

Abstract
Unleashing the immune system with immune checkpoint inhibitors (ICi) has significantly improved overall survival for subsets of Stage III/IV cancer patients. However, many tumors are non-responsive to ICi, in part due to a lack of tumor-infiltrating lymphocytes (TIL). Converting these “immune cold” tumors to “hot” tumors and, thus, more likely to respond to ICi is a major obstacle for cancer treatment. Triggering inflammatory forms of cell death, necroptosis and pyroptosis, may alter the tumor immune microenvironment and the influx of TIL. We present an emerging view that promoting tumor-localized necroptosis and pyroptosis may ultimately enhance responses to ICi.
Keywords: apoptosis, necroptosis, pyroptosis, immune checkpoint therapy
1. Introduction
A hallmark feature of cancer cells is their resistance to cell death. Delineating the molecular mechanisms that mediate resistance to cell death has led to the development and FDA-approval of the Bcl-2 family protein inhibitor, venetoclax. Bcl-2 proteins serve pro-survival functions in apoptosis, a form of programmed cell death that is typically considered to be immunologically silent. Mechanisms of apoptosis and their translation have been reviewed previously (1,2) and will not be discussed at length here. The non-inflammatory feature of apoptosis is thought to be due to the containment of cell destruction within an intact cell membrane, as well as the engulfment and the rapid clearance of dying cells by phagocytic innate immune cells in a process termed efferocytosis (3). The mechanisms underlying apoptosis are well understood and overall the evidence suggests the rapid clearance of apoptotic cancer cells promotes immune evasion and the secretion of anti-inflammatory mediators by phagocytes (4,5). Indeed, blocking efferocytosis by inhibition of the phagocytic receptor, MerTK, can enhance anti-tumor immune responses and improve the efficacy of immune checkpoint inhibitors (ICi) (6). However, it is important to mention that there is emerging evidence that apoptosis is not always immunologically silent. There is a great deal of cross-talk between apoptosis, necroptosis, and pyroptosis, with key molecular switches. Notably, apoptosis can influence inflammatory cell death through the inhibition of apoptotic caspases 3, 7 and 9 (7–10).
Unlike apoptosis, other forms of programmed cell death such as necroptosis and pyroptosis, are defined by the release of inflammatory mediators. Altering the inflammatory state of the tumor microenvironment has implications for the response to ICi therapy. Agents including anti-PD1 (nivolumab and pembrolizumab), anti-PD-L1 (atezolizumab) and anti-CTLA4 (ipilimumab) release T cell killing activity (11). These inhibitors alone and in combination have elicited remarkable effects in many cancers including skin melanoma, lung cancer, head and neck squamous cell carcinoma (HNSCC) and renal cell cancer (11). Many tumors, especially ones with low levels of tumor-infiltrating lymphocytes (TIL) termed immunologically “cold”, remain non-responsive to ICi and other tumors acquire resistance to ICi; therefore, the development of new approaches to boost anti-tumor immunity is critical. Understanding the mechanisms of inflammatory cell death and how they alter the tumor immune microenvironment may have profound effects on future therapeutic strategies in both the up-front and salvage treatment settings.
The role of inflammation in cancer is complex and may either promote or inhibit tumorigenesis and metastasis. It follows that forms of cell death that promote inflammation may have contrasting effects on tumor growth and metastasis depending on the context. A process termed ‘immunogenic cell death’ was defined by the Nomenclature Committee on Cell Death as a form of regulated cell death that is capable of activating an adaptive immune response (12). Given that the adaptive immune system is necessary for effective anti-tumor immunity, the study of immunogenic cell death has important implications in cancer. However, the release of inflammatory factors from innate immune cells can also contribute to anti-tumor immunity. In this review, we will broadly discuss forms of cell death capable of releasing inflammatory mediators and their impact on cancer and therapeutic strategies.
2. Inflammatory Cell Death
Over the last few years, there has been significant insight into the mechanisms of necroptosis and pyroptosis. These two inflammatory forms of cell death lead to the production and secretion of damage-associated molecular patterns (DAMPs) and cytokines into the tumor milieu, which in turn have the ability to dramatically alter the tumor immune microenvironment and the influx of TIL. In this section, we discuss mechanisms of necroptosis (Figure 1a) and pyroptosis (Figure 1b) and how they may be altered in cancer cells.
Figure 1: Major forms of inflammatory cell death.
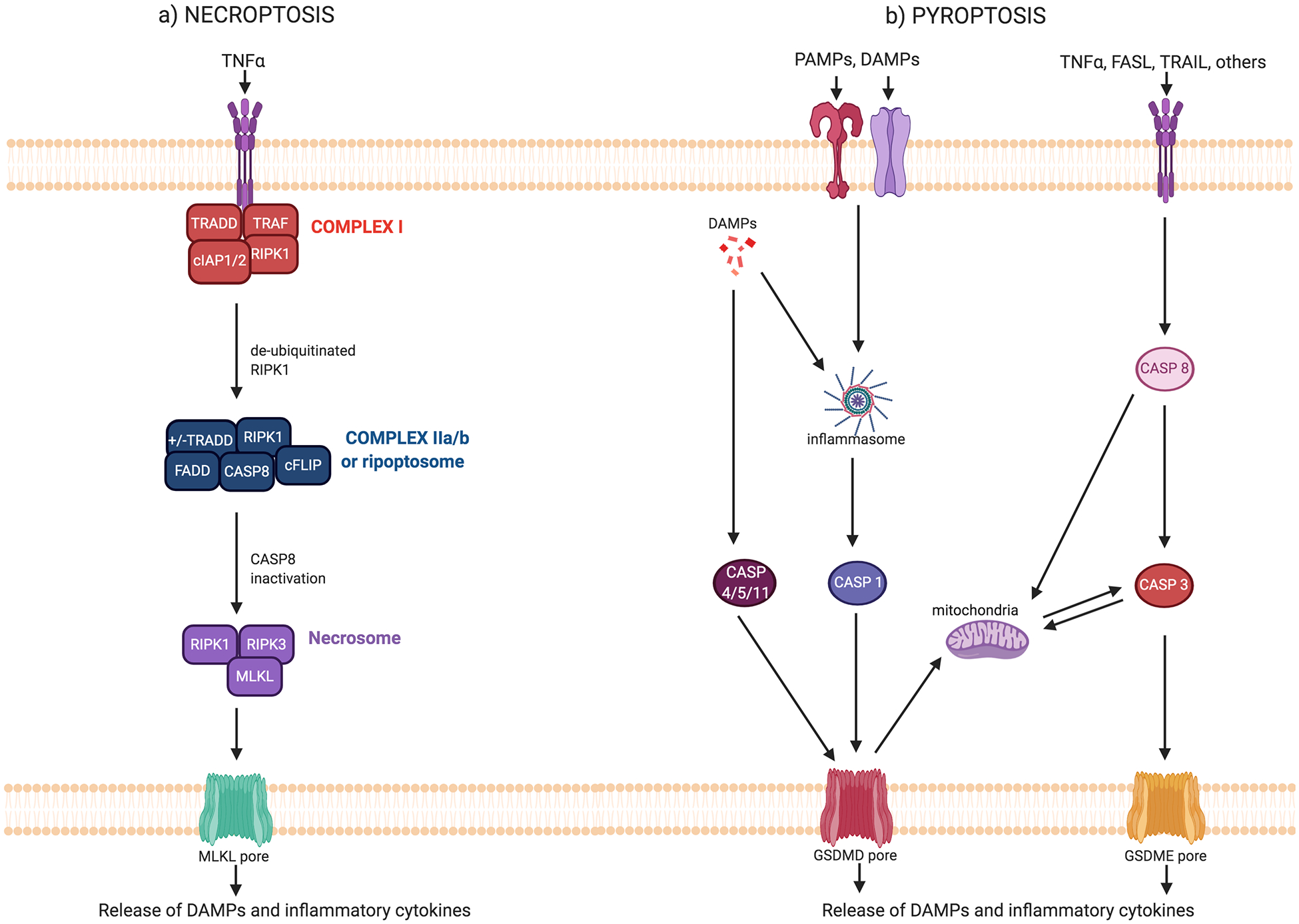
a) Necroptosis is activated through engagement of death receptors for TNFα, Fas-L, and TRAIL. At the membrane, TNFα receptor engagement triggers the assembly of Complex I. Within this complex, RIPK1 is regulated by ubiquitination and phosphorylation. When RIPK1 is deubiquitinated, it dissociates from Complex I and translocates to the cytosol. Additional factors are recruited to assemble Complex IIa or Complex IIb/ripoptosome. Complex IIa forms when cells are treated with cycloheximide or when NF-κB signaling is inhibited, promotes RIPK1-independent apoptosis when caspase-8 is active, and is inhibited by cFLIP. Complex IIb/ripoptosome forms when cIAP1/2 are deleted or inhibited. When caspase-8 is active, this complex promotes RIPK1-dependent apoptosis. Conversely, when caspase-8 is deleted or inhibited by cFLIP, this complex promotes necroptosis. Inactivation of caspase-8 allows for RIPK1 to associate with RIPK3 to form the necrosome, where autophosphorylation and transphosphorylation events activate RIPK1 and RIPK3. Activated RIPK3 binds to and phosphorylates MLKL, driving its oligomerization. Oligomerized MLKL forms pores in the plasma membrane, allowing for the release of inflammatory mediators. b) Receptor binding or intracellular recognition of PAMPs and DAMPs leads to activation of various inflammasomes, depending on the danger stimuli. Inflammasome activation leads to the cleavage of caspase-1, which can then cleave GSMD. Non-canonical inflammasome activation involves activation of caspases-4 and −5 (or the murine homolog caspase-11) which can cleave GSDMD. GSDMD oligomerizes and forms pores in the plasma membrane, allowing for the release of inflammatory mediators. Alternatively, death receptor engagement leads to the activation of the apoptosis pathway, leading to caspase-8 and caspase-3 activation. Caspase-3 can cleave GSDME, which oligomerizes and forms pores in the plasma membrane to allow for the release of inflammatory mediators. GSDMD and GSDME also form pores in the mitochondria, which can amplify the cell death pathway.
Necroptosis
Early studies in the death receptor signaling field demonstrated that tumor necrosis factor (TNF) could induce apoptosis as well as a programmed form of necrotic inflammatory cell death termed necroptosis. It is now known that necroptosis is canonically triggered by the activation of death receptors such as tumor necrosis factor receptor 1 (TNFR1), Fas, and tumor necrosis factor-related apoptosis-inducing ligand receptor (TRAIL-R). Death receptor stimuli induce the assembly of subsequent cell death signaling platforms, the formation of which are highly context dependent, tightly regulated, and engage in complex crosstalk that is still not yet fully understood. While Fas ligand and TRAIL can activate pathways leading to apoptosis or necroptosis (13–17), TNF signaling is the best characterized cell death cascade, and here we will utilize this pathway to introduce the major players in necroptosis.
