

Abstract
Gram-positive bacteria colonize mucosal tissues against large mechanical perturbations, such as coughing, which generate shear forces that exceed the ability of non-covalent bonds to remain attached. To overcome these challenges, the pathogen Streptococcus pyogenes utilizes the protein Cpa, a pilus tip-end adhesin equipped with a Cys-Gln thioester bond. The reactivity of this bond towards host surface ligands enables covalent anchoring; however, colonization also requires cell migration and spreading over surfaces. The molecular mechanisms underlying these seemingly incompatible requirements remain unknown. Here, we demonstrate a magnetic tweezers force spectroscopy assay that resolves the dynamics of Cpa thioester bond under force. While folded at forces < 6 pN, Cpa thioester bond reacts reversibly with amine ligands, that are a common occurrence in inflammation sites; however, mechanical unfolding and exposure to forces > 6 pN block thioester reformation. We hypothesize that this folding-coupled reactivity switch—“smart covalent bond”—could allow the adhesin to undergo binding and unbinding to surface ligands under low force and remain covalently attached under mechanical stress.
Main text
In the ancient arms race between host and pathogen, bacteria have evolved novel adhesion strategies such as biofilm formation1,2, non-covalent catch bond binding3,4, and direct covalent binding to host substrates5,6. In particular, Gram-positive bacteria express a class of protein adhesins that contain internal Cys-Gln thioester bonds5-7. The thioester bond functions as an electrophilic substrate to draw a nucleophilic ligand, creating a covalent crosslink between a ligand and the adhesin of the bacterium6. Thioester bonds have evolved to permit bacterial adherence under large mechanical stresses8; however, bacterial colonization also benefits from cell rolling and spreading over surfaces9,10, and the molecular mechanisms reconciling the interplay between mobility and covalent anchoring are not known.
We have recently demonstrated a novel assay to study the reactivity of the pilus-tip thioester adhesin Cpa from the Gram-positive pathogen Streptococcus pyogenes11 (Figure 1a), the causative agent of strep throat and necrotizing fasciitis12. Similar to our assays for disulfide bond mechano-chemistry13-15, our Atomic Force Microscopy (AFM) force spectroscopy assay directly measured the presence or absence of the thioester bond in unfolding Cpa adhesins. However, due to technical limitations in AFM force spectroscopy16, our assay could not probe how force regulated Cpa folding and its coupling with thioester mechano-chemistry.
Figure 1. Mechano-chemistry of S. pyogenes Cpa adhesin.
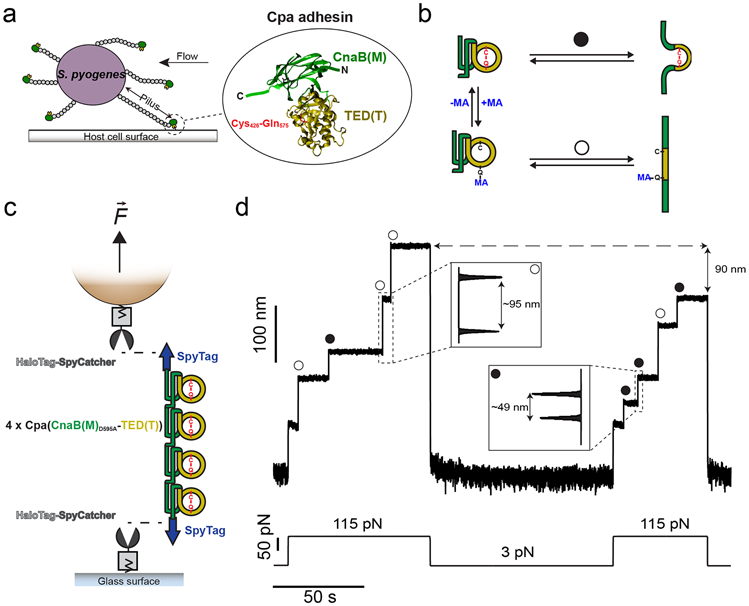
a) S. pyogenes attach to host cell surfaces through the Cpa protein, present in the tip-end of the pili. Cpa main core comprises the CnaB(M) domain (green), in whose fold the TED(T) domain is inserted (yellow). The TED(T) domain contains a thioester bond formed between the residues Cys426 and Gln575 (red), which mediates the attachment to cell-surface molecules. b) In the folded state, nucleophiles like methylamine (MA) can cleave the thioester bond and bind covalently to the Gln side chain (+MA); however, thioester bond reformation and ligand uncoupling (−MA) can occur. After mechanical extension, the presence (circle pathway) or absence (empty circle pathway) of the thioester bond can be assessed as a difference in the extension of the protein. c) Double-covalent magnetic tweezers experimental assay. Protein anchors SpyCatcher-HaloTag are covalently immobilized both to the surface of the glass and the paramagnetic bead. A chimeric polyprotein made of four copies of Cpa and flanked by SpyTag peptides is covalently linked to the glass and the bead through the reaction of the SpyCatcher/SpyTag split protein system. On the top of the scheme (not shown), the position of a pair of magnets is controlled for the application of calibrated forces to the tethered molecule. d) Magnetic tweezers recording of a Cpa polyprotein exposed to 100 mM methylamine, where the extension of the molecule is registered along time. A force pulse of 115 pN leads to the mechanical unfolding of the four Cpa domains, which is detected as stepwise increases in the extension. Here, three of the domains lack their internal thioester bond (empty circles) yielding an extension of ~95 nm, while one of the domains preserves its thioester bond (circle) and yields an unfolding extension of ~49 nm. Following a 100 s-long quench force pulse at 3 pN, which favors both folding and bond reformation, a second 115 pN pulse reveals that two Cpa domains reformed their thioester bonds (circles), decreasing the final extension of the polyprotein by 90 nm, as a consequence of the polypeptide sequence trapped by the newly formed bonds.
Here, we demonstrate a novel magnetic tweezers force spectroscopy approach to resolve in detail the force-dependency of Cpa thioester bond reactivity in the 3-115 pN force range. Unlike AFM, magnetic tweezers possesses an incomparable stability that grants access to days-long recordings on the same molecule, with millisecond and sub-pN resolution17,18. Cpa is a mechanically stable protein11, and, to apply high forces for long times, we design a novel double-covalent anchoring strategy based on HaloTag chemistry and SpyCatcher/SpyTag split protein technique19-21, which allows for the end-to-end covalent immobilization of single Cpa polyprotein molecules. This technical advance enables us to explore different conditions on the same molecule without probe detachment, a limiting factor in force spectroscopy experiments22-25. With these improvements, we now determine the force-dependency of Cpa folding and its relation to thioester bond cleavage by the nucleophile methylamine. We find that methylamine-induced cleavage is inhibited at forces >35 pN, while thioester reformation and ligand uncoupling occur at forces <6 pN. Our observations indicate that protein folding is a prerequisite for thioester reformation, which suggests an allosteric role of folding on the reactivity of this bond. We hypothesize that the force ranges over which thioester reformation and Cpa folding occur could indicate a novel mechanism to respond to varying levels of shear stress. Under high force conditions, the adhesin-ligand covalent interaction can withstand forces over 1000 pN. When the mechanical stress eases up, the folding of the Cpa parent protein, at 6 pN or less, reestablishes the thioester bond reactivity by enabling its cyclic reformation and ligand-induced cleavage by surface ligands. We dub such folding-controlled covalent reactivity: “smart covalent bond”. In the current context of antibiotic resistance26, targeting the bacterial adhesion molecules stands out as a promising strategy to battle infections27, especially considering the difficulties for treating those caused by Gram-positive pathogens28. In such an effort, we identify a mechanism for the abrogation of Cpa thioester bond reactivity towards surface ligands, based on the oxidation of the side chain thiol of the Cys residue involved in the thioester bond. A better understanding of the adhesive chemistries of Gram-positive pathogens will permit the rational development of novel classes of antibiotics and vaccines, of great significance to society.
Results
Double-covalent magnetic tweezers anchoring
To explore Cpa thioester bond mechano-chemistry, we use a polyprotein of the domains CnaBD595A(M)-TED(T) of this adhesin (Fig. 1a, b), as we previously described11. The Cys426-Gln575 thioester bond resides within the TED domain (thioester domain), whose fold is contained inside the fold of the CnaB domain (Fig. 1a). The D595A mutation prevents the formation of the native isopeptide bond present in the CnaB(M) domain. With its abrogation, the CnaB(M) domain can be mechanically unfolded when pulled from its N and C termini, which allows us to apply force to the TED(T) domain and evaluate the presence, absence, or real-time ligand-induced rupture of the thioester bond. Despite the absence of the native isopeptide bond, this protein still exhibits high mechanical stability and requires the application of high forces for unfolding. To solve this problem, we develop a strategy to covalently anchor Cpa polyproteins both to the glass surface and the magnetic probes of a magnetic tweezers setup. Both glass and probe surfaces are independently functionalized with the HaloTag ligand, which permits the covalent immobilization of HaloTag proteins17,19,20. First, we immobilize the chimeric protein SpyCatcher-HaloTag on the glass and on the bead surface. Then, we add the chimeric polyprotein SpyTag-(CnaBD595A-TED)4-SpyTag to the glass surface, allowing the reaction with the SpyCatcher-HaloTag present on the surface. The SpyCatcher/SpyTag split protein system reacts to form an intermolecular isopeptide bond between the SpyTag and the SpyCatcher counterpart21,29,30, covalently connecting both chimeric proteins. Finally, we close this assembly by adding functionalized paramagnetic beads, whose surface-bound SpyCatcher-HaloTag protein reacts with the SpyTag peptide present on the free end of the Cpa polyprotein (Fig. 1c). After capping, the Cpa polyprotein becomes covalently tethered both to the glass and bead surfaces.
