

Abstract
Parallelism, the evolution of similar traits in populations diversifying in similar conditions, provides strong evidence of adaptation by natural selection. Many studies of parallelism focus on comparisons of different ecotypes or contrasting environments, defined a priori, which could upwardly bias the apparent prevalence of parallelism. Here, we estimated genomic parallelism associated with components of environmental and phenotypic variation at an intercontinental scale across four freshwater adaptive radiations (Alaska, British Columbia, Iceland, Scotland) of the three-spined stickleback (Gasterosteus aculeatus). We combined large-scale biological sampling and phenotyping with RAD-sequencing data from 73 freshwater lake populations and four marine ones (1,380 fish) to associate genome-wide allele frequencies with continuous distributions of environmental and phenotypic variation. Our three main findings demonstrate: 1) quantitative variation in phenotypes and environments can predict genomic parallelism; 2) genomic parallelism at the early stages of adaptive radiations, even at large geographic scales, is founded on standing variation; and 3) similar environments are a better predictor of genome-wide parallelism than similar phenotypes. Overall, this study validates the importance and predictive power of major phenotypic and environmental factors likely to influence the emergence of common patterns of genomic divergence, providing a clearer picture than analyses of dichotomous phenotypes and environments.
Introduction
Adaptive radiations are rapid branchings on the tree of life, associated with adaptation to distinct ecological niches (1). As major sources of biodiversity, their study has revealed much about the evolution of phenotypic variation (1,2). Adaptive radiations also highlight what is unknown about biodiversity evolution. For example, despite abundant phenotypic diversity, not all trait combinations evolve in every radiation, yet organisms in different places sometimes arrive at very similar endpoints (3,4). The latter has been observed in cichlid fish (5), Anolis lizards (6) and Darwin’s finches (7); famous examples of parallel phenotypic evolution. This suggests Stephen Jay Gould’s contention that evolution is contingent and unrepeatable (8) cannot be completely true. Extensive prior work has examined the role genetic correlations between traits might play in constraining diversity, but the answers provided have not been entirely satisfactory (3,9,10). Alongside these processes, it is probable that repeatable patterns of evolution are channelled in predictable ways by common environments and selection regimes. Often termed parallel evolution (distinguishable from convergence by shared evolutionary ‘start’ as well as ‘end’ points, but see (11,12)), this process results from environmental similarities within and between radiations. Striking, natural examples of phenotypic parallelism (1,6) support this hypothesis, and the persistent appearance of familiar forms in similar ecological niches demonstrates the significance of selection.
However, a limitation with studies on phenotypic parallelism is that they have concentrated largely on the comparison of pairs of strongly different ecotypes or environments (13–15). This approach might upwardly bias the prevalence of parallelism with comparisons known a priori to occur repeatedly, effectively constraining the evolutionary ‘end’ point. Further, similar environments are typically assumed on the basis of comparable, typically morphological or life history, phenotypes, concealing the role of individual components of environmental variation in driving parallelism. This compromises our ability to understand adaptation, much of which is likely to be broadly physiological. Such drawbacks highlight the importance of combining measures of phenotype, environment and genomics in studies of parallelism. Addressing this gap is needed for a complete understanding of adaptation (16).
Highlighting consistent signatures of adaptation in the genomes of multiple, independent natural populations has also proven to be a valuable tool for studies on the genetic basis of adaptation (17–19). However, again our comprehension of the relationship between genomic parallelism and continuous phenotypic or environmental variation is surprisingly poor. A major barrier to combining genotype, environment and phenotype has been achieving the necessary biological replication across all three to make broad inferences and shift from description to hypothesis testing. This has rarely been applied (but see 20–22), and it remains to be shown whether signals of parallelism obtained from continuous measures are comparable to those from ecotypes and previous studies. Here we make use of such methods to test for environmentally and phenotypically associated genomic parallelism across radiations of three-spined stickleback fish (Gasterosteus aculeatus, hereafter ‘stickleback’).
Stickleback provide a powerful natural experiment to test parallelism. They are ancestrally marine but, following colonisation of freshwater across the northern hemisphere, are in the early stages of replicated adaptive radiations (which we define as sets of geographically proximal, closely-related populations). Comparing multiple populations derived from the marine ancestor provides a model for exploring both phenotypic parallelism and its genetic basis (23,24) in response to environmental variation. Phenotypic parallelism is well established (25,26), and whilst often considered in dichotomous pairings of marine-freshwater, benthic-limnetic or lake-stream ecotypes, there is substantial continuous phenotypic variation among freshwater populations that has rarely been explored (27) in this context. Parallel genomic loci under selection have been identified across the contrasting ecotype pairs (19,28,29), but the combination of phenotypic and genomic parallelism in one study is rare (although see 20,30,31) and has not been done at the scale of replicated adaptive radiations across continents.
For this study we sampled 73 freshwater lake populations (1,304 fish) and four marine populations (76 fish) from four adaptive radiations: two from the Pacific coast of North America (Alaska and British Columbia [‘BC’]), and two from Atlantic Europe (Iceland and the island of North Uist [‘Scotland’]) (Fig. 1, Supplementary Table 1). We consider the variation between lakes, within geographic regions, as adaptive radiations. Assessing groups of lakes as adaptive radiations distinguishes the stickleback system from other notable systems of adaptive radiations, such as the comparisons of within-lake cichlid radiations (5, 14,17). We quantify parallelism between phenotypes, environments, and genomic loci under selection (using RAD-sequencing data) in each adaptive radiation and examine how they are associated. Rewards to be gained by connecting the evolution of parallelism more explicitly to the environmental and phenotypic variation include a better grasp of why some traits evolve in concert and a predictive understanding of parallelism and repeatability (4). This new understanding is essential if we are to reach a consensus on how biodiversity is altered by adaptation.
Fig. 1.