TNFR1 activation leads to the recruitment of proteins into Complex I, consisting of TNFR1-associated death domain protein (TRADD), receptor-interacting serine/threonine protein kinase 1 (RIPK1), and TNF-receptor associated factor 2 (TRAF2) (Figure 1a) (18). In turn, Complex I promotes either cell survival, apoptosis, or necroptosis. The specific fate is tightly regulated with RIPK1 modification being an important factor in this decision. Upon TNF stimulation, RIPK1 is ubiquitinated by cellular inhibitor of apoptosis 1 and 2 (cIAP1/2), and further ubiquitinated by components of the linear ubiquitin chain assembly complex (LUBAC) (Figure 2) (18–21). RIPK1 ubiquitination allows for subsequent recruitment of TGFβ-activated kinase 1 binding protein 2 and 3 (TAB2/3), TGFβ-activated kinase 1 (TAK1) and the IκB kinase (IKK) complex (21–25). TAK1 phosphorylation of the IKK complex allows for the activation of NF-κB, which promotes the transcription of inflammatory and pro-survival genes (26). LUBAC-mediated ubiquitination is important since depletion of LUBAC components leads to dysregulation of NF-κB signaling and promotion of cell death (20,21,27–30). Ubiquitination of RIPK1 can also lead to its degradation by the proteasome (31) and preventing it from interacting with subsequent cell death signaling platforms. Conversely, RIPK1 can be deubiquitinated by the enzymes, CYLD or A20, to allow for cell death signaling to proceed (32–35).
Figure 2: Cross talk between cell death pathways.
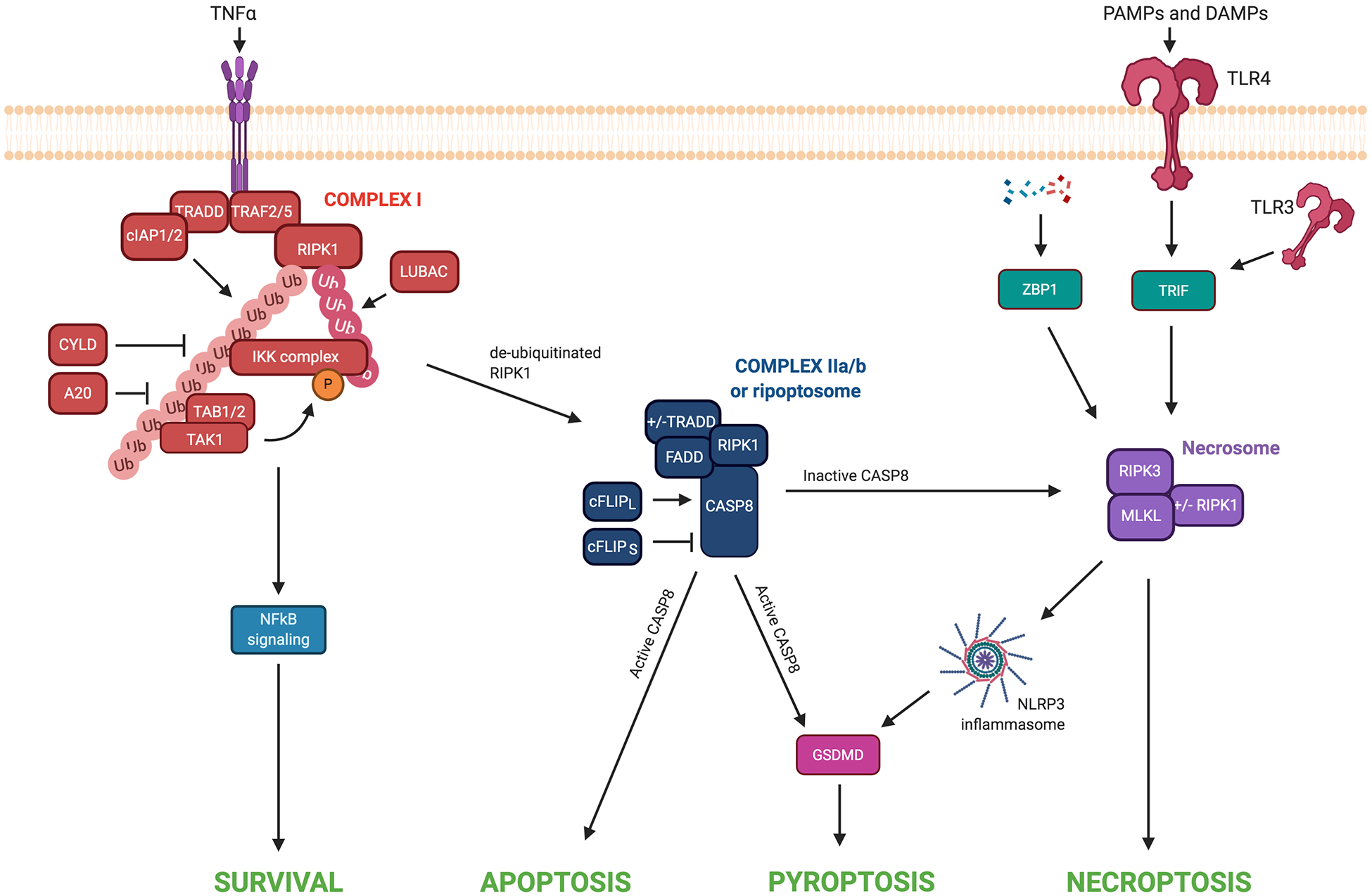
TNFα receptor engagement triggers the formation of Complex I. At this complex, cIAP1/2 and LUBAC ubiquitinate RIPK1, allowing for the recruitment and binding of TAK1, TAB1/2, and IKK complex. Phosphorylation of proteins within the IKK complex by TAK1-TAB2/3 promotes the activation of NF-κB signaling and cell survival. CYLD and A20 can deubiquitinate RIPK1 within Complex I. When RIPK1 is insufficiently ubiquitinated and has not been inactivated by inhibitory phosphorylation events, it translocates to the cytosol where it assembles with additional factors to form Complex II. Within this Complex II, the long isoform of cFLIP activates caspase-8 to prevent necroptotic cell death. Activated caspase-8 can promote activation of the apoptotic signaling cascade and/or cleavage of GSDMD and pyroptotic cell death. Alternatively, the short isoform of cFLIP inhibits caspase-8 activity, allowing for necrosome formation and necroptosis. Alternatively, RIPK3/MLKL can activate the NLRP3 inflammasome to induce pyroptosis. The detection of PAMPs and DAMPs by TLR3/4 leads to the activation of TRIF. When caspase-8 is inhibited, TRIF can promote RIPK3-dependent necroptosis. Intracellular sensing of nucleic acids by ZBP1 promotes necroptotic cell death in specific contexts when RIPK1, FADD, or caspase-8 are deleted or inhibited.
In addition to ubiquitination, RIPK1 is also regulated by phosphorylation in Complex I. Phosphorylation of RIPK1 by the IKK complex, TANK-binding kinase 1 (TBK1), MAP kinase-activated protein kinase 2 (MK2), or TAK1 prevents RIPK1-dependent cell death (36–39) . Of note, the role of TAK1 is complicated; its deletion sensitizes cells to apoptosis while its hyperactivation can promote necroptosis in specific contexts, possibly through hyperphosphorylation of RIPK1 (38,40,41). Additionally, RIPK1 autophosphorylation is important for its activation, and phosphorylation at serine-166 is often used as a marker for activation (42–44). Altogether, these studies show that modification of RIPK1 is a critical point of regulation at Complex I.
TNF signaling proceeds with the dissociation of RIPK1 from Complex I, and the recruitment of additional factors to form a secondary aggregate, Complex IIa or Complex IIb. The decision to form Complex IIa versus IIb is context-dependent and not comprehensively understood. When NF-κB signaling is inhibited or cells are treated with cycloheximide, TNFα treatment promotes the formation of Complex IIa, consisting of RIPK1, TRADD, FAS-associated death domain (FADD), caspase-8, and cellular FLICE inhibitor protein (cFLIP), and this complex is capable of inducing apoptotic cell death (33,45). In this case, apoptosis is not dependent on the kinase activity of RIPK1 and is inhibited by the expression of cFLIPL. Alternatively, when cIAP1/2 are depleted by genetic deletion or treatment with second mitochondria-derived activator of caspases (SMAC) mimetics, RIPK1 associates with FADD, caspase-8, and caspase-10 in Complex IIb also known as the ripoptosome (33,46). While TNF signaling can lead to the formation of Complex IIb/ripoptosome, mechanisms independent of death receptor signaling have also been described in response to chemotherapy (18,47). Once formed, Complex IIb induces either RIPK1-dependent necroptosis or apoptosis depending on the activity of cFLIP and caspase-8 (18,33,46,47).
cFLIP-mediated inhibition of caspase-8 or caspase-8 depletion allows for RIPK1 to associate via RIP homology interaction motifs with the related RIPK3 to form a complex termed the necrosome (48). In this manner, caspase inhibition is an important factor in the cell’s decision to undergo necroptosis over apoptosis. Necrosome formation leads to activation of both RIPK1 and RIPK3 (49). In turn, activated RIPK3 binds to and phosphorylates the pseudokinase mixed lineage kinase domain-like (MLKL), driving its oligomerization (50). Oligomerized MLKL translocates to plasma membranes, forming pores and disrupting membrane integrity to allow for the influx of calcium ions and the release of pro-inflammatory intracellular contents, including DAMPs (50). Because RIPK3 is also capable of promoting NLRP3 inflammasome activation, leading to pyroptotic inflammatory cell death (discussed below) (51,52), the requirement for MLKL is becoming regarded as the defining feature of necroptosis.
Necroptosis can also be triggered by the detection of PAMPs or DAMPs. PAMP binding to TLR3/4 leads to the subsequent activation of the adaptor protein TIR-domain-containing adapter-inducing interferon-β (TRIF). TRIF is then able to recruit and activate RIPK1 and RIPK3 (53). When RIPK3 is present and caspase-8 is inhibited, TRIF activation of RIPK3 can subsequently promote necroptosis; while in other contexts TRIF activation of RIPK3 can promote NF-κB signaling or apoptosis (53–56). DAMPs also trigger necroptosis through binding to sensor proteins such as Z-DNA-binding protein 1 (ZBP1). ZBP1 was first identified as an interferon (IFN)-inducible mediator of innate immune responses in response to cytosolic DNA. Upon activation, ZBP1 promotes type I IFN gene induction through interaction with TBK1 and IRF3 (57). Recent studies have shown that intracellular sensing of nucleic acids by ZBP1 can also trigger the recruitment and activation of RIPK1 and RIPK3, which can subsequently lead to NF-κB signaling (58,59). In other contexts when RIPK1, FADD, or caspase-8 are deleted or inhibited, ZBP1 activation by nucleic acid sensing or IFN stimulation triggers necroptosis (60,61). Interestingly, in response to type A influenza virus, ZBP1 was found to simultaneously activate apoptotic, necroptotic, and pyroptotic cell death pathways (62). Thus, ZBP1 may be involved in the induction of multiple forms of cell death, though its precise role in cell death crosstalk requires further investigation.
The expression level of RIPK3 may represent a determinant for whether necroptosis is initiated (63). Loss of RIPK3 expression has been observed in multiple cancer types, highlighting the potential importance of this pathway in tumorigenesis (64,65). In an analysis of pan-cancer cell lines, RIPK3 expression correlated with sensitivity to necroptosis and, in some cancers, RIPK3 loss was shown to be driven by oncogenic BRAF and AXL (66). Notably, the expression of RIPK3 may be important for eliciting immune responses. RIPK3 knockout in syngeneic models of lung carcinoma and lymphoma reduced the efficacy of chemotherapeutic agents in vivo, an effect that was associated with decreased infiltration of CD8+ T cells (67). Furthermore, injection of RIPK3-activated, necroptotic tumor cells into a pre-existing tumor enhanced anti-tumor immunity in syngeneic models of melanoma and lung adenocarcinoma (68).