The magnetic tweezers experiment starts when the protein-bound paramagnetic bead is exposed to a magnetic field17. The presence or absence of the thioester bonds in the Cpa polyprotein can be easily detected as a difference in the unfolding extensions (Fig. 1b). Fig. 1d shows a magnetic tweezers trajectory of a Cpa polyprotein which has been previously exposed to a solution containing 100 mM methylamine (Hepes 50 mM pH 8.5, NaCl 150 mM, ascorbic acid 10 mM, EDTA 1 mM). The application of a constant force of 115 pN leads to the sequential unfolding of the Cpa polyprotein, yielding stepwise increases in length of different sizes: one corresponding to thioester bond-intact proteins (~49 nm), and three corresponding to thioester bond-cleaved proteins (~95 nm). Subsequently, the force was reduced to 3 pN to allow the folding of Cpa and also the reformation of the thioester bonds. A second 115 pN pulse reveals that two more Cpa domains reformed their bonds (~49 nm steps), and the total extension of the polyprotein decreases by 90 nm, since the formation of these two thioester bonds prevents the full extension of the protein. The different extensions of Cpa, depending on the presence or absence of its internal thioester bond, serve to clearly identify the status of the bond.
Force-dependency of the thioester bond cleavage and reformation
The mechanical unfolding of the CnaBD595A-TED domains with the thioester bond intact remains limited to the polypeptide sequence not-trapped by the bond. This accounts for a total of 164 residues located before the Cys426 and after the Gln575, which corresponds to the ~49 nm steps observed on Fig. 1d. In a nucleophile-free solution, the polyprotein unfolding at 115 pN reveals stepwise increases in length of 48.8 ± 3.8 nm (mean±SD), as it can be seen in the trajectory of Fig. 2a. In these unfolding extensions, the entire CnaB fold and a small region of the TED domain—TED fold spans from residues Ala393 to Gly579, and the sequence sequestered by the thioester bond spans from Cys426 to Gln575—unfold as an unique step; however, pulling at lower forces allows to separate the unfolding of these two regions, revealing a short-lived intermediate state as we previously reported11 (Supplementary Fig. 1). Due to the exquisite force resolution of magnetic tweezers, we can explore not only the Cpa polyprotein unfolding at high forces, but also the reversible process of folding at low forces (Fig. 2b). As it can be seen on Fig. 2a, a quench for 100 s at 6 pN and a subsequent pulling pulse at 115 pN shows no evidence of protein folding (Pf=0.0). On the contrary, holding the protein at 4 pN for the same amount time is enough to completely fold the thioester-intact Cpa polyprotein (Pf=1.0), while at 5.5 pN only half of the domains could fold (Pf=0.5). In this manner, we determine the folding probability of the thioester-intact polyprotein, which shows a sharp transition from fully folded at 4.5 pN, to completely unfolded at 6.5 pN (Fig. 2c). Therefore, Cpa mechanical unfolding in the absence of nucleophiles yields a homogeneous population of steps of ~49 nm, which confirms that over the explored range of forces the thioester bond remains inert.
Figure 2. Dynamics of the thioester-intact Cpa polyprotein under force.
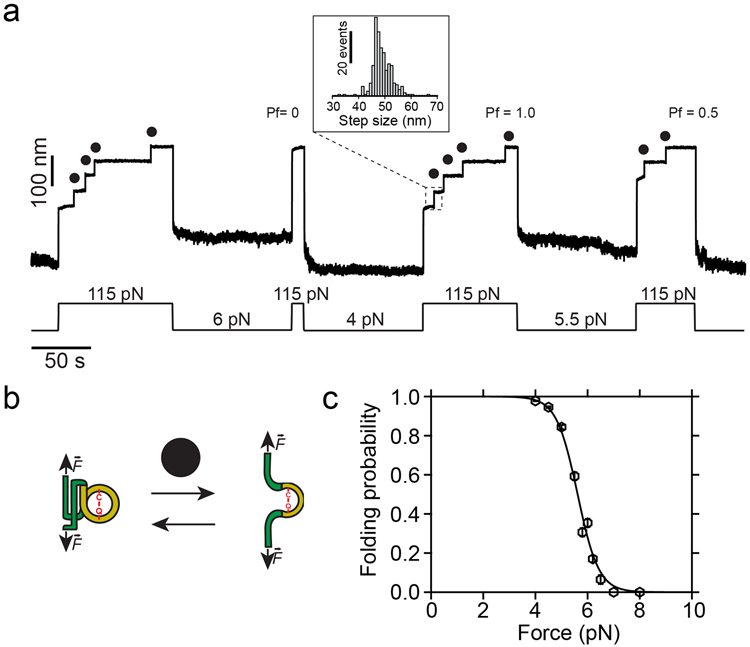
a) Magnetic tweezers trajectory of the Cpa polyprotein. High force pulses at 115 pN unfold the thioester-intact Cpa domains, which show 48.8 ± 3.8 nm (mean±SD, n=272) stepwise extensions (inset histogram). Low force pulses of 100 seconds long allow Cpa refolding, enabling us to determine the folding probability (Pf) at different forces. As an example, a quench at 6 pN does not allow folding of any of the domains, while the four-fold at 4 pN (Pf=1.0), and only two-fold at 5.5 pN (Pf=0.5). b) Cartoon representation of the folding-unfolding of the Cpa domain. The thioester bond between Cys426 and Gln575 clamps the TED domain (yellow), limiting its extensibility. c) Folding probability of thioester-intact Cpa. Data points are fitted to a sigmoidal function and they represent the probability at each of the forces tested for 100 s (n=54 at 4 pN; n=30 at 4.5 pN; n=18 at 5 pN; n=16 at 5.5 pN; n=16 at 5.8 pN; n=23 at 6 pN; n=14 at 6.2 pN; n=10 at 6.5 pN; n=9 at 7 pN; n=5 at 8 pN). Data points are the mean and the bars are the SD calculated using a jackknife analysis.
The stability of magnetic tweezers and the double-covalent anchoring of the protein allow us to exchange the solution in the experimental fluid chamber, enabling us to expose a single molecule to different conditions. Hence, to explore the thioester bond reactivity under force, we change to a solution containing 100 mM methylamine, which we add after the mechanical unfolding of the thioester-intact Cpa, as shown in Fig. 3a. At 115 pN, the addition of methylamine does not yield any additional extension increase, indicating the lack of reactivity of the thioester bond at high forces. Taking advantage of the magnetic tweezers force resolution, we apply a protocol with consecutive decreasing force pulses of 100 s to elucidate the force-range reactivity of this bond in real time. Initially, decreasing the force to 30 pN does not alter the thioester bond state, as it can be seen from the following 115 pN pulse where the same final extension of the molecule is reached. By contrast, applying a pulse of 28 pN, reveals one discrete step originating from the bond cleavage of one of the four Cpa proteins. When we stretch again at 115 pN, the final extension of the molecule increases by 45 nm, which confirms this observation. This additional length comes from the release of the polypeptide sequence sequestered by the Cys426-Gln575, which scales with the number of residues previously trapped by the bond and also with the applied force following the freely-jointed chain model for polymer elasticity31 (Supplementary Fig. 2). Finally, dropping the force to 20 pN leads to the rapid cleavage of the three remaining bonds in the polyprotein, yielding three steps of 38 ± 3.1 nm (mean±SD, inset histogram #2). Exploring the range from 10 to 35 pN, we determine that thioester bond cleavage does not occur over a 100 s time-window if Cpa is exposed to forces >35 pN. When held at lower forces, stepwise increases in length occur due to thioester bond cleavage, reaching completion in 100 s at forces <23 pN (Fig. 3b), which indicates a negative force-dependency in the ligand-induced cleavage. We sought to delve into the kinetics of thioester bond cleavage by measuring the rates of bond cleavage as a function of force (no time window limited) in the range spanning from 10 to 30 pN, as we show in Fig. 3c. We observe that the rate of cleavage experiences an optimum at ~20 pN, after which it decreases, as we expected from our observations in 100 s time windows (Fig. 3b). Interestingly, at lower forces this tendency is reversed. This behavior can be explained in the context of two sequential processes with opposite force-dependencies: the chemical cleavage of the bond and the mechanical unfolding of the protein. In these experiments, our observational event is the unfolding of the TED domain after the cleavage of its thioester bond by methylamine. In order to observe the extension of the TED domain, the chemical cleavage of the thioester bond has to occur before. Assuming the Bell model for bond lifetimes under force32, we elaborate a model (see Methods for a detailed description) which accounts for the rates of protein unfolding and the chemical cleavage of the bond. Between 10 and 20 pN the chemical cleavage is favored in comparison with higher forces; however, the mechanical unfolding of the TED domain becomes limiting and, hence, it increases with the mechanical load. On the contrary, at forces above 20 pN, the mechanical unfolding of the TED domain is increasingly favored but the chemical cleavage process is hindered and, therefore, slowed down. This model correctly describes the behavior observed, and predicts a positive distance to the transition state of ~0.9 nm for the unfolding of the TED domain, and a negative distance to the transition state for the chemical cleavage of the thioester bond of ~0.4 nm. The latter negative trend indicates that thioester bond lysis requires a structural shortening of the protein conformation, a transition which becomes less favorable as the mechanical load increases.
Figure 3. Cpa thioester bond cleavage is negatively force-dependent.
