

SUMMARY
Hepatic stellate cells (HSCs) are resident non-parenchymal liver pericytes whose plasticity enables them to regulate a remarkable range of physiologic and pathologic responses. To support their functions in health and disease, HSCs engage pathways regulating carbohydrate, mitochondrial, lipid, and retinoid homeostasis. In chronic liver injury, HSCs drive hepatic fibrosis and are implicated in inflammation and cancer. To do so, the cells activate, or transdifferentiate, from a quiescent state into proliferative, motile myofibroblasts that secrete extracellular matrix, which demands rapid adaptation to meet a heightened energy need. Adaptations include reprogramming of central carbon metabolism, enhanced mitochondrial number and activity, endoplasmic reticulum stress, and liberation of free fatty acids through autophagy-dependent hydrolysis of retinyl esters that are stored in cytoplasmic droplets. As an archetype for pericytes in other tissues, recognition of the HSCs’ metabolic drivers and vulnerabilities offer the potential to target these pathways therapeutically to enhance parenchymal growth and modulate repair.
INTRODUCTION
Hepatic stellate cells (HSCs) are a resident non-parenchymal liver cell population that first gained attention because they are the primary fibrogenic cell type in liver injury (Friedman, 2008). Yet, stellate cells (previously termed lipocytes, fat-storing, perisinusoidal, or Ito cells) are now appreciated as a remarkably plastic cell type that regulates hepatic growth, immunity, and inflammation, as well as energy and nutrient homeostasis. These functions rely on metabolic versatility and tight regulation of energy expenditure. Recent studies have brought to light the pathways underlying the cell’s plasticity, and their contributions to both liver homeostasis and the organ’s response to injury. Given the role of the liver in regulating whole-body energy metabolism, HSCs are also sensitive to systemic metabolic regulation and disruption. Moreover, since HSCs are liver-specific pericytes akin to similar cells with fibrogenic potential in other organs, they are a paradigm for metabolic regulation of tissue homeostasis outside the liver. Importantly, HSCs are also an overlooked determinant of immunometabolism in supporting liver function and the response to injury.
Stellate cells are derived from the septum transversum mesenchyme during development. Mesothelial and submesothelial cells of mesenchymal origin migrate inward from the embryonic liver surface, giving rise to HSCs and other perivascular mesenchymal cells (Asahina et al., 2009, 2011). In normal liver, HSCs have best been recognized for their storage of vitamin A as retinyl esters within perinuclear droplets; the cells are the primary depot for the body’s retinoid stores (Blomhoff and Wake, 1991) (see below).
The expanding application of single-cell transcriptomic methods to characterize HSCs has yielded a nuanced and deeper appreciation of their heterogeneity (Dobie et al., 2019; Ramachandran et al., 2019, 2020; Xiong et al., 2019). Single-cell RNA sequencing has thereby uncovered zonally distributed subpopulations of HSCs in the healthy liver—portal-vein-associated HSCs and central-vein associated HSCs (Dobie et al., 2019). During liver injury HSCs transdifferentiate, or “activate” into proliferative, fibrogenic myofibroblasts (MFBs) that acquire a range of feature that collectively perpetuate injury and fibrosis (Figure 1) (Tsuchida and Friedman, 2017); as liver injury subsides the number of HSC-derived MFBs decline through either apoptosis or reversion to an “inactivated” phenotype. These inactivated cells harbor unique epigenetic signatures, and these HSCs re-activate more readily upon re-injury (Kisseleva et al., 2012; Liu et al., 2020; Troeger et al., 2012).
Figure 1. Features of HSC Activation and Resolution.

Normally quiescent vitamin-A-rich pericytic cells, upon liver injury the cells initiate activation, rendering them responsive to many cytokines and soluble signals that trigger a full cellular transdifferentiation to myofibroblasts, whose features include enhanced proliferation, contractility, fibrogenesis, as well as altered matrix degradation properties, chemotaxis (directed migration), and the capacity to modify the inflammatory and immune milieu. Once injury resolves, the fate of these cells includes apoptosis, progressive senescence and/or deactivation to a more quiescent, intermediate cell phenotype. The relative distribution among these three fates during fibrosis resolution has not been determined, however.
Genes related to zonation are preserved following liver injury, and lineage tracing reveals that the dominant source of pathogenic MFBs are pericentral HSCs when the injury begins in that region (Dobie et al., 2019), underscoring the concept that patterns of activation overlap with the areas of injury. Moreover, in pericentral injury, periportal HSCs respond with increased proliferative capacity, but they may not transdifferentiate into collagen-producing MFBs (Dobie et al., 2019). Apart from their zonal distribution activated HSCs can also be subdivided into other subpopulations that are pro-regenerative (high growth factor expression), anti-regenerative (profibrogenic), and mixed (Krenkel et al., 2019). While the paradigm of “quiescent,” “activated,” and “inactivated” cellular states provides a valuable conceptual construct, it may not fully reflect intermediate or hybrid states. HSC heterogeneity might be manifested as different subtypes with variable capacity for activation, or with divergent contributions to regeneration, fibrosis, cancer, and immunomodulation, among others. For example, during regeneration, HSCs contribute to the initiation and termination of liver regeneration and are a major source of hepatocyte growth factor (HGF) in the liver (Bansal, 2016; Maher, 1993). They also support sinusoidal endothelial cell integrity through paracrine interactions, which in turn are critical for angiogenesis during repair and regeneration (DeLeve, 2013, 2015; Desroches-Castan et al., 2019).
Chronic liver injury promotes ongoing activation of HSCs, leading to accumulation of extracellular matrix proteins that disrupt the liver’s architecture and undermine its function (Henderson et al., 2013; Mederacke et al., 2013). HSC activation is triggered by a number of signals that serve as markers of cellular injury, including proinflammatory cytokines produced by infiltrating immune cells, hepatocyte apoptotic bodies, endothelial-cell-mediated growth factor activation, and increased reactive oxygen species (ROS) burden. The cells’ response to injury is potentiated by an array of paracrine and autocrine loops (Higashi et al., 2017), including fibrogenic signals such as transforming growth factor beta 1 (TGF-β1) and connective tissue growth factor (CTGF).
Activation of HSCs requires energy to support the cell’s ability to proliferate; secrete extracellular matrix, proteases, and cytokines; and migrate toward regions of cellular injury. To meet these energy demands, the cells harness a number of metabolic pathways similar to cancer cells, which also have high energy demands (Lane et al., 2020), although the latter have unregulated growth driven by major genomic and epigenomic disruptions not described in HSCs.
The rapidly evolving breadth of the HSC’s functions and the highly contextual nature of its responses make understanding its metabolic regulation a new priority, with broader implications for pericyte metabolism in other tissues. In this review, we integrate emerging insights into HSC energetics, its crosstalk with cellular stress responses, and non-cell autonomous effects on liver homeostasis and immunometabolism to demonstrate how metabolic regulation of HSCs underlies their functions in health and disease, revealing a rich and wide-ranging biology.
HSC BIOENERGETICS
Carbohydrate Metabolism
Glycolysis and Gluconeogenesis
The reprogramming of HSC central carbon metabolism plays a major role in the cell’s activation, as they upregulate glycolysis to meet the energy demands of transdifferentiation into a myofibroblast (MFB) phenotype (Figure 2). Importantly, changes in carbon metabolism are not only a hallmark of the MFB phenotype, but they also contribute to activation. Studies interrogating glucose homeostasis regulation in HSCs rely almost entirely on cultured cells, and thus future studies will need to validate these findings in disease models in vivo.
Figure 2. Glucose and Mitochondrial Metabolism in Activated HSCs.
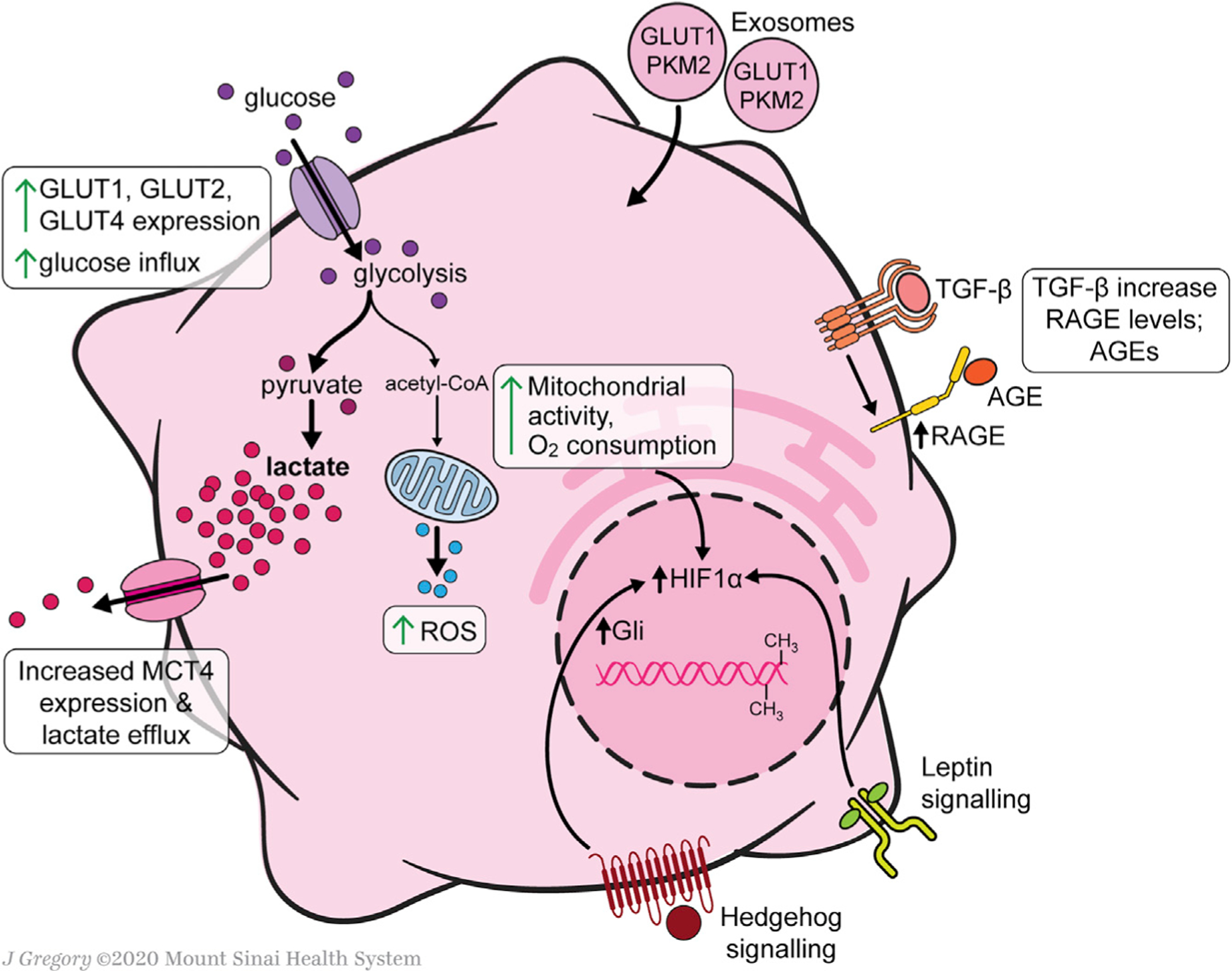
Exosomes from aHSCs, which contain GLUT1 and pyruvate kinase muscle isozyme 2 (PKM2), induce activation of qHSCs. Overall glycolytic flux is increased in activated HSCs relative to quiescent HSCs. Glucose influx capacity increases through upregulation of the glucose transporters GLUT1, GLUT2, and GLUT4. The pyruvate byproduct of glycolysis is shunted primarily toward lactate production, which accumulates in the cells despite increased expression of the lactate transporter MCT4 and increased lactate efflux. Lactate accumulation is a contributor to HSC activation. Despite a preferential shunting toward lactate production, overall mitochondrial activity is increased when HSCs activate. Additionally, there is an increase in mitochondrial ROS production. Major pro-fibrotic signals that contribute to the reprogramming of glucose metabolism include Hh signaling through Gli and leptin, both of which converge upon the transcription factor HIF-1α, an inducer of glucose transporters and glycolytic enzymes. DNA methyltransferase and histone methyltransferases induce epigenetic modifications that shift HSC metabolism toward increased glycolysis. TGF-β signaling increases HSC expression of RAGE, thereby making activated HSCs sensitive to global glucose metabolism, as AGEs are a result of hyperglycemia.
Compared with their quiescent counterparts, immortalized human HSCs (aHSCs) and culture-activated rat HSCs have increased glucose utilization rates (Gajendiran et al., 2018). Primary culture-activated cells have enhanced glucose transport capacity and glycolytic activity (Chen et al., 2012). Glucose transporter proteins, including GLUT1 (Chen et al., 2012). GLUT2 (Lin and Chen, 2011), and GLUT4 (Chandrashekaran et al., 2016), are expressed in primary culture-activated or immortalized rat HSCs and can be regulated under these conditions by high extracellular glucose (Chen et al., 2012) or purinergic signaling (Chandrashekaran et al., 2016). Interestingly, GLUT1 also is highly overexpressed in cancer cells, which primarily rely on anaerobic metabolism for ATP production; in cancer cells, glucose transport constitutes a rate-limiting step for ATP production (Amann and Hellerbrand, 2009).
