin and enrichment score of YAP signature (left) or mesenchymal signature (right) in lung cancer cell lines. (D) SERPINE1 messenger (m)RNA expression upon indicative concentration of doxorubicin (Doxo) treatment. (E) AXL (left) and SERPINE1 (right) mRNA expression upon indicative concentration of etoposide (Eto) treatment.
Fig. S3. (A) Representative images of zymography assay of A549 and TD cells after AXL siRNA treatment. (B) AXL mRNA expression in A549 and TD cells. (C,D) Immunoblotting analysis for PARP‐1 in TD cells, introducing of control (siNC) or SERINE1 (siSERPINE1) after treatment of doxorubicin (C: Doxo, 2 μm) or, etoposide (D: Eto, 40 μm). (E) Representative images of two‐chamber invasion assay of A549 and TD cells with control (siNC) or SERINE1 (siSERPINE1) (left) and graphical presentation of invaded area (% of Area) (right).
Fig. S4. (A) Distribution of AXL expression levels of 181 lung cancer cell lines in CCLE data. Cell lines were divided into tertiles of AXL expression (high, intermediate and low AXL groups). (B) Realtime PCR analysis for SMAD4 mRNA expression in A549 and TD cells with control (siNC) or SMAD4 (siSMAD4) siRNA. (C) Real‐time PCR analysis for ZEB1 mRNA expression in A549 and TD cells after indicative time of TGFβ treatment (5 ng/mL). (D) Real‐time PCR analysis for SERPINE1 mRNA expression with control (siNC) or SMAD4 (siSMAD4) siRNA, NS, not significant.
Fig. S5. Graphical presentation of Reporter activity of GTIIC and SBE with wild type (YAP WT) or constitutively active mutant (YAP8SA) of YAP.
Fig. S6. (A) Correlation between AXL expression and TGFB1, ZEB2, CTGF and YAP1 expression levels in 515 lung adenocarcinoma patients in TCGA data. (B) The Kaplan–Meier curves showing recurrence‐free survival time of lung adenocarcinoma patients. Patients were divided into tertiles (high, intermediate, low) by the expression levels of VIM, ZEB1, SNAI2 and SERPINE1. High and low groups were used for comparison. (C) Distribution of AXL expression levels of lung adenocarcinoma patients in TCGA data. Patients were divided into high, intermediate, and low AXL groups based on the basal expression level of AXL.
Fig. S7. Uncut immunoblotting data used in these studies.
Table S1. Correlation coefficient between cell‐line mesenchymal score (Taube et al., Grojer et al. and Byers et al.) and cell‐line oncogenic score calculated by using ssGSEA with 189 oncogenic signature gene sets.
Table S2. Correlation coefficient between cell‐line response (AUC) to doxorubicin and cell‐line oncogenic score.
Acknowledgements
This work was supported by a grant from the National Research Foundation of Korea (NRF‐2020R1A2C2005914 and 2017M3C9A5028691) and the Global Core Research Center (GCRC, #2011‐0030001), as well as by the Creative‐Pioneering Researchers Program through Seoul National University (SNU).
Jeong‐Yun Choi, Heaseung Lee, and Eun‐Ji Kwon contributed equally to this work.
Data accessibility
The datasets used and/or analyzed herein can be obtained from the corresponding author upon request.