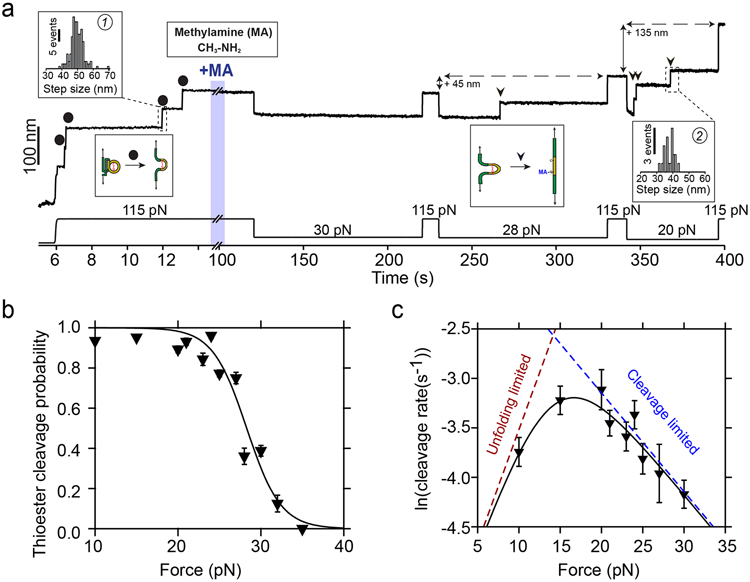
a) Magnetic tweezers trajectory of the Cpa polyprotein. After the unfolding of the thioester-intact Cpa domains at 115 pN (circles; histogram inset #1: 49.6±4.1 nm, mean±SD, n=164), the buffer is exchanged to a Hepes solution containing 100 mM methylamine (+MA). At high force, no additional steps are registered as it would be expected from a thioester bond cleavage event. Thereafter, we apply a protocol with subsequent pulses of decreasing mechanical load to investigate the force-dependency of the reaction. While at 30 pN no cleavage is observed, 100 s at 28 pN reveal one step that comes from the methylamine-induced cleavage of the thioester bond of one of the four Cpa domains (triangle). At 115 pN, the final extension of the molecule has increased by 45 nm, which originates from the polypeptide sequence released after thioester bond lysis. When held at 20 pN, the three remaining thioester bonds are cleaved (triangles; histogram inset #2: 38± 3.1 nm, mean±SD, n=21) and the final extension of the molecule increases for another 135 nm. b) Thioester bond cleavage probability as a function of force measured over a 100 s time-window. Data points are the mean and the error bars are the SD calculated using a jackknife analysis. The line represents a sigmoidal fit to the data (n=12 at 10 pN; n=20 at 15 pN; n=15 at 20 pN; n=9 at 21 pN; n=10 at 23 pN; n=9 at 24 pN; n=15 at 25 pN; n=15 at 27 pN; n=7 at 28 pN; n=15 at 30 pN; n=5 at 32 pN; n=6 at 35 pN). c) Rate of thioester bond cleavage as a function of force. Data points show the natural logarithm of the cleavage rate and the bars show the standard error of the mean. The curve represents a fit to the data described by a model that takes into account the effect of two sequential reactions: the rate of protein unfolding, which increases with force, and the rate of thioester bond cleavage, which decreases with the force. From this fit, we obtain a distance to the transition state for TED protein unfolding (x†U) of 0.9 nm, while the thioester bond cleavage exhibits a negative distance to the transition state (x†C = −0.4 nm), which suggests a requirement of a contraction of the Cpa polypeptide substrate to proceed with the cleavage of the bond, explaining its negative force-dependence. The dotted lines represent the individual unfolding and cleavage rates as obtained from the fit to the proposed model (Eq. S3, see Methods) (n=30 at 10 pN; n=38 at 15 pN; n=24 at 20 pN; n=23 at 21 pN; n=23 at 23 pN; n=24 at 24 pN; n=21 at 25 pN; n=37 at 27 pN; n=15 at 30 pN). Rate vs force dependency data was obtained in unrestricted time windows experiments.
Our results indicate that thioester bond cleavage is hindered when forces >35 pN are directly applied to the bond, and that the kinetics of this reaction are steeply affected by the mechanical load. Methylamine-induced cleavage leads to the covalent binding of this nucleophile to the Gln side chain, but the backwards reaction involving thioester reformation and ligand uncoupling can occur in the folded state of Cpa. To explore this opposite reaction, we design the force protocol shown on Fig. 4a. After mechanical unfolding of the Cpa polyprotein, and the cleavage of the thioester bonds with methylamine (see Fig. 4b), we wash the nucleophile out of the reaction buffer and reduce the force on the protein, to favor both the bond reformation and folding of the protein. These conditions allow us to observe a sharp increase in the reformation probability once the Cpa protein is exposed to forces <6 pN (Fig. 4c). The number of reformation events—detected as thioester-intact Cpa unfolding steps at 115 pN—scales with the number of cleavage events observed before the methylamine washout (Extended Fig. 1). Interestingly, the bond reformation force range closely tracks that of the folding of thioester-intact Cpa proteins. Given that the Cys and the Gln residues are moved away after cleavage, the force must be decreased to bring close the Cys thiol to attack the Gln carbonyl group and reform the thioester. The fact that Cpa folding occurs at higher forces entails that folding precedes the thioester bond reformation, as it has been also described for the formation of disulfide bonds33,34.
Figure 4. Protein folding drives thioester bond reformation.
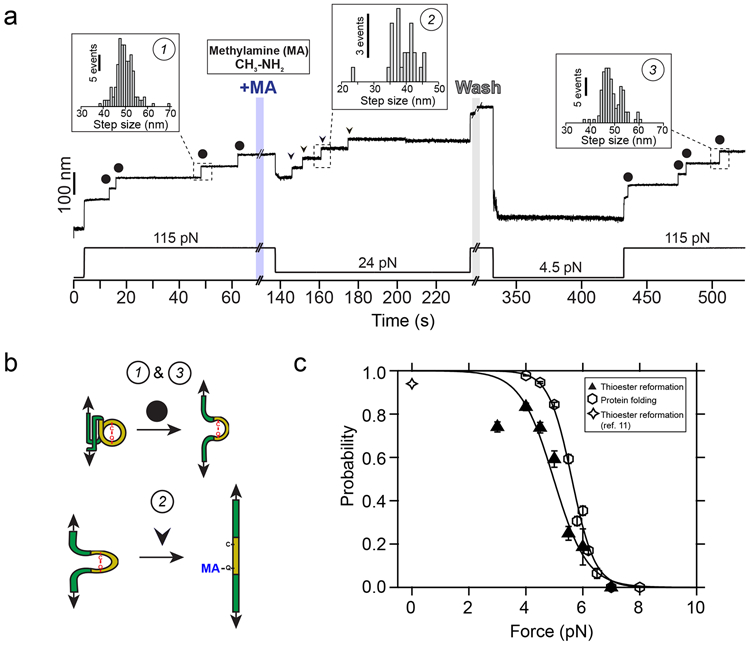
a) Magnetic tweezers trajectory of the Cpa polyprotein. After the unfolding of the thioester-intact Cpa domains at 115 pN (circles; inset histogram #1: 49.6 ± 4.1 nm, mean±SD, n=164), the buffer is exchanged and the polyprotein is exposed to a solution containing 100 mM methylamine (+MA). As expected, we do not observe cleavage at this high force, but a drop to 24 pN permits the full cleavage of the four candidate thioester bonds (arrows, inset histogram #2; 38.8 ± 4.4 nm for 24 pN, mean±SD, n=25). To study the reformation of the bond, we remove the nucleophile-containing buffer at high force, and quench the force to 4.5 pN for 100 s to favor bond reformation and protein folding. We stretch again the polyprotein at 115 pN and identify four thioester-intact Cpa domains, which indicates that the four cleaved candidates were able to fold and to reform their bonds (circles; inset histogram #3: 48.8 ± 4.1 nm, mean±SD, n=117). b) Cartoon representation of the extension events registered on the Cpa trajectory shown in a). Events #1 and #3 show the mechanical extension at 115 pN of thioester-intact Cpa, before cleavage and after reformation, respectively. Event #2 shows the extension after methylamine (MA) cleavage at 24 pN. c) Comparison between the thioester bond reformation (upwards triangles and sigmoidal fit) and the thioester-intact Cpa folding probability (hexagons and sigmoidal fit, from Figure 2c) as a function of the mechanical load. Star symbol indicates the reformation probability obtained at 0 pN from our previous work with AFM11. Data points for reformation are the mean and the error bars are the SD calculated using a jackknife analysis. Reformation registered as the amount of thioester-intact domains after methylamine washout and after a 100 s time-window at the folding/reformation force range (n=13 at 3 pN; n=16 at 4 pN; n=15 at 4.5 pN; n=12 at 5 pN; n=6 at 5.5 pN; n=7 at 6 pN; n=6 at 7 pN).