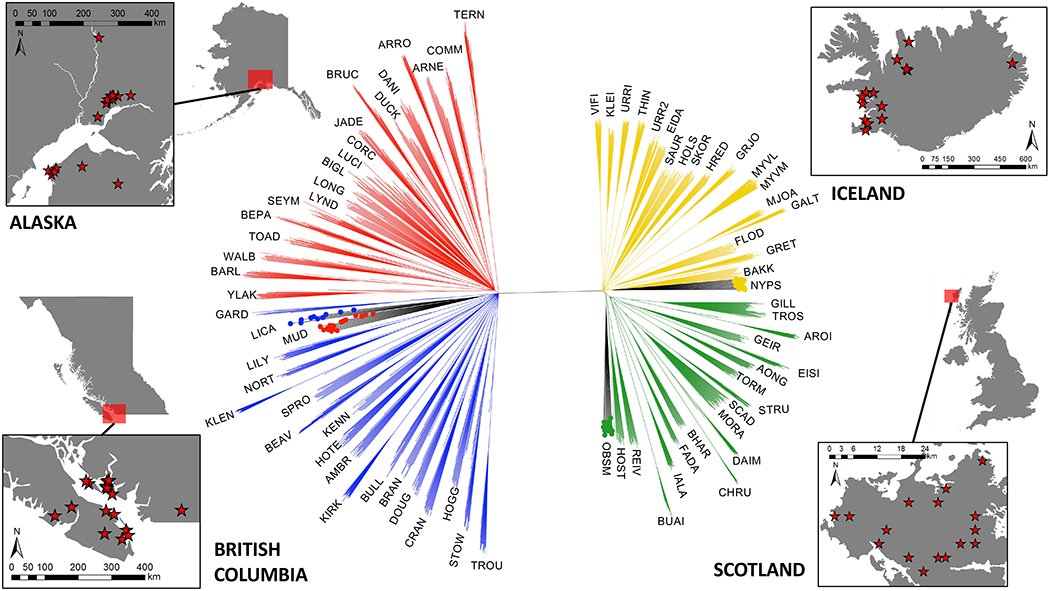
Sampling sites and bootstrapped unrooted NJ tree for stickleback from 73 freshwater lake populations and 4 marine ones from four countries on two continents, based on 11,266 genetic markers for 1,380 individuals. All nodes shown have bootstrap support of at least 80 (other nodes were collapsed). Freshwater branches are coloured by the radiation to which they belong. Marine branches are black with the tips coloured according to radiation. Tips represent individual fish, which were generally tightly clustered by population (small labels). Stars represent lakes sampled.
RESULTS & DISCUSSION
Environmental and phenotypic similarity across radiations
We first quantified environmental and phenotypic parallelism across four adaptive radiations to provide an indication of how much genomic parallelism associated with environments and phenotypes to expect. For environments, we quantified lake area, parasite prevalence (Gyrodactylus spp [Gyro] and Schistocephalus spp [Schisto]) and water chemistry (pH and metal cation concentrations of calcium [Ca], sodium [Na], zinc [Zn]). For phenotypes, we collected 12 variables associated with shape (3), armour (7) and tropic (2) morphology (Supplementary Table 2). See methods for detailed information on sampling and measures.
I). Environment.
A Principal Component Analysis (PCA) on all seven variables across all lakes revealed that the first axis of environmental variance (EnvPC1) separated lakes along a predominant gradient of pH, with additional minor loadings reflecting Ca and Gyro (Fig. 2a; Supplementary Table 3). This axis did not separate radiations but emphasised the variation from alkaline to acid present in all of them (the most acidic environments were absent in Iceland). EnvPC2 separated lakes with high and low zinc, with additional minor loadings of Schisto (positive) and Ca (negative). This axis separated European and North American clusters. Interestingly, BC lakes were completely subsumed within the more environmentally variable Alaskan lake cluster (Fig 2a,b).
Fig. 2.
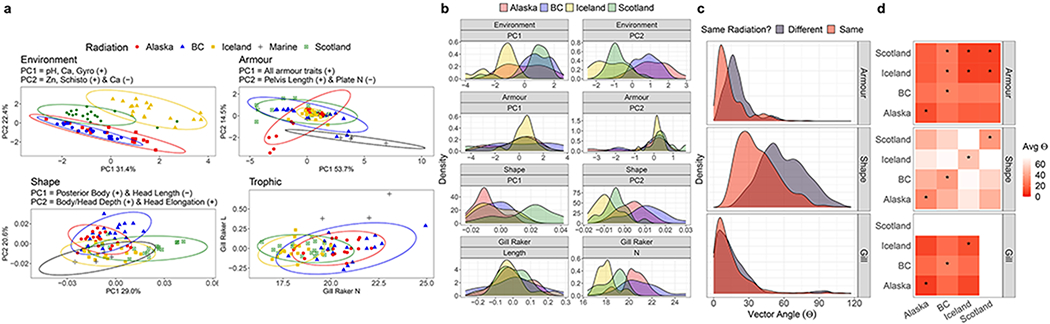
Comparisons and analyses of environmental and phenotypic parallelism across adaptive radiations: (a) Principal Component Analyses of environmental variables (Environment); regression residuals of Procrustes coordinates against log centroid of body shape (Shape); armour traits; and trophic traits gill raker numbers and length. Each point represents a population and ellipses are 95% confidence ellipses. Names of variables with the highest positive (+) or negative (−) loadings in subtitle. Marine populations (+) are projected where data was available using PC loadings calculated with freshwater populations only; (b) Density distributions of populations along each PC axis grouped by adaptive radiation; (c) Angles between marine-freshwater vectors for armour, shape, and gill raker traits. For each category of traits, density distributions are coloured according to whether vectors were compared within, or between different radiations; (d) Heatmaps of comparisons of vectors between specific radiation pairings coloured inversely according to average angle. Each panel includes the p-value denoting whether between-radiation vectors differ significantly from within-radiation vectors (defined as radiation on y-axis). If between-radiation vectors do not differ significantly from within-radiation vectors, a common trajectory of marine-freshwater phenotypes is assumed.
II). Phenotypes - Armour.