In either immortalized human or primary murine HSCs, there is also increased mRNA expression of genes related to intracellular processing of glucose, hexokinase 2 (HK2), fructose-2,6-bi-sphosphatase-3 (PFKFB3), and pyruvate kinase (PK) (Mejias et al., 2020). Upregulation of PFKFB3 is mediated by stabilization of its mRNA through binding its 3-untranslated region by the cytoplasmic polyadenylation-element-binding protein 4 (CEBP) (Mejias et al., 2020). Enhanced glycolysis attributable to these enzyme inductions is accompanied by downregulation of genes involved in gluconeogenesis, including phosphoenolpyruvate carboxykinase 1 (PCK1) and fructose bisphosphatase 1 (FBP1) (Chen et al., 2012).
Increased glycolysis in cultured HSCs is accompanied by shunting of central carbon metabolic products away from the citric acid cycle. Activated HSCs have increased expression of pyruvate dehydrogenase kinase 3 (PDK3), which inhibits the conversion of pyruvate to acetyl coenzyme A, thereby channeling pyruvate toward lactate production. As noted above, a similar phenomenon, known as the Warburg effect, is observed in cancer cells (Liberti and Locasale, 2016; Vander Heiden et al., 2009). Like in cancer cells, this response may confer an evolutionary advantage by allowing them to prioritize ATP production rate over efficiency (i.e., ATP yield per glucose molecule) (Liberti and Locasale, 2016), or by diverting resources away from ATP production and toward proliferation (Vander Heiden et al., 2009). By analogy, both possibilities support the enhanced energy demands of aHSCs to secrete extracellular matrix (ECM) protein, migrate, and proliferate. These features are likely to be relevant to myofibroblasts in other tissues, but it is unclear whether the Warburg-like effect is more dominant in myofibroblasts than other proliferating cell types during injury. However, the enzyme pyruvate kinase M2 (PKM2) may represent a unique, shared link between aHSCs and cancer cells (Christofk et al., 2008), since it is induced in both cell types to promote aerobic glycolysis (Zheng et al., 2020). A PKM2 antagonist is antifibrotic in an animal model by promoting tetramerization of the enzyme, which attenuates its activity; in contrast, the activating effect of PKM2 is mediated by histone modifications, albeit in cultured HSCs but not yet established in vivo (Zheng et al., 2020).
Interestingly, lactate does not play a bystander role in HSC metabolism; rather, it is directly implicated in HSC activation and the perpetuation of the MFB phenotype. Despite upregulation of expression of the lactate export pump monocarboxylate transporter 4 (MCT4), intracellular levels of lactate remain elevated in aHSCs. Importantly, blocking the intracellular accumulation of lactate inhibits proliferation, suppresses genes associated with the MFB signature, and induces lipid accumulation and lipogenic genes (Chen et al., 2012).
Exosomes provide a mechanism for the rapid induction of glycolysis to support the metabolic reprogramming from quiescent HSCs (qHSCs) to activated HSCs (aHSCs), allowing for a synchronization of stromal cell injury response. Exosomes from aHSCs, which contain GLUT1 and PKM2, induce activation of qHSCs (Wan et al., 2019). The production of exosomes by aHSCs is potentiated by hypoxia-inducible factor 1-alpha (HIF-1α) signaling, which stimulates HSC activation in hypoxic and inflammatory conditions (Wang et al., 2013).
Epigenetic regulation also contributes to shifts in glucose metabolism in HSCs. DNA methyltransferase DNMT1 and histone methyltransferase G9a induce epigenetic modifications that shift HSC metabolism toward increased glycolysis (Barcena-Varela et al., 2020). The profibrotic cytokine TGF-β triggers recruitment of these enzymes to chromatin in cultured human HSCs. Furthermore, combined inhibition of DNMT1 and G9a returns glycolytic rates to those of quiescent HSCs in in vitro models of hypoxia-driven and TGF-β1-driven activation (Barcena-Varela et al., 2020).
Glycosylation
In addition to changes in carbohydrate catabolism, aHSCs display altered protein glycosylation profiles (Qin et al., 2012; Wu et al., 2017). The cell-surface glycome of aHSCs contains glycans that are favorable for galectin-1 (Gal-1) binding. Gal-1, which is upregulated in aHSCs relative to qHSCs, binds to neuropilin-1 (NRP-1), a receptor that induces angiogenesis, vascular permeability, and wound healing (Troeger and Schwabe, 2011). The interaction of Gal-1 and NRP-1 induces TGF-β- and PDGF-like signaling cascades (Wu et al., 2017) (TGF-β signaling is discussed further in the redox metabolism section, below).
Hyperinsulinemia and Hyperglycemia
Hyperinsulinemia and hyperglycemia, key features of the metabolic syndrome, can activate HSCs in culture but their specific contribution to HSC activation in the context of NAFLD and NASH is unknown. For example, HSCs grown in insulin-containing media undergo activation, as evidenced by a loss of lipid droplets (Lin et al., 2009), and have increased expression of CTGF, a key fibrogenic signaling molecule (Gao and Brigstock, 2004; Paradis et al., 2001). Primary murine HSCs cultured in high-glucose media have increased production of collagen type I, which is mediated by the SMAD3 transcription factor. Exposure to high glucose results in HSC upregulation of protein inhibitor of Stat4 (PIAS4), which represses transcription of SIRT1. Reduced SIRT1 leads to lysine hyperacetylation of SMAD3, thereby increasing affinity of SMAD3 for its profibrotic DNA-binding targets (Sun et al., 2016). Like many studies interrogating HSC metabolism; however, as noted, the links between hyperglycemia and hyperinsulinemia and HSC activation are based on culture studies, rather than in vivo studies. The only in vivo studies linking hyperinsulinemia to HSC responses describes enhanced mitochondrial volume but not typical features of HSC activation (Evert et al., 1998)
Advanced glycation end products (AGEs), which are generated by non-enzymatic reactions between proteins and sugars, also activate HSCs. In culture, murine HSC activation driven by TGF-β induces expression of the receptor for advanced glycation end product (RAGE), an effect that is abrogated through MAPK/ERK pathway blockade (Fehrenbach et al., 2001). Medium containing AGEs increases fibrogenic gene expression and ROS by cultured human HSCs (Iwamoto et al., 2008). In vivo, rats injected intraperitoneally with AGEs, develop more severe liver fibrosis following bile-duct ligation (BDL) compared with those not receiving AGEs. AGE-exposure also increased a-smooth muscle actin protein levels but does not induce fibrosis (Goodwin et al., 2013). This finding suggests that AGEs and hyperglycemia alone may not be sufficient to induce fibrosis; however, in the presence of tissue injury they may amplify fibrogenesis driven by other factors.
Mitochondrial Metabolism
Despite a preferential increase in glycolysis, the demands of maintaining a myofibroblast-like phenotype still require major energy contributions from oxidative phosphorylation, reflected in an increase in mitochondrial number (Chen et al., 2012) and activity (Gajendiran et al., 2018) during cellular activation. However, changes in mitochondrial metabolism from activation may not be limited to ATP production, as studies point to a role in ROS production as well (Jain et al., 2013) (the contributions of ROS to HSC activation are detailed in subsequent sections).
Compared with freshly isolated qHSCs in which mitochondria are sparse, culture-activated HSCs have abundant mitochondria (Chen et al., 2012; Friedman et al., 1985). This increased number is accompanied by heightened mitochondrial activity, as measured by elevated oxygen consumption rate, mitochondrial membrane potential, and expression of F1-Fo ATPase (Gajendiran et al., 2018). Mitochondrial uncoupling inhibits HSC activation in vitro. Treatment with the chemical uncouplers FCCP and valinomycin reduce HSC expression of activation markers and blunt the response to the profibrotic cytokine TGF-β (Guimarães et al., 2012). Uncoupling also reduces ATP production by 30% (Guimarães et al., 2012), suggesting that despite upregulation of glycolysis, a significant and necessary portion of ATP production is still generated through oxidative phosphorylation. In vivo, enhanced mitochondrial volume in HSCs has been described in hyperinsulinemic mice (Evert et al., 1998), similar to changes described in in-culture-activated HSCs (Friedman et al., 1992, 1989, 1985). Interestingly, there are no studies to date that document similar features of mitochondria in aHSCs in fibrotic liver in vivo.
Fibroblast populations from other tissues reinforce a broader context for these findings. For example, in kidney fibroblasts TGF-β activity increases ROS production through inhibition of the mitochondrial protein complex III (Jain et al., 2013), a member of the electron transport chain. In cardiac fibroblasts, TGF-β stimulation increases mitochondrial ROS through NLRP3, a pattern recognition receptor implicated in the pathogenesis of inflammatory diseases (Bracey et al., 2014).
Vitamin A and Lipid Metabolism
Retinol Storage and Lipid Droplets
Regulation of vitamin A homeostasis is a defining characteristic of HSCs in healthy and injured liver. HSCs store 50%–95% of the body’s vitamin A (Blomhoff and Blomhoff, 2006; Saeed et al., 2017), which is comprised retinol and its metabolites. Over 95% of these retinoids are stored as retinyl esters (REs), which account for 30%–50% of HSC cytoplasmic lipid droplet content (Senoo et al., 2010). While RE may be a precursor for retinoic acid (RA) and hence increase retinoid signaling, they may also function as buffer that limits the amount of active retinoids. Retinoids in HSCs are associated with several perilipins, proteins that associate with the surface of lipid droplets. These include adipose differentiation-related protein (ADRP) (Fukushima et al., 2005; Lee et al., 2010), adipophilin (Straub et al., 2013), perilipin 5 (Lin et al., 2018), ATGL, and CGI-58 (Eichmann et al., 2015), as well as liver fatty-acid-binding protein (L-FABP) (Chen et al., 2013). Knowledge about the functional roles of perilipins in this cell type is limited, but in general greater perilipin expression reduces HSC activation, possibly by stabilizing retinoid droplets to attenuate their catabolism to fatty acids that provide energy (Lee et al., 2010; Zhou et al., 2020).
Blood vitamin A levels are tightly regulated, but specific mechanisms of vitamin A uptake and release in the healthy liver are not fully clarified (Saeed et al., 2017). Retinol uptake by HSCs occurs both as free retinol and as holo-RBP, the combination of retinol-binding protein (RBP) and retinol, although the latter is preferentially esterified (Ottonello et al., 1987; Trøen et al., 1994). Retinol esterification is primarily under the enzymatic control of lecithin:- retinol acyltransferase (LRAT) (Ajat et al., 2017; Blaner et al., 1985, 1990; Blomhoff et al., 1985; Matsuura et al., 1997; Trøen et al., 1994), although acyl-CoA:retinol acyltransferase (ARAT) also plays a minor role (Ajat et al., 2017; Ross, 1982).
As with many features of HSCs, the cells are heterogenous with respect to retinoid content (D’Ambrosio et al., 2011; Krenkel et al., 2019). Differential FACS sorting of primary HSCs—sorting for retinol autofluorescence versus collagen-promoter driven GFP expression—reveals at least two major subpopulations within the healthy liver (D’Ambrosio et al., 2011). Sorting for autofluorescence reveals a subpopulation with a higher capacity for retinol storage, while sorting for collagen-promoter driven GFP expression uncovers a subpopulation with a more MFB-like phenotype, suggesting that these HSCs may be “primed” to respond to liver injury. The upregulation of genes related to retinol catabolism and smaller lipid droplet size in the “primed” HSCs reveals some early changes to HSC lipid metabolism that may occur in vivo in the setting of liver injury (D’Ambrosio et al., 2011).
Retinol is thought to contribute to maintenance of the qHSC phenotype by its interaction with the retinoic acid receptor β (RARβ) and retinoid X receptor α (RXRα) nuclear receptors (Wang et al., 2002). Vitamin A treatment of HSCs prevents culture activation and can partially maintain qHSC marker expression (GFAP and PPAR-γ) while suppressing the expression of MFB markers (αSMA, Col1a1, and HSP-47) (Yoneda et al., 2016). Additionally, retinol exposure causes a partial reversion of aHSCs to a quiescent state (El Taghdouini et al., 2015; Hong et al., 2018; Lee et al., 2010). RA activates the nuclear receptors RARβ and RXRα, allowing them to bind to RA responsive elements located in the Col1α2 promoter (Wang et al., 2002), thereby providing a mechanism for endogenous RA-mediated potentiation of a quiescence phenotype in physiologic conditions (Allenby et al., 1993). The exact relationship between retinyl ester catabolism and signaling through RARβ and RXRα is not defined, however.
The loss of lipid droplets occurs histologically in a biphasic manner (Testerink et al., 2012). The first phase involves the breakdown of lipid droplets into smaller units, which are then redistributed from the perinuclear region to newly formed cellular extensions. Within the aHSC population, retinoid content is relatively homogeneous, with similar enzymatic activity and lipid composition regardless of size or localization. In this initial stage, the retinyl ester content of the lipid droplet declines, while relative triglyceride levels increase. In the second phase the lipid droplets have reduced size, until none are present in the fully transdifferentiated MFB.