Blocking the thioester bond reformation
Our experiments with methylamine demonstrate the full reversibility of the cleavage reaction when Cpa is held at low forces and allowed to fold. These experimental conditions resemble the kind of interactions that Cpa adhesin could establish with the host ligands, binding and unbinding depending on the mechanical load experienced at the bond interface. From a therapeutic perspective, the irreversible thioester bond cleavage by a ligand analog would prevent bacterial adhesion, easing the bacterial removal from the tissues by the host’s clearance mechanisms—mucus flow, coughing, etc. Taking into account the Cys residue side chain, we explored the cleavage and reformation of Cpa thioester bond after the treatment with cystamine, another primary-amine nucleophile which contains a disulfide bond in its structure. Following the same protocol as with methylamine, we first unfold thioester-intact Cpa proteins (Fig. 5a, inset histogram #1) and then introduce a solution containing 100 mM cystamine. Upon force reduction to 25 and 20 pN for 100 s, the cleavage steps appear as it occurred with methylamine (inset histogram #2). After cystamine removal from the solution, the protein is allowed to refold and to reform the thioester bonds at 4 pN. If bond reformation occurs, at 115 pN we should detect the same ~49 nm steps registered before cystamine treatment. However, 97.1 ± 5.2 nm single steps (mean±SD, inset histogram #3) appear, which account for the full extension of thioester-cleaved Cpa proteins. Despite attempts to reform the bonds by reducing the force for several cycles (Extended Fig. 2), we can only detect full Cpa unfolding steps after cystamine. This nucleophile’s disulfide bond can be attacked by Cys426 free thiol to generate an intermolecular disulfide bond (diagram on Fig. 5b). Cys426 thiol oxidation would prevent thioester reformation, which could explain our observations where bond reformation is never observed after cystamine intervention. To further test this hypothesis, we add the reducing agent TCEP, to reduce disulfide bonds and liberate the Cys426 thiol. After solution exchange and force reduction, we observe again at high force the unfolding steps of thioester-intact Cpa proteins (Extended Fig. 3). Fig. 5c compares the cleavage and the reformation probability of the thioester bond after treatment with methylamine and cystamine. While both nucleophiles exhibit the same cleavage behavior at 20 and 25 pN, reformation at 4 pN is completely abolished after cystamine treatment. However, if cystamine-blocked proteins are treated with a solution containing 10 mM TCEP, the thioester bond recovery reaches the same values as with methylamine. Notably, the mechanical resistance of Cpa is significantly reduced when cystamine or methylamine are bound, suggesting a destabilizing role of these molecules on the protein. After the treatment with TCEP, the unfolding kinetics of Cpa are restituted, which indicates that disulfide bond reduction and thioester bond reformation occurred and the cystamine has been expelled from the catalytic pocket of the TED domain (Supplementary Fig. 3). These findings strongly support the idea that cystamine blocking activity relies on the formation of an intermolecular disulfide bond with Cpa Cys426 side chain, which prevents thioester bond reformation and which can only be rescued after the action of a reducing agent.
Figure 5. Cystamine-mediated abrogation of Cpa thioester bond reformation.
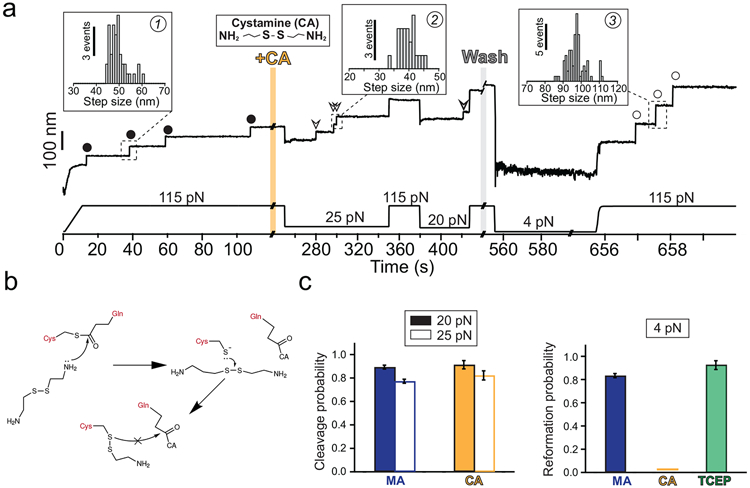
a) Magnetic tweezers trajectory of the Cpa polyprotein. After the unfolding of the thioester-intact Cpa domains at 115 pN (circles, inset histogram #1; 49.3 ± 3.8 nm, mean±SD, n=42), the buffer is exchanged and the polyprotein is exposed to a solution containing 100 mM cystamine (+CA). At 115 pN, no additional extensions are registered, but a drop in the force to 25 pN for 100 s leads to the appearance of three steps which account for the release of the polypeptide sequence trapped by the thioester bonds (empty arrows, inset histogram #2; 39.6 ± 2.7 nm for 25 pN, mean±SD, n=23). After the cleavage of all the bonds and after CA washout, force is quenched to 4 pN for 100 s to favor folding and reformation of the thioester. The final 115 pN pulse reveals three steps corresponding to thioester bond-cleaved Cpa domains (empty circles, inset histogram #3; 97.1 ± 5.2 nm, mean±SD, n= 78). b) Chemical scheme depicting the reformation blocking effect of CA. After the thioester bond nucleophilic cleavage by one of the CA primary amines, the free Cys thiol can attack CA disulfide bond (from the bound CA, or from another CA molecule). As a result, an intermolecular disulfide bond between Cpa Cys426 and CA is formed, preventing the thioester bond reformation. This disulfide reshuffling breaks the CA molecule and generates one free CA molecule (not shown in the scheme), and a Cys426-bound CA. c) Left panel compares the thioester bond cleavage probability by methylamine (MA) and CA at 20 and at 25 pN (MA, n=15 at 20 pN, n=15 at 25 pN; CA, n=8 at 20 pN, n=9 at 25 pN). Right panel compares the thioester bond reformation probability after 100 s at 4 pN after the treatment with MA, CA, and after the treatment with CA followed by TCEP (MA, n=16; CA, n=17; TCEP, n=6). Histogram bars are the mean and the error bars are the SD calculated using a jackknife analysis
Discussion
Bacterial pathogens possess molecular traits that enable host colonization under mechanical stress. Among these, isopeptide bonds stand out by conferring high mechanical and thermal stability to the adhesive proteins and pili of Gram-positive bacteria35-37. These bonds preserve the mechanical integrity of the bacterial anchors38-40, but ultimately the adhesion lifetime relies on the properties and the strength of the bacteria receptor-host ligand interaction. Gram-positive adhesin-ligand binding has evolved to withstand nanoNewton-scale mechanical loads, like Staphylococcus epidermidis SdrG adhesin41, but also to respond to force in a putative catch bond-like manner, such as Staphylococcus aureus ClfA and Clfb adhesins42,43, or Streptococcus pneumoniae pilin RrgB44. In the catch bond mechanism, force triggers conformational changes on the adhesin structure that increase the bond lifetime with the ligand, enabling the bacteria to respond to force thresholds45. Most of these adhesins interact with extracellular matrix proteins—such as fibrinogen and collagen46,47—and establish non-covalent bonds with their ligands. In addition to these, it was recently discovered the existence of thioester bond-adhesins in some Gram-positive organisms5-7. These adhesins can form a covalent bond with the substrate through the nucleophilic attack of its thioester bond by a primary-amine ligand, like the ε-amino group of a Lys residue5. Nevertheless, the establishment of an irreversible covalent anchoring would impose a sessile strategy on the cell, hindering its spreading and colonization48. Experiments with S. pyogenes Cpa adhesin revealed that in the absence of force the thioester bond cleavage by soluble nucleophiles and its reformation existed in equilibrium; however, the application of tensile stress to the thioester bond prevented both its cleavage and reformation, indicating that force modulates the reactivity of this bond11. Intramolecular thioester bonds are uncommon in the structure of proteins, having been only identified in the immune complement proteins, in α2-macroglobulin anti-protease49-51, and in Gram-positive adhesins6,7. In the case of non-activated complement proteins, nucleophilic cleavage and reformation can occur52, but the proteolytic activation of these proteins leads to a rapid and irreversible binding to its target substrates53, which contrasts with the reversible and force-modulated reactivity of S. pyogenes adhesin.
Here, using magnetic tweezers force spectroscopy and a new protocol for the covalent anchoring and assembly of polyproteins, we identify the force range for Cpa thioester bond reactivity. Our results indicate that ligand-induced cleavage occurs when the thioester bond is held at forces <35 pN. This impaired reactivity under force contrasts with the positive effect of force on the mechano-chemical cleavage of disulfide bonds by small reducing agents. These disulfide reductions proceed via an SN2 mechanism that experiences a ~0.3 - 0.4 Å elongation to the transition state13,14. On the contrary, enzymatically-catalyzed disulfide reductions by thioredoxin exhibit a negative force-dependency, where the substrate polypeptide must contract under force in order to align with key catalytic residues of the enzyme15,54,55. A similar mechanism of polypeptide contraction may underlie the observed force-dependency of the Cpa thioester bond, as it can be inferred from the negative distance to the transition state we have observed. At lower forces, where thioester bond cleavage is less hindered, the rate of TED unfolding increases exponentially with force; however, as the load increases, the rate of bond cleavage decreases and the TED unfolding rate is slowed down because of the detrimental effect of force on the cleavage reaction, leading to a negative force-dependency. We explain the negative force-dependency of thioester cleavage as an autocatalytic mechanism that facilitates the nucleophilic attack, as it has been reported in a close Cpa homolog in S. pyogenes8; the mechanical load would disrupt the thioester active site and inhibit bond cleavage, deforming the spatial arrangement of key catalytic residues placed in the vicinity of the Cys-Gln bond. Notably, we observe small stepwise fluctuations at forces below 35 pN, which precede thioester cleavage and disappear after the reaction occurs (Supplementary Fig. 4). However, no single discrete step size population is apparent and we cannot assign a specific structural transition to these fluctuations. Nevertheless, the close temporal relationship of these fluctuations to thioester cleavage events suggests a requirement of some structural contraction, and in turn a negative force-dependency to the reaction rate. In the backward reaction, the Cys426 and Gln575 residues must be in close proximity for thioester reformation, which is most probable at or close to the native folded state. Supporting this mechanism, we measured the folding force-dependency of thioester-intact Cpa (Fig. 2c), which closely tracks the profile of thioester reformation (Fig. 4c). This pathway of refolding followed by reformation can explain the sharp transition in the force-dependency from 4 pN to 6 pN, with reformation restricted to the force range of protein folding. This behavior shows an analogy with the process of enzymatic-assisted oxidative folding, where protein folding brings in close proximity the Cys residues involved before disulfide bond formation can proceed33,56.