All armour traits loaded positively onto ArmourPC1 (Fig. 2a; Supplementary Table 3). All radiations overlapped on ArmourPC1 (Fig. 2b), but there were also significant differences in ArmourPC1 values between radiations (Supplementary Table 4). Scotland and Alaska had populations with extreme armour reduction (low ArmourPC1), but in Scotland these populations also had complete loss of armour plates (high ArmourPC2) which were retained in Alaska (low ArmourPC2). These deviations produce the anomalous relationship between ArmourPC1 and ArmourPC2 in Alaska (Fig. 2a). Iceland exhibited minimal variation in armour traits compared to other radiations. Aside from a few populations from BC, there was no overlap in armour traits between marine and freshwater populations. Marine populations have a higher number of lateral plates and generally more exaggerated armour traits. Importantly however, projection of marine armour phenotypes suggests they fall on the same, parallel, axis, but the marine morpho space is beyond freshwater space (Fig. 2a).
Phenotypic change vectors analysis (PCVA - see methods) highlighted phenotypic constraint along a common axis for armour (mean angle (θ) within radiations = 14.5°, between = 19.5°) (Fig. 2c, Supplementary Table 5). Indeed, θ values between vectors from Iceland and Scotland were not significantly larger than θ values between vectors within Iceland or within Scotland (p > 0.05), suggesting a highly parallel axis for marine to freshwater transition in Europe. There was some divergence away from this axis, particularly in Alaska, with significant deviations (p < 0.001) in trajectory through trait space (Fig. 2d). This likely comes as a result of Alaskan ArmourPC2 deviations, but these results suggest some non-parallelism between North America and Europe. Despite significant variation among vector trajectories, average θ remained low (≤ 28.3°), suggesting these significant differences in vector angle represent relatively minor idiosyncrasy along an otherwise conserved, parallel axis.
III). Phenotypes - Body shape.
Body shape differed significantly between lakes and radiations (Supplementary Table 4), despite overlap within morphospace (Fig. 2a,b). Scotland exhibited the most extreme body shapes (high ShapePC1: elongated, slender bodies, small heads). BC populations had particularly deep bodies, and long, deep heads (high ShapePC2). Surprisingly Scotland, the smallest region sampled (303 km2), had the most shape variation. Marine populations had low ShapePC1 and ShapePC2 scores, overlapping with some freshwater populations mostly from Iceland (Fig. 2a). Shape PCVA highlighted greater non-parallelism compared with armour (mean θ within radiations = 38.3°, between = 59.3°), with numerous orthogonal comparisons (Fig. 2c). All between-radiation comparisons of θ were significantly greater than within-radiation (p < 0.001) (Fig. 2d), demonstrating marine-freshwater shape phenotype transitions have occurred along variable, nonparallel trajectories.
IV). Phenotypes - Trophic morphology.
Generally, gill raker length increased with gill raker number. Alaskan trophic morphology was the most variable, and BC contained a subset of that variation (Fig. 2a,b). Marine-freshwater trophic PCVA highlighted parallel changes (mean θ within radiations = 15.7°, between = 16.6°), although we lacked marine trophic data from Scotland (Fig. 2c, Supplementary Table 5). Significant deviations among Alaska, BC and Iceland (Fig. 2d), however suggest some idiosyncrasy among radiations. Freshwater populations had shorter gill rakers for their size relative to marine populations.
V). Relationship between environmental and phenotypic similarity.
We used GLMs to test for parallel associations between each environmental variable and morphology (full results in Supplementary Table 6). Between armour traits and environmental variables, only ArmourPC1 was significantly associated with pH in a parallel way across all radiations (F1,71 = 9.20, FDR = 0.006, σ2 explained = 11.3%). Although there were also nonparallel, radiation-specific associations (slopes varying between radiations) between armour and several other environmental variables, such as the North-American specific associations between ArmourPC2 and both Ca and Na. This is consistent with previously reported nonparallel associations between armour and Ca between North American and European radiations (32). These results highlight parallel reductions of skeletal traits (ArmourPC1) with lower pH, and suggest pH is a predictor of phenotypic armour parallelism globally. Given Iceland lacks more acidic freshwater habitats, this may explain why Iceland has comparably limited armour variation.
ShapePC1 and Na were associated in parallel across our dataset (F1,71 = 6.51, FDR = 0.018, σ2 explained = 46.2%), whereby fish at lower salinity levels were elongated and had small heads. ShapePC1 and Gyro were associated but only in Europe (F3,65 = 3.01, FDR = 0.045, σ2 explained = 48.7%). ShapePC2 varied in a parallel manner with Schisto (F1,71 = 46.4, FDR = <0.001, σ2 explained = 55.5%), potentially because of body shape distortion that can occur as a result of S. solidus infection. Although the causative factors driving the link between body shape and water chemistry are largely unknown, similar relationships between salinity and stickleback body shape have been found in other lakes (33).
We expected trophic morphology to evolve in response to zooplankton communities, which can vary in response to water chemistry (34,35), particularly Zn. In agreement, we found parallel, inverse associations between gill raker number and Zn (F1,71 = 63.6, FDR = <0.001, σ2 explained = 53.4%).
The parallel environment - phenotype associations described here (either globally or between specific radiations) might thus be expected to be underwritten by parallel genetic variation. This is particularly true for environmental variables that also overlap between radiations (such as pH and Ca) and phenotypes associated with them. These predictions are contingent on traits having a simple genetic basis however (28,36,37), and may not hold for phenotypes with more complex, polygenic architectures.
Phylogenetic relationship among radiations.
Older lineages, or lineages not experiencing gene flow, are expected to share less ancestral variation as a function of common ancestry, which may constrain parallelism (18,38,39).