Retinyl ester hydrolases (REHs) liberate REs from lipid droplets during HSC activation (Saeed et al., 2017). A number of potential enzymes have also been implicated, including adipose triglyceride lipase (ATGL/PNPLA2) (Taschler et al., 2015), PNPLA3 (Pirazzi et al., 2014; Taschler et al., 2015), hormone-sensitive lipase (Mello et al., 2008; Pang et al., 2011), and carboxyl ester lipase (Weng et al., 1999). However, other uncharacterized REHs may contribute to hydrolysis as well (Schreiber et al., 2012). Culture studies suggest retinol release from HSCs occurs independently of RBP or binding partners, as inhibition of protein secretion does not modulate retinol secretion (Friedman et al., 1993; Sauvant et al., 2001). However, the primary modality of retinol secretion is controversial and could be characterized by either the direct release of free retinol into the blood by HSCs, or holo-RBP secretion by hepatocytes (Senoo et al., 2010).
HSC activation appears to require loss of lipid droplets through autophagy, which may be critical to fueling this metabolically demanding cellular response (Figure 3) (Hernández-Gea and Friedman, 2012). HSC activation increases intracellular REH activity and subsequently provokes release of retinol without an increase in the release of REs. The lack of extracellular REH activity in aHSCs further implicates intracellular hydrolysis in mediating retinol loss (Friedman et al., 1993). Bile salt-independent REHs (Friedman et al., 1993), PNPLA3 (Pirazzi et al., 2014), ATGL (Taschler et al., 2015), and lysosomal acid lipase (LAL) (Grumet et al., 2016; Tuohetahuntila et al., 2017) have been implicated as enzymatic mediators of RE release from lipid droplets. Interestingly, hormone-sensitive lipase, which is an important REH in liver adipocytes (Mello et al., 2008), is not highly expressed by HSCs and does not affect HSC lipid mobilization (Taschler et al., 2015). In parallel to increased hydrolase activity, aHSCs also have a diminished capacity for retinol esterification, as HSC activation results in a rapid loss of LRAT expression (Kluwe et al., 2011).
Figure 3. Lipid Metabolism during HSC Activation.
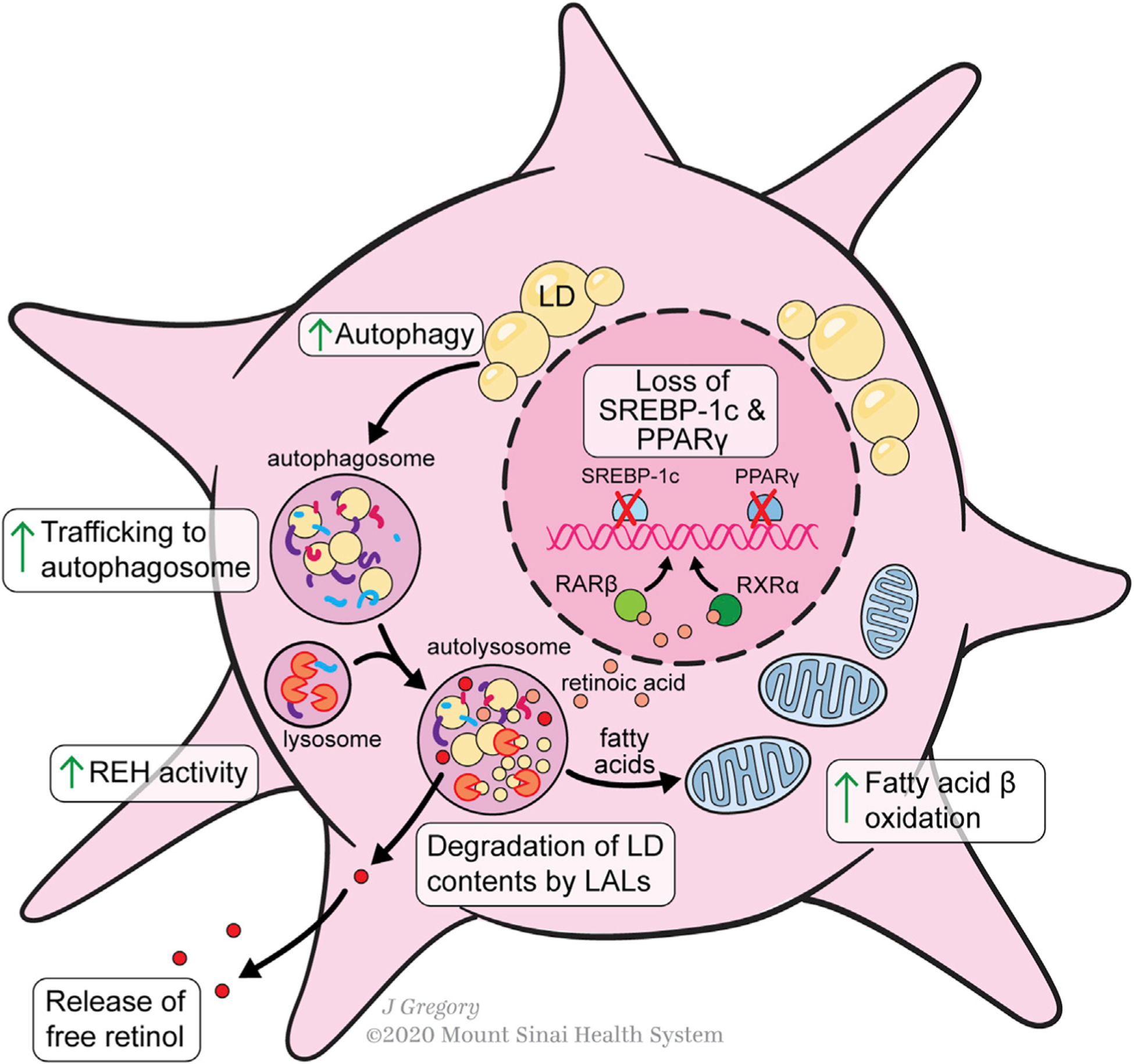
Lipid metabolism in activated HSCs is characterized by a loss of retinyl ester-containing cytoplasmic droplets. The transcription factors PPAR-γ and SREBP-1c, adipogenic master regulators, are markers of HSC quiescence that are downregulated during activation. Lipid droplets are trafficked to the autosome for degradation. Vitamin A de-esterification also occurs under the control of lysosomal acid lipase REH activity is also increased, liberating free retinol to the extracellular space. Lipid droplet contents from the autolysosome are further metabolized by the cell through increased fatty acid β-oxidation. Conversion of some intracellular retinol to retinoic acid promotes increased RARβ and RXRβ transcription.
Consistent with engagement of autophagy, retinyl ester processing occurs partly within lysosomes. REs are trafficked to the lysosome, likely through the autophagy pathway, where they are broken down by LAL into retinol (Tuohetahuntila et al., 2017). The inhibition of LAL, which also catabolizes cholesterol esters and triglycerides, decreases HSC activation. Notably, despite inhibition of lysosomal RE degradation, culture activation of HSCs still results in a 75% loss of REs, implying a significant role of other pathways in processing of lipid droplets (Tuohetahuntila et al., 2017).
Adipogenic Phenotype and Fatty Acids
Regulators of fatty acid content and metabolism control HSC activation. Peroxisome proliferator-activated receptor gamma (PPAR-γ) (Shao et al., 2016) and sterol regulatory-element-binding protein-1 (SREBP-1c) (Eberlé et al., 2004), two adipogenic transcriptional master regulators, are markers of HSC quiescence that are lost upon activation (She et al., 2005; Tsukamoto, 2005). Furthermore, ectopic induction of PPAR-γ and SREBP-1c can reverse HSC activation, demonstrating that an adipogenic transcriptional program maintains HSC quiescence (She et al., 2005; Tsukamoto, 2005), along with a growing list of transcriptional activators (Liu et al., 2020; Nakano et al., 2020; Wang and Friedman, 2020). Fatty acid synthesis-related genes under transcriptional control of PPAR-γ and SREBP-1c include acyl-CoA oxidase, solute carrier family 27 (fatty acid transporter) member 4, acetyl-CoA carboxylase, and fatty acid synthase (She et al., 2005). Fatty acids provide an important substrate for the esterification of retinol in qHSCs (Tsukamoto et al., 2006).
During activation, contents of HSC lipid droplets are metabolized to provide fatty acid species for oxidation, generating energy for activation/transdifferentiation under the control of PPAR-β (She et al., 2005), a known activator of fatty acid oxidation (Wang et al., 2003) (see above). PPAR-γ and SREBP-1c downregulation occurs through activation of the MAPK/ERK signaling cascade (Yan et al., 2012; Zhou et al., 2010), which is a nexus for several profibrogenic cytokines.
In early stages of activation, the most abundant fatty acids—palmitic, oleic, palmitoleic, and stearic acid—reach peak concentrations. This is followed by a decline in the total amount of free fatty acids in the later stages of activation, with a relative enrichment of arachidonic (C20:4) and docosahexaenoic (C22:6) acid and an upregulation of fatty acid elongases (Elovl5, Elovl6) and desaturases (Scd1) (Shmarakov et al., 2019). The overall decline in HSC fatty acid content is consistent with loss of an adipogenic phenotype. Fatty acid β-oxidation is an important energy source for HSCs undergoing activation, as inhibition of mitochondrial fatty acid catabolism blocks HSC activation (Hernández-Gea and Friedman, 2012). Interestingly, β-oxidation inhibition has analogous effects to autophagy inhibition, reinforcing the importance of autophagy to HSC lipolysis (Hernández-Gea and Friedman, 2012).
Acyl-CoA carboxylase (ACC), a regulator of fatty acid β-oxidation and de novo lipogenesis (DNL), has been implicated in HSC metabolic reprogramming during activation. ACC inhibition reduces HSC activation as measured by reduced αSMA expression and collagen production. The antifibrotic activity of ACC inhibitors is through inhibition of DNL, which is necessary for the induction of glycolysis and oxidative phosphorylation during HSC activation. While the molecular mechanism through which DNL regulates HSC metabolism has yet to be elucidated, ACC inhibition is being evaluated as a treatment for NASH, as it both reduces lipotoxicity in hepatocytes and inhibits HSC fibrogenesis (Bates et al., 2020); however, effects of ACC inhibition in vivo have not been directly ascribed to its effects on HSCs.
Cholesterol
Cholesterol contributes to the pathogenesis of fibrogenesis in NASH, as HSC activation can be induced by free cholesterol (FC) accumulation (Teratani et al., 2012; Tomita et al., 2014). Moreover, murine diets high in cholesterol are more effective in inducing NASH (Tsuchida et al., 2018; Wang et al., 2020). In cultured HSCs, FC accumulation increases Toll-like receptor 4 (TLR4) levels, sensitizing the HSCs to TGF-β-induced activation (Seki et al., 2007; Teratani et al., 2012). FC accumulation in HSCs is under the control of low-density lipoprotein receptor (LDLR) and miR-33a, a microRNA regulator of cholesterol metabolism, both of which are upregulated during HSC activation (Tomita et al., 2014). LDLR and miR-33a-mediated signaling impairs endosomal-lysosomal degradation of TLR4, which, in turn, downregulates bone morphogenetic protein and activin membrane-bound inhibitor (BAMBI) levels, a pseudoreceptor for TGF-β, thereby increasing HSC sensitivity to TGF-β (Onichtchouk et al., 1999; Teratani et al., 2012).
Oxidized LDL (ox-LDL) can directly activate HSCs, at least in culture. The scavenger receptors CD36 and lectin-like oxidized LDL receptor-1 (ox-LOX receptor-1, or LOX-1) mediate HSC ox-LDL uptake, resulting in an increased expression of profibrogenic genes (Kang and Chen, 2009; Schneiderhan et al., 2001). LOX-1 expression is upregulated by canonical Wnt signaling (Kang and Chen, 2009), which has been implicated as an HSC activator (Cheng et al., 2008; Myung et al., 2007).
Glutaminolysis
The immense metabolic demands of ECM production are met not only through reprogramming of carbohydrate and lipid metabolism but also protein metabolism. A comparison of the differentially expressed metabolic genes between qHSCs and aHSCs reveals that only 6% of such genes involve carbohydrate metabolism, while 38% are involved in protein metabolism, signified by a shift toward glutaminolysis (Du et al., 2018). Glutaminolysis is the conversion of the amino acid glutamine into α-ketoglutarate, a TCA cycle intermediate, that is typically observed in cancer cells. Activated HSCs have upregulated expression levels of the glutaminase GLS1, the rate-limiting enzyme of glutamine utilization in cancer cells. Increased glutaminase tGLS1 is abrogated by blocking yes-associated protein (YAP) signaling, which is a downstream effector of the fibrogenic agonist Hh. Glutamine deprivation of HSCs provokes lipid accumulation, increased PPAR-γ expression, and decreased collagen I expression, underscoring the importance of glutaminolysis as an energy source for aHSC (Du et al., 2018). In vivo, glutaminolysis by HSCs marks active fibrogenesis and its cell-specific antagonism represents a potential therapeutic target by depriving the cells of glutamine (Du et al., 2020). In contrast to glutamine, glucose deprivation does not have an antifibrogenic effect, despite increased glycolytic gene expression and glucose consumption by aHSCs (Chen et al., 2012; Du et al., 2018).
LINKING HSC METABOLISM TO STRESS RESPONSES
Oxidative Stress
ROS
Production of ROS is an important characteristic of aHSCs, as knockdown or inhibition of ROS-producing enzymes attenuate HSC activation. Enhanced ROS production corresponds with increased mitochondrial activity (Gajendiran et al., 2018).