While some studies have demonstrated that necroptosis can promote anti-tumor immune responses, others show a pro-tumorigenic role. In a mouse model of pancreatic cancer, deletion of RIPK3 or inhibition of RIPK1 slowed oncogenic progression. Effects of RIPK3 deletion were associated with decreased infiltration of tumor-associated macrophages and myeloid-derived suppressive cells, and reduced expression of the chemokine attractant CXCL1 and the inflammatory mediator splicesome-associated protein 130 (69). In breast cancer models, deletion of MLKL in tumor cells reduced spontaneous metastasis to the lungs (70). There is also some evidence that the induction of necroptosis in stromal cells can also affect tumorigenesis. The intratumoral injection of necroptotic fibroblast cells reduced tumor growth, while apoptotic fibroblast cells had no effect (68). Conversely, studies in multiple cancer types showed the ability of tumor cells, via expression of amyloid precursor protein, to induce necroptosis in endothelial cells, leading to trans-endothelial migration and metastasis of tumor cells (71). The implication is that activation of the necrosome may enhance cancer progression in some settings, although it is not clear whether effects on metastasis are dependent on necroptosis itself or the destruction of endothelial barriers. Consistent with this notion, RIPK1 inhibitors can prevent the ability of cancer cells to colonize the lung, or induce reprogramming of intra-tumoral macrophages for control of established tumors and metastases (72). Importantly, different RIPK1 inhibitors have shown contrasting results and, thus, require further study (73). These findings reveal a more complicated interplay between the induction of inflammatory forms of cell death in different cell types and early-disease outcomes.
Pyroptosis
Pyroptosis (pyro- from the Greek word for fire) was first observed when bacterial infection of macrophages led to cell death. It was later linked to the activation of inflammatory caspase-1 (74). After mechanistic studies, pyroptosis is now defined as an inflammatory form of cell death characterized by the formation of pores in the plasma membrane by gasdermin (GSDM) protein family members. GSDM proteins are widely expressed (75), with GSDMD and GSDME being the most studied.
Pyroptosis is triggered by the binding of pattern-associated molecular patterns (PAMPs) and DAMPs by pattern recognition receptors (PRRs) and inflammasomes. Inflammasome complexes (NLRP3, NLRP1, NAIP-NLRC4, AIM2 and Pyrin) recognize various ligands including viral components and cytosolic DNA (74). Canonical inflammasome activation leads to the cleavage and activation of caspase-1, which promotes pyroptosis via two actions (Figure 1b). Caspase 1 cleaves inflammatory cytokines, IL-1β and IL-18, into their mature forms, and also cleaves GSDMD (76). Alternative signals, such as cytosolic lipopolysaccharide can lead to non-canonical inflammasome signaling involving the autoactivation of caspases-4 and −5 (or its murine homolog caspase-11), which subsequently cleave GSDMD (77).
A defining feature of pyroptosis is the formation of GSDM-based pores. GSDMD cleavage releases its active N-terminal domain from the autoinhibitory C-terminal domain, allowing GSDMD N-termini to oligomerize and form pores in the plasma membrane (76). Efflux of inflammatory cytokines through GSDM pores leads to disruption of membrane integrity, and cell rupture. While release of IL-1β is characteristic of pyroptotic cell death, it can also occur in a sub-lytic phase. For example, GSDMD pore formation can lead to IL-1β release from viable macrophages (78,79). The ability of macrophages to avoid cell lysis and death following a pyroptotic stimulus may be due to cell membrane repair by ESCRT proteins (78,79). Furthermore, RIPK3-MLKL activation can lead to inflammasome activation and IL-1β release, independent of GSDMD (80,81). IL-1β and IL-18 are critical inflammatory mediators that are produced as danger signals in response to microbial products/damage, and are responsible for driving an innate immune response ((82) and detailed below). The key role for GSDM proteins in pyroptotic cell death is supported by studies showing that, in the absence of GSDMD, caspase-1-initiated cell death switches to apoptosis (83,84). Expression of GSDMD is transcriptionally regulated by interferon regulatory transcription factor-2 (IRF2), one of a family of transcription factors that is involved in the regulation of IFN and IFN-inducible genes (85). Given that loss of IRF2 expression in cancer leads to immune evasion and resistance to immunotherapy, IRF2 regulation of GSDMD levels may represent a critical regulatory axis in cancer cell death (86).
In addition to GSDMD, pyroptosis can also be activated by the cleavage of GSDME (also known as DFNA5). Unlike inflammasome-mediated pyroptosis, GSDME cleavage is mediated by the apoptotic executioner, caspase-3 (87,88). Through this mechanism, GSDME serves as the nexus in the switch between apoptosis and secondary pyroptosis modes of cell death. GSDMD- and GSDME-mediated pyroptosis have been analyzed to date in different cellular contexts, and much remains to be understood about the two mechanisms. GSDME also forms pores and leads to the release of DAMPs and cytokines. To date, little is known about the regulation of its expression, although it may be expressed in a p53-dependent manner (89) and silenced in some cancers. The loss of GSDME accelerates tumor growth in melanoma models (90,91), suggesting that GSDME-mediated pyroptosis may play an important role in regulating the tumor immune microenvironment and the ability of immune cells to attack tumors. It was also recently observed that granzyme B, a serine protease released by cytotoxic T cells to induce cell death of their targets, is capable of cleaving GSDME (90). Granzyme B cleavage of GSDME resulted in enhanced anti-tumor immunity and reduced tumor growth (90). These findings are of particular interest given that immunotherapeutic strategies often seek to improve T cell responses to cancer.
In addition to the plasma membrane, both GSDMD and GSDME are capable of forming pores in the mitochondrial membrane. Through this mechanism, GSDMD/E enhance the intrinsic apoptosis pathway and act in a positive feedback loop to amplify the cell death signal (91,92). One study found that mitochondrial pores formed by GSDMD could lead to the release of mitochondrial DNA, and subsequent activation of the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway (92). Thus, GSDMD and GSDME may further alter the state of the local tumor microenvironment via proinflammatory pathways.
Proteins that mediate cell death pathway crosstalk: IAPs, cFLIP and caspase-8
The key characteristics that distinguish one form of programmed cell death from another are well delineated (12), yet many of the same proteins are involved in multiple forms of cell death and there is significant crosstalk between pathways. Here, we present the emerging research on cell death signaling proteins and their roles in mediating the switch between inflammatory and non-inflammatory modes of cell death.
a). Inhibitor of apoptosis (IAP) proteins:
A major class of proteins involved in a cell’s decision to die in an inflammatory or non-inflammatory manner are IAPs, including X-linked inhibitor of apoptosis (XIAP), cIAP1, and cIAP2. Extrinsic cell death signals, such as TNFα, trigger the recruitment of cIAP1/2 via TRAF2 to Complex I, which forms at TNFRI (18). Polyubiquitination of RIPK1 by cIAP1/2 simultaneously prevents RIPK1 from activation while also creating a scaffold that promotes NFκB pro-survival signaling (Figure 2) (18). In this way, cIAP1/2 prevents necroptotic inflammatory cell death and targeting of cIAP and TRAF2 sensitizes melanoma cells to TNFα-induced death (93). XIAP is also an E3 ubiquitin ligase and blocks cell death by inhibiting caspases-3, −7 and −9 (94). Based on these findings, IAP proteins have been targets of interest for anti-cancer therapeutics with several agents including the SMAC mimetic, birinapant, reaching clinical trials (94).
b). Cellular FLICE-inhibitory protein (cFLIP):
cFLIP is a catalytically inactive protein resembling caspase-8. Its expression is regulated by NF-κB signaling and it acts to modulate caspase-8 activity through the formation of heterodimers. Within the ripoptosome, the induction of necroptosis or apoptosis is regulated by cFLIP-caspase-8 dimers (46,94). Short and long isoforms of cFLIP differentially modulate caspase-8 activity (Figure 2), although this is a complex and potentially controversial topic. Dimerization with the short isoform of cFLIP (cFLIPS) prevents catalytic activation of caspase-8, inhibiting apoptosis, and promotes ripoptosome formation, promoting necroptosis (18,46). The role of the predominantly expressed long isoform of cFLIP (cFLIPL) is more controversial. Elevated levels of cFLIPL prevent the recruitment and activation of caspase-8 to inhibit apoptosis (18,94). However, other studies demonstrate that caspase-8 forms heterodimers more readily with cFLIPL than homodimers, and that cFLIPL is a potent activator of caspase-8 catalytic activity (18,46,48,95,96). Of note, this heterodimer may act on different substrates (97). These apparent contradictions highlight the complexity of cFLIP-caspase-8 interactions in regulating cell death, which has been extensively reviewed elsewhere (98,99). Thus, while both isoforms can inhibit apoptosis, cFLIPL and cFLIPS may play different roles in the regulation of necroptosis.
c). Caspase-8:
Caspase-8 is predominantly known for its role as an initiator caspase in the extrinsic apoptosis cascade, but it is also a critical mediator of the cell’s decision between apoptosis and necroptosis. When caspase-8 catalytic activity is inhibited or absent, cells can be diverted to undergo necroptosis or, when MLKL is additionally inhibited, to undergo pyroptosis (100,101). Caspase-8 also cleaves RIPK1, RIPK3, and CYLD to inhibit necroptosis (48,95,96,102). In this way, caspase-8 is positioned at a critical juncture between apoptosis and necroptosis, acting as a molecular switch to promote non-inflammatory over inflammatory forms of cell death. Inactivating mutations and epigenetic loss of caspase-8 expression have been observed across several cancers, implying that cancer cells which are resistant to apoptosis may be driven to inflammatory cell death in the right contexts (103).
In other scenarios, caspase-8 can regulate pyroptosis and inflammation. When combined with inhibitors of pro-survival signaling, TNFα can trigger caspase-8 mediated inflammatory cell death that is associated with GSDMD cleavage and inflammasome activation (104). Inhibition of the serine/threonine kinase transforming growth factor beta-activated kinase 1 (TAK1/MAP3K7) can also induce caspase-8 cleavage of GSDMD and pyroptosis, a process that is inhibited by cFLIPL (105,106). While these studies were performed in macrophages, they can have important implications for cancer. Indeed, TAK1 inhibitors have been investigated as a potential cancer therapy and their effect on inflammatory tumor cell death requires further investigation (107). Additionally, in tumor cells treated with the apoptotic stimulus TRAIL, caspase-8 can function as a scaffolding protein to mediate the production of cytokine release and subsequent pro-tumorigenic macrophage recruitment (108,109). These data demonstrate that while caspase-8 has important roles in apoptosis, it can also behave as a mediator of inflammatory cell death when other cell death mechanisms are inhibited.
Overall, these studies highlight the complex interplay that exists between cell death pathways. The presence, level, and activation of key molecular switches such as cIAPs, cFLIP isoforms, RIPK3, and caspases can dictate whether cell death will be inflammatory or immunologically silent. Future studies addressing the regulation and potential exploitation of these molecular switches in cancer will help us to better understand cell death in this context and therapeutic strategies.