Recent studies have highlighted this as a probable limitation of parallelism in adaptive radiations across continental scales (32,40–42). Knowing the genetic relationship across all populations is therefore important to interpret patterns of genomic parallelism. A Neighbour Joining (NJ) tree based on 11,266 unlinked SNPs showed that the four geographic locations form four well-resolved radiations (Fig. 1). TERN in Alaska is slightly anomalous, sitting at a shorter evolutionary distance from European lakes compared with the rest of Alaska, suggesting more recent colonisation by the common ancestor. Interestingly, Icelandic and Scottish marine populations clustered separately with their respective freshwater populations, but both North American marine populations clustered together. This may indicate stronger structuring of marine populations in the Atlantic relative to the Pacific, or a re-invasion of Alaskan marine regions by BC marine populations. PCA on freshwater populations confirmed that radiations form independent clusters, with the dominant axis of variation (PC1 = 36.0%) separating Pacific/North American and Atlantic/European radiation pairs (Supplementary Fig. 1). PC2 (7.0%) and PC3 (5.8%) separated North American and European radiations respectively. Geographically adjacent radiations were also the most genetically similar (Supplementary Table 7, Alaska and BC: mean pairwise FST = 0.198), Scotland and Iceland: FST = 0.194), suggesting that despite independent clustering, lineage splits may be relatively recent, or that gene flow may be occurring within the Atlantic and Pacific groups. Divergence across continents was stronger and deeper (0.314 ⩽ FST ⩽ 0.338) than within oceans/continents, consistent with previous studies that have estimated the time of divergence between Atlantic and Pacific stickleback at approximately 200,000 years (43,44 but see 45).
Shared polymorphisms among adaptive radiations were structured predictably (Supplementary Fig. 2), with most shared sites found between Alaska and BC (N = 11,524), and Iceland and Scotland (N = 8,789). A considerable number of SNP polymorphisms were common to all four radiations however (SNP was polymorphic in all radiations, N = 6,339), suggesting some global retention of ancestral variation. Not including globally shared polymorphisms, sharing among inter-continental comparisons was minimal (N ≤ 933). In agreement, between-continent structuring accounts for the largest proportion of molecular variance in our data (AMOVA: σ = 889.7, 34.7%). Within continents, populations within radiations (σ = 366.5, 14.3%) were more genetically variable than radiations themselves (σ = 142.3, 5.6%) (Supplementary Table 8). Molecular variance then is not structured according to geographic scale (Continent > Radiation > Populations). This may be due to intra-continental gene flow, bottlenecks at the founding of ancestral Pacific/Atlantic marine lineages (52), or older Pacific/Atlantic divergence times relative to modern freshwater, but it is not possible to differentiate between the two scenarios without demographic modelling.
Phenotypically and environmentally associated SNPs and genomic regions within radiations.
We associated allele frequency changes with each environmental/phenotypic variable (N = 19) within each radiation (18-19 populations) using Bayenv2 (46) (see methods), and compared outlier genome windows across radiations to identify parallel genome changes.
Several thousand SNPs for each radiation were highly associated (high Bayesfactor [>log10(1.5)] and top 5% of Spearman’s ρ, see methods) with environmental and phenotypic variables (Supplementary Table 9). We mapped SNPs onto non-overlapping 50kb windows, consistent with approximate linkage disequilibrium within the stickleback genome (47,48), but also examined 75kb, 100kb, 200kb windows (Supplementary Dataset 1) and windows of equivalent genetic distance (0.1 cM), which confirmed our results were robust and not influenced by variable linkage across the genome (Supplementary Information 1).
We found 1836 unique 50kb windows associated with an environmental variable or a phenotypic trait (Supplementary Dataset 1), ranging from 146 windows associated with pelvic spine length in BC, to 11 associated with lake area variation in Scotland. These strong signals of association, even across modest variation, support the adaptive nature of these radiations. Across unique windows, 454 were associated with both an environmental and phenotypic variable in the same radiation, suggesting some phenotypically-associated regions are also responsible for local adaptation to environments (Supplementary Dataset 1). This result also suggests measuring important aspects of the environment may provide profitable ways of identifying candidate regions for adaptation and cryptic phenotypes, such as physiology.
Genomic parallelism associated with environmental and phenotypic variation across radiations.
To assess genomic parallelism, we compared observed windows associated in two or more radiations (parallel windows) against randomly permuted (10,000 iterations) null distributions. We first quantified the overall level of genomic parallelism associated with groups of environmental or phenotypic variables. There were no environmentally- or phenotypically- associated 50kb windows parallel in all four radiations for individual variables (Randomised permutations NExpected-Environmental= 0, NExp-Pheno = 0.0002), but one window was parallel in a group of three radiations (chrIV: 14400000-14450000 associated with length of pelvis in BC, Iceland and Scotland) (NExp-Env = 0.05, NExp-Pheno = 0.149, p = 0.133). Many windows however exhibited parallelism between pairs of radiations. A total of 39 environmentally-associated windows (pooled across all seven variables) (NExp = 12.4, 95% Upper limit (UL) = 18, p < 0.001) and 65 phenotypically-associated windows (pooled across all 12 variables) (NExp = 30.7, 95% UL = 40, p < 0.001) were parallel between two radiations (Supplementary Table 10). Parallelism was disproportionately greater for armour (46/65) and gill raker traits (12/65) (number mostly) than for shape (7/65) (χ2= 6.506, P = 0.04). This is consistent with skeletal traits, with simple genetic architectures (28,36,37), being more likely to show evidence of phenotypic parallelism. Interestingly, parallel associated windows (mean SNP N = 7.13, sd = 4.4) had on average more SNPs per window than nonparallel windows (mean SNP N = 6.27, sd = 4.3) (GLM, LRT1,3867 = 22.4, p < 0.001) and exhibited slightly stronger signals of association with variables (mean residual SNPs above expected = 1.82 parallel; 1.60 non-parallel; GLM, LRT1,3867 = 5.48, p = 0.019).
We next explored parallelism associated with individual environmental/phenotypic variables. We observed significantly more parallel windows than expected for two environmental variables (Ca and pH) (Fig. 3; Supplementary Table 10). Reflecting expectations, these were the same variables that load onto the shared EnvPC1 across all radiations (Fig. 2b), and were involved in parallel environment x phenotype interactions in all (pH) or some (Ca) radiations. Further, we did not detect significant genomic parallelism associated with variables that varied between radiations, such as salinity, zinc and S. solidus prevalence. These results highlight that common environmental axes, such as the shared acid-alkali axis, promote signals of parallelism in the genome, although parallelism appears limited to specific pairs rather than extending to all radiations. However, not all environmental variables with parallel environment x phenotype interactions produced signals of genomic parallelism, such as Na-ShapePC1 and Zn-Gill Raker number.