Several classic activators of HSCs converge on ROS-mediated activation, especially through upregulation of NAPDH oxidase (NOX) enzymes (Figure 4). Generation of ROS is viewed as a “feed forward” mechanism of fibrogenic cell activation, particularly through TGF-β (Hellerbrand et al., 1999). Redox imbalances activate the latent form of TGF-β, while TGF-β signaling generates redox imbalances (Liu and Desai, 2015). TGF-β acts primarily through SMAD transcription factors (Breitkopf et al., 2006) and the MAPK/ERK pathway to activate a fibrogenic transcriptional program (Tsukada et al., 2005). TGF-β signaling upregulates several NAPDH oxidase enzymes—NOX1, NOX2, NOX4, and NOX5 (Andueza et al., 2018; Sancho et al., 2012) — which increase H2O2 generation leading to enhanced collagen production. Other inducers of NOX in HSCs include platelet-derived growth factor (PDGF) (Adachi et al., 2005), leptin (De Minicis et al., 2008), angiotensin II (Ang II) (Bataller et al., 2003), and advance glycation end products (Iwamoto et al., 2008). Like TGF-β signaling, PDGF (Adachi et al., 2005), leptin (De Minicis et al., 2008), Ang II (Bataller et al., 2003), and AGEs (Chang et al., 2004) act through the phosphorylation and activation of the MAPK/ERK signaling pathway. The knockdown or inhibition of NOX enzymes blunt the potency of stimuli to fibrogenesis, underscoring their importance to HSC activation in liver fibrosis (Adachi et al., 2005; Bataller et al., 2003; De Minicis et al., 2008; Hanafusa et al., 1999; Iwamoto et al., 2008; Tsukada et al., 2005). H2O2 facilitates the binding of the transcription factor C/EBPβ to its cognate element on the alpha-1 type I collagen (col1α1) promoter, thereby increasing its expression (García-Trevijano et al., 1999). The TGF-β promoter also contains C/EBPβ responsive elements (Abraham et al., 2009), supporting the “feed forward” element of TGF-β/ROS signaling. However, SMADs, which are downstream effectors of TGF-β, attenuate C/EBPβ-mediated TGF-β upregulation (Abraham et al., 2009), suggesting that the TGF-β/ROS circuit is tightly regulated in HSCs.
Figure 4. Redox Metabolism in HSC Activation.
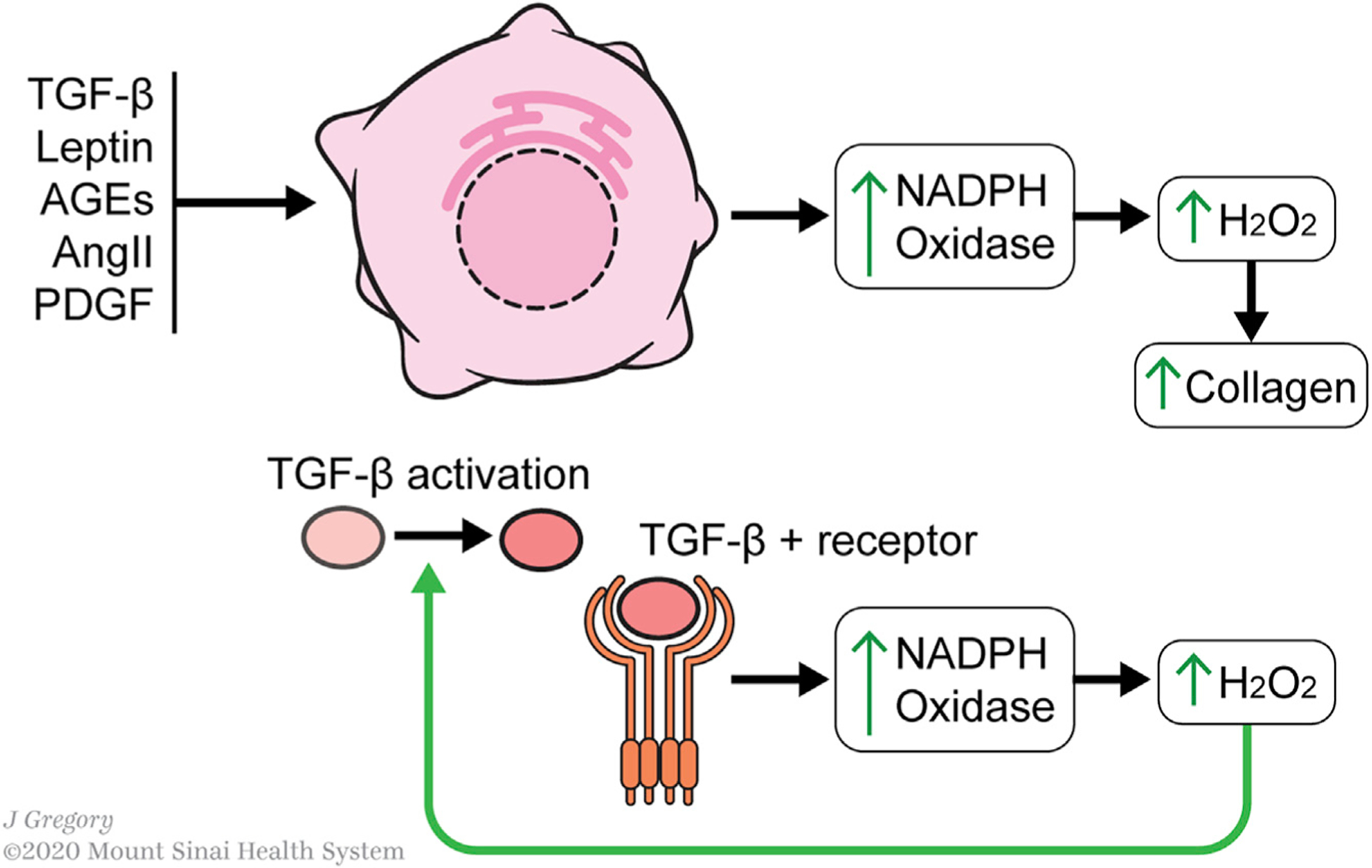
Profibrotic stimuli (TGFb, leptin, AGEs, angiotensin II, and PDGF) converge upon a MAPK/ERK signaling pathway that leads to increased NADPH oxidase (NOX) expression. Increased NOX expression increases H2O2 levels, which facilitates the finding of the transcription factor C/EBPβ to its cognate element on the Col1α1 promoter, thereby increasing collagen expression. The increase in H2O2 levels is accompanied by an increased capacity of HSCs to handle oxidative stress, such as through increased glutathione and superoxide dismutase levels. Combined with responsiveness of collagen expression to H2O2, this points toward a signaling role for H2O2 in activated HSC, which is traditionally considered to be a marker of cell stress.
Redox metabolism in HSCs also acts a feedforward mechanism for activation; TGF-β signaling increased H2O2 levels through induction of NOX expression, while TGF-β itself converted from its latent from to its active form in the presence of H2O2
While the profibrogenic contributions of NOX-generated ROS are well characterized, a definitive role for mitochondrial ROS in HSC activation has not been established. Studies conducted in embryonic (Ranganathan et al., 2001), kidney (Jain et al., 2013), and heart fibroblasts (Bracey et al., 2014) demonstrate increased mitochondrial ROS production during cellular activation. Furthermore, mitochondrial ROS metabolized to H2O2 by manganese superoxide dismutase (Sod2) upregulate matrix metalloproteinase transcription in embryonic fibroblasts, which increases Sod2 activity resulting in increased MMP-1, MMP-2, MMP-3, and MMP-7 expression (Ranganathan et al., 2001). Conversely, an endocannabinoid-mediated increase in mitochondrial ROS induces HSC cell death (Siegmund et al., 2007), consistent with the classical role of mitochondrial ROS as a regulator of apoptosis (Fleury et al., 2002; Giorgio et al., 2005; Li et al., 2003). Differential regulation of these two major axes of ROS production may explain the more variable contributions of mitochondrial ROS, in contrast to the definitively profibrogenic nature of NOX enzymes (Brunati et al., 2010).
RNS
The functions of reactive nitrogen species (RNS) in HSC biology are not as well clarified as those of ROS. Nearly all liver cell types, including HSC, can produce nitric oxide (NO·) (Feder et al., 1993). NO· has been implicated as a promoter of HSC apoptosis (Dong et al., 2015; Jin et al., 2017; Langer et al., 2008) and a downstream effector of adiponectin (Langer et al., 2008), an adipokine with antifibrotic properties (Urtasun et al., 2008). Furthermore, NO· production by Kupffer cells (resident hepatic macrophages) and activated neutrophils can antagonize the stimulatory activity of ROS on collagen expression, potentially by acting as a free radical scavenger of ROS species (Casini et al., 1997; Friedman, 2003).
Antioxidant Responses
The endogenous antioxidant activity of HSCs is not well characterized. In vitro, TGF-β-activated HSCs have increased glutathione content (De Bleser et al., 1999), an observation that is consistent with studies in culture-activated HSCs (Proell et al., 2007). Additionally, varying degrees of HSC activation correspond to different redox states. In a study of late-stage cellular activation using the M1–4HSC cell line, which is considered an “activated HSC” based on loss of lipid droplets and expression of α-SMA, TGF-β promotes further activation, leading to a fully transdifferentiated myofibroblastoid M-HT cell (Proell et al., 2007). In culture, M1–4HSCs contain higher intracellular ROS content despite having lower levels of NOX activity compared with M-HTs (Proell et al., 2007). The fully transdifferentiated M-HTs have enhanced superoxide dismutase activity and glutathione levels (Proell et al., 2007), which may be necessary to curb ROS-induced cellular damage, while still allowing the cells to function as intracellular signaling molecules. Studies of HSCs isolated from animal models of fibrotic disease report conflicting data about the increase in HSC glutathione content with activation (De Bleser et al., 1999; Maher et al., 1997). Additionally, increased antioxidant availability attenuates the effects of profibrotic signals such as TGF-β and PDGF (Foo et al., 2011) and reduces fibrosis in animal models of disease (Jia et al., 2015; Serviddio et al., 2014).
ER Stress
Quiescent HSCs display moderately developed rough ER, which becomes enlarged and more abundant when they transdifferentiate to a MFB phenotype, consistent with increased capacity for ECM protein synthesis (Friedman, 2008; Friedman et al., 1985; Minato et al., 1983). The resultant accumulation of proteins in the ER initiates the unfolded protein response (UPR), which is an early marker of HSC activation (Mannaerts et al., 2019). Activated HSCs develop ER stress due to a combination of high-volume production of ECM proteins and exposure to ROS.
The UPR is a homeostatic response to protein folding stress that coordinates multiple functions to preserve ER function. The UPR-associated transcription factors initiate a number of ER stress-alleviating actions, including increased chaperone and foldase gene expression, activation of the autophagy pathway to direct damaged ER portions and protein aggregates for lysosomal degradation, and regulation of glucose metabolism. Patients with more severe fibrosis have higher levels of ER stress markers, such as GRP78, CHOP, and p-PKR-like endoplasmic reticulum kinase (PERK), compared with patients with milder fibrosis (Koo et al., 2016). Notably, the UPR alone does not activate qHSCs, as chemical induction of ER stress using tunicamycin is not sufficient to induce activation (Mannaerts et al., 2019). However, amelioration of ER stress through overexpression of Grp78 reduces the expression of profibrotic genes, including TGF-β, α-SMA, Col1α1, and Timp1, suggesting that the induction of ER stress through increased biosynthetic load is a driver of HSC activation (Koo et al., 2016). SMAD2, a member of the SMAD family of TGF-β signaling effectors (Breitkopf et al., 2006), provides a direct link between ER stress and HSC transcriptional reprogramming. ER stress activates PERK, which increases degradation of MIR18A, a microRNA inhibitor of SMAD2 expression (Koo et al., 2016).
ER stress in HSCs can also result from increased ROS burden, which is characteristic of aHSCs. HSC treatment with H2O2, as well as ethanol-feeding of rats, induce the UPR, as demonstrated by an upregulation of ER stress markers ATF4, ATF6, IRE1, and XBP1 (Hernández-Gea et al., 2013; Kim et al., 2016). The roles of IRE1 and XBP1 as mediators of autophagy are described in further detail below.
Autophagy
Macroautophagy, (“autophagy”), is a cellular stress response that engages auto-lysosomal digestion of macromolecules and organelles to generate intracellular nutrients and energy (Mehrpour et al., 2010; Mizushima et al., 2008). Several activation-related metabolic changes described above converge on autophagy, including increased lipolysis (Hernández-Gea et al., 2012; Tuohetahuntila et al., 2017), ROS production (Zhang et al., 2017), and ER stress (Hernández-Gea et al., 2013; Kim et al., 2016).
Lipid droplet mobilization, a key feature of HSC activation, is driven by autophagy, as HSCs from mice deficient in ATG7, a key autophagy mediator, fail to lose lipid droplets or activate following liver injury (Hernández-Gea et al., 2012). While early efforts examining the impact of lipid droplet mobilization focused on the release of free retinol outside the cell following intracellular retinyl ester hydrolysis (Friedman et al., 1993), it is now clear that a critical consequence of hydrolysis is the liberation of free fatty acids to “fuel” cellular activation. In support of this model, addition of endogenous oleic acid can rescue activation in autophagy-deficient HSCs, whereas inhibition of fatty acid oxidation has the opposite effect (Hernández-Gea et al., 2012). On the other hand, HSCs lacking lipid droplets in mice due to deficiency of lecithin-retinol acyltransferase (LRAT) still can fully activate but do not undergo spontaneous activation (Kluwe et al., 2011). This paradox—that lack of retinoid content due to LRAT deficiency does not alter activation, yet retinoid loss in other models accompanies HSC activation—is not easily reconciled, but one possibility is that these LRAT-deficient cells utilize an alternative energy source other than fatty acids.