3. Immune Response to Inflammatory Cell Death
Immune response to DAMP and cytokine release
Pyroptosis and necroptosis cause inflammation through release of DAMPs and inflammatory cytokines and inducing changes in the antigen presenting cells (APCs) responsible for phagocytosing the dying cells. Well-characterized factors released directly from cells undergoing pyroptosis include high-mobility group box-1 (HMGB1), calreticulin, ATP, and IL-1 family cytokines. In addition, inflammatory factors secreted by necrotic cells include heat shock proteins, BCL-2, cyclophilin A, F-actin, histones and non-histone chromosomal proteins, mitochondrial and genomic DNA, monosodium urate, N-formyl peptides, and S100 proteins (110) but many of these are not fully characterized in the context of pyroptosis. While many of the secreted DAMPs and cytokines directly modulate the innate immune response, enhanced recruitment of adaptive immune cells along with increased antigen presentation and toll-like receptor (TLR) activation lead to broader immune activation (111–114). Here, we review the actions of well-recognized DAMPs and cytokines (summarized in Table 1) and discuss the role of APCs in the amplification of cell death signals.
Table 1:
Examples of DAMPs and cytokines released during inflammatory cell death
| Inflammatory factor | Form of cell death | Effect on tumor cells and anti-tumor immunity |
|---|---|---|
| Calreticulin (CRT) | Apoptosis, Necroptosis, Pyroptosis |
|
| HMGB1 | Necroptosis, Pyroptosis |
|
| ATP | Necroptosis, Pyroptosis |
|
| IL-1β | Pyroptosis |
|
| IL-18 | Pyroptosis |
|
| IL-6 | Necroptosis, Pyroptosis |
|
| IL-33 | Necroptosis, Pyroptosis |
|
| Prostaglandin E2 |
|
DAMPs
a). Calreticulin:
Phagocytosis is tightly regulated by a balance of “eat me” and “don’t eat me” signals. During cell death, “don’t eat me” signals such as CD31, CD200, and CD47 are downregulated or relocated from the plasma membrane while “eat me” signals such as calreticulin and phosphatidylserine are exposed at the membrane (115,116). Calreticulin may also be released by cells undergoing all forms of programmed cell death. Soluble calreticulin acts as a DAMP by increasing IL-8 production in macrophages (117) and promoting dendritic cell (DC) maturation and increased expression of CD83, CD86, and HLA-DR, IL-6, IL-8, and TNFα (118,119).
b). High-mobility group box-1 (HMGB1):
It is well characterized that the DNA binding protein, HMGB1, is released during both necroptosis and pyroptosis. After release, HMGB1 binds to RAGE receptors on tumor and immune cells, or TLR-2, 4, and 9 in the immune compartment (111,112,120). Importantly, signaling through each of these receptors has a differential effect on tumor growth. HMGB1 binding to RAGE activates ERK1/2 signaling leading to increased cell migration through activation of Rac1 and Cdc42 (121). Elevated HMGB1 has been described in many cancer types and is often associated with invasion and metastasis (122). Blocking HMGB1 and RAGE signaling in a model of murine lung cancer suppressed tumor growth and metastasis (123). In contrast, HMGB1 signaling through TLR2 and TLR4 receptors on the surface of neutrophils, monocytes, macrophages, DCs, and lymphocytes, leads to activation of transcription factors NF-κB and AP-1, production of inflammatory cytokines such as IL-6, TNFα, IL-8, and IL-1 (111), and an increase in co-stimulatory molecules needed for CD8+ T cell priming. In response to chemotherapy and radiotherapy-induced cell death, HMGB1 signaling through TLR4 on DCs increases antigen processing and cross-presentation (124). Whether HMGB1 alternatively regulates tumor growth by signaling through RAGE or TLRs remains unclear.
c). ATP:
During pyroptosis and necroptosis, ATP is passively released from cells and acts in both an autocrine and paracrine manner. ATP primarily binds to the P2X and P2Y purinergic receptors (125,126) and can elicit different anti-tumor effects when signaling through each receptor. P2YR signaling stimulates production of the pro-inflammatory cytokine IL-8 (127), which is responsible for increased neutrophil recruitment and phagocytic potential (128). Alternatively, signaling through P2X7R can trigger NLRP3 inflammasome activation in macrophages which can further release IL-18, induce IFNγ production, and promote type I T helper cell (Th1) responses (126,129).
Inflammatory cytokines
a). IL-1β:
When released, IL-1β binds and activates IL1R1, which is expressed on most leukocytes as well as epithelial cells (130). Upon IL1R1 activation, MyD88 is recruited intraceullarly and acts as an adaptor to recruit the IL-1R associated kinase (IRAK) family of proteins. IRAKs signal downstream via IKK and/or mitogen-activated protein kinase kinase (MEK) to regulate gene expression through the NFκB pathway or MAPK cascade, respectively. In the myeloid compartment, IL-1β signaling leads to maturation of DCs and differentiation of monocytes into DCs and inflammatory macrophages. Additionally, IL-1β can directly act on cells in the lymphoid compartment to drive antigen-specific cytotoxic CD8+ T cell responses, increase Th1 CD4+ T cell numbers, and inhibit differentiation of immunosuppressive T regulatory cells (113,114). Although rampant IL-1β (and IL-1α) production has been shown to contribute to the formation of some cancer types such as multiple myeloma, IL-1 production has largely been shown to produce anti-tumor effects in murine models. Consistent with this, intratumoral injections of IL-1 are sufficient to cause CD8+ T cell mediated tumor regression in models of melanoma, adenocarcinoma, and sarcoma (130).
b). IL-18:
Released IL-18 can bind to two receptors, IL-18Rα and IL-18Rβ, albeit with varying affinities. IL-18Rα is expressed on most cells and binds IL-18 with low affinity while IL-18Rβ is only expressed on T cells and DCs and is the high affinity binding partner for IL-18 (131). Once bound to the IL-18 receptor, signal transduction is nearly identical to that described above for IL-1β, with MyD88 recruitment to the IL-18 receptor and subsequent signaling through IRAKs. However, the active form of the cytokine can also elicit much different effects than IL-1β when combined with other inflammatory cytokines. In combination with IL-12 or IL-15, which both upregulate the IL-18Rβ receptor, IL-18 can strongly induce IFNγ and, thus, plays a major role in Th1 polarization, natural killer (NK) cell recruitment, and NK cell activation (82). Additionally, IL-18 can promote nitric oxide synthesis, chemokine production, and expression of adhesion molecules.
Unlike IL-1β, the IL-18 precursor is constitutively expressed in blood monocytes and epithelial cells even in the absence of cell death or disease (131). It is also constitutively produced and released from gastric cancer cells (132). Paradoxically, in the absence of other inflammatory cytokines, IL-18 can promote Th2 responses and angiogenesis which lead to increased migration and invasion in tumors (133). Thus, IL-18 may have a differential effect on the tumor depending on the makeup of the cytokine milieu.
Additional factors released
While IL-1β, IL-18 and HMGB1 are often thought of as classical markers of inflammatory forms of cell death, this is an incomplete picture of the full repertoire of factors released. IL-6 and IL-33 are also directly released from necroptotic and/or pyroptotic cells (110,134). IL-6 binds to the IL-6R and signaling through JAK/STAT to activate either the MAPK or PI3K pathway (135,136) and leads to a broad range of effects in both the hematopoietic and non-hematopoietic compartments. In the immune compartment, IL-6 has a profound impact on the adaptive response by increasing CD8+ T cell trafficking, cytotoxic CD8+ T cell differentiation, increasing antibody production, and decreasing Treg differentiation (137,138). Tumor cells can also express and/or respond to IL-6 leading to increasing proliferation and invasion (138). IL-33, is cleaved by apoptotic caspases; however, processing decreases its biologic activity (139) compared to the non-cleaved form released during inflammatory cell death. IL-33 signals through IL-1R4 and initiates a similar signaling cascade to IL-1 and IL-18. Also similar to IL-18, IL-33 acts as an immunomodulatory cytokine and, depending on the cytokine milieu, may promote either IFNγ production and inflammatory responses or anti-inflammatory responses. Interestingly, inhibitory DAMPs such as prostaglandin E2 can also be released during inflammatory cell death (140). As prostaglandin E2 has particularly high expression in colon cancer and breast cancer, the balance between immune stimulation and immune suppression may be an important factor to consider when evaluating inflammatory cell death in the context of solid tumors. As the field develops, differences in the timing and context-dependencies of factors release will certainly become more apparent. It is already evident that by predominantly studying IL-1β, IL-18, and HMGB1 production, we are overlooking many of the ways cell death mechanisms can profoundly alter the immune compartment.
The role of APCs in cell death signal amplification
In addition to the factors secreted directly by cells undergoing pyroptosis and necroptosis, the immune response to inflammatory cell death is further amplified when the cells are cleared by APCs such as DCs and macrophages. Clearance of dying cells and debris is a critical role of APCs and, depending on the mode of cell death, can lead to either anti-inflammatory responses (when clearing apoptotic cells) or pro-inflammatory responses (when clearing pyroptotic or necroptotic cells). Upon engulfment of apoptotic cells, phagocytes secrete anti-inflammatory factors such as IL-10 and TGF-β and inhibit secretion of pro-inflammatory cytokines and chemokines such as IL-6, IL-1β, CCL2, CCL3, CCL4, CXCL10 (4,5,141,142). Conversely, when APCs are recruited to pyroptotic and necroptotic cells through release of ATP (143,144), phagocytic uptake of necroptotic colon carcinoma cells leads to maturation of DCs and efficient cross-presentation to CD8+ T cells. These actions ultimately promote CD8+ T cell activation and IFNγ production (145). Thus, the release of inflammatory mediators directly from the cells undergoing inflammatory cell death in combination with DC maturation and CD8+ T cell activation upon phagocytosis of the cells leads to a potent immune response.
4. Inflammatory cell death in cancer
Resistance to cell death is a hallmark of cancer, and Bcl-2 family proteins are the target of cancer therapies. Given the many pro-survival features of neoplastic cells, it is no surprise that mechanisms to down-regulate inflammatory cell death have also been reported. Multiple cancers types reduce expression of key proteins involved in necroptosis and pyroptosis, such as AIM2, GSDMD and GSDME (74). Decreased expression of calreticulin, HMGB1, and PRRs has also been observed (146). The anti-tumor effects of inflammatory cell death are supported by studies demonstrating that induction of pyroptosis or necroptosis by inflammasome agonists or DAMP-mediated activation of RIPK3 enhance immune cell responses and decrease tumor growth (147,148). In certain contexts, inflammatory cell death may also be pro-tumorigenic (74). This is consistent with the complex role of inflammation in both enhancing and inhibiting tumor growth, and suggests that the timing, levels, and components of induction need to be closely monitored. Detection reagents and biomarkers of the different forms of inflammatory cell death is a developing area.
Intra- and extra-cellular stresses including DNA damage, endoplasmic reticulum stress, and reactive oxygen species generate danger signals that can induce cell death associated with the release of inflammatory mediators (146). Tumor cells are adept at surviving harsh conditions, and stress-induced activation of the unfolded protein response is particularly important for their survival (149). There is likely to be some overlap in cancer cell stress responses, resistance to inflammatory cell death, and immune evasion. Nonetheless, numerous therapeutic agents used in the clinic to treat cancer cause cellular stress and/or inflammatory cell death. Beyond killing malignant cells, induction of inflammatory cell death can promote long-lasting immune responses. Therapies capable of inducing pyroptosis and/or necroptosis are of particular interest, especially in consideration of combination strategies with immunotherapy. Such therapies include ionizing radiation, chemotherapy, and targeted therapies, and will be discussed here. Various combinations of these therapies are already being tested in the clinic, with combinations of immune checkpoint therapy and chemotherapy predominating. Though not an exhaustive list, we provide examples of these clinical trials in Table 2.