Significantly, our experimental pulling axis from the N and C ends of Cpa is not the physiological one. In our system, the introduction of the D595A mutation abrogates the formation of the native isopeptide bond present in the CnaB domain of Cpa, which in the native protein shields the TED domain from experiencing force when pulled from the N and the C termini. This pulling configuration has enabled us to explore in real time the thioester bond dynamics in the presence of nucleophiles with unprecedented resolution. Although the thioester bond in the native folded TED domain is not expected to be under tensile stress, we can expect that its in vivo reactivity will be modulated by force. In our previous work, the simulation of the in vivo pulling axis between the C-terminus residue—connected to the pilus—and Gln575—linked to the surface ligand—revealed mechanical deformation of the thioester active site11, suggesting that the TED domain experiences force upon surface ligand-induced cleavage of the thioester bond and binding to the Gln575 side chain. This mechanical deformation would alter the position of key residues involved in the thioester bond reactivity, hindering the reformation process and extending the lifetime of the adhesin-ligand bond. This in vivo geometry indicates that thioester bond reformation is affected by force and, although we cannot access experimentally the exact force pathway, the force-dependency of reformation is not expected to be dependent on the pulling axis. The effect of different pulling geometries on the unfolding of proteins has been widely explored in the force spectroscopy field and, while the specific unfolding forces can change depending on how the force is applied, the force-dependency of this reaction remains unaltered57-60. In the case of the Cpa adhesin, although the native pulling configuration would probably affect quantitatively the force range at which reformation occurs, our approach clearly identifies and underpins the force-dependent modulation of this bond. Based on our observations, we hypothesize that the bond reactivity modulation by folding could have implications in the bacterium adhesion strategy, reconciling the mobility and the anchoring problem (Fig. 6). Non-covalent catch bonds show an increased lifetime at certain levels of mechanical load, but exceeding forces terminate the binding45. In the absence of force, thioester bonds would operate as catch bonds, where surface ligands cleave them but reformation can occur, as long as the protein remains in the unperturbed folded state. Under these conditions, soluble ligands like histamine—which is released at infection sites61—can also bind to Cpa and compete with the surface targets of the adhesin. However, the lack of tensile stress in the Gln575-histamine interface would allow the Cys-Gln thioester bond reformation, which would release the histamine and reset Cpa ready for another incoming ligand. By contrast, increasing mechanical loads on those bonds established with surface-bound ligands at low force would prevent reformation due to the mechanical deformation and partial unfolding of Cpa—mechanical allostery—, inducing a long-lived covalent bond able to survive nanoNewton-scale perturbations62,63. Only after the mechanical challenge is finished and the force is reduced, Cpa folding and bond reformation can occur to favor cell rolling again. Given the folding-modulated reactivity of this adhesin thioester bond, and its possible implications for Gram-positive adhesion, we dub these adhesin-ligand interactions as “smart covalent bonds”. This mechanism could provide the bacterium with a balanced strategy: to switch from a nomadic phase at low shear stress that optimizes cell spreading (cleavage-binding and reformation-unbinding states coexist), to a locked phase under harsh mechanical conditions that induce dislodgement (bound state). The demonstration of the existence of such a mechanism would add another adhesion strategy class to the repertoire observed in bacterial attachment.
Figure 6. Bacterium mobility strategy model based on the allosteric modulation of the Cpa thioester bond by protein folding.
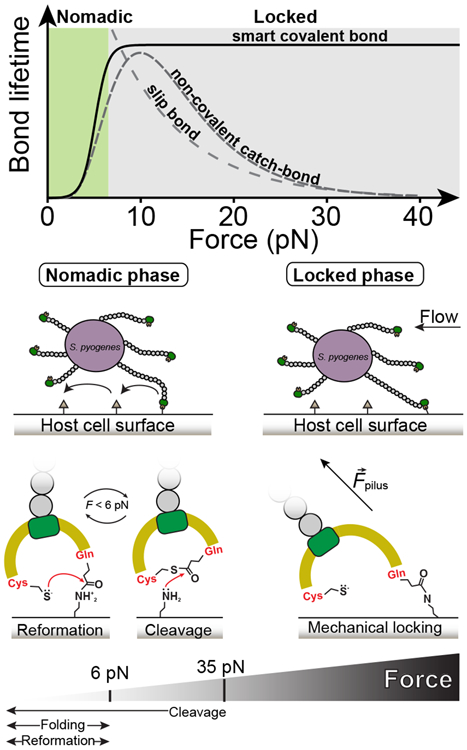
Top graph compares bond lifetimes as a function of the mechanical load for slip bonds, non-covalent catch bonds, and smart covalent bonds (slip bond and catch bond data adapted from64, plotted in arbitrary units). The smart covalent bond lifetime (plotted as the inverse of the thioester bond reformation probability from Figure 4c) is defined as the lifetime of the bond made between the surface ligand and the Gln575 side chain after the nucleophilic cleavage of the thioester bond. While higher loads decrease exponentially the lifetime of slip bonds, in non-covalent catch bonds it increases; however, loads above certain threshold decrease the lifetime. The adhesin-ligand smart covalent bond is allosterically modulated by force, establishing short-lived bonds with surface ligands at low mechanical stress—where thioester bond reformation and cleavage coexist—when the protein is folded, but turning into a long-lived bond that permits the bacterium to remain attached under large mechanical challenges, where thioester bond reformation is prevented. We hypothesize that these smart covalent bonds could allow bacteria to switch between a nomadic mobility phase at low force to a mechanically locked phase at larger loads.
Our results indicate that protein folding can modulate the binding activity of S. pyogenes Cpa, and they also indicate that chemically targeting the Cys-Gln thioester bond can be of potential interest for the development of antiadhesive drugs. The inhibitory effect observed after the treatment with cystamine indicates that, after nucleophilic cleavage, disulfide bond exchange occurs between Cys426 free thiol and cystamine disulfide bond, arresting the reformation. This conclusion is supported by the regenerative effect registered after TCEP treatment, which reduces the cystamine-Cpa intermolecular disulfide bond and frees the Cys426 thiol, enabling the reformation reaction. While methylamine and histamine transiently cleave the thioester bond, bifunctional soluble ligands with nucleophilic and thiol oxidation activities could permanently bind to Cpa to disable its adhesin function, establishing a new therapeutic path to tackle the antibiotic-resistance problem.
Methods
Protein engineering and expression
All the reagents employed in this research were from Sigma-Aldrich, unless otherwise specified. S. pyogenes Cpa gene was kindly provided by Mark Banfield (John Innes Centre, Norwich, UK). The gene was modified to include a 5’-end BamHI restriction site, a point mutation D595A to abolish CnaB(M) intramolecular isopeptide bond formation, and 3’-end BglII and KpnI restriction sites, as described previously11. A polyprotein containing four copies of Cpa—CnaB(M)-TED(T)—was assembled through successive cloning steps involving BamHI, BglII, and KpnI restriction sites, using pT7Blue (Novagen) as the cloning plasmid. The construct was then digested with BamHI/BglII and cloned into the expression plasmid pQE80L (Qiagen), which carries a N-terminal His tag. This plasmid was previously modified to contain two copies of the SpyTag sequence with a BamHI restriction site in between, which was digested to allow the insertion of the construct, generating the SpyTag-(Cpa)4-SpyTag construct (pQE80L-SpyTag-(CnaBD595A)4-SpyTag). All the cloning and amplification steps were done in XL10-Gold E. coli cells (Agilent Technologies). The probe and surface anchor protein, SpyCatcher-HaloTag, was cloned using this same protocol of digestion and ligation of restriction enzyme sites, and finally transferred to an empty pQE80L expression plasmid (pQE80L-SpyCatcher-HaloTag). The C-terminal HaloTag protein version was used for this construct20.
Protein expression and purification was done as described elsewhere36. In brief, E. coli ERL cells (kindly provided by R.T. Sauer from Massachusetts Institute of Technology) were transformed with the pQE80L-SpyTag-(Cpa)4-SpyTag plasmid or pQE80L-SpyCatcher-HaloTag, and protein expression was induced with 1 mM Isopropyl β-D-1-thiogalactopyranoside overnight at 25°C or 37°C, respectively. Cells were lysed in a French press (Sim-Aminco), and then the proteins were purified from the lysate with the His60 Ni Superflow Resin (Clontech). An additional purification step was done through size exclusion chromatography in a Superdex 200 FPLC column (GE Healthcare), eluting the proteins in 10 mM Hepes (pH 7.2), 150 mM NaCl, 1 mM EDTA (Hepes buffer). In the case of SpyCatcher-HaloTag protein, Hepes buffer additionally contained 10% v/v of glycerol. Purified proteins were aliquoted and frozen at −20°C until their use.
Cpa protein sequence
CpaD595A sequence. In green is highlighted the sequence of the CnaB domain. In yellow, the sequence of the TED domain, which spans from A393 to G579. In red are indicated the C426 and the Q575, the residues which form the thioester bond between their side chains. In bold is represented the residue A595, which is a mutation from the native D595 residue.