Other studies have further defined cellular interactions that support autophagy. For example, PI3K-CIII, a kinase regulator of autophagy engages Rab25, a small GTPase, to direct the recognition of lipid droplets by the autophagy pathway (Zhang et al., 2017). Interestingly, Rab25 activity in HSCs is partially regulated by ROS, which activates Rab25 to increase autophagic flux (Zhang et al., 2017).
ER stress is functionally upstream of autophagy in the sequence of HSC activation (Kim et al., 2016). IRE1, which promotes HSC activation and fibrogenesis, signals through the p38-MAPK pathway to activate autophagy, as evidenced by IRE1 mediated accumulation of the autophagy marker LC3II (Hernández-Gea et al., 2013). Furthermore, the profibrogenic effects of XBP1, the downstream effector of IRE1, are abrogated through suppression of ATG7 expression, a critical autophagy mediator (Hernández-Gea et al., 2013).
NON-CELL-AUTONOMOUS EFFECTS OF HSC METABOLISM
Hepatic Stellate Cell-Hepatocyte Metabolic Crosstalk
Hepatocytes, which makes up >80% of liver cells and represent the main metabolic workhorse of the body, interact with HSC metabolism through the exchange of retinoid metabolites and signaling modalities. During HSC activation, free retinol released by HSCs (Friedman et al., 1993; Sauvant et al., 2001) is rapidly taken up in a contact-dependent fashion by hepatocytes (Ikeda et al., 1997; Sauvant et al., 2001), which release it into circulation as holo-RBP (Bellovino et al., 1999, 1996; Sauvant et al., 2001). Apart from loss of retinoid during HSC activation, it is not known if hepatic metabolism of vitamin A directly contributes to liver injury and fibrosis. The total liver content of retinoid declines with progression of human liver disease (Leo et al., 1993), and in two murine models of NASH there is depletion of retinol-RBP4 with redistribution of retinoid to hepatocytes rather than HSCs, the significance of which is not yet clear (Saeed et al., 2020).
Signaling pathways downstream of Hedgehog (Hh) and leptin receptor binding regulate glucose metabolism in HSCs (Chandrashekaran et al., 2016; Chen et al., 2012). Hh signaling has been broadly implicated in wound healing, and Hh ligands are produced by hepatocytes during liver injury (Jung et al., 2010; Wang et al., 2016). These ligands act upon neighboring HSCs to induce activation through the HIF-1α transcription factor (Chen et al., 2012), a master regulator of energy metabolism that activates a repertoire of target genes, including glucose transporters and glycolytic enzymes (Lee et al., 2004). Since hypoxia upregulates oxygen-independent ATP production pathways, HIF-1α mediated regulation of glucose metabolism upon activation is concordant with the Warburg-effect-like phenomenon observed in aHSCs. In addition, interactions between hepatocytes and HSCs contribute to steatohepatitis through neuropilin1-mediated regulation of lgfbp3 and SerpinA12 (Arab et al., 2020), as well as signaling by glutamate (Choi et al., 2019), Taz/Hedgehog (Wang et al., 2016), and Notch (Schwabe et al., 2020; Zhu et al., 2018).
While these examples of paracrine interactions between HSCs and hepatocytes are limited, they hint at a rich repertoire of bidirectional signaling yet to be uncovered, governing not only metabolism but also injury, inflammation, regeneration, and cancer. The maturation of technologies for single-cell analysis, coupled with powerful informatics to predict and validate cell-cell interactions, will yield accelerating progress.
HSCs as Effectors of Immunometabolism
While emerging concepts of immunometabolism focus primarily on immune cells (Hotamisligil, 2017; Lee et al., 2018), HSCs also contribute significantly to convergent pathways of metainflammation and tissue injury, especially in alcoholic and non-alcoholic steatohepatitis (NASH) (Schwabe et al., 2020). Through their control of retinoid metabolism, they regulate immune cells function in liver as well as in systemic immunity (Lee and Jeong, 2012; Wang et al., 2002; Weiskirchen and Tacke, 2014) (see section on Retinol Storage and Lipid Droplets, above). Stellate cells produce and respond to inflammatory mediators including cytokines and chemokines that can amplify the liver’s response to injury (Roh and Seki, 2018). Conversely, they are often overlooked determinants of the liver’s unique immune tolerance (Weiskirchen and Tacke, 2014) through induction of T regulatory cells (Heine et al., 2016; Jiang et al., 2008) and myeloid-derived suppressor cells (Heine et al., 2016). HSCs are also proposed as antigen-presenting cells (Winau et al., 2007), and they can respond to innate immune ligands through Toll-like receptor signaling and inflammasome activation (Guo et al., 2009; Miura et al., 2012; Seki et al., 2007; Watanabe et al., 2007). Interactions between HSCs and inflammatory cells can drive inflammation and injury through paracrine signaling. For example, HSC interactions with macrophages through the MER-TK receptor contribute to fibrosis in experimental NASH (Cai et al., 2020), as well as crosstalk with NK, NKT (Notas et al., 2009), and gamma delta T cells (Seo et al., 2016).
There is fragmentary knowledge about specific immunometabolites from HSCs that engage with immune cells, but several examples are notable: (1) release of all-trans RA by HSCs promotes a tolerogenic phenotype of dendritic cells through induction of arginase-1 and inducible nitric oxide synthase (Bhatt et al., 2014); (2) HSCs can expand FoxP3+ regulatory T cells in response to LPS by inducing the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), an immunoregulatory molecule, which catabolizes tryptophan to generate kynurenine (Kumar et al., 2017). Interestingly, inhibition of IDO reduces proliferation in cultured HSCs (Oh et al., 2017); (3) asymmetric dimethylargine (ADMA) is catabolized by dimethylarginine dimethylaminohydrolase (DDAH), which reduces TGFβ1-mediated activation in primary rat HSCs (Liu et al., 2018); and (4) the bacterial product microcystin-leucine arginase (MC-LR) can activate HSCs through Hedgehog signaling, and administration of this molecule induces hepatic fibrosis in vivo, but its impact on others cell types might account in part for this effect (Gu et al., 2020)
THERAPEUTIC IMPLICATIONS AND FUTURE DIRECTIONS
Although therapies based on metabolic regulation of HSCs are limited to date, clarifying metabolic pathways provides a clear opportunity for new and potentially targeted interventions to treat both liver disease and systemic illnesses that engage the liver. While a comprehensive review of therapeutics is beyond the scope of this review, currently those drugs targeting metabolic pathways to attenuate fibrosis are being investigated in NASH, including: (1) bile acids, which have anti-apoptotic and ER stress relieving properties (Golan-Gerstl et al., 2017), (2) FXR agonists, which regulate glucose, lipid, and bile acid metabolism (Younossi et al., 2019), (3) thyroid hormone receptor-beta agonists, which moderate lipid and glucose homeostasis (Harrison et al., 2018), (4) PPAR agonists, which are adipogenic master regulators (Ratziu et al., 2016), and (5) vitamin A coupling for HSC targeting, among others (Lemoinne and Friedman, 2019; Sato et al., 2008).
A broad array of cell injury signals, including proinflammatory cytokines, apoptotic bodies, endothelial-cell-mediated growth factors, and ROS, conspire to induce HSC activation. As the key event that leads to hepatic fibrogenesis, HSC activation to amplify ECM production exacts a heavy bioenergetic toll on HSCs, demanding broad metabolic reprogramming that mirrors findings from cancer metabolism, including a Warburg-like effect, a reliance on glutaminolysis, and increased autophagic flux.
At the same time, HSCs also contribute to liver and immune homeostasis through functions that do not necessarily require their activation. This concept is reinforced by analyzing recently published single-cell RNA sequencing data to define expression of metabolism-associated genes in both murine and human HSCs from normal and diseased liver. Interestingly, expression of many of these genes is not altered with activation, suggesting that perhaps there is post-transcriptional regulation of some of the metabolic pathways they support, and/or their activity is unchanged with activation (Figure 5). In fact, there has been no systematic clarification of which metabolic functions of the cells are strictly dependent on their transdifferentiation/activation and which are associated with basal cellular homeostasis. As noted above, the original concept of HSCs as either “activated,” “quiescent,” or “inactivated” may be oversimplified, and the emergence of different subtypes of HSCs in different contexts is increasingly apparent. Single-cell-based approaches, coupled with advances in “-omic” technologies (metabolomics, proteomics, and epigenomics), could reveal greater HSC heterogeneity within specific homeostatic and disease contexts, yielding further insight into how metabolic regulation supports these varied cellular phenotypes. In leveraging these new technologies, novel paradigms of cellular energetics in fibrogenic cells, and new therapeutic vulnerabilities may emerge.
Figure 5. Metabolism-Related Gene Expression by HSCs from Healthy and Diseased Liver Based on Published Single-Cell RNA Sequencing Studies.
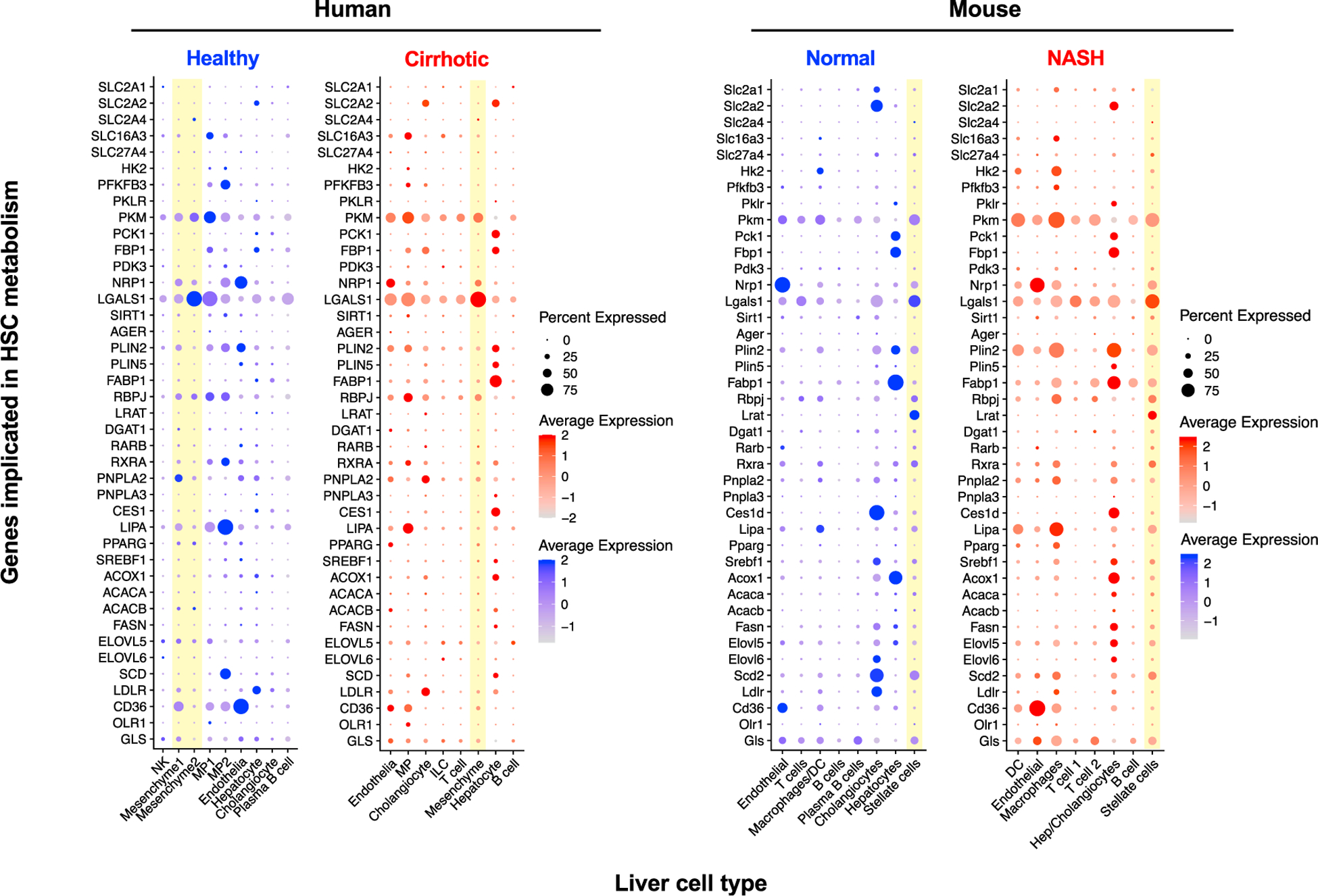
Human healthy and cirrhotic liver single-cell RNA sequencing datasets were extracted from Ramachandran et al. (2019) and from a study analyzing health and NASH mouse liver (Xiong et al., 2019). Data were processed using Seurat 3.0 (Butler et al., 2018; Stuart et al., 2019) using default parameters with cell clusters assigned using gene markers reported in the original publications. Mesenchymal cell clusters that likely represent HSCs are highlighted in yellow. “Percent Expression” refers to percentage of cells from each cell type that express the gene of interest. “NK,” natural killer cells; “MP,” mononuclear phagocytes; “ILC,” innate lymphoid cells; and “DC,” dendritic cells.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health (R01DK56621) to S.L.F., the American Gastroenterological Association to S.W. (AGA2020-13-03), and the Mount Sinai, MD/MSCR Patient-Oriented Research Training and Leadership (PORTAL) program to P.T.