Table 2:
Examples of clinical trials testing combinations of immune modulatory agents with drugs capable of inducing inflammatory cell death
| Drug combination | Trial examples (Cancer type) |
|---|---|
| Immune checkpoint inhibitor, chemotherapy −/+ radiation *addition of an angiogenesis inhibitor |
NCT04238988, NCT03612791 (Cervical cancer) NCT04262687, NCT04262687*, NCT04446091* (Colorectal cancer) NCT04444921 (Anal cancer) NCT03394885 (Ovarian cancer) NCT04282109, NCT03532737 (HNSCC) NCT04447612, NCT03984357 (Nasopharyngeal carcinoma) NCT04247165 (Pancreatic cancer) NCT02918162 (Gastric cancer) NCT04062708, NCT04137588*, NCT04517526* (Non-small cell lung cancer) NCT04393506* (Oral squamous cell carcinoma) NCT03937830* (Hepatocellular carcinoma) NCT04472806* (Mucosal melanoma) NCT03409458 (Multiple solid tumors) |
| TLR agonist alone or in combination |
NCT03734692 (Ovarian cancer) NCT02126579, NCT04364230, NCT04401995 (Melanoma) NCT02668770, NCT04270864, NCT04101357, NCT04116320, NCT02643303, NCT03416335 (Advanced malignancies) NCT04062721 (Colorectal cancer) NCT03906526 (HNSCC) NCT02452697 (Myeloid malignancies) NCT02927964, NCT01976585, NCT03410901 (Lymphomas) NCT04387071, NCT04050085 (Pancreatic cancer) NCT04338685 (Hepatocellular carcinoma or solid tumors with hepatic metastases) |
| SMAC mimetic, chemotherapy −/+ radiation | NCT03803774, NCT04459715, NCT02022098 (HNSCC) |
| SMAC mimetic, immune checkpoint inhibitor |
NCT04122625, NCT03270176 (Advanced solid malignancies) NCT03871959 (Pancreatic cancer and colorectal adenocarcinoma) |
| Antibody drug-conjugates | NCT02980341 (HER3+ Breast cancer) |
| Inhibitor of extracellular ATP degradation, alone or in combination |
NCT04336098, NCT04306900 (Advanced solid tumors) NCT03884556 (Advanced solid tumors and lymphoma) |
Ionizing radiation and chemotherapy
Ionizing radiation and chemotherapy target rapidly dividing cells and provoke cancer cells to undergo cell death. Many advanced-stage tumors are eliminated with a combination of chemotherapy and radiation, and radiotherapy is utilized in approximately half of all cancer treatment strategies for localized tumors. Only recently, it was discovered that pyroptosis plays a role in the anti-tumor effects of radiotherapy and chemotherapy.
Ionizing radiation causes DNA damage, which elicits immunogenic effects. In cancer cells, DNA fragments generated by ionizing radiation are recognized by intracellular DNA sensors, AIM2 or ZBP1, which in turn activate inflammasome signaling and pyroptosis (150). ZBP1 is also capable of inducing apoptosis and necroptosis in response to viral infection or IFN stimulation, but its precise role in cancer is unclear (61,150). Cytosolic DNA can also promote activation of the cGAS-STING pathway, which provides additional immune stimulation through the production of type I interferon. As an additional effect within the tumor microenvironment, ionizing radiation also induces pyroptosis of tumor-recruited macrophages, which increases the release of pro-inflammatory cytokines into the tumor milieu (151). Thus, radiation elicits alterations on the tumor immune microenvironment through effects on both tumor and stromal compartments.
Chemotherapy can trigger inflammatory cell death through a variety of different mechanisms, leading to increased immune cell infiltration. Several chemotherapeutic agents induce endoplasmic reticulum stress leading to the unfolded protein response (146). This subsequently promotes the exposure of calreticulin and release of DAMPs. Cancer cells have developed various mechanisms of surviving under stress (149), and one potential targeting strategy is to surpass the threshold for stress management in order to induce inflammatory cell death. Importantly, some chemotherapy agents such as cisplatin are not effective at inducing the unfolded protein response and, as a result, do not promote powerful immune responses. Chemotherapy drugs can also induce pyroptosis through activation of the apoptotic cascade, leading to caspase-3 dependent cleavage of GSDME (87,88). The effects of inflammatory cell death on the tumor microenvironment suggest that these treatments may synergize with other immune stimulating therapeutic agents, such as PD-1 blockers. Indeed, many of these therapies are already being combined in the clinic across multiple cancer types (152–154) (Table 2).
Targeted inhibitors
Targeted inhibitors block the activity of key oncogenic proteins and several agents are FDA-approved to treat select cancer types. Through inhibition of signaling pathways essential for cancer cell proliferation and survival, targeted therapies decrease tumor growth but are also capable of promoting inflammatory cell death. Treatment of BRAF mutant melanoma cells with BRAF inhibitors plus MEK inhibitors induced HMGB1 release associated with cleavage of GSDME, and dependent on GSDME expression (155). In lung cancer, targeted inhibition of MEK, EGFR, or ALK in cell lines driven by these pathways led to pyroptotic cell death, associated with GSDME cleavage (156). Third, inhibition of cyclin-dependent kinases in syngeneic mouse models promoted features characteristic of inflammatory cell death, including calreticulin surface exposure and release of ATP and HMGB1 (157). Consequently, mice showed improved anti-tumor immune responses and increased response to ICi (157). These studies support the premise that targeted inhibition of driver pathways in cancer may be sufficient to induce inflammatory cell death with immunogenic consequences. Based on this premise, multiple clinical trials are underway to evaluate the efficacy of MAPK pathway inhibition in combination with ICi. Many of these trials are testing the different FDA-approved BRAF inhibitors and MEK inhibitors in combination with either anti-PD-1 or anti-PD-L1 in BRAF mutant melanoma. Results from two trials evaluating the safety and efficacy of these combinations showed promising results, although the precise timing and combinations needed for improved responses remains an open question (158–160). Additional trials such as SECOMBIT are testing the optimal sequencing and combination of targeted inhibitors and ICi.
Other targeted therapies can promote inflammatory cell death by directly modulating the activity of death receptor signaling proteins. SMAC mimetics bind to IAP proteins and cause their degradation. While IAP proteins are involved in both apoptosis and necroptosis, SMAC mimetics often promote apoptosis in the absence of additional inhibitors. In acute myeloid leukemia, the effects of the SMAC mimetic, birinapant, were augmented when combined with a caspase inhibitor to induce necroptosis (161). Another study completed in acute lymphocytic leukemia observed a heterogeneous cell death response to birinapant, possibly indicating simultaneous engagement of both apoptotic and necroptotic cell death pathways (162). In HNSCC models, birinapant sensitized cells to TNFα and TRAIL-mediated cell death that was RIPK1-dependent in some cell lines and caspase-dependent in others (163). Furthermore, more durable anti-tumor responses were observed in vivo when mice bearing tumors were treated with a combination of birinapant and radiotherapy. In melanoma, birinapant sensitized cells to TNFα-mediated T cell killing, and improved responses to immune checkpoint blockade (93). These data are further supported by studies in glioblastoma demonstrating synergy when SMAC mimetics were combined with immune checkpoint inhibitors or innate immunostimulants (164). It is important to note that SMAC mimetics have been demonstrated to exert effects directly on immune cells; these actions are reviewed elsewhere (165). Taken together, these studies provide evidence that the induction of necroptotic inflammatory cell death through IAP inhibition can promote anti-tumor immune responses. While SMAC mimetics have previously shown limited efficacy in the clinic, combinations strategies are currently being tested. Notably, an ongoing clinical trial (NCT02022098) in HNSCC recently reported enhanced disease control when the SMAC mimetic, Debio 1143, was combined with chemotherapy (166).
Additional therapies that induce inflammatory cell death
Radiation, chemotherapy, and targeted inhibitors are widely used in the clinic, but additional cancer therapeutic strategies under scientific and clinical investigation are also capable of eliciting inflammatory cell death. Oncolytic viruses have been tested as therapeutic agents in a variety of cancers, and work by infecting and killing tumor cells along with promoting an anti-tumor immune response. Viral infection triggers necroptosis or activates the inflammasome to induce pyroptosis through PRR signaling or through direct effects of viral proteins (167,168). Additionally, successful oncolytic viruses can induce the release of inflammatory cytokines, as well as tumor antigens to further stimulate immune responses. Well-characterized oncolytic viruses such as Semliki Forest virus, Vaccinia virus, and Adenovirus can all induce forms of inflammatory cell death, which ultimately may impact the extent of the immune response (168). These are likely to be important considerations for therapeutic strategies utilizing oncolytic viruses in cancer.
Targeting the ectoenzyme CD39 presents an additional therapeutic strategy for induction of inflammatory cell death. ATP is released into the tumor microenvironment by dying cells, and is capable of activating the NLRP3 inflammasome and eliciting an inflammatory response. CD39 is primarily expressed on the plasma membrane of macrophages and other monocytic lineage cells. It functions to convert exported ATP into adenosine causing both the removal of pro-inflammatory ATP from the microenvironment, and increasing the levels of the immunosuppressant, adenosine (169). Antibodies against CD39 are able to improve anti-tumor immunity and promote response to immune checkpoint inhibitors in syngeneic mouse tumor models (169,170). The mechanism of action for CD39 blockade was shown to depend on NLRP3 inflammasome activation in macrophages, and the subsequent release of inflammatory cytokines (170). Importantly, CD39-targeting agents, SRF617 and TTX-030, have recently entered clinical trials (Table 2).
A third approach is to utilize antibody drug conjugates with agents that trigger inflammatory cell death. In syngeneic mouse tumor models, U3–1402, an anti-ErbB3/HER3 antibody conjugated to the topoisomerase I inhibitor DXd, was used to treat ErbB3-expressing melanomas (171). DXd induced HMGB1 release from cells and U3–1402 promoted both innate and adaptive immune cell infiltration. This agent is currently being tested in the clinic for efficacy in treating HER3+ breast cancer (NCT02980341). Together, these studies highlight potential therapeutic strategies for direct induction of inflammatory cell death within the tumor microenvironment and demonstrate potential avenues for combination therapies with immune checkpoint blockade.
5. The Potential for Synergy Between Immune Checkpoint Blockade and Inflammatory Cell Death-Inducing Therapies
Shortcomings of ICi therapy
While ICi and other immunotherapies have revolutionized the cancer treatment landscape, limitations exist. Although ICi therapy elicits durable responses in some tumor types, the majority of cancer patients fail to respond across multiple cancer types (172,173); hence, a major goal in the cancer therapy field is to determine rational combination approaches that will increase the percentage of patients with durable responses. As success of ICi is dependent on the presence of intratumoral CD8+ T cells, these therapies have shown the most success in immune replete solid tumor types such as melanoma and lung cancer. In general, response highly correlates with the number of CD8+ T cells present in the tumor, with ICi therapy often being insufficient on its own to increase numbers of CD8+ TIL (174). The response to ICi may also be dependent on the production of CXCL9 and CXCL10 by tumor-associated macrophages, which recruit critical numbers of CD8+ T cell necessary for an effect (175). Thus, ICi may be most effective when combined with therapies that can increase the number of CD8+ T cells and/or the inflammatory macrophages recruited to the tumor, and we speculate that there may be strong potential for synergy with inflammatory cell death-inducing agents.