References
- 1. Chang A (2011) Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer 71, 3–10. [DOI] [PubMed] [Google Scholar]
- 2. Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, Choi H, El Rayes T, Ryu S, Troeger J et al (2015) Epithelial‐to‐mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, Snel MH, Chresta CM, Rosenberg R, Nitsche U et al (2014) Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial‐to‐mesenchymal transition. Int J Cancer 134, 552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dang CV, Reddy EP, Shokat KM & Soucek L (2017) Drugging the ‘undruggable’ cancer targets. Nat Rev Cancer 17, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwon OS, Kim W, Cha HJ & Lee H (2019) In silico drug repositioning: from large‐scale transcriptome data to therapeutics. Arch Pharm Res 42, 879–889. [DOI] [PubMed] [Google Scholar]
- 6. Kwon OS, Lee H, Kong HJ, Kwon EJ, Park JE, Lee W, Kang S, Kim M, Kim W & Cha HJ (2020) Connectivity map‐based drug repositioning of bortezomib to reverse the metastatic effect of GALNT14 in lung cancer. Oncogene 39, 4567–4580. [DOI] [PubMed] [Google Scholar]
- 7. Kwon OS, Hong SK, Kwon SJ, Go YH, Oh E & Cha HJ (2017) BCL2 induced by LAMTOR3/MAPK is a druggable target of chemoradioresistance in mesenchymal lung cancer. Cancer Lett 403, 48–58. [DOI] [PubMed] [Google Scholar]
- 8. Bae GY, Hong SK, Park JR, Kwon OS, Kim KT, Koo J, Oh E & Cha HJ (2016) Chronic TGFbeta stimulation promotes the metastatic potential of lung cancer cells by Snail protein stabilization through integrin beta3‐Akt‐GSK3beta signaling. Oncotarget 7, 25366–25376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H et al (2014) An integrin beta(3)‐KRAS‐RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol 16, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hong SK, Lee H, Kwon OS, Song NY, Lee HJ, Kang S, Kim JH, Kim M, Kim W & Cha HJ (2018) Large‐scale pharmacogenomics based drug discovery for ITGB3 dependent chemoresistance in mesenchymal lung cancer. Mol Cancer 17, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma S, Meng Z, Chen R & Guan KL (2019) The Hippo pathway: biology and pathophysiology. Annu Rev Biochem 88, 577–604. [DOI] [PubMed] [Google Scholar]
- 12. Moroishi T, Hansen CG & Guan KL (2015) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK et al (2013) An epithelial‐mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, Jones DS II, Kapur S, Mathews II, Koong AC et al (2017) Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J Clin Invest 127, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schoumacher M & Burbridge M (2017) Key roles of AXL and MER receptor tyrosine kinases in resistance to multiple anticancer therapies. Curr Oncol Rep 19, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calses PC, Crawford JJ, Lill JR & Dey A (2019) Hippo pathway in cancer: aberrant regulation and therapeutic opportunities. Trends Cancer 5, 297–307. [DOI] [PubMed] [Google Scholar]
- 17. Park HW & Guan KL (2013) Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci 34, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elisi GM, Santucci M, D’Arca D, Lauriola A, Marverti G, Losi L, Scalvini L, Bolognesi ML, Mor M & Costi MP (2018) Repurposing of drugs targeting YAP‐TEAD functions. Cancers (Basel) 10, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bum‐Erdene K, Zhou D, Gonzalez‐Gutierrez G, Ghozayel MK, Si Y, Xu D, Shannon HE, Bailey BJ, Corson TW, Pollok KE et al (2019) Small‐molecule covalent modification of conserved cysteine leads to allosteric inhibition of the TEADYap protein‐protein interaction. Cell Chem Biol 26, 378–389, e313. [DOI] [PubMed] [Google Scholar]
- 20. Gibault F, Coevoet M, Sturbaut M, Farce A, Renault N, Allemand F, Guichou JF, Drucbert AS, Foulon C, Magnez R et al (2018) Toward the discovery of a novel class of YAP(‐)TEAD interaction inhibitors by virtual screening approach targeting YAP(‐)TEAD protein(‐)protein interface. Cancers (Basel) 10, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J, Poon RT, Zender L, Lowe SW, Hong W et al (2011) AXL receptor kinase is a mediator of YAP‐dependent oncogenic functions in hepatocellular carcinoma. Oncogene 30, 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu C, Wei Y & Wei X (2019) AXL receptor tyrosine kinase as a promising anti‐cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 18, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rankin EB & Giaccia AJ (2016) The receptor tyrosine kinase AXL in cancer progression. Cancers (Basel) 8, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Antony J & Huang RY (2017) AXL‐driven EMT state as a targetable conduit in cancer. Cancer Res 77, 3725–3732. [DOI] [PubMed] [Google Scholar]
- 25. Taniguchi H, Yamada T, Wang R, Tanimura K, Adachi Y, Nishiyama A, Tanimoto A, Takeuchi S, Araujo LH, Boroni M et al (2019) AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun 10, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, Abdel‐Rahman M, Wang X, Levine AD, Rho JK et al (2012) Activation of the AXL kinase causes resistance to EGFR‐targeted therapy in lung cancer. Nat Genet 44, 852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zuo Q, Liu J, Huang L, Qin Y, Hawley T, Seo C, Merlino G & Yu Y (2018) AXL/AKT axis mediated‐resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 37, 3275–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, Armstrong A & Zhu JG (2017) Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 8, 41064–41077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ludwig KF, Du W, Sorrelle NB, Wnuk‐Lipinska K, Topalovski M, Toombs JE, Cruz VH, Yabuuchi S, Rajeshkumar NV, Maitra A et al (2018) Small‐molecule inhibition of Axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res 78, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quinn JM, Greenwade MM, Palisoul ML, Opara G, Massad K, Guo L, Zhao P, Beck‐Noia H, Hagemann IS, Hagemann AR et al (2019) Therapeutic inhibition of the receptor tyrosine kinase AXL improves sensitivity to platinum and taxane in ovarian cancer. Mol Cancer Ther 18, 389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore‐Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ et al (2017) Dependency of a therapy‐resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P & Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Xu X, Maglic D, Dill MT, Mojumdar K, Ng PK, Jeong KJ, Tsang YH, Moreno D, Bhavana VH et al (2018) Comprehensive molecular characterization of the Hippo signaling pathway in cancer. Cell Rep 25, 1304–1317, e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Diepenbruck M, Waldmeier L, Ivanek R, Berninger P, Arnold P, van Nimwegen E & Christofori G (2014) Tead2 expression levels control the subcellular distribution of Yap and Taz, zyxin expression and epithelial‐mesenchymal transition. J Cell Sci 127, 1523–1536. [DOI] [PubMed] [Google Scholar]