REFERENCES
- Abraham S, Sweet T, Khalili K, Sawaya BE, and Amini S (2009). Evidence for activation of the TGF-beta1 promoter by C/EBPbeta and its modulation by Smads. J. Interferon Cytokine Res 29, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi T, Togashi H, Suzuki A, Kasai S, Ito J, Sugahara K, and Kawata S (2005). NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 41, 1272–1281. [DOI] [PubMed] [Google Scholar]
- Ajat M, Molenaar M, Brouwers JFHM, Vaandrager AB, Houweling M, and Helms JB (2017). Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 176–187. [DOI] [PubMed] [Google Scholar]
- Allenby G, Bocquel MT, Saunders M, Kazmer S, Speck J, Rosenberger M, Lovey A, Kastner P, Grippo JF, and Chambon P (1993). Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proc. Natl. Acad. Sci. USA 90, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann T, and Hellerbrand C (2009). GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 13, 1411–1427. [DOI] [PubMed] [Google Scholar]
- Andueza A, Garde N, García-Garzón A, Ansorena E, López-Zabalza MJ, Iraburu MJ, Zalba G, and Martínez-Irujo JJ (2018). NADPH oxidase 5 promotes proliferation and fibrosis in human hepatic stellate cells. Free Radic. Biol. Med 126, 15–26. [DOI] [PubMed] [Google Scholar]
- Arab JP, Cabrera D, Sehrawat TS, Jalan-Sakrikar N, Verma VK, Simonetto D, Cao S, Yaqoob U, Leon J, Freire M, et al. (2020). Hepatic stellate cell activation promotes alcohol-induced steatohepatitis through Igfbp3 and SerpinA12. J. Hepatol 73, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K, Tsai SY, Li P, Ishii M, Maxson RE Jr., Sucov HM, and Tsukamoto H (2009). Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 49, 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahina K, Zhou B, Pu WT, and Tsukamoto H (2011). Septum transversum-derived mesothelium gives rise to hepatic stellate cells and perivascular mesenchymal cells in developing mouse liver. Hepatology 53, 983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal MB (2016). Hepatic stellate cells: fibrogenic, regenerative or both? Heterogeneity and context are key. Hepatol. Int 10, 902–908. [DOI] [PubMed] [Google Scholar]
- Barcena-Varela M, Paish H, Alvarez L, Uriarte I, Latasa MU, Santamaria E, Recalde M, Garate M, Claveria A, Colyn L, et al. (2020). Epigenetic mechanisms and metabolic reprogramming in fibrogenesis: dual targeting of G9a and DNMT1 for the inhibition of liver fibrosis. Gut. [DOI] [PubMed] [Google Scholar]
- Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, et al. (2003). NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J. Clin. Invest 112, 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates J, Vijayakumar A, Ghoshal S, Marchand B, Yi S, Kornyeyev D, Zagorska A, Hollenback D, Walker K, Liu K, et al. (2020). Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol 73, 896–905. [DOI] [PubMed] [Google Scholar]
- Bellovino D, Lanyau Y, Garaguso I, Amicone L, Cavallari C, Tripodi M, and Gaetani S (1999). MMH cells: an in vitro model for the study of retinol-binding protein secretion regulated by retinol. J. Cell. Physiol 181, 24–32. [DOI] [PubMed] [Google Scholar]
- Bellovino D, Morimoto T, Tosetti F, and Gaetani S (1996). Retinol binding protein and transthyretin are secreted as a complex formed in the endoplasmic reticulum in HepG2 human hepatocarcinoma cells. Exp. Cell Res 222, 77–83. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Qin J, Bennett C, Qian S, Fung JJ, Hamilton TA, and Lu L (2014). All-trans retinoic acid induces arginase-1 and inducible nitric oxide synthase-producing dendritic cells with T cell inhibitory function. J. Immunol 192, 5098–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaner WS, Hendriks HF, Brouwer A, de Leeuw AM, Knook DL, and Goodman DS (1985). Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J. Lipid Res 26, 1241–1251. [PubMed] [Google Scholar]
- Blaner WS, van Bennekum AM, Brouwer A, and Hendriks HF (1990). Distribution of lecithin-retinol acyltransferase activity in different types of rat liver cells and subcellular fractions. FEBS Lett. 274, 89–92. [DOI] [PubMed] [Google Scholar]
- Blomhoff R, and Blomhoff HK (2006). Overview of retinoid metabolism and function. J. Neurobiol 66, 606–630. [DOI] [PubMed] [Google Scholar]
- Blomhoff R, Rasmussen M, Nilsson A, Norum KR, Berg T, Blaner WS, Kato M, Mertz JR, Goodman DS, and Eriksson U (1985). Hepatic retinol metabolism. Distribution of retinoids, enzymes, and binding proteins in isolated rat liver cells. J. Biol. Chem 260, 13560–13565. [PubMed] [Google Scholar]
- Blomhoff R, and Wake K (1991). Perisinusoidal stellate cells of the liver: important roles in retinol metabolism and fibrosis. FASEB J. 5, 271–277. [DOI] [PubMed] [Google Scholar]
- Bracey NA, Gershkovich B, Chun J, Vilaysane A, Meijndert HC, Wright JR Jr., Fedak PW, Beck PL, Muruve DA, and Duff HJ (2014). Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem 289, 19571–19584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkopf K, Godoy P, Ciuclan L, Singer MV, and Dooley S (2006). TGF-beta/Smad signaling in the injured liver. Z. Gastroenterol 44, 57–66. [DOI] [PubMed] [Google Scholar]
- Brunati AM, Pagano MA, Bindoli A, and Rigobello MP (2010). Thiol redox systems and protein kinases in hepatic stellate cell regulatory processes. Free Radic. Res 44, 363–378. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Dongiovanni P, Corey KE, Wang X, Shmarakov IO, Zheng Z, Kasikara C, Davra V, Meroni M, Chung RT, et al. (2020). Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metab 31, 406–421.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, Foschi M, Caligiuri A, Pinzani M, and Surrenti C (1997). Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology 25, 361–367. [DOI] [PubMed] [Google Scholar]
- Chandrashekaran V, Das S, Seth RK, Dattaroy D, Alhasson F, Michelotti G, Nagarkatti M, Nagarkatti P, Diehl AM, and Chatterjee S (2016). Purinergic receptor x7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. BBA Mol. Basis Dis 1862, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, Chen TH, Chang CJ, Hou CC, Chan P, and Lee HM (2004). Advanced glycosylation end products induce inducible nitric oxide synthase (iNOS) expression via a p38 MAPK-dependent pathway. Kidney Int 65, 1664–1675. [DOI] [PubMed] [Google Scholar]
- Chen A, Tang Y, Davis V, Hsu FF, Kennedy SM, Song H, Turk J, Brunt EM, Newberry EP, and Davidson NO (2013). Liver fatty acid binding protein (L-Fabp) modulates murine stellate cell activation and diet-induced nonalcoholic fatty liver disease. Hepatology 57, 2202–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, Xie G, Moylan CA, Garibaldi F, Premont R, et al. (2012). Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology 143, 1319–1329.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JH, She H, Han YP, Wang J, Xiong S, Asahina K, and Tsukamoto H (2008). Wnt antagonism inhibits hepatic stellate cell activation and liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol 294, G39–G49. [DOI] [PubMed] [Google Scholar]
- Choi WM, Kim HH, Kim MH, Cinar R, Yi HS, Eun HS, Kim SH, Choi YJ, Lee YS, Kim SY, et al. (2019). Glutamate signaling in hepatic stellate cells drives alcoholic steatosis. Cell Metab. 30, 877–889.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, and Cantley LC (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio DN, Walewski JL, Clugston RD, Berk PD, Rippe RA, and Blaner WS (2011). Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS One 6, e24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bleser PJ, Xu G, Rombouts K, Rogiers V, and Geerts A (1999). Glutathione levels discriminate between oxidative stress and transforming growth factor-beta signaling in activated rat hepatic stellate cells. J. Biol. Chem 274, 33881–33887. [DOI] [PubMed] [Google Scholar]
- De Minicis S, Seki E, Oesterreicher C, Schnabl B, Schwabe RF, and Brenner DA (2008). Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology 48, 2016–2026. [DOI] [PubMed] [Google Scholar]
- DeLeve LD (2013). Liver sinusoidal endothelial cells and liver regeneration. J. Clin. Invest 123, 1861–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeve LD (2015). Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 61, 1740–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desroches-Castan A, Tillet E, Ricard N, Ouarné M, Mallet C, Belmudes L, Couté Y, Boillot O, Scoazec JY, Bailly S, et al. (2019). Bone morphogenetic protein 9 is a paracrine factor controlling liver sinusoidal endothelial cell fenestration and protecting against hepatic fibrosis. Hepatology 70, 1392–1408. [DOI] [PubMed] [Google Scholar]
- Dobie R, Wilson-Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, Wallenborg K, Picelli S, Zagorska A, Pendem SV, et al. (2019). Single-cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep. 29, 1832–1847.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Su L, Esmaili S, Iseli TJ, Ramezani-Moghadam M, Hu L, Xu A, George J, and Wang J (2015). Adiponectin attenuates liver fibrosis by inducing nitric oxide production of hepatic stellate cells. J. Mol. Med. (Berl.) 93, 1327–1339. [DOI] [PubMed] [Google Scholar]
- Du K, Chitneni SK, Suzuki A, Wang Y, Henao R, Hyun J, Premont RT, Naggie S, Moylan CA, Bashir MR, et al. (2020). Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell. Mol. Gastroenterol. Hepatol 10, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, et al. (2018). Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology 154, 1465–1479.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberlé D, Hegarty B, Bossard P, Ferré P, and Foufelle F (2004). SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848. [DOI] [PubMed] [Google Scholar]
- Eichmann TO, Grumet L, Taschler U, Hartler J, Heier C, Woblistin A, Pajed L, Kollroser M, Rechberger G, Thallinger GG, et al. (2015). ATGL and CGI-58 are lipid droplet proteins of the hepatic stellate cell line HSC-T6. J. Lipid Res 56, 1972–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Taghdouini A, Najimi M, Sancho-Bru P, Sokal E, and van Grunsven LA (2015). In vitro reversion of activated primary human hepatic stellate cells. Fibrogenesis Tissue Repair 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evert M, Dombrowski F, Schirmacher P, and Pfeifer U (1998). Nonparenchymal cells in chronically hyperinsulinemic liver acini of diabetic rats, with special regard to hepatic stellate cells. J. Hepatol 28, 709–716. [DOI] [PubMed] [Google Scholar]
- Feder LS, Todaro JA, and Laskin DL (1993). Characterization of interleukin-1 and interleukin-6 production by hepatic endothelial cells and macrophages. J. Leukoc. Biol 53, 126–132. [DOI] [PubMed] [Google Scholar]
- Fehrenbach H, Weiskirchen R, Kasper M, and Gressner AM (2001). Upregulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology 34, 943–952. [DOI] [PubMed] [Google Scholar]
- Fleury C, Mignotte B, and Vayssière JL (2002). Mitochondrial reactive oxygen species in cell death signaling. Biochimie 84, 131–141. [DOI] [PubMed] [Google Scholar]
- Foo NP, Lin SH, Lee YH, Wu MJ, and Wang YJ (2011). Alpha-lipoic acid inhibits liver fibrosis through the attenuation of ROS-triggered signaling in hepatic stellate cells activated by PDGF and TGF-beta. Toxicology 282, 39–46. [DOI] [PubMed] [Google Scholar]
- Friedman SL (2003). Liver fibrosis - from bench to bedside. J. Hepatol 38 (suppl 1), 38–53. [DOI] [PubMed] [Google Scholar]
- Friedman SL (2008). Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 88, 125–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL, Rockey DC, McGuire RF, Maher JJ, Boyles JK, and Yamasaki G (1992). Isolated hepatic lipocytes and Kupffer cells from normal human liver: morphological and functional characteristics in primary culture. Hepatology 15, 234–243. [DOI] [PubMed] [Google Scholar]
- Friedman SL, Roll FJ, Boyles J, Arenson DM, and Bissell DM (1989). Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J. Biol. Chem 264, 10756–10762. [PubMed] [Google Scholar]
- Friedman SL, Roll FJ, Boyles J, and Bissell DM (1985). Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc. Natl. Acad. Sci. USA 82, 8681–8685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman SL, Wei S, and Blaner WS (1993). Retinol release by activated rat hepatic lipocytes: regulation by Kupffer cell-conditioned medium and PDGF. Am. J. Physiol 264, G947–G952. [DOI] [PubMed] [Google Scholar]
- Fukushima M, Enjoji M, Kohjima M, Sugimoto R, Ohta S, Kotoh K, Kuniyoshi M, Kobayashi K, Imamura M, Inoguchi T, et al. (2005). Adipose differentiation related protein induces lipid accumulation and lipid droplet formation in hepatic stellate cells. In Vitro Cell. Dev. Biol. Anim 41, 321–324. [DOI] [PubMed] [Google Scholar]
- Gajendiran P, Vega LI, Itoh K, Sesaki H, Vakili MR, Lavasanifar A, Hong K, Mezey E, and Ganapathy-Kanniappan S (2018). Elevated mitochondrial activity distinguishes fibrogenic hepatic stellate cells and sensitizes for selective inhibition by mitotropic doxorubicin. J. Cell. Mol. Med 22, 2210–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, and Brigstock DR (2004). Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J. Biol. Chem 279, 8848–8855. [DOI] [PubMed] [Google Scholar]
- García-Trevijano ER, Iraburu MJ, Fontana L, Domínguez-Rosales JA, Auster A, Covarrubias-Pinedo A, and Rojkind M (1999). Transforming growth factor beta1 induces the expression of alpha1(I) procollagen mRNA by a hydrogen peroxide-C/EBPbeta-dependent mechanism in rat hepatic stellate cells. Hepatology 29, 960–970. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, et al. (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122, 221–233. [DOI] [PubMed] [Google Scholar]