Combination potential of ICi and inflammatory cell death-inducing therapies
Both ICi and inflammatory cell death-inducing therapies have major strengths and weaknesses when used as monotherapies but may elicit additive or synergistic effects when combined (Figure 3). While ICi therapy is greatly limited by the degree of CD8+ T cell infiltration (174), inflammatory cell death-inducing therapies are successful at recruiting CD8+ T cells as well as other lymphocytes and myeloid cells to the lesion. Indeed, preclinical evidence suggests that increased immune infiltration through pyroptosis enhances response to therapy. For example, BRAF inhibitor and MEK inhibitor-resistant melanomas tend to be immunologically cold; however, inducing pyroptosis in pre-clinical models of resistant melanomas with the DNA topoisomerase inhibitor etoposide was able to reduce tumor growth (155). Additionally, treatment with anti-ErbB3-antibody drug conjugate, U3–1402, renders ICi-unresponsive ErbB3/HER3+ murine melanoma sensitive to anti-PD-1, likely through induction of inflammatory cell death mechanisms (171).
Figure 3: Effects of inflammatory cell death-inducing therapy, immune checkpoint blockade therapy, and potential synergy.
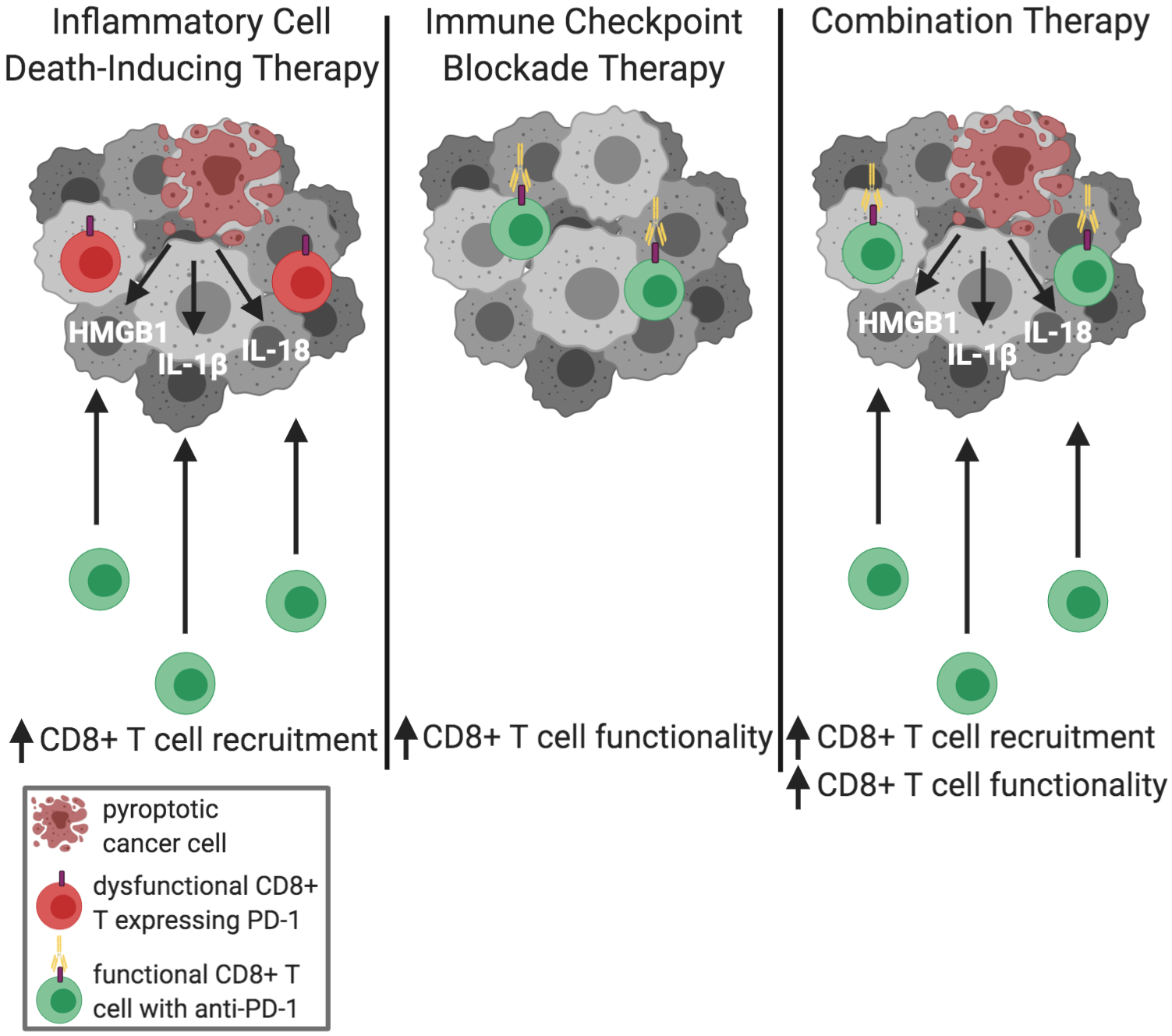
Therapies that induce inflammatory cell death recruit new CD8+ T cells to the tumor through release of DAMPs and cytokines. The long-term functionality of the CD8+ T cells in the tumor immune microenvironment, however, has not been fully characterized. Immune checkpoint inhibitors such as anti PD-1 therapy increase functionality in CD8+ T cells in addition to other tumor immune-specific effects but are not always sufficient to recruit additional CD8+ T cells needed for full therapeutic efficacy. Combining inflammatory cell death-inducing therapy and immune checkpoint blockades has the potential to potently increase CD8+ T cell numbers and functionality.
Most studies to date have used murine melanoma models to test the combination of ICi and inflammatory cell-death inducing therapies. However, this strategy may be broadly applicable as a strategy to increase responses to ICi in other models by creating an early increase in functional tumor infiltrating CD8+ T cells that could mediate tumor cell killing. As this area of research is still in its infancy, there are likely to be several obstacles to adapting this work for the clinic (176). It is important to consider the tolerability of inducing inflammatory cell death, especially in combination with ICi. By concurrently increasing the immune local response and the release of self-antigen, generating autoimmunity becomes a major consideration. Notably, autoimmune conditions such as colitis, dermatitis, hypophysitis, thyroiditis, hepatitis, and pneumonitis are relatively common side effects in patients treated with ICi (177). ICi have also been reported to induce cytokine release syndrome (CRS) in a subset of patients and combining ICi with pyroptosis-inducing therapies may increase this risk (178,179). Although complications are possible in some patients, combination strategies have the potential to promote tumor cell killing, creating a durable and sustained response that is unable to be elicited by either therapy alone.
Concluding remarks
The study of inflammatory cell death is a broad and rapidly evolving field. An emerging view is that triggering necroptosis and/or pyroptosis in a tumor localized manner may have profound effects on the tumor immune microenvironment and response to ICi. While we have focused this review on recent advances in the understanding of necroptosis and pyroptosis, we also acknowledge that other forms of cell death such as ferroptosis and NETosis, may also influence the tumor immune milieu. Ferroptosis is induced by iron-dependent lipid peroxidation, and can cause the release of DAMPs (12), which may also enhance the efficacy of ICi (180). Cancer cell dependency on iron highlights a potential vulnerability of these cells to ferroptosis-inducing agents, and is the focus of many therapeutic strategies under investigation (181). NETosis can occur when neutrophils release a neutrophil extracellular trap (NET), consisting of chromatin, various proteins, and proteases to trap foreign microbes, resulting in programmed cell death. The study of NETosis is controversial and ongoing with evidence that NET formation also occurs independently of cell death through release of mitochondrial DNA, as well as in other cell types in addition to neutrophils (182).
While substantial progress has been made in understanding mechanisms of inflammatory cell death and its effect on the immune system, there are still several open questions. GSDMD and GSDME are the major gasdermin proteins studied thus far. However, additional gasdermin proteins GSDMA and GSDMB also contain a pore-forming domain, and a recent study demonstrated GZMA-driven cleavage of GSDMB was capable of inducing pyroptosis (183). The role of other gasdermin proteins in inflammatory cell death remains unclear and requires further investigation. Furthermore, cytokines released from inflammatory cell death exert effects on the immune compartment, the tumor cells, and the stroma. While the effect on the immune compartment generally leads to an anti-tumor response, cytokine signaling in the tumor cells can lead to proliferation and invasion. At present, the net effect of inflammatory cytokine signaling on tumor growth and anti-tumor immunity remains unclear and may be context dependent. Additional studies on the role of cytokine signaling in cancer are required to gain a complete picture of how inflammatory cell death might affect anti-tumor immunity.
Further understanding the interplay between mechanisms of cell death and how they may impact the tumor microenvironment will be critical when optimizing combination therapy approaches. Moving forward, integrated studies on inflammatory cell death induction will enhance our understanding of how to develop successful therapeutic strategies. Pre-clinical testing of the order/timing of combinations and schedules will likely be critical to maximize potential benefits within the tumor while minimizing the effects of cytokine storm.
Statement of significance.
Many tumors types respond poorly to immune checkpoint inhibitors (ICi) or respond but subsequently acquire resistance. Effective therapies for ICi non-responsive tumors are lacking and should be guided by evidence from pre-clinical studies. Promoting inflammatory cell death mechanisms within the tumor may alter the local immune microenvironment towards an ICi-responsive-state.
Acknowledgements
The authors thank Drs. Maureen Murphy (The Wistar Institute) and Jianke Zhang (Thomas Jefferson University) for insightful feedback on the article. All figures were created with BioRender.com.
Funding: This work was supported by grants from the NCI: R01 CA182635 and project 4 of P01 CA114046-11A1 to A.E. Aplin (lead PIs are Drs. M. Herlyn and A. Weeraratna). S.R. Rosenbaum is funded by an NCI F99 grant, CA245552. N. Wilski is supported by an NCI T32 training grant in Cancer Biology, CA236736.
Footnotes
Conflict of interest: A.E. Aplin has ownership interest in patent number 9880150 and reports receiving a commercial research grant from Pfizer Inc. (2013-2017).