NQPQTTSVLIRKYAIGDYSKLLEGATLQLTGDNVNSFQARVFSSNDIGERIELSDGTYTLTELNSPAGYSIAEPITFKVEAGKVYTIIDGKQIENPNKEIVEPYSVEAYNDFEEFSVLTTQNYAKFYYAKNKNGSSQVVYCFNADLKSPPDSEDGGKTMTPDFTTGEVKYTHIAGRDLFKYTVKPRDTDPDTFLKHIKKVIEKGYREKGQAIEYSGLTETQLRAATQLAIYYFTDSAELDKDKLKDYHGFGDMNDSTLAVAKILVEYAQDSNPPQLTDLDFFIPNNNKYQSLIGTQWHPEDLVDIIRMEAKKEV
Bead surface functionalization
108 amine coated Dynabeads M270 (Thermo Fisher Scientific) were washed in PBS buffer, pH 7.4 and incubated in a PBS solution containing 1% v/v glutaraldehyde for 1 h in a rotator at 18 rpm (Labnet). After extensive washing, the beads were incubated in a PBS solution containing 25 μg/mL of the HaloTag ligand O4 (Promega) for at least 4 h at constant rotation. After washing, beads were treated with blocking buffer, which contains Tris-HCl pH 7.4, NaCl 150 mM, NaN3 0.001% w/v, and 1% w/v of sulfhydryl-blocked BSA (Lee Biosolutions), overnight at 4°C and at constant rotation. Optimal bead protein functionalization was achieved with a 15:5 μM ratio of HaloTag protein and SpyCatcher-HaloTag, respectively, for at least 12 hours at 4°C and at constant rotation. This 3:1 proportion results in an optimal bead surface coverage that prevents the formation of multiple tethers, since only the SpyCatcher-HaloTag molecules will serve as anchors for the glass surface-bound proteins. Beads were stored under this condition until use, moment in which they were extensively washed to remove unbound protein.
Fluid chamber functionalization
Magnetic tweezers experiments were conducted on fluid chambers made of two sandwiched glasses (Ted Pella) of 24x40 mm (bottom) and 22x22 (top) separated by a thin parafilm template cut with a laser cutting machine (Superland). The templates have a bow tie-like shape that allows the immobilization of the top glass over the bottom glass, and the formation of one well on each end of the bottom glass, which permits the exchange of buffer along the experiments. Prior to fluid chamber assembly, bottom glasses were washed and sonicated for 20 minutes in Hellmanex 1% (Helma), acetone, and ethanol. After the wash, the glasses were dried and exposed to air plasma for 15 minutes. Then, glasses were silanized for 20 minutes with an ethanol solution containing 0.1 % v/v of (3-aminopropyl)-trimethoxysilane, followed by several washes in ethanol. Finally, the glasses were dried with air, baked at 100°C for more than 20 minutes, and stored in a desiccator until further use. Top glasses were sonicated for 20 minutes in Hellmanex 1%, washed with ethanol, dried with air, and dried at 100°C for 10 minutes. Then, the top glasses were placed inside of a glass beaker and immersed in repel silane (Sigma) for 30 minutes at room temperature to make them hydrophobic. After, the glasses were dried with air, baked for 20 minutes at 100°C, and stored in a desiccator until use.
Fluid chambers assembly was done sandwiching the parafilm bow tie templates between the bottom and the top glasses, placed over a hot plate at 85°C, and with a flat 1 kg aluminum block pressing it. After 10 minutes, the fluid chambers were removed from the plate and a solution of PBS pH 7.4 with glutaraldehyde 1% v/v was flowed into the chambers and let to react for 1 h. After, a PBS solution containing 0.02 % w/v of 3.5-3.9 μm amine-coated polystyrene beads (Spherotech) was flowed and incubated for 20 minutes. After washing extensively, a PBS solution containing 25 μg/mL of the HaloTag ligand O4 was incubated overnight at room temperature. Finally, the fluid chambers were washed, blocked with blocking buffer overnight at room temperature, and stored at 4°C until further use.
Double covalent and molecular assembly
Fluid chambers were incubated with 5 μM SpyCatcher-HaloTag for 30 minutes. After an extensive rinse with Hepes buffer, the chambers were incubated with 5 μM SpyTag-(CnaB-TED)4-SpyTag for at least 1 h, and then extensively rinsed again. Once the fluid chamber was placed on the microscope, 20 μL of a 1:10 dilution of HaloTag:SpyCatcher-HaloTag functionalized beads were added to the fluid chamber and recirculated twice. Then, beads were allowed to react with the surface-bound molecules for 5 minutes before approaching the magnets and starting the experiment.
Magnetic tweezers force spectroscopy
Force spectroscopy experiments were conducted on a custom-built magnetic tweezers apparatus, as previously described17. The experimental fluid chambers are placed on the top of an inverted microscope (Olympus IX-71/Zeiss Axiovert S100) and illuminated with a collimated cold white LED (ThorLabs). The reference beads and the protein-bound paramagnetic beads are visualized employing a 100X oil-immersion objective (Zeiss/Olympus), which is mounted on a nanofocusing piezo actuator (P-725; Physik Instrumente). Image acquisition was done using a CMOS Ximea MQ013MG-ON camera, and image processing was done with custom-written C++/Qt software. Data acquisition and piezo position control were done using a multifunction DAQ card (NI USB-6289, National Instruments). Proteins were exposed to calibrated forces using a pair of magnets mounted on the top of a voice-coil (Equipment Solutions) placed above the experimental fluid chamber. Magnets position was maintained under electronic feedback with a PID controller.
Single molecule magnetic tweezers experiments on thioester bond cleavage and reformation
All the experiments were started applying a force of 4 pN, which lifts the protein-bound beads from the surface and prevents nonspecific interactions. The unfolding pulses were done at 115 pN, until the complete unfolding of the thioester-intact Cpa domains (~49 nm steps). Only molecules showing the initial unfolding of 3 or 4 domains were considered. Buffer exchange to add or remove nucleophile molecules was done at 115 pN. Upon nucleophile addition to the fluid chamber, thioester bond cleavage was monitored on 100 s time windows at forces ranging from 10 to 35 pN. Then, a 115 pN pulse was applied to monitor and compare the final extension of the molecule before and after the nucleophile treatment. At this high force, the nucleophile-containing buffer is washed out and then the force is quenched for 100 s at forces ranging from 3 to 7 pN, to favor refolding and thioester bond reformation. After, the folding and the thioester bond status of the domains are evaluated with a 115 pN pulse. In the case of folding and thioester bond reformation, thioester-intact Cpa domains are detected (~49 nm steps); in the case of having only folding, the full extension of the Cpa domain is observed (~95 nm steps). The buffer used along the experiments contained 50 mM Hepes pH 8.5, 150 mM NaCl, 1 mM EDTA, 10 mM L-ascorbic acid (to prevent oxidative damage65), and was supplemented with 100 mM of methylamine or cystamine for the thioester bond cleavage. To induce thioester bond reformation after cystamine treatment, the same buffer but supplemented with 10 mM of TCEP was added and the force quenched to 4 pN to favor folding and reformation. At least 3 different molecules were used for each data point collected.
Analysis
Analysis was done with Igor Pro 8.0 software (Wavemetrics). Recordings were smoothed using a 4th order Savitzky-Golay filter with a box size of 51 points. Step sizes were determined measuring the distance between the peaks of Gaussian fits done on the unfolding steps. Folding probability was calculated as the ratio between the number of unfolded domains and the number of domains able to fold after 100 s at each of the forces tested. Thioester bond cleavage probability on 100 s time windows was calculated as the ratio between detected cleavage steps at any of the forces tested, and the number of thioester-intact Cpa domains susceptible to be cleaved. Reformation probability was calculated as the ratio between thioester-intact Cpa domains detected and the number of cleaved domains registered before. For folding, cleavage and reformation probabilities, a jackknife estimator was used for the calculation of the average probability and the standard deviation.
Thioester bond cleavage kinetics
In Figure 3c we show the kinetics of thioester bond-cleavage/TED domain unfolding under force in the presence of methylamine. In these experiments, we detect the unfolding of the previously trapped sequence of the TED domain by the thioester bond along unrestricted time windows. Direct observation of thioester bond cleavage prior to unfolding is not possible under these experimental conditions, since the cleavage does not produce an extension signature detectable by our technique. Between 10 and 20 pN, force reduces the time to detect the mechanical extension of the TED domain. On the contrary, between 20 and 30 pN this process is slowed down and TED domain extension requires longer exposure times as the force is increased. We rationalize this change in the kinetics of this process under force as the result of two sequential processes with opposite force-dependencies. At forces <20 pN, the cleavage of the bond occurs, but the mechanical unfolding of the TED domain becomes limiting, increasing the waiting time for the detection of its unfolding step. At forces >20 pN, the mechanical unfolding of the TED domain is increasingly favored, but the negative effect of the force on the thioester bond geometry and the neighboring residues of the catalytic pocket of the protein impairs the nucleophilic attack by methylamine, resulting in slower kinetics of TED domain extension.
This picture can be formalized as a two-step kinetic process. Schematically, it can be represented as:
where TED is the folded and bonded TED state, TED* the folded and cleaved TED state, and TED*U the cleaved and unfolded TED state. The thioester bond cleavage process (kC) must occur before TED domain unfolding (kU) can be detected. Here, we assume that the rates of folding (kF) and bond reformation (kR) are negligible in the force range tested, as Figure 4c suggests. Since we cannot observe the transition from TED to TED* (kC), what we observe is the transition (kobs) from the initial to the final state (TED to TEDU*):
These assumptions lead to the next set of linear differential equations:
| (1) |
Which have as boundary condition:
And these initial conditions:
where, PTED, PTED*, and PTEDu* are the occupation probabilities of the folded and bonded TED state, the folded and cleaved TED state, and the cleaved and unfolded TED state, respectively. In our experiments, we measure the time (tobs) required to reach the cleaved and unfolded state (TEDU*), and we calculate the observed rate as:
Where <tobs> is the average time. Hence, from this expression we can calculate the observational rate (kobs) which contains the rates of cleavage (kC) and unfolding (kU).
| (2) |
Assuming the Bell model for bond lifetimes32, we use an expression that accounts for both opposing processes and explains the tendency we report in this study:
Where kC is the rate of thioester bond cleavage as a function of force, kC0 is the rate of thioester bond cleavage in the absence of force, -xC† is the negative distance to the transition state, F is the force, kB is the Boltzmann constant and T is the temperature. In the case of unfolding, kU is the rate of TED unfolding as a function of force, kU0 is the rate of protein unfolding in the absence of force, and xU† is the distance to the transition state. Finally, the expression used to fit the data in Figure 3c is:
| (3) |
From this fit, we obtain for the cleavage reaction kC0 = 0.32 ± 0.26 s−1 and xC† = −0.41 ± 0.26 nm, and for the unfolding reaction kU0 = (2.94 ± 3.58)x10−3 s−1 and xU† = 0.94 ± 0.51 nm. The dotted lines showed in Figure 3c correspond to the individual rates kC(F) and kU(F) as obtained from the fit to Eq. 3.