- Golan-Gerstl R, Oren R, Brazovski E, Hayardeny L, and Reif S (2017). Anti-fibrotic effect of aramchol on fibrosis in TAA animal model. J. Hepatol 66, S655–S656. [Google Scholar]
- Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, and Angus P (2013). Advanced glycation end products augment experimental hepatic fibrosis. J. Gastroenterol. Hepatol 28, 369–376. [DOI] [PubMed] [Google Scholar]
- Grumet L, Eichmann TO, Taschler U, Zierler KA, Leopold C, Moustafa T, Radovic B, Romauch M, Yan C, Du H, et al. (2016). Lysosomal acid lipase hydrolyzes retinyl ester and affects retinoid turnover. J. Biol. Chem 291, 17977–17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Yan M, Wang C, Meng X, Xiang Z, Qiu Y, and Han X (2020). Microcystin-leucine-arginine induces liver fibrosis by activating the Hedgehog pathway in hepatic stellate cells. Biochem Biophys Res Commun. 10.1016/j.bbrc.2020.09.075. [DOI] [PubMed] [Google Scholar]
- Guimarães EL, Best J, Dollé L, Najimi M, Sokal E, and van Grunsven LA (2012). Mitochondrial uncouplers inhibit hepatic stellate cell activation. BMC Gastroenterol. 12, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Loke J, Zheng F, Hong F, Yea S, Fukata M, Tarocchi M, Abar OT, Huang H, Sninsky JJ, et al. (2009). Functional linkage of cirrhosis-predictive single nucleotide polymorphisms of toll-like receptor 4 to hepatic stellate cell responses. Hepatology 49, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H, Matsumoto K, and Nishida E (1999). Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem 274, 27161–27167. [DOI] [PubMed] [Google Scholar]
- Harrison S, Moussa S, Bashir M, Alkhouri N, Frias J, Baum S, Tetri B, Bansal M, and Taub R (2018). MGL-3196, a selective thyroid hormone receptor-beta agonist significantly decreases hepatic fat in NASH patients at 12 weeks, the primary endpoint in a 36-week serial liver biopsy study. J. Hepatol 68, S38. [Google Scholar]
- Heine A, Schilling J, Grünwald B, Krüger A, Gevensleben H, Held SAE, Garbi N, Kurts C, Brossart P, Knolle P, et al. (2016). The induction of human myeloid derived suppressor cells through hepatic stellate cells is dose-dependently inhibited by the tyrosine kinase inhibitors nilotinib, dasatinib and sorafenib, but not sunitinib. Cancer Immunol. Immun 65, 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, and Brenner DA (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J. Hepatol 30, 77–87. [DOI] [PubMed] [Google Scholar]
- Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. (2013). Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med 19, 1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Gea V, and Friedman SL (2012). Autophagy fuels tissue fibrogenesis. Autophagy 8, 849–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, and Friedman SL (2012). Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 142, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, Devi LA, and Friedman SL (2013). Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J. Hepatol 59, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi T, Friedman SL, and Hoshida Y (2017). Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev 121, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong Y, Li S, Wang J, and Li Y (2018). In vitro inhibition of hepatic stellate cell activation by the autophagy-related lipid droplet protein ATG2A. Sci. Rep 8, 9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2017). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kawada N, Sato E, Inoue M, and Kaneda K (1997). In vitro evidence of retinol transfer from stellate cells to hepatocytes. Cells Hepatic Sinusoid 6, 127–130. [Google Scholar]
- Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, Tazuma S, and Chayama K (2008). Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol 43, 298–304. [DOI] [PubMed] [Google Scholar]
- Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR, and Chandel NS (2013). Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J. Biol. Chem 288, 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia D, Duan F, Peng P, Sun L, Ruan Y, and Gu J (2015). Pyrroloquinoline-quinone suppresses liver fibrogenesis in mice. PLoS One 10, e0121939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Yang HR, Wang L, Wildey GM, Fung J, Qian S, and Lu L (2008). Hepatic stellate cells preferentially expand allogeneic CD4+ CD25+ FoxP3+ regulatory T cells in an IL-2-dependent manner. Transplantation 86, 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Gao H, Wang J, Yang S, Wang J, Liu J, Yang Y, Yan T, Chen T, Zhao Y, et al. (2017). Role and regulation of autophagy and apoptosis by nitric oxide in hepatic stellate cells during acute liver failure. Liver Int. : Off. J. Int. Assoc. Study Liver 37, 1651–1659. [DOI] [PubMed] [Google Scholar]
- Jung Y, Witek RP, Syn WK, Choi SS, Omenetti A, Premont R, Guy CD, and Diehl AM (2010). Signals from dying hepatocytes trigger growth of liver progenitors. Gut 59, 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Q, and Chen A (2009). Curcumin eliminates oxidized LDL roles in activating hepatic stellate cells by suppressing gene expression of lectin-like oxidized LDL receptor-1. Lab. Investig 89, 1275–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RS, Hasegawa D, Goossens N, Tsuchida T, Athwal V, Sun X, Robinson CL, Bhattacharya D, Chou HI, Zhang DY, et al. (2016). The XBP1 arm of the unfolded protein response induces fibrogenic activity in hepatic stellate cells through autophagy. Sci. Rep 6, 39342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, Iwaisako K, Moore-Morris T, Scott B, Tsukamoto H, et al. (2012). Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 109, 9448–9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe J, Wongsiriroj N, Troeger JS, Gwak G-Y, Dapito DH, Pradere J-P, Jiang H, Siddiqi M, Piantedosi R, O’Byrne SM, et al. (2011). Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 60, 1260–1268. [DOI] [PubMed] [Google Scholar]
- Koo JH, Lee HJ, Kim W, and Kim SG (2016). Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology 150, 181–193.e8. [DOI] [PubMed] [Google Scholar]
- Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, and Tacke F (2019). Single cell RNA sequencing identifies subsets of hepatic stellate cells and myofibroblasts in liver fibrosis. Cells 8, 503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wang J, Thomson AW, and Gandhi CR (2017). Hepatic stellate cells increase the immunosuppressive function of natural Foxp3+ regulatory T cells via IDO-induced AhR activation. J. Leukoc. Biol 101, 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AN, Higashi RM, and Fan TW (2020). Metabolic reprogramming in tumors: contributions of the tumor microenvironment. Genes Dis. 7, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer DA, Das A, Semela D, Kang-Decker N, Hendrickson H, Bronk SF, Katusic ZS, Gores GJ, and Shah VH (2008). Nitric oxide promotes caspase-independent hepatic stellate cell apoptosis through the generation of reactive oxygen species. Hepatology 47, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Bae SH, Jeong JW, Kim SH, and Kim KW (2004). Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp. Mol. Med 36, 1–12. [DOI] [PubMed] [Google Scholar]
- Lee TF, Mak KM, Rackovsky O, Lin YL, Kwong AJ, Loke JC, and Friedman SL (2010). Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by adipose differentiation-related protein (ADRP). J. Cell. Physiol 223, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, and Jeong WI (2012). Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol 27 (suppl 2), 75–79. [DOI] [PubMed] [Google Scholar]
- Lee YS, Wollam J, and Olefsky JM (2018). An integrated view of immunometabolism. Cell 172, 22–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemoinne S, and Friedman SL (2019). New and emerging anti-fibrotic therapeutics entering or already in clinical trials in chronic liver diseases. Curr. Opin. Pharmacol 49, 60–70. [DOI] [PubMed] [Google Scholar]
- Leo MA, Rosman AS, and Lieber CS (1993). Differential depletion of carotenoids and tocopherol in liver disease. Hepatology 17, 977–986. [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, and Robinson JP (2003). Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem 278, 8516–8525. [DOI] [PubMed] [Google Scholar]
- Liberti MV, and Locasale JW (2016). The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, and Chen A (2011). Curcumin diminishes the impacts of hyperglycemia on the activation of hepatic stellate cells by suppressing membrane translocation and gene expression of glucose transporter-2. Mol. Cell. Endocrinol 333, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zheng S, Attie AD, Keller MP, Bernlohr DA, Blaner WS, Newberry EP, Davidson NO, and Chen A (2018). Perilipin 5 and liver fatty acid binding protein function to restore quiescence in mouse hepatic stellate cells. J. Lipid Res 59, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Zheng S, and Chen A (2009). Curcumin attenuates the effects of insulin on stimulating hepatic stellate cell activation by interrupting insulin signaling and attenuating oxidative stress. Lab. Investig 89, 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu RM, and Desai LP (2015). Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 6, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Xu J, Rosenthal S, Zhang LJ, McCubbin R, Meshgin N, Shang L, Koyama Y, Ma HY, Sharma S, et al. (2020). Identification of lineage-specific transcription factors that prevent activation of hepatic stellate cells and promote fibrosis resolution. Gastroenterology 158, 1728–1744.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wang J, Xing W, Peng Y, Huang Y, and Fan X (2018). Role of DDAH/ADMA pathway in TGF-beta1-mediated activation of hepatic stellate cells. Mol. Med. Rep 17, 2549–2556. [DOI] [PubMed] [Google Scholar]
- Maher JJ (1993). Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J. Clin. Invest 91, 2244–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher JJ, Saito JM, and Neuschwander-Tetri BA (1997). Glutathione regulation in rat hepatic stellate cells. Comparative studies in primary culture and in liver injury in vivo. Biochem. Pharmacol 53, 637–641. [DOI] [PubMed] [Google Scholar]
- Mannaerts I, Thoen LFR, Eysackers N, Cubero FJ, Batista Leite S, Coldham I, Colle I, Trautwein C, and van Grunsven LA (2019). Unfolded protein response is an early, non-critical event during hepatic stellate cell activation. Cell Death Dis. 10, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura T, Gad MZ, Harrison EH, and Ross AC (1997). Lecithin:retinol acyltransferase and retinyl ester hydrolase activities are differentially regulated by retinoids and have distinct distributions between hepatocyte and nonparenchymal cell fractions of rat liver. J. Nutr 127, 218–224. [DOI] [PubMed] [Google Scholar]
- Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, and Schwabe RF (2013). Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun 4, 2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrpour M, Esclatine A, Beau I, and Codogno P (2010). Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am. J. Physiol. Cell Physiol 298, C776–C785. [DOI] [PubMed] [Google Scholar]
- Mejias M, Gallego J, Naranjo-Suarez S, Ramirez M, Pell N, Manzano A, Suñer C, Bartrons R, Mendez R, and Fernandez M (2020). CPEB4 increases expression of PFKFB3 to induce glycolysis and activate mouse and human hepatic stellate cells, promoting liver fibrosis. Gastroenterology 159, 273–288. [DOI] [PubMed] [Google Scholar]
- Mello T, Nakatsuka A, Fears S, Davis W, Tsukamoto H, Bosron WF, and Sanghani SP (2008). Expression of carboxylesterase and lipase genes in rat liver cell-types. Biochem. Biophys. Res. Commun 374, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y, Hasumura Y, and Takeuchi J (1983). The role of fat-storing cells in Disse space fibrogenesis in alcoholic liver disease. Hepatology 3, 559–566. [DOI] [PubMed] [Google Scholar]
- Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, and Seki E (2012). TLR2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation. Hepatology 57, 577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, and Klionsky DJ (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung SJ, Yoon JH, Gwak GY, Kim W, Lee JH, Kim KM, Shin CS, Jang JJ, Lee SH, Lee SM, et al. (2007). Wnt signaling enhances the activation and survival of human hepatic stellate cells. FEBS Lett. 581, 2954–2958. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kamiya A, Sumiyoshi H, Tsuruya K, Kagawa T, and Inagaki Y (2020). A deactivation factor of fibrogenic hepatic stellate cells induces regression of liver fibrosis in mice. Hepatology 71, 1437–1452. [DOI] [PubMed] [Google Scholar]
- Notas G, Kisseleva T, and Brenner D (2009). NK and NKT cells in liver injury and fibrosis. Clin. Immunol 130, 16–26. [DOI] [PubMed] [Google Scholar]
- Oh JE, Shim KY, Lee JI, Choi SI, Baik SK, and Eom YW (2017). 1-Methyl-L-tryptophan promotes the apoptosis of hepatic stellate cells arrested by interferon-gamma by increasing the expression of IFN-γRβ, IRF-1 and FAS. Int. J. Mol. Med 40, 576–582. [DOI] [PubMed] [Google Scholar]
- Onichtchouk D, Chen YG, Dosch R, Gawantka V, Delius H, Massagué J, and Niehrs C (1999). Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 401, 480–485. [DOI] [PubMed] [Google Scholar]
- Ottonello S, Petrucco S, and Maraini G (1987). Vitamin A uptake from retinol-binding protein in a cell-free system from pigment epithelial cells of bovine retina. Retinol transfer from plasma retinol-binding protein to cytoplasmic retinol-binding protein with retinyl-ester formation as the intermediate step. J. Biol. Chem 262, 3975–3981. [PubMed] [Google Scholar]