- 35. Park J, Kim DH, Shah SR, Kim HN, Kshitiz, Kim P, Quinones‐Hinojosa A & Levchenko A (2019) Switch‐like enhancement of epithelial‐mesenchymal transition by YAP through feedback regulation of WT1 and Rho‐family GTPases. Nat Commun 10, 2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hong SK, Park JR, Kwon OS, Kim KT, Bae GY & Cha HJ (2016) Induction of integrin beta3 by sustained ERK activity promotes the invasiveness of TGFbeta‐induced mesenchymal tumor cells. Cancer Lett 376, 339–346. [DOI] [PubMed] [Google Scholar]
- 37. Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K & Derynck R (2019) Chronic TGF‐beta exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal 12, eaau8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dupont S (2019) Luciferase reporter assays to determine YAP/TAZ activity in mammalian cells In The Hippo Pathway: Methods and Protocols (Hergovich A, ed.), pp. 121–135. Springer, New York, NY. [DOI] [PubMed] [Google Scholar]
- 39. Foroutan M, Cursons J, Hediyeh‐Zadeh S, Thompson EW & Davis MJ (2017) A transcriptional program for detecting TGFbeta‐induced EMT in cancer. Mol Cancer Res 15, 619–631. [DOI] [PubMed] [Google Scholar]
- 40. Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM et al (2008) TEAD mediates YAP‐dependent gene induction and growth control. Genes Dev 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Si Y, Ji X, Cao X, Dai X, Xu L, Zhao H, Guo X, Yan H, Zhang H, Zhu C et al (2017) Src inhibits the Hippo tumor suppressor pathway through tyrosine phosphorylation of Lats1. Cancer Res 77, 4868–4880. [DOI] [PubMed] [Google Scholar]
- 43. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- 44. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A et al (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147, 759–772. [DOI] [PubMed] [Google Scholar]
- 45. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y & Guan KL (2008) TAZ promotes cell proliferation and epithelial‐mesenchymal transition and is inhibited by the Hippo pathway. Mol Cell Biol 28, 2426–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pickup M, Novitskiy S & Moses HL (2013) The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer 13, 788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Massague J (2008) TGFbeta in cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Varelas X, Sakuma R, Samavarchi‐Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW & Wrana JL (2008) TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem‐cell self‐renewal. Nat Cell Biol 10, 837–848. [DOI] [PubMed] [Google Scholar]
- 49. Luo K (2017) Signaling cross talk between TGF‐beta/Smad and other signaling pathways. Cold Spring Harb Perspect Biol 9, a022137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saito A & Nagase T (2015) Hippo and TGF‐beta interplay in the lung field. Am J Physiol Lung Cell Mol Physiol 309, 756–767. [DOI] [PubMed] [Google Scholar]
- 51. Nistico P, Bissell MJ & Radisky DC (2012) Epithelial‐mesenchymal transition: general principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol 4, a011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Qin Z, Xia W, Fisher GJ, Voorhees JJ & Quan T (2018) YAP/TAZ regulates TGF‐beta/Smad3 signaling by induction of Smad7 via AP‐1 in human skin dermal fibroblasts. Cell Commun Signal 16, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pefani DE, Pankova D, Abraham AG, Grawenda AM, Vlahov N, Scrace S & O’Neill E (2016) TGF‐beta targets the Hippo pathway scaffold RASSF1A to facilitate YAP/SMAD2 nuclear translocation. Mol Cell 63, 156–166. [DOI] [PubMed] [Google Scholar]
- 54. Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP & Brabletz T (2016) ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun 7, 10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hiemer SE, Szymaniak AD & Varelas X (2014) The transcriptional regulators TAZ and YAP direct transforming growth factor beta‐induced tumorigenic phenotypes in breast cancer cells. J Biol Chem 289, 13461–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kurowska‐Stolarska M, Alivernini S, Melchor EG, Elmesmari A, Tolusso B, Tange C, Petricca L, Gilchrist DS, Di Sante G, Keijzer C et al (2017) MicroRNA‐34a dependent regulation of AXL controls the activation of dendritic cells in inflammatory arthritis. Nat Commun 8, 15877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, Deng Y, Zhao J, Jiang S, Yuan Y et al (2012) TGF‐beta‐miR‐34a‐CCL22 signaling‐induced Treg cell recruitment promotes venous metastases of HBV‐positive hepatocellular carcinoma. Cancer Cell 22, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) CCLE lung cancer cell lines ranked by mesenchymal score. Distribution of mesenchymal score is shown in the right panel. Cell lines were divided into tertiles by mesenchymal score (mesenchymal, intermediate, epithelial groups). (B) Distribution of mesenchymal score of 78 lung cancer cell lines in GSE4824. Cell lines were divided into three groups according to mesenchymal score (mesenchymal, intermediate, epithelial groups). (C) ZEB2 mRNA expression level in A549, TD, H358 and H1299 cells. (D) Immunostaining of YAP in A549 and TD cells.