Fig. 3.
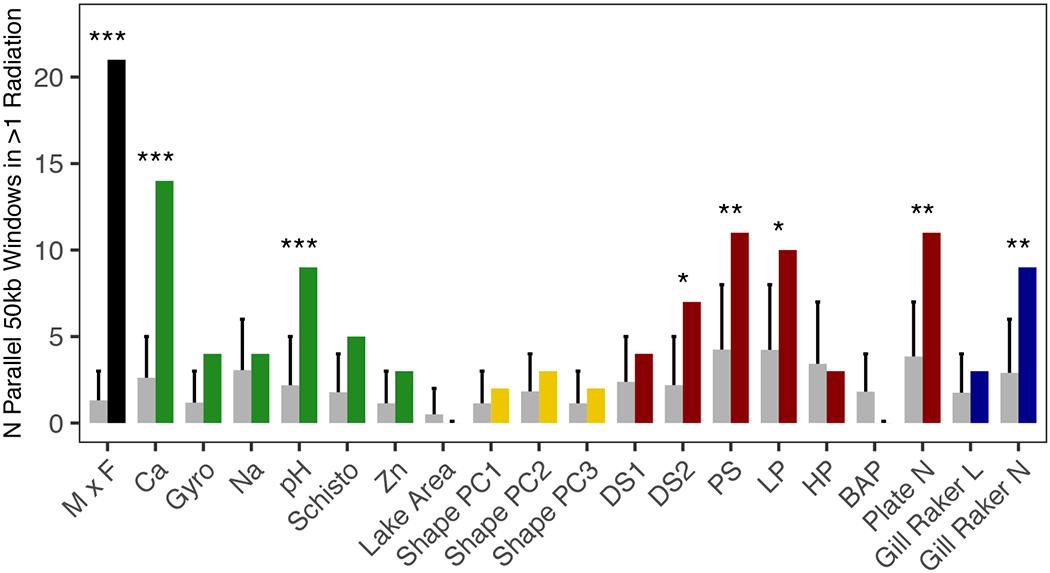
Expected and observed counts of 50kb windows containing an above 99% binomial expectation number of SNPs associated with marine x freshwater (MxF), environmental variables and phenotypic traits in at least 2 radiations. Expected bars (grey) represent mean counts across 10,000 simulated outcomes with 95% confidence intervals per a one-tailed hypothesis. Asterisks denote significance of FDR-corrected one-tailed tests between the observed counts and the 100,000 simulated counts at the <0.05 (*), <0.01 (**) and <0.001 (***) levels.
Five phenotypic variables were associated with more genomic parallelism than expected by chance (Fig. 3; Supplementary Table 10): four armour traits (2nd dorsal spine, pelvic spine length, length of pelvis and armour plate number) and gill raker number. These results are consistent with the observed, constrained marine-freshwater armour phenotype trajectory. This also suggests that variation in armour trajectories (e.g. in Alaska) are the result of different genotypes being selected in different environments. Additionally, parallel QTLs have been described for gill raker number (49), but not length, which exhibits more plasticity (50). Shape traits were not associated with any significant genomic parallelism, despite parallel environment x phenotype interactions. The probable polygenic architecture of shape phenotypes, as has similarly been described in cichlids (51), may result in greater redundancy in the genotype-phenotype map, reducing the likelihood of genetic parallelism. Moreover, the partly plastic nature of body shape (52,53) may lead to environment x shape associations via the reaction norm rather than genomic re-use.
Marine-freshwater (MxF) associated windows were more parallel than those associated with specific variables (Fig. 3), and overlapped well with previously identified MxF QTLs (proportions overlapping: Alaska = 0.85, BC = 0.85, Iceland = 0.81, Scotland = 0.69) (19,47,54) (Supplementary Table 11). Several parallel MxF windows were also parallel for Ca, pH, Na, armour traits and gill raker number (Supplementary Table 12). These results suggest our methods and sequencing coverage are robust enough to recover known parallel regions, and that, unsurprisingly, genomic parallelism associated with freshwater variables is more modest than marine-freshwater parallelism. The latter may reflect subtler variation between lakes compared with stark marine-freshwater contrasts but demonstrates that reduced parallelism for individual fitness components is likely biological rather than methodological. Marine-freshwater comparisons lump together many selective agents without being able to discern which are parallel and which are not. Overall, our results suggest that across a number of comparisons involving two or three (but not all four) freshwater adaptive radiations analysed, evolution of these phenotypes and environmentally associated traits are disproportionately linked to the same genomic regions.
Finally, we explored genomic parallelism between specific comparisons, to identify pairs of radiations with the highest levels of parallelism. We found the greatest number of significantly parallel windows in the comparison between Alaska and BC (Ca, Gyro, pelvic spine length, plate number and gill raker number), followed by Iceland and Scotland (Ca, pH, dorsal spine length) (Supplementary Table 10). Inter-continental parallelism was weaker: Alaska-Iceland (Ca, pelvis length); BC-Iceland (Schisto); BC-Scotland (pelvic spine length); Alaska-Scotland (none). Indeed, restricting permutations exclusively to Alaska-BC and Iceland-Scotland was enough to recover the significant parallelism observed when all radiations were analysed together (Supplementary Fig. 3). Phylogenetic patterns strongly support the notion that radiations within continents share similar genetic variation, making parallelism through shared standing variation the most parsimonious explanation for these intra-continental biases. Marginal evidence for this was observed as parallel associated regions had a greater enrichment of shared sites (mean sum of log10 enrichment scores = 1.3) compared to random expectations relative to associated regions that did not overlap among radiations (mean = 1.16, p = 0.14) and neutral regions (mean = 1.02, p < 0.001). However, comparisons between parallel and outlier regions were not significant. This suggests that radiations may be exploiting pools of ancestral variation for adaptation, but the same ancestral variation is not always adapted in parallel.