- Pang W, Zhang Y, Wang S, Jia A, Dong W, Cai C, Hua Z, and Zhang J (2011). The mPlrp2 and mClps genes are involved in the hydrolysis of retinyl esters in the mouse liver. J. Lipid Res 52, 934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, Conti M, Huet S, Ba N, Buffet C, et al. (2001). High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 34, 738–744. [DOI] [PubMed] [Google Scholar]
- Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, Burza MA, Indiveri C, Ferro Y, Montalcini T, et al. (2014). PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet 23, 4077–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proell V, Carmona-Cuenca I, Murillo MM, Huber H, Fabregat I, and Mikulits W (2007). TGF-beta dependent regulation of oxygen radicals during transdifferentiation of activated hepatic stellate cells to myofibroblastoid cells. Comp. Hepatol 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Zhong Y, Dang L, Zhu M, Yu H, Chen W, Cui J, Bian H, and Li Z (2012). Alteration of protein glycosylation in human hepatic stellate cells activated with transforming growth factor-beta1. J. Proteomics 75, 4114–4123. [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, Portman JR, Matchett KP, Brice M, Marwick JA, et al. (2019). Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575, 512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, and Henderson NC (2020). Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol 17, 457–472. [DOI] [PubMed] [Google Scholar]
- Ranganathan AC, Nelson KK, Rodriguez AM, Kim KH, Tower GB, Rutter JL, Brinckerhoff CE, Huang TT, Epstein CJ, Jeffrey JJ, et al. (2001). Manganese superoxide dismutase signals matrix metalloproteinase expression via H2O2-dependent ERK1/2 activation. J. Biol. Chem 276, 14264–14270. [DOI] [PubMed] [Google Scholar]
- Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, et al. (2016). Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 150, 1147–1159.e5. [DOI] [PubMed] [Google Scholar]
- Roh YS, and Seki E (2018). Chemokines and chemokine receptors in the development of NAFLD. Adv. Exp. Med. Biol 1061, 45–53. [DOI] [PubMed] [Google Scholar]
- Ross AC (1982). Retinol esterification by rat liver microsomes. Evidence for a fatty acyl coenzyme A: retinol acyltransferase. J. Biol. Chem 257, 2453–2459. [PubMed] [Google Scholar]
- Saeed A, Bartuzi P, Heegsma J, Dekker D, Kloosterhuis N, de Bruin A, Jonker JW, van de Sluis B, and Faber KN (2020). Impaired hepatic vitamin A metabolism in NAFLD mice leading to vitamin A accumulation in hepatocytes. Cell. Mol. Gastroenterol. Hepatol 10.1016/j.jcmgh.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A, Dullaart RPF, Schreuder TCMA, Blokzijl H, and Faber KN (2017). Disturbed vitamin A metabolism in non-alcoholic fatty liver disease (NAFLD). Nutrients 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernández-Rodriguez CM, Pinedo F, Huber H, Eferl R, Mikulits W, and Fabregat I (2012). NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One 7, e45285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, Takimoto R, Takada K, Miyanishi K, Matsunaga T, et al. (2008). Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol 26, 431–442. [DOI] [PubMed] [Google Scholar]
- Sauvant P, Sapin V, Alexandre-Gouabau MC, Dodeman I, Delpal S, Quadro L, Partier A, Abergel A, Colantuoni V, and Rock E (2001). Retinol mobilization from cultured rat hepatic stellate cells does not require retinol binding protein synthesis and secretion. Int. J. Biochem. Cell Biol 33, 1000–1012. [DOI] [PubMed] [Google Scholar]
- Schneiderhan W, Schmid-Kotsas A, Zhao J, Grünert A, Nüssler A, Weidenbach H, Menke A, Schmid RM, Adler G, and Bachem MG (2001). Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology 34, 729–737. [DOI] [PubMed] [Google Scholar]
- Schreiber R, Taschler U, Preiss-Landl K, Wongsiriroj N, Zimmermann R, and Lass A (2012). Retinyl ester hydrolases and their roles in vitamin A homeostasis. Biochim. Biophys. Acta 1821, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Tabas I, and Pajvani UB (2020). Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, and Schwabe RF (2007). TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med 13, 1324–1332. [DOI] [PubMed] [Google Scholar]
- Senoo H, Yoshikawa K, Morii M, Miura M, Imai K, and Mezaki Y (2010). Hepatic stellate cell (vitamin A-storing cell) and its relative-past, present and future. Cell Biol. Int 34, 1247–1272. [DOI] [PubMed] [Google Scholar]
- Seo W, Eun HS, Kim SY, Yi HS, Lee YS, Park SH, Jang MJ, Jo E, Kim SC, Han YM, et al. (2016). Exosome-mediated activation of toll-like receptor 3 in stellate cells stimulates interleukin-17 production by gammadelta T cells in liver fibrosis. Hepatology 64, 616–631. [DOI] [PubMed] [Google Scholar]
- Serviddio G, Bellanti F, Stanca E, Lunetti P, Blonda M, Tamborra R, Siculella L, Vendemiale G, Capobianco L, and Giudetti AM (2014). Silybin exerts antioxidant effects and induces mitochondrial biogenesis in liver of rat with secondary biliary cirrhosis. Free Radic. Biol. Med 73, 117–126. [DOI] [PubMed] [Google Scholar]
- Shao X, Wang M, Wei X, Deng S, Fu N, Peng Q, Jiang Y, Ye L, Xie J, and Lin Y (2016). Peroxisome proliferator-activated receptor-gamma: master regulator of adipogenesis and obesity. Curr. Stem Cell Res. Ther 11, 282–289. [DOI] [PubMed] [Google Scholar]
- She H, Xiong S, Hazra S, and Tsukamoto H (2005). Adipogenic transcriptional regulation of hepatic stellate cells. J. Biol. Chem 280, 4959–4967. [DOI] [PubMed] [Google Scholar]
- Shmarakov IO, Jiang H, Liu J, Fernandez EJ, and Blaner WS (2019). Hepatic stellate cell activation: a source for bioactive lipids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1864, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund SV, Qian T, de Minicis S, Harvey-White J, Kunos G, Vinod KY, Hungund B, and Schwabe RF (2007). The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J. 21, 2798–2806. [DOI] [PubMed] [Google Scholar]
- Straub BK, Gyoengyoesi B, Koenig M, Hashani M, Pawella LM, Herpel E, Mueller W, Macher-Goeppinger S, Heid H, and Schirmacher P (2013). Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology 62, 617–631. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Fan Z, Chen J, Tian W, Li M, Xu H, Wu X, Shao J, Bian Y, Fang M, et al. (2016). Transcriptional repression of SIRT1 by protein inhibitor of activated STAT 4 (PIAS4) in hepatic stellate cells contributes to liver fibrosis. Sci. Rep 6, 28432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschler U, Schreiber R, Chitraju C, Grabner GF, Romauch M, Wolinski H, Haemmerle G, Breinbauer R, Zechner R, Lass A, et al. (2015). Adipose triglyceride lipase is involved in the mobilization of triglyceride and retinoid stores of hepatic stellate cells. Biochim. Biophys. Acta 1851, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teratani T, Tomita K, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Tominaga S, Hiroi S, Irie R, Okada Y, et al. (2012). A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 142, 152–164.e10. [DOI] [PubMed] [Google Scholar]
- Testerink N, Ajat M, Houweling M, Brouwers JF, Pully VV, van Manen HJ, Otto C, Helms JB, and Vaandrager AB (2012). Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation. PLoS One 7, e34945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Teratani T, Suzuki T, Shimizu M, Sato H, Narimatsu K, Okada Y, Kurihara C, Irie R, Yokoyama H, et al. (2014). Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 59, 154–169. [DOI] [PubMed] [Google Scholar]
- Troeger JS, Mederacke I, Gwak GY, Dapito DH, Mu X, Hsu CC, Pradere JP, Friedman RA, and Schwabe RF (2012). Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 143, 1073–1083.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeger JS, and Schwabe RF (2011). Neuropilin and liver fibrosis: hitting three birds with one stone? Hepatology 54, 1091–1093. [DOI] [PubMed] [Google Scholar]
- Trøen G, Nilsson A, Norum KR, and Blomhoff R (1994). Characterization of liver stellate cell retinyl ester storage. Biochem. J 300, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, and Friedman SL (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol 14, 397–411. [DOI] [PubMed] [Google Scholar]
- Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S, Fiel MI, Goossens N, Chou HI, Hoshida Y, et al. (2018). A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol 69, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada S, Westwick JK, Ikejima K, Sato N, and Rippe RA (2005). SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J. Biol. Chem 280, 10055–10064. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H (2005). Adipogenic phenotype of hepatic stellate cells. Alcohol. Clin. Exp. Res 29 (Supplement), 132S–133S. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, She H, Hazra S, Cheng J, and Miyahara T (2006). Anti-adipogenic regulation underlies hepatic stellate cell transdifferentiation. J. Gastroenterol. Hepatol 21 (suppl 3), S102–S105. [DOI] [PubMed] [Google Scholar]
- Tuohetahuntila M, Molenaar MR, Spee B, Brouwers JF, Wubbolts R, Houweling M, Yan C, Du H, VanderVen BC, Vaandrager AB, et al. (2017). Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation. J. Biol. Chem 292, 12436–12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urtasun R, Conde de la Rosa L, and Nieto N (2008). Oxidative and nitrosative stress and fibrogenic response. Clin. Liver Dis 12, 769–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Xia T, Du Y, Liu J, Xie Y, Zhang Y, Guan F, Wu J, Wang X, and Shi C (2019). Exosomes from activated hepatic stellate cells contain GLUT1 and PKM2: a role for exosomes in metabolic switch of liver nonparenchymal cells. FASEB J. 33, 8530–8542. [DOI] [PubMed] [Google Scholar]
- Wang L, Tankersley LR, Tang M, Potter JJ, and Mezey E (2002). Regulation of the murine alpha(2)(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys 401, 262–270. [DOI] [PubMed] [Google Scholar]
- Wang S, and Friedman SL (2020). Hepatic fibrosis: a convergent response to liver injury that is reversible. J. Hepatol 73, 210–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, Shi H, Valenti L, Pajvani UB, Sandhu J, et al. (2020). Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab. 31, 969–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, et al. (2016). Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 24, 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang Y, Guan F, Xiao Y, Deng J, Chen H, Chen X, Li J, Huang H, and Shi C (2013). Hypoxia-inducible factor-1alpha and MAPK co-regulate activation of hepatic stellate cells upon hypoxia stimulation. PLoS One 8, e74051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, and Evans RM (2003). Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 113, 159–170. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, and Mehal WZ (2007). Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 46, 1509–1518. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, and Tacke F (2014). Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr 3, 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng W, Li L, van Bennekum AM, Potter SH, Harrison EH, Blaner WS, Breslow JL, and Fisher EA (1999). Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry 38, 4143–4149. [DOI] [PubMed] [Google Scholar]
- Winau F, Hegasy G, Weiskirchen R, Weber S, Cassan C, Sieling PA, Modlin RL, Liblau RS, Gressner AM, and Kaufmann SH (2007). Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26, 117–129. [DOI] [PubMed] [Google Scholar]
- Wu MH, Chen YL, Lee KH, Chang CC, Cheng TM, Wu SY, Tu CC, and Tsui WL (2017). Glycosylation-dependent galectin-1/neuropilin-1 interactions promote liver fibrosis through activation of TGF-beta- and PDGF-like signals in hepatic stellate cells. Sci. Rep 7, 11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, et al. (2019). Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Deng X, Zhai X, Zhou M, Jia X, Luo L, Niu M, Zhu H, Qiang H, and Zhou Y (2012). p38 mitogen-activated protein kinase and liver X receptor-alpha mediate the leptin effect on sterol regulatory element binding protein-1c expression in hepatic stellate cells. Mol. Med 18, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Sakai-Sawada K, Niitsu Y, and Tamura Y (2016). Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp. Cell Res 341, 8–17. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, Bedossa P, Geier A, Beckebaum S, Newsome PN, et al. (2019). Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zhao S, Yao Z, Wang L, Shao J, Chen A, Zhang F, and Zheng S (2017). Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 11, 322–334. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zheng D, Jiang Y, Qu C, Yuan H, Hu K, He L, Chen P, Li J, Tu M, Lin L, et al. (2020). Pyruvate kinase M2 tetramerization protects against hepatic stellate cell activation and liver fibrosis. Am. J. Pathol 190, 2267–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Cui S, He Q, Guo Y, Pan X, Zhang P, Huang N, Ge C, Wang G, Gonzalez FJ, et al. (2020). SUMOylation inhibitors synergize with FXR agonists in combating liver fibrosis. Nat. Commun 11, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Jia X, Qin J, Lu C, Zhu H, Li X, Han X, and Sun X (2010). Leptin inhibits PPARgamma gene expression in hepatic stellate cells in the mouse model of liver damage. Mol. Cell. Endocrinol 323, 193–200. [DOI] [PubMed] [Google Scholar]
- Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, Meroni M, Graham MJ, Yates KP, Diehl AM, et al. (2018). Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med 10, eaat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]