Fig. S2. (A) Association between cell‐line enrichment scores of oncogenic signatures and drug sensitivity (AUC) to doxorubicin, topotecan and gemcitabine in 181 lung cancer cell lines in the CTD2 database. (B) Lung cancer cell lines ranked by doxorubicin sensitivity. Distribution of area under the curve (AUC) is shown in the right panel. Cell lines were divided into tertiles of AUC (doxorubicin‐resistant, ‐intermediate and ‐sensitive groups). (C) Correlation between cell‐line sensitivity (AUC) to doxorubicin and enrichment score of YAP signature (left) or mesenchymal signature (right) in lung cancer cell lines. (D) SERPINE1 messenger (m)RNA expression upon indicative concentration of doxorubicin (Doxo) treatment. (E) AXL (left) and SERPINE1 (right) mRNA expression upon indicative concentration of etoposide (Eto) treatment.
Fig. S3. (A) Representative images of zymography assay of A549 and TD cells after AXL siRNA treatment. (B) AXL mRNA expression in A549 and TD cells. (C,D) Immunoblotting analysis for PARP‐1 in TD cells, introducing of control (siNC) or SERINE1 (siSERPINE1) after treatment of doxorubicin (C: Doxo, 2 μm) or, etoposide (D: Eto, 40 μm). (E) Representative images of two‐chamber invasion assay of A549 and TD cells with control (siNC) or SERINE1 (siSERPINE1) (left) and graphical presentation of invaded area (% of Area) (right).
Fig. S4. (A) Distribution of AXL expression levels of 181 lung cancer cell lines in CCLE data. Cell lines were divided into tertiles of AXL expression (high, intermediate and low AXL groups). (B) Realtime PCR analysis for SMAD4 mRNA expression in A549 and TD cells with control (siNC) or SMAD4 (siSMAD4) siRNA. (C) Real‐time PCR analysis for ZEB1 mRNA expression in A549 and TD cells after indicative time of TGFβ treatment (5 ng/mL). (D) Real‐time PCR analysis for SERPINE1 mRNA expression with control (siNC) or SMAD4 (siSMAD4) siRNA, NS, not significant.
Fig. S5. Graphical presentation of Reporter activity of GTIIC and SBE with wild type (YAP WT) or constitutively active mutant (YAP8SA) of YAP.
Fig. S6. (A) Correlation between AXL expression and TGFB1, ZEB2, CTGF and YAP1 expression levels in 515 lung adenocarcinoma patients in TCGA data. (B) The Kaplan–Meier curves showing recurrence‐free survival time of lung adenocarcinoma patients. Patients were divided into tertiles (high, intermediate, low) by the expression levels of VIM, ZEB1, SNAI2 and SERPINE1. High and low groups were used for comparison. (C) Distribution of AXL expression levels of lung adenocarcinoma patients in TCGA data. Patients were divided into high, intermediate, and low AXL groups based on the basal expression level of AXL.
Fig. S7. Uncut immunoblotting data used in these studies.
Table S1. Correlation coefficient between cell‐line mesenchymal score (Taube et al., Grojer et al. and Byers et al.) and cell‐line oncogenic score calculated by using ssGSEA with 189 oncogenic signature gene sets.
Table S2. Correlation coefficient between cell‐line response (AUC) to doxorubicin and cell‐line oncogenic score.
Data Availability Statement
The datasets used and/or analyzed herein can be obtained from the corresponding author upon request.