Experimental studies of parallelism have increasingly implicated standing genetic variation in the genesis of parallelism in stickleback (55) and other species (56,57,58). Coancestry patterns, centred at the focal, causative loci, can discern between parallelism via de novo mutations, standing variation, or introgressed alleles (59), however we lack the sequencing resolution to make these comparisons here. Further, the source of parallelism is likely to vary locus-by-locus and trait-by-trait, making it difficult to assess with a genome-wide approach. Indeed, all three modes of repeated gene-reuse have been observed in the radiation of a wild tomato clade (60).
Linkage and the genomic location of parallel regions.
As a reduced-representation approach, selection scans with RAD-sequencing depend on linkage disequilibrium (LD) between markers and functional loci (47,61,62). LD varies widely across organisms and within genomes but has been relatively well-characterized in stickleback (63,64), and RAD-sequencing has been used successfully in this species to test for genomic parallelism (36,47,59,63–65). LD associates inversely with recombination rate across the genome, so we estimated how recombination may affect our results using a previously published genetic map (65). Recombination was significantly reduced in associated windows and parallel windows compared with non-associated windows (Supplementary Fig. 4; Kruskal-Wallis, χ2 = 122.21, p < 0.001), but did not differ significantly between associated and parallel windows (p = 0.55). Reduced recombination can be an important mechanism in adaptation through maintaining adaptive alleles, as seen in stickleback (66) and cichlids (67).
These patterns may also reflect an increased ability to detect selection in low-recombination windows through increased linkage with causative SNPs. If true, our estimates of association, and by extension parallelism, may be conservative if false-negatives are pervasive in high-recombination regions. Importantly, our signatures of parallelism cannot be explained by variable recombination. Background selection can produce false-positive signatures of parallelism by reducing local diversity in shared low-recombination regions (68). This is less of an issue however when associating allele frequencies with environmental/phenotypic clines as done here.
Genetic distance (0.1 cM) windows corroborated 50kb results, returning significant parallelism for Ca, pH, pelvic spine length, pelvis length, plate number and gill raker number. Interestingly, we also recovered weakly significant parallelism for several other environmental and armour variables (Supplementary Fig. 5, Supplementary Information 1), suggesting our 50kb results may be conservative.
We plotted parallel 50kb windows (Supplementary Fig. 6) and merged adjacent windows (Supplementary Table 13) to inspect putative causative genes (Supplementary Dataset 2). Merging adjacently prior to permutations did not change which variables were significantly parallel (Fig. 3). Doing this we identified some wider genomic regions with parallelism across multiple radiations. One example was the pooling of plate number-associated windows in three radiations around the well-known Eda gene, with known functional role in armour plate evolution (19,28,29), which emerged despite the limited plate number variation across freshwater populations.
We also observed adjacent windows around a known inversion (69) region (250kb) on chromosome I, which contains the genes igfbp2a, stk11ip and atp1a1, and was strongly associated with calcium, sodium and pH in several radiations (Supplementary Data 2). Removing these windows prior to permutations did not change which variables were significantly parallel (Fig. 3). Inversions can be beneficial for adaptive haplotypes by reducing local recombination, and have been implicated in genomic parallelism in other systems such as parallel crab/wave ecotypes of Littorina saxatillis (70). Within this region atpa1a1 is particularly interesting, given its previously detected association with the major ecological transition from marine to freshwater (71) and functional role in metal ion management (19,69).
Extensive LD in freshwater populations could result from drift, but is consistent with strong directional selection after freshwater invasion and has been reported for stickleback populations from Alaska (47). It had not however previously been observed for the same regions across several independent adaptive radiations. These results are also consistent with many diverged marine-freshwater SNPs aggregating in just 19 short genomic regions, including three known inversions (69). Overall these results suggest physically linked genomic regions are hitch-hiking in separate radiation pairs, which may contain parallel genes across all radiations but are undetected by our methods.
Relationships between genomic parallelism and phylogenetic, phenotypic and environmental similarity
Based on the assumption that freshwater populations radiated from common marine ancestors (1,18), and to leverage statistical power from our biological sampling, we also compared parallel FST outliers between all marine-freshwater comparisons to examine the relative effects of genetic, phenotypic, and environmental similarity on genome-wide parallelism at large geographic scale. To do this, we calculated the top 5% of 50kb windows based on FST for each freshwater population and its relevant marine population and assessed all pairwise overlaps (N = 2,628) (Fig. 4a). Genome-wide FST outliers are more susceptible to random parallelism through recurrent drift or background selection although, as discussed above, these processes are unlikely to influence previous results obtained by comparing allele frequency gradients across all populations within a radiation. Nevertheless, broad patterns inferred over all 2,628 comparisons should be apparent despite noise.
Fig. 4.