References
- 1.Leverson JD, Sampath D, Souers AJ, Rosenberg SH, Fairbrother WJ, Amiot M, et al. Found in Translation: How Preclinical Research Is Guiding the Clinical Development of the BCL2-Selective Inhibitor Venetoclax. Cancer Discov 2017;7:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hata AN, Engelman JA, Faber AC. The BCL2 Family: Key Mediators of the Apoptotic Response to Targeted Anticancer Therapeutics. Cancer Discov 2015;5:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boada-Romero E, Martinez J, Heckmann BL, Green DR. The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 2020;21:398–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-Associated Phagocytosis and Inflammation. Journal of Molecular Biology 2017;429:3561–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao YQ, Freire-de-Lima CG, Schiemann WP, Bratton DL, Vandivier RW, Henson PM. Transcriptional and Translational Regulation of TGF-β Production in Response to Apoptotic Cells. J Immunol 2008;181:3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y, Fei M, Zhang G, Liang W-C, Lin W, Wu Y, et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 2020;52:357–73.e9 [DOI] [PubMed] [Google Scholar]
- 7.White Michael J, McArthur K, Metcalf D, Lane Rachael M, Cambier John C, Herold Marco J, et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 2014;159:1549–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rongvaux A, Jackson R, Harman Christian CD, Li T, West AP, de Zoete Marcel R, et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 2014;159:1563–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 2017;19:1116–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nature Immunol 2020;21:546–54 [DOI] [PubMed] [Google Scholar]
- 11.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359:1350–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death & Differentiation 2018;25:486–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nature Immunol 2000;1:489–95 [DOI] [PubMed] [Google Scholar]
- 14.Meurette O, Rebillard A, Huc L, Le Moigne G, Merino D, Micheau O, et al. TRAIL Induces Receptor-Interacting Protein 1–Dependent and Caspase-Dependent Necrosis-Like Cell Death under Acidic Extracellular Conditions. Cancer Res 2007;67:218. [DOI] [PubMed] [Google Scholar]
- 15.Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W, et al. Dual Signaling of the Fas Receptor: Initiation of Both Apoptotic and Necrotic Cell Death Pathways. Journal of Experimental Medicine 1998;188:919–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death & Differentiation 2012;19:2003–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: Decisions between life and death. The International Journal of Biochemistry & Cell Biology 2007;39:1462–75 [DOI] [PubMed] [Google Scholar]
- 18.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 2015;15:362–74 [DOI] [PubMed] [Google Scholar]
- 19.Bertrand MJM, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, et al. cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Mol Cell 2008;30:689–700 [DOI] [PubMed] [Google Scholar]
- 20.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 2011;471:591–96 [DOI] [PubMed] [Google Scholar]
- 21.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the Linear Ubiquitin Chain Assembly Complex Stabilizes the TNF-R1 Signaling Complex and Is Required for TNF-Mediated Gene Induction. Mol Cell 2009;36:831–44 [DOI] [PubMed] [Google Scholar]
- 22.Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB2 and TAB3 Activate the NF-κB Pathway through Binding to Polyubiquitin Chains. Mol Cell 2004;15:535–48 [DOI] [PubMed] [Google Scholar]
- 23.Tang Y, Tu H, Zhang J, Zhao X, Wang Y, Qin J, et al. K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nature Communications 2019;10:4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ea C-K, Deng L, Xia Z-P, Pineda G, Chen ZJ. Activation of IKK by TNFα Requires Site-Specific Ubiquitination of RIP1 and Polyubiquitin Binding by NEMO. Mol Cell 2006;22:245–57 [DOI] [PubMed] [Google Scholar]
- 25.Wu C-J, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation. Nat Cell Biol 2006;8:398–406 [DOI] [PubMed] [Google Scholar]
- 26.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-Induced Apoptosis by NF-κB. Science 1996;274:787. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011;471:637–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peltzer N, Darding M, Montinaro A, Draber P, Draberova H, Kupka S, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 2018;557:112–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B, et al. HOIP Deficiency Causes Embryonic Lethality by Aberrant TNFR1-Mediated Endothelial Cell Death. Cell Rep 2014;9:153–65 [DOI] [PubMed] [Google Scholar]
- 30.Witt A, Vucic D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death & Differentiation 2017;24:1160–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J, Liccardi G, et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol Cell 2018;69:566–80.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003;424:801–05 [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Du F, Wang X. TNF-alpha Induces Two Distinct Caspase-8 Activation Pathways. Cell 2008;133:693–703 [DOI] [PubMed] [Google Scholar]
- 34.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a Molecular Signaling Network that Regulates a Cellular Necrotic Cell Death Pathway. Cell 2008;135:1311–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 2004;430:694–99 [DOI] [PubMed] [Google Scholar]
- 36.Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough Peter J, et al. NF-κB-Independent Role of IKKα/IKKβ in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell 2015;60:63–76 [DOI] [PubMed] [Google Scholar]
- 37.Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol 2018;20:1389–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nature Communications 2017;8:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell 2017;66:698–710.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mihaly SR, Ninomiya-Tsuji J, Morioka S. TAK1 control of cell death. Cell Death & Differentiation 2014;21:1667–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morioka S, Broglie P, Omori E, Ikeda Y, Takaesu G, Matsumoto K, et al. TAK1 kinase switches cell fate from apoptosis to necrosis following TNF stimulation. J Cell Biol 2014;204:607–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 2008;4:313–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, et al. Cutting Edge: RIP1 Kinase Activity Is Dispensable for Normal Development but Is a Key Regulator of Inflammation in SHARPIN-Deficient Mice. J Immunol 2014;192:5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurien L, Nagata M, Schünke H, Delanghe T, Wiederstein JL, Kumari S, et al. Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation. Nature Communications 2020;11:1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003;114:181–90 [DOI] [PubMed] [Google Scholar]
- 46.Feoktistova M, Geserick P, Kellert B, Dimitrova Diana P, Langlais C, Hupe M, et al. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol Cell 2011;43:449–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell 2011;43:432–48 [DOI] [PubMed] [Google Scholar]
- 48.Feng S, Yang Y, Mei Y, Ma L, Zhu D-e, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007;19:2056–67 [DOI] [PubMed] [Google Scholar]
- 49.Cho Y, Challa S, Moquin D, Genga R, Ray TD, Guildford M, et al. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009;137:1112–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014;16:55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death & Differentiation 2017;24:1184–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nature Communications 2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3–induced NF-κB activation. Nature Immunol 2004;5:503–07 [DOI] [PubMed] [Google Scholar]
- 54.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 2013;288:31268–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaiser WJ, Offermann MK. Apoptosis Induced by the Toll-Like Receptor Adaptor TRIF Is Dependent on Its Receptor Interacting Protein Homotypic Interaction Motif. J Immunol 2005;174:4942. [DOI] [PubMed] [Google Scholar]
- 56.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3–mediated pathway. Proc Natl Acad Sci U S A 2011;108:20054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007;448:501–05 [DOI] [PubMed] [Google Scholar]
- 58.Rebsamen M, Heinz LX, Meylan E, Michallet M-C, Schroder K, Hofmann K, et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO reports 2009;10:916–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaiser WJ, Upton JW, Mocarski ES. Receptor-Interacting Protein Homotypic Interaction Motif-Dependent Control of NF-κB Activation via the DNA-Dependent Activator of IFN Regulatory Factors. J Immunol 2008;181:6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 2020;580:391–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, et al. ZBP1 mediates interferon-induced necroptosis. Cell Mol Immunol 2020;17:356–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 2016;1:aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He S, Wang L Fau - Miao L, Miao L Fau - Wang T, Wang T Fau - Du F, Du F Fau - Zhao L, Zhao L Fau - Wang X, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137:1100–11 [DOI] [PubMed] [Google Scholar]
- 64.Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer 2019;18:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geserick P, Wang J, Schilling R, Horn S, Harris PA, Bertin J, et al. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis 2015;6:e1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Najafov A, Zervantonakis IK, Mookhtiar AK, Greninger P, March RJ, Egan RK, et al. BRAF and AXL oncogenes drive RIPK3 expression loss in cancer. PLoS Biol 2018;16:e2005756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016;5:e1149673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol 2019;4:eaaw2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seifert L, Werba G, Tiwari S, Giao Ly NN, Alothman S, Alqunaibit D, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016;532:245–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiao D, Cai Z, Choksi S, Ma D, Choe M, Kwon H-J, et al. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Research 2018;28:868–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016;536:215–18 [DOI] [PubMed] [Google Scholar]
- 72.Hou J, Ju J, Zhang Z, Zhao C, Li Z, Zheng J, et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis 2019;10:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patel S, Webster JD, Varfolomeev E, Kwon YC, Cheng JH, Zhang J, et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death & Differentiation 2020;27:161–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis 2019;10:650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol 2017;27:673–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015;526:660–65 [DOI] [PubMed] [Google Scholar]
- 77.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–71 [DOI] [PubMed] [Google Scholar]
- 78.Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018;48:35–44.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018;362:956–60 [DOI] [PubMed] [Google Scholar]
- 80.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, Núñez G, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci U S A 2017;114:E961–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vince James E, Wong WW-L, Gentle I, Lawlor Kate E, Allam R, O’Reilly L, et al. Inhibitor of Apoptosis Proteins Limit RIP3 Kinase-Dependent Interleukin-1 Activation. Immunity 2012;36:215–27 [DOI] [PubMed] [Google Scholar]
- 82.Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 2018;281:8–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, et al. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nature Communications 2019;10:2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep 2017;21:3846–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, Rourke K, Mirrashidi KM, et al. IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal 2019;12:eaax4917. [DOI] [PubMed] [Google Scholar]
- 86.Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019;35:559–72.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017;547:99–103 [DOI] [PubMed] [Google Scholar]
- 88.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nature Communications 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Masuda Y, Futamura M, Kamino H, Nakamura Y, Kitamura N, Ohnishi S, et al. The potential role of DFNA5, a hearing impairment gene, in p53-mediated cellular response to DNA damage. J Hum Genet 2006;51:652–64 [DOI] [PubMed] [Google Scholar]
- 90.Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020;579:415–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nature Communications 2019;10:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, et al. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020;52:475–86.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting Immunotherapy Impact by Lowering Tumor TNF Cytotoxicity Threshold. Cell 2019;178:585–99.e15 [DOI] [PubMed] [Google Scholar]
- 94.Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lin Y, Devin A, Rodriguez Y, Liu Z-g. Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes Dev 1999;13:2514–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 2011;471:363–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pop C, Oberst A, Drag M, Van Raam Bram J, Riedl Stefan J, Green Douglas R, et al. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochemical Journal 2011;433:447–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by flip. Nat Rev Immunol 2001;1:50–58 [DOI] [PubMed] [Google Scholar]