Extended Data
Extended Data Fig. 1. Cleavage-reformation-cleavage sequence.
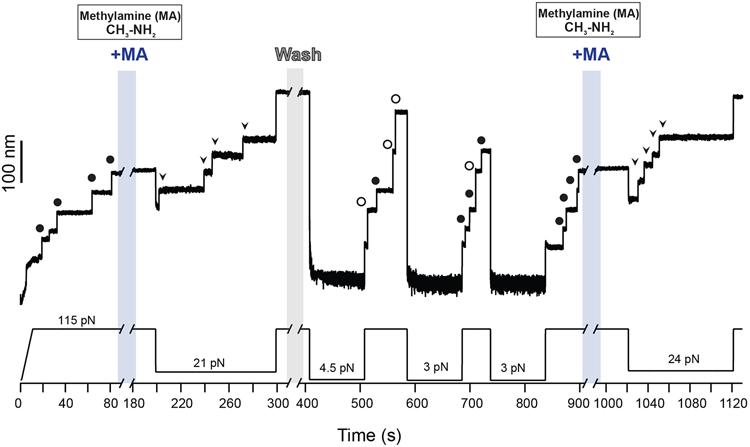
Magnetic tweezers force-clamp trajectory of the Cpa polyprotein. After the unfolding of four thioester-intact Cpa domains at 115 pN (circles, ~49 nm), the buffer is exchanged and the polyprotein is exposed to a solution containing 100 mM methylamine (+MA). At 21 pN, four steps appear which account for the release of the polypeptide sequence trapped by the thioester bonds (arrows). Then, the force is increased again to 115 pN, revealing the complete extension of the molecule. Immediately after, MA is washed out from the fluid chamber and the polyprotein is allowed to fold and reform the thioester bonds for 100 s at 4.5 pN. A 115 pN pulse reveals three ~95 nm steps (empty circles) which correspond with the full extension of Cpa, and one Cpa domain with its thioester bond reformed (circles, ~49 nm). Two more quenches at 3 pN are applied to completely recover the thioester-reformed state in all the four domains, as it can be seen in the 115 pN pulse applied approximately after 800 s of experiment (circles). Then, MA is added again and the force quenched to 24 pN to trigger again the cleavage of the thioester bonds of the polyprotein (arrows).
Extended Data Fig. 2. Cystamine permanent blocking of Cpa thioester bond reformation.
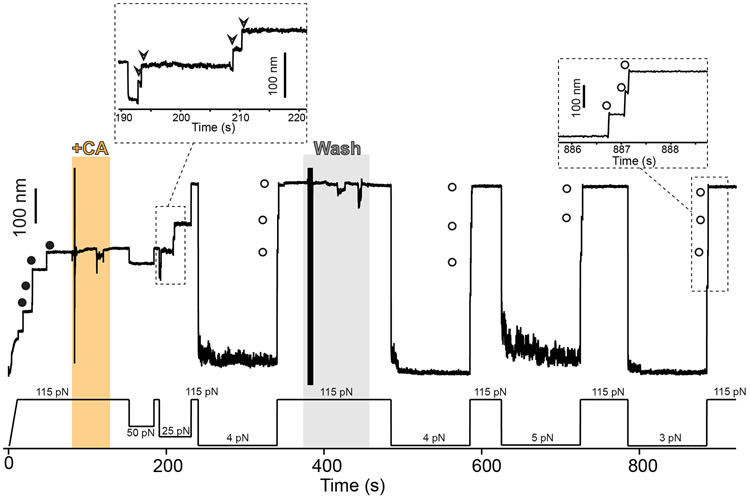
Magnetic tweezers force-clamp trajectory of the Cpa polyprotein. After the unfolding of the thioester-intact Cpa domains at 115 pN (circles), the buffer is exchanged and the polyprotein is exposed to a solution containing 100 mM cystamine (+CA). At 115 pN and at 50 pN, no additional extensions are registered as a consequence of thioester bond cleavage, but a drop in force to 25 pN leads to the appearance of four steps which account for the release of the polypeptide sequence trapped by the thioester bonds (empty arrows in the inset). Then, the force is increased again to 115 pN, revealing the complete extension of the molecule. After 100 s at 4 pN and in the presence of CA, a 115 pN pulse reveals three ~95 nm steps (empty circles) which correspond with the full extension of Cpa. CA is then removed from the solution, and several consecutive 100 s force quenches (at 4, 5, and 3 pN) followed by 115 pN pulses are applied. These cycles reveal that, after CA treatment, Cpa is able to fold but not to reform its thioester bond, as it can be observed from the ~95 nm steps observed (empty circles). After the first 300 s of the experiment, one of the Cpa domains stops folding back as a consequence of oxidative damage65. The disturbances observed in the extension during +CA addition (orange block) and washing (gray block) are originated from the movement of buffer volumes in the liquid cell used in the experiments, which transiently alter the measurement
Extended Data Fig. 3. TCEP rescues Cpa thioester bond reformation.
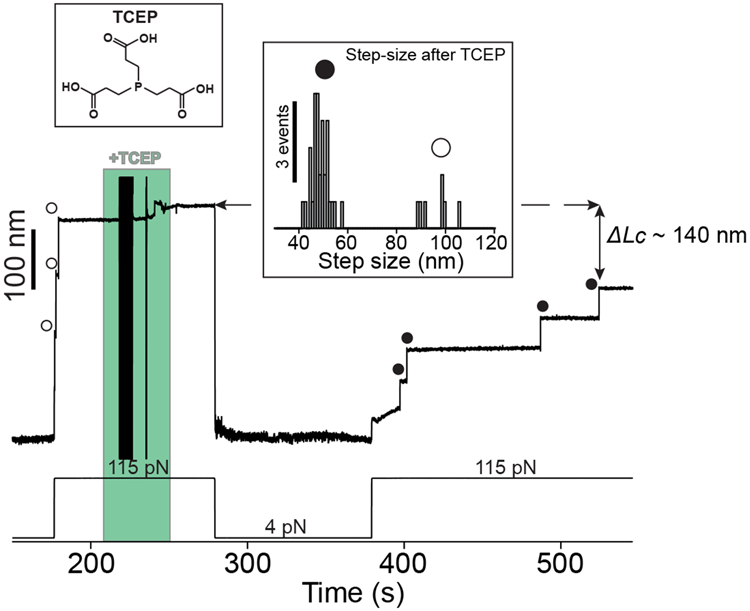
A Cpa polyprotein previously treated with cystamine shows three ~95 nm steps at 115 pN corresponding with the full extension of each of the domains (empty circles). The addition of 10 mM TCEP and 100 s at 4 pN is enough to trigger thioester bond reformation, as it can be observed in the ~49 nm thioester-intact Cpa steps (circles) registered at 115 pN. The fourth domain not observed at the beginning was probably unfolded and its thioester bond intact, since the difference in the final extension between the first 115 pN pulse and the last is ~140 nm, which matches with the expected final extension decrease from three reformation events. Inset histogram shows the two populations of steps observed after TCEP treatment, thioester-intact Cpa (circles, 48.3 ± 3.5 nm, mean±SD, n=32) and thioester-cleaved Cpa (empty circles, 95.7 ± 6.4 nm mean±SD, n=7). The latter full length steps of Cpa after TCEP treatment could be due to cleavage events induced by remaining cystamine which was not completely washed from the experimental liquid cell. The disturbances observed in the extension during +TCEP addition (green block) are originated from the movement of buffer volumes in the liquid cell used in the experiments, which transiently alter the measurement.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health grant R35129962 (J.M.F). A.A-C. and R.T-R. express their gratitude to Fundación Ramón Areces (Madrid, Spain) for financial support. We thank Carmen L. Badilla for assistance in molecular biology procedures, and for reading and reviewing the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability statement
All data supporting the results and conclusions are available within this paper and the Supplementary Information.