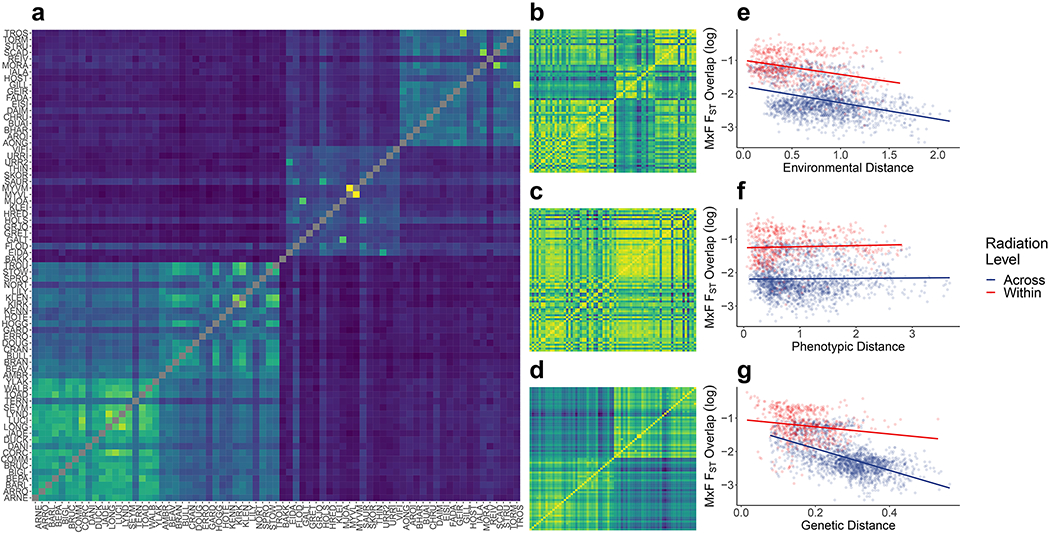
Associations between genome-wide marine - freshwater FST and environmental, phenotypic and genetic distance across all pairwise comparisons of 73 freshwater populations: a) Proportion of MxF FST 50kb outlier windows that overlap among freshwater replicates. Freshwater populations are ordered as Alaska, British Columbia, Iceland and Scotland, with these location distinguishable as four clear clusters; b) Environmental distances between freshwater populations, recorded as Euclidean distances in PCA space for all 7 environmental variables; c) Phenotypic (Euclidean) distances between freshwater populations for the 12 phenotypic variables; d) Genetic distances between freshwater populations, recorded as genome-wide pairwise FST based on 8,395 unlinked SNPs; e-g) Associations between environmental (e), phenotypic (f) and genetic (g) distances and MxF FST overlap (log-transformed). Points are coloured according to whether pairwise comparison is being made within a radiation or across radiations.
Parallel windows were more common in intra- rather than inter-continental comparisons, again highlighting importance of these pairings as the major contributors towards pairwise genomic parallelism signals, as discussed previously. This strongly suggests that genomic parallelism at large geographic scales must be partly contingent on shared genetic variation, although exceptions exist such as recurrent de novo mutation at Pitx1 (72) during freshwater pelvis evolution. There is also the possibility of haplotype sharing between radiations within continents by gene flow through marine populations, which may be facilitated in North America, despite the greater geographic distance, by a shared coastline connecting Alaska and BC (38).
We used Mantel tests to compare a parallel FST overlap matrix with genetic, environmental, and phenotypic distance matrices (Fig. 4b–d). Across all comparisons the number of parallel windows was strongly negatively correlated with genetic (r = −0.67, p < 0.001) and environmental dissimilarity (r = −0.45, p < 0.001), but only weakly with phenotypic dissimilarity (r = −0.07, p = 0.060) (Fig. 4e–g). Thus, genomic parallelism increases in populations that are more genetically or environmentally similar, but not phenotypically similar. Associations between environmental dissimilarity and FST parallelism were still strongly negative (r = −0.41, p < 0.001), after correcting for genetic similarity in partial Mantel tests, suggesting correlations between local environment and local ancestry do not drive this effect. Phenotypic dissimilarity remained un-associated with FST parallelism after controlling for genetic similarity (r = −0.026, p = 0.341), suggesting environmental similarity is a better predictor of genomic parallelism than phenotypic similarity, at least in terms of our measured environmental variables and the observable morphological phenotypes in this system. The genotype-phenotype map may be simpler than the equivalent genotype-environment map, potentially leading to over-correction when including genetic distance matrices, but this would not explain why phenotypic distance associations were weaker prior to correction.
Early studies of marine and freshwater stickleback (19,29,47) were some of the first to demonstrate the repeatability of the genetic basis of adaptation in natural populations, and that it might be pervasive. These results drove researchers to examine other systems for genomic parallelism, such as cichlids (67,73), periwinkles (74), stick insects (15) and Arabidopsis (75). These diverse systems have highlighted that genomic parallelism is highly variable, and more recent studies of stickleback from outside the original Eastern Pacific populations have similarly shown variability within the system itself (69).The field has since matured to question how and why this variability persists, and the stickleback system remains at the forefront of this research. Studies on 16 lake-stream stickleback pairs from British Columbia demonstrated that continuous phenotypic and environmental variation predicts genomic parallelism (FST regions) (20) and suggested that ecotype genomic parallelism may be stronger for certain phenotypic traits than others (76). These findings are recapitulated and extended here, to demonstrate that similarity of specific environmental and phenotypic variables is a good predictor for signatures of genomic parallelism. Our results also confirm these results extend beyond only BC. Further, our data provides additional statistical power (73 marine-freshwater comparisons vs 16 lake-stream, albeit with reduced independence) to elucidate that environmental similarity is a better predictor of genomic parallelism than phenotypic parallelism.
A major question of interest concerns the geographic scale to which patterns of genomic parallelism extend. Whilst we found marine-freshwater genomic parallelism to be constrained across all four radiations, we do observe genomic parallelism at a continental scale, and within freshwater populations founded from both Eastern Pacific and Atlantic marine populations. Agreeing with our results, a comparison of ‘regional’ lake-stream parallelism in BC to ‘global’ lake-stream parallelism in a collection of lake-stream pairs from Europe and North America (one pair per region) highlighted a global constraint on parallelism at the genetic level and for some phenotypes (40). However, comparable watersheds of multiple lake-stream ecotype pairs are less common beyond BC (40, but see 77,78), restricting global comparisons of lake-stream adaptive radiations such as those presented here for marine-freshwater.
A recent comparison of levels of genomic parallelism in North American (Pacific-founded) and European populations suggested that a founding event of Atlantic marine populations limited standing freshwater variation, leading to lower parallelism in European populations (42). Consistent with the idea that even minor differences in selection may limit genomic parallelism of standing genetic variation (38), this study also speculated that differences in selection homogeneity between North American and European environments could explain variable genomic parallelism. The data to test this hypothesis however has been hitherto lacking. Our results on segregating variants and molecular variance confirm distinct North American and European clusters of standing genetic variation, which are consistent with a founding bottleneck, or older divergence time between inter-continental radiations compared to within each ocean. However, genetic variation within North American and European radiations was broadly comparable, suggesting similar potential within Europe to produce freshwater parallelism as in North America. This result, combined with our stronger observed environmental homogeneity in North America vs Europe, and strong associations between environmental distance and genome-wide parallelism (Fig. 4b,e), suggests environmental homogeneity is a valid explanation for some of the differences in parallelism observed between the two continents. Overall, our study thus highlights that variable levels of genomic parallelism observed within marine-freshwater stickleback comparisons at the global scale are likely the result of an interplay between environmental heterogeneity (but not physical distance) at continental scales, and a history of founding bottlenecks and segregating genetic variation among founding populations at a global scale. Further, genomic parallelism of specific phenotypes is predictable on the basis of common phenotypic trajectories, likely underwritten by simple vs complex genetic architectures.