- 99.Oberst A, Green DR. It cuts both ways: reconciling the dual roles of caspase 8 in cell death and survival. Nat Rev Mol Cell Biol 2011;12:757–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fritsch M, Günther SD, Schwarzer R, Albert M-C, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019;575:683–87 [DOI] [PubMed] [Google Scholar]
- 101.Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019;575:679–82 [DOI] [PubMed] [Google Scholar]
- 102.O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol 2011;13:1437–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Stupack DG. Caspase-8 as a therapeutic target in cancer. Cancer Lett 2013;332:133–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 2019;38:e101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, et al. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 2020;367:1379–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018;362:1064–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mukhopadhyay H, Lee NY. Multifaceted roles of TAK1 signaling in cancer. Oncogene 2020;39:1402–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Henry CM, Martin SJ. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol Cell 2017;65:715–29 [DOI] [PubMed] [Google Scholar]
- 109.Hartwig T, Montinaro A, von Karstedt S, Sevko A, Surinova S, Chakravarthy A, et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol Cell 2017;64:730–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012;12:860–75 [DOI] [PubMed] [Google Scholar]
- 111.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006;26:174–9 [DOI] [PubMed] [Google Scholar]
- 112.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 2007;8:487–96 [DOI] [PubMed] [Google Scholar]
- 113.Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med 2013;210:491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jain A, Song R, Wakeland EK, Pasare C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun 2018;9:3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krysko DV, Ravichandran KS, Vandenabeele P. Macrophages regulate the clearance of living cells by calreticulin. Nat Commun 2018;9:4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng M, Marjon KD, Zhu F, Weissman-Tsukamoto R, Levett A, Sullivan K, et al. Programmed cell removal by calreticulin in tissue homeostasis and cancer. Nature Communications 2018;9:3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Osman R, Tacnet-Delorme P, Kleman JP, Millet A, Frachet P. Calreticulin Release at an Early Stage of Death Modulates the Clearance by Macrophages of Apoptotic Cells. Front Immunol 2017;8:1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007;13:54–61 [DOI] [PubMed] [Google Scholar]
- 119.Bajor A, Tischer S, Figueiredo C, Wittmann M, Immenschuh S, Blasczyk R, et al. Modulatory role of calreticulin as chaperokine for dendritic cell-based immunotherapy. Clin Exp Immunol 2011;165:220–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gong H, Zuliani P, Komuravelli A, Faeder JR, Clarke EM. Analysis and verification of the HMGB1 signaling pathway. BMC Bioinformatics 2010;11 Suppl 7:S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, et al. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 2008;283:34457–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. HMGB1 and RAGE in inflammation and cancer. Annu Rev Immunol 2010;28:367–88 [DOI] [PubMed] [Google Scholar]
- 123.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nature 2000;405:354–60 [DOI] [PubMed] [Google Scholar]
- 124.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev 2007;220:47–59 [DOI] [PubMed] [Google Scholar]
- 125.Pandolfi F, Altamura S, Frosali S, Conti P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin Ther 2016;38:1017–28 [DOI] [PubMed] [Google Scholar]
- 126.Gombault A, Baron L, Couillin I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol 2012;3:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Save S, Persson K. Extracellular ATP and P2Y receptor activation induce a proinflammatory host response in the human urinary tract. Infect Immun 2010;78:3609–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Beste MT, Lomakina EB, Hammer DA, Waugh RE. Immobilized IL-8 Triggers Phagocytosis and Dynamic Changes in Membrane Microtopology in Human Neutrophils. Ann Biomed Eng 2015;43:2207–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Amores-Iniesta J, Barbera-Cremades M, Martinez CM, Pons JA, Revilla-Nuin B, Martinez-Alarcon L, et al. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep 2017;21:3414–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bent R, Moll L, Grabbe S, Bros M. Interleukin-1 Beta-A Friend or Foe in Malignancies? Int J Mol Sci 2018;19:2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol 2013;4:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim KE, Song H, Hahm C, Yoon SY, Park S, Lee HR, et al. Expression of ADAM33 is a novel regulatory mechanism in IL-18-secreted process in gastric cancer. J Immunol 2009;182:3548–55 [DOI] [PubMed] [Google Scholar]
- 133.Park S, Cheon S, Cho D. The dual effects of interleukin-18 in tumor progression. Cell Mol Immunol 2007;4:329–35 [PubMed] [Google Scholar]
- 134.Neumann K, Schiller B, Tiegs G. NLRP3 Inflammasome and IL-33: Novel Players in Sterile Liver Inflammation. Int J Mol Sci 2018;19:2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 2003;374:1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luo Y, Zheng SG. Hall of Fame among Pro-inflammatory Cytokines: Interleukin-6 Gene and Its Transcriptional Regulation Mechanisms. Front Immunol 2016;7:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 2014;6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol 2014;26:38–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009;31:84–98 [DOI] [PubMed] [Google Scholar]
- 140.Hangai S, Ao T, Kimura Y, Matsuki K, Kawamura T, Negishi H, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci U S A 2016;113:3844–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kim S, Elkon KB, Ma X. Transcriptional Suppression of Interleukin-12 Gene Expression following Phagocytosis of Apoptotic Cells. Immunity 2004;21:643–53 [DOI] [PubMed] [Google Scholar]
- 142.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998;101:890–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang Q, Ju X, Zhou Y, Chen K. Necroptotic cells release find-me signal and are engulfed without proinflammatory cytokine production. In Vitro Cellular & Developmental Biology - Animal 2015;51:1033–39 [DOI] [PubMed] [Google Scholar]
- 144.Wang Q, Imamura R, Motani K, Kushiyama H, Nagata S, Suda T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. International Immunology 2013;25:363–72 [DOI] [PubMed] [Google Scholar]
- 145.Aaes Tania L, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep 2016;15:274–87 [DOI] [PubMed] [Google Scholar]
- 146.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97–111 [DOI] [PubMed] [Google Scholar]
- 147.Elion DL, Jacobson ME, Hicks DJ, Rahman B, Sanchez V, Gonzales-Ericsson PA-O, et al. Therapeutically Active RIG-I Agonist Induces Immunogenic Tumor Cell Killing in Breast Cancers. Cancer Res 2018;78:6138–95 [DOI] [PubMed] [Google Scholar]
- 148.Takemura R, Takaki H, Okada S, Shime H, Akazawa T, Oshiumi H, et al. PolyI:C-Induced, TLR3/RIP3-Dependent Necroptosis Backs Up Immune Effector-Mediated Tumor Elimination In Vivo. Cancer Immunol Res 2015;3:902–14 [DOI] [PubMed] [Google Scholar]
- 149.Vanacker H, Vetters J, Moudombi L, Caux C, Janssens S, Michallet M-C. Emerging Role of the Unfolded Protein Response in Tumor Immunosurveillance. Trends Cancer 2017;3:491–505 [DOI] [PubMed] [Google Scholar]
- 150.McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer 2020;20:203–17 [DOI] [PubMed] [Google Scholar]
- 151.Liu Y-g, Chen J-k, Zhang Z-t, Ma X-j, Chen Y-c, Du X-m, et al. NLRP3 inflammasome activation mediates radiation-induced pyroptosis in bone marrow-derived macrophages. Cell Death Dis 2017;8:e2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. New England Journal of Medicine 2018;379:2220–29 [DOI] [PubMed] [Google Scholar]
- 153.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. New England Journal of Medicine 2018;379:2108–21 [DOI] [PubMed] [Google Scholar]
- 154.Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum–etoposide versus platinum–etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. The Lancet 2019;394:1929–39 [DOI] [PubMed] [Google Scholar]
- 155.Erkes DA, Cai W, Sanchez IM, Purwin TJ, Rogers C, Field CO, et al. Mutant BRAF and MEK inhibitors regulate the tumor immune microenvironment via pyroptosis. Cancer Discov 2020;10:254–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lu H, Zhang S, Wu J, Chen M, Cai M-C, Fu Y, et al. Molecular targeted therapies elicit concurrent apoptotic and GSDME-dependent pyroptotic tumor cell death. Clin Cancer Res 2018;24:6066–77 [DOI] [PubMed] [Google Scholar]
- 157.Hossain Dms Fau - Javaid S, Javaid S Fau - Cai M, Cai M Fau - Zhang C, Zhang C Fau - Sawant A, Sawant A Fau - Hinton M, Hinton M Fau - Sathe M, et al. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. Journal of Clinical Investigation 2018;128:644–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rozeman EA, Blank CU. Combining checkpoint inhibition and targeted therapy in melanoma. Nature Medicine 2019;25:879–82 [DOI] [PubMed] [Google Scholar]
- 159.Ribas A, Lawrence D, Atkinson V, Agarwal S, Miller WH, Carlino MS, et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nature Medicine 2019;25:936–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nature Medicine 2019;25:929–35 [DOI] [PubMed] [Google Scholar]
- 161.Brumatti G, Ma C, Lalaoui N, Nguyen N-Y, Navarro M, Tanzer MC, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Science Translational Medicine 2016;8:339ra69. [DOI] [PubMed] [Google Scholar]
- 162.McComb S, Aguadé-Gorgorió J, Harder L, Marovca B, Cario G, Eckert C, et al. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Science Translational Medicine 2016;8:339ra70. [DOI] [PubMed] [Google Scholar]
- 163.Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res 2016;76:5442–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Beug ST, Beauregard CE, Healy C, Sanda T, St-Jean M, Chabot J, et al. Smac mimetics synergize with immune checkpoint inhibitors to promote tumour immunity against glioblastoma. Nature Communications 2017;8:14278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Michie J, Kearney CJ, Hawkins ED, Silke J, Oliaro J. The Immuno-Modulatory Effects of Inhibitor of Apoptosis Protein Antagonists in Cancer Immunotherapy. Cells 2020;9:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sun X-S, Tao Y, Le Tourneau C, Pointreau Y, Sire C, Kaminsky M-C, et al. Debio 1143 and high-dose cisplatin chemoradiotherapy in high-risk locoregionally advanced squamous cell carcinoma of the head and neck: a double-blind, multicentre, randomised, phase 2 study. The Lancet Oncology 2020;21:1173–87 [DOI] [PubMed] [Google Scholar]
- 167.van Vloten JP, Workenhe ST, Wootton SK, Mossman KL, Bridle BW. Critical Interactions between Immunogenic Cancer Cell Death, Oncolytic Viruses, and the Immune System Define the Rational Design of Combination Immunotherapies. J Immunol 2018;200:450–58 [DOI] [PubMed] [Google Scholar]
- 168.Ma J, Ramachandran M, Jin C, Quijano-Rubio C, Martikainen M, Yu D, et al. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis 2020;11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Perrot I, Michaud H-A, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep 2019;27:2411–25.e9 [DOI] [PubMed] [Google Scholar]
- 170.Li X-Y, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, et al. Targeting CD39 in cancer reveals an extracellular ATP and inflammasome driven tumor immunity. Cancer Discov 2019;9:1754–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Haratani K, Yonesaka K, Takamura S, Maenishi O, Kato R, Takegawa N, et al. U3–1402 sensitizes HER3-expressing tumors to PD-1 blockade by immune activation. J Clin Invest 2020;130:374–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol 2018;62:29–39 [DOI] [PubMed] [Google Scholar]
- 173.Wan N, Ji B, Li J, Jiang J, Yang C, Zhang T, et al. A pooled meta-analysis of PD-1/L1 inhibitors incorporation therapy for advanced non-small cell lung cancer. Onco Targets Ther 2019;12:4955–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, et al. Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res 2020;26:487–504 [DOI] [PubMed] [Google Scholar]
- 176.Gaiha GD, McKim KJ, Woods M, Pertel T, Rohrbach J, Barteneva N, et al. Dysfunctional HIV-specific CD8+ T cell proliferation is associated with increased caspase-8 activity and mediated by necroptosis. Immunity 2014;41:1001–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kähler KC, Hassel JC, Heinzerling L, Loquai C, Thoms K-M, Ugurel S, et al. Side effect management during immune checkpoint blockade using CTLA-4 and PD-1 antibodies for metastatic melanoma – an update. JDDG: Journal der Deutschen Dermatologischen Gesellschaft 2020;18:582–609 [DOI] [PubMed] [Google Scholar]
- 178.Rotz SJ, Leino D, Szabo S, Mangino JL, Turpin BK, Pressey JG. Severe cytokine release syndrome in a patient receiving PD-1-directed therapy. Pediatric Blood & Cancer 2017;64:e26642. [DOI] [PubMed] [Google Scholar]
- 179.Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA: A Cancer Journal for Clinicians 2020;70:86–104 [DOI] [PubMed] [Google Scholar]
- 180.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019;569:270–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019;35:830–49 [DOI] [PubMed] [Google Scholar]
- 182.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death & Differentiation 2009;16:1438–44 [DOI] [PubMed] [Google Scholar]
- 183.Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020:eaaz7548. [DOI] [PubMed] [Google Scholar]