References
- 1.Hall-Stoodley L, Costerton JW & Stoodley P Bacterial biofilms: from the Natural environment to infectious diseases. Nat. Rev. Microbiol 2, 95–108 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Yan J & Bassler BL Surviving as a Community: Antibiotic Tolerance and Persistence in Bacterial Biofilms. Cell Host Microbe 26, 15–21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forero M, Yakovenko O, Sokurenko EV, Thomas WE & Vogel V Uncoiling mechanics of Escherichia coli type I fimbriae are optimized for catch bonds. PLoS Biol. 4, 1509–1516 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauer MM et al. Catch-bond mechanism of the bacterial adhesin FimH. Nat. Commun 7, 10738 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pointon JA et al. A Highly Unusual Thioester Bond in a Pilus Adhesin Is Required for Efficient Host Cell Interaction. J. Biol. Chem 285, 33858–33866 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walden M et al. An internal thioester in a pathogen surface protein mediates covalent host binding. Elife 4, 1–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller OK, Banfield MJ & Schwarz-Linek U A new structural class of bacterial thioester domains reveals a slipknot topology. Protein Sci. 22, 1–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linke-Winnebeck C et al. Structural model for covalent adhesion of the Streptococcus pyogenes pilus through a thioester bond. J. Biol. Chem 289, 177–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson BN et al. Weak Rolling Adhesion Enhances Bacterial Surface Colonization. J. Bacteriol 189, 1794–1802 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall KC Planktonic Versus Sessile Life of Prokaryotes in The Prokaryotes 191–201 (Springer Berlin; Heidelberg, 2013). doi: 10.1007/978-3-642-30123-0_49 [DOI] [Google Scholar]
- 11.Echelman DJ, Lee AQ & Fernández JM Mechanical forces regulate the reactivity of a thioester bond in a bacterial adhesin. J. Biol. Chem 292, 8988–8997 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brouwer S, Barnett TC, Rivera-Hernandez T, Rohde M & Walker MJ Streptococcus pyogenes adhesion and colonization. FEBS Lett. 590, 3739–3757 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Koti Ainavarapu SR, Wiita AP, Dougan L, Uggerud E & Fernandez JM Single-Molecule Force Spectroscopy Measurements of Bond Elongation during a Bimolecular Reaction. J. Am. Chem. Soc 130, 6479–6487 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Wiita AP, Ainavarapu SRK, Huang HH & Fernandez JM Force-dependent chemical kinetics of disulfide bond reduction observed with single-molecule techniques. Proc. Natl. Acad. Sci 103, 7222–7227 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiita AP et al. Probing the chemistry of thioredoxin catalysis with force. Nature 450, 124–127 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schönfelder J, De Sancho D & Perez-Jimenez R The Power of Force: Insights into the Protein Folding Process Using Single-Molecule Force Spectroscopy. J. Mol. Biol 428, 4245–4257 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Popa I et al. A HaloTag Anchored Ruler for Week-Long Studies of Protein Dynamics. J. Am. Chem. Soc 138, 10546–10553 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tapia-Rojo R, Eckels EC & Fernández JM Ephemeral states in protein folding under force captured with a magnetic tweezers design. Proc. Natl. Acad. Sci 116, 7873–7878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi Y & Kawakami M Application of Halotag protein to covalent immobilization of recombinant proteins for single molecule force spectroscopy. Langmuir 26, 10433–10436 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Popa I et al. Nanomechanics of HaloTag Tethers. J. Am. Chem. Soc 135, 12762–12771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zakeri B et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci 109, E690–E697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janissen R et al. Invincible DNA tethers: Covalent DNA anchoring for enhanced temporal and force stability in magnetic tweezers experiments. Nucleic Acids Res. 42, e137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brujić J, Hermans RIZ, Garcia-Manyes S, Walther KA & Fernandez JM Dwell-Time Distribution Analysis of Polyprotein Unfolding Using Force-Clamp Spectroscopy. Biophys. J 92, 2896–2903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Durner E, Ott W, Nash MA & Gaub HE Post-Translational Sortase-Mediated Attachment of High-Strength Force Spectroscopy Handles. ACS Omega 2, 3064–3069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng Y et al. Enzymatic biosynthesis and immobilization of polyprotein verified at the single-molecule level. Nat. Commun 10, 2775 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.CDC. Antibiotic resistance threats in the United States, 2013. Current 114 (2013). doi:CS239559-B [Google Scholar]
- 27.Cascioferro S, Cusimano MG & Schillaci D Antiadhesion agents against Gram-positive pathogens. Future Microbiol. 9, 1209–1220 (2014). [DOI] [PubMed] [Google Scholar]
- 28.ECDC. The bacterial challenge : time to react. The bacterial challenge: time to react 6 July 201, (2009). [Google Scholar]
- 29.Veggiani G et al. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci 113, 1202–1207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min D, Arbing MA, Jefferson RE & Bowie JU A simple DNA handle attachment method for single molecule mechanical manipulation experiments. Protein Sci. 25, 1535–1544 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flory PJ Theory of elastic mechanisms in fibrous proteins. J. Am. Chem. Soc 78, 5222–5235 (1956). [Google Scholar]
- 32.Bell G Models for the specific adhesion of cells to cells. Science (80-. ). 200, 618–627 (1978). [DOI] [PubMed] [Google Scholar]
- 33.Kosuri P et al. Protein folding drives disulfide formation. Cell 151, 794–806 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eckels EC, Haldar S, Tapia-Rojo R, Rivas-Pardo JA & Fernández JM The Mechanical Power of Titin Folding. Cell Rep. 27, 1836–1847.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang HJ & Baker EN Intramolecular Isopeptide Bonds Give Thermodynamic and Proteolytic Stability to the Major Pilin Protein of Streptococcus pyogenes. J. Biol. Chem 284, 20729–20737 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alegre-Cebollada J, Badilla CL & Fernández JM Isopeptide bonds block the mechanical extension of pili in pathogenic Streptococcus pyogenes. J. Biol. Chem 285, 11235–11242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Echelman DJ et al. CnaA domains in bacterial pili are efficient dissipaters of large mechanical shocks. Proc. Natl. Acad. Sci. U. S. A 113, 2490–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivas-Pardo JA, Badilla CL, Tapia-Rojo R, Alonso-Caballero Á & Fernández JM Molecular strategy for blocking isopeptide bond formation in nascent pilin proteins. Proc. Natl. Acad. Sci 115, 9222–9227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang B, Xiao S, Edwards SA & Gräter F Isopeptide bonds mechanically stabilize Spy0128 in bacterial pili. Biophys. J 104, 2051–2057 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang HJ & Baker EN Intramolecular isopeptide bonds: Protein crosslinks built for stress? Trends Biochem. Sci 36, 229–237 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Milles LF, Schulten K, Gaub HE & Bernardi RC Molecular mechanism of extreme mechanostability in a pathogen adhesin. Science (80-. ). 359, 1527–1533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herman-Bausier P et al. Staphylococcus aureus clumping factor A is a force-sensitive molecular switch that activates bacterial adhesion. Proc. Natl. Acad. Sci 115, 5564–5569 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vitry P et al. Force-Induced Strengthening of the Interaction between Staphylococcus aureus Clumping Factor B and Loricrin. MBio 8, 1–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becke TD et al. Pilus-1 Backbone Protein RrgB of Streptococcus pneumoniae Binds Collagen I in a Force-Dependent Way. ACS Nano 13, 7155–7165 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Thomas WE, Vogel V & Sokurenko E Biophysics of Catch Bonds. Annu. Rev. Biophys 37, 399–416 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Sridharan U & Ponnuraj K Isopeptide bond in collagen- and fibrinogen-binding MSCRAMMs. Biophys. Rev 8, 75–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreikemeyer B et al. Streptococcus pyogenes collagen type I-binding Cpa surface protein: Expression profile, binding characteristics, biological functions, and potential clinical impact. J. Biol. Chem 280, 33228–33239 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Stewart PS Biophysics of biofilm infection. Pathog. Dis 70, 212–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Law SK & Dodds AW The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 6, 263–74 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia-Ferrer I et al. Structural and functional insights into Escherichia coli α 2 -macroglobulin endopeptidase snap-trap inhibition. Proc. Natl. Acad. Sci 112, 8290–8295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wong SG & Dessen A Structure of a bacterial α2-macroglobulin reveals mimicry of eukaryotic innate immunity. Nat. Commun 5, 4917 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Dodds AW, Ren X-D, Willis AC & Law SKA The reaction mechanism of the internal thioester in the human complement component C4. Nature 379, 177–179 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Nilsson B & Nilsson Ekdahl K The tick-over theory revisited: Is C3 a contact-activated protein? Immunobiology 217, 1106–1110 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Alegre-Cebollada J, Perez-Jimenez R, Kosuri P & Fernandez JM Single-molecule force spectroscopy approach to enzyme catalysis. J. Biol. Chem 285, 18961–18966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang J & Fernández JM Mechanochemistry: One bond at a time. ACS Nano 3, 1628–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kahn TB, Fernández JM & Perez-Jimenez R Monitoring oxidative folding of a single protein catalyzed by the disulfide oxidoreductase DsbA. J. Biol. Chem 290, 14518–14527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carrion-Vazquez M et al. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol 10, 738–743 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Jagannathan B, Elms PJ, Bustamante C & Marqusee S Direct observation of a force-induced switch in the anisotropic mechanical unfolding pathway of a protein. Proc. Natl. Acad. Sci. U. S. A 109, 17820–17825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dietz H, Berkemeier F, Bertz M & Rief M Anisotropic deformation response of single protein molecules. Proc. Natl. Acad. Sci. U. S. A 103, 12724–12728 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brockwell DJ et al. Pulling geometry defines the mechanical resistance of a β-sheet protein. Nat. Struct. Biol 10, 731–737 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Stone KD, Prussin C & Metcalfe DD IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol 125, S73–S80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grandbois M, Beyer M, Rief M, Clausen-Schaumann H & Gaub HE How strong is a covalent bond. Science. 283, 1727–1730 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Pill MF, East ALL, Marx D, Beyer MK & Clausen-Schaumann H Mechanical Activation Drastically Accelerates Amide Bond Hydrolysis, Matching Enzyme Activity. Angew. Chemie - Int. Ed 58, 9787–9790 (2019). [DOI] [PubMed] [Google Scholar]
- 64.B.T. M et al. Direct observation of catch bonds involving cell-adhesion molecules. Nature 423, 190–193 (2003). [DOI] [PubMed] [Google Scholar]
References (Methods)
- 65.Valle-Orero J et al. Mechanical Deformation Accelerates Protein Ageing. Angew. Chemie Int. Ed 56, 9741–9746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data supporting the results and conclusions are available within this paper and the Supplementary Information.