Methods
Sampling and environmental data collection.
We sampled 18 (19 in Alaska) freshwater lakes and one marine coastal population in North Uist, Scotland (April and June 2013), Iceland (May and June 2014), British Columbia (BC) (April and May 2015) and the Cook Inlet basin, Alaska (June 2015). Lake names, geographic coordinates and numbers of samples used in the study are shown in Supplementary Table 1. Each adaptive radiation analysed comprises a variety of different adaptive forms that most likely evolved from closely related ancestral marine lineages in each region. We measured a set of seven biotic and abiotic environmental variables and a set of 12 phenotypic traits (measures of body shape, armour traits and gill rakers). We measured the pH, concentrations of metallic cation concentrations sodium (“Na”), calcium (“Ca”) and zinc (“Zn”) of each lake, lake area, and calculated population prevalence of Gyrodactylus spp. and Schistochephalus solidus. Concentrations of cations, pH, lake area and parasite prevalence per lake are shown in Supplementary Table 2. The environmental variables included in our analyses were selected on the basis of presumed fitness effects and that could be precisely measured (Supplementary Table 2). For abiotic environmental variables we chose pH, ionic concentrations of calcium (Ca), sodium (Na) and zinc (Zn), and lake area (Area) which have been associated previously with the evolution of body shape, size and armour in stickleback (27,33,79,80). Many biotic variables are difficult to quantify precisely so we used the prevalence of two parasites Gyrodactylus sp. (ectoparasitic trematodes) (Gyro) and Schistocephalus solidus (endoparasitic cestode) (Schisto) that are likely to have an impact on the reproduction and life cycle of stickleback (81–83). It is important to note that measured variables might actually be proxies for other, unmeasured variables and not the primary causes of selection. Details of the fish collection and quantification of abiotic and abiotic variables can be found in Supplementary Methods. As marine-freshwater parallelism is well-documented (19,29,84), we compared our results for parallelism across freshwater radiations with well-studied marine-freshwater parallelism in this species, and used the results as a positive control for the methods used (Supplementary Information 2). Some marine-freshwater associated regions detected by our use of Bayenv2 could be the result of differences in allele frequencies between only a few freshwater populations and marine populations (within-freshwater variation rather than explicit marine-freshwater divergence). Such false-positives are however unlikely as a combination of high Bayes factor and high Spearman’s ρ requires many freshwater populations to display consistent allele frequency changes relative to marine populations. Some variables that vary among freshwater populations may not vary consistently between marine and freshwater, such that our parallel MxF regions are not expected to be a sum of our single variable, parallel, freshwater regions.
DNA extractions, RAD library preparation and sequencing.
Genomic DNA was purified from 10 to 21 individuals from each of the populations, chosen to represent a widely distributed subset of the most environmentally and phenotypically variable lakes (Supplementary Dataset 3). Extracted genomic DNA was normalized to a concentration of 25 ng /µL in 96-well plates.
In 2014 we conducted RAD sequencing on samples from Scotland and from Iceland. Sequencing libraries were prepared and processed into RAD following the modified libraries according to (85). In 2016 we conducted RAD sequencing on samples from British Columbia and from Alaska. Sequencing libraries were prepared following the modified single-digest RAD protocol of (86). The two RADseq protocols interrogate the same set of loci across the genome, so that the SNP data are compatible across all four radiations. See Supplementary Methods for details of the RAD library preparation and sequencing.
Population genetics statistics and phylogenetic tree.
Raw sequence reads were demultiplexed using Stacks – 1.35 (87). Number of reads per individual are shown in Supplementary Dataset 3 (see Supplementary Methods for details on the alignment of reads and Stacks pipeline used). For Bayenv2 and association analyses, autosomal SNPs were called using the Populations within Stacks as being present in >7 populations (-p 8), >50% of the individuals within a population (-r 0.5), and with a MAF-filter of 0.05 (--min-maf 0.05). After filtering we retained 26,169, 29,111, 26,937, 26,990 SNPs for Alaska, BC, Iceland, and Scotland, Iceland, British Columbia and respectively.
For analyses of population structure and phylogenetics across all radiations, a subset of unlinked SNPs were generated. Here, autosomal SNPs were called that were present in all radiations (-p 4) and in >50% of individuals within a radiation (-r 0.5), with a MAF-filter of 0.05 (within a radiation) (--min-maf 0.05). Only the first SNP per RAD locus was retained (--write-single-snp). FST was bootstrapped and calculated in POPULATIONS. This set of SNPs were then pruned for linkage disequilibrium in plink using indep-pairwise 50 5 0.2. We also produced an additional linkage-pruned dataset with marine populations (-p 5) with 11,266 SNPs. The unlinked SNP dataset with marine fish was used to construct a neighbour-joining tree for all fish in the R package ‘ape’, using a distance matrix (bionj) computed from the SNP data (88). The tree was bootstrapped 100 times and nodes with less than 80% support were collapsed. PCA analysis of population structure was conducted using plink (89).
Phenotypic and environmental variation - body shape, armour, gill rakers and environmental data analyses.
All morphological measurements (body shape, body armour and gill raker traits) were done following (27). Details of the quantification of phenotypic traits can be found in Supplementary Methods.
We performed three Principal Component Analyses (PCAs): one on the armour traits, another on body shape, and another on the 6 environmental variables. Body shape and armour PCAs were performed on regression residuals of all individuals from all radiations pooled together to extract the common PCs of body shape, armour, and environmental variation, and retained axes that explained more than 10% of the total variance. Armour and environmental PCAs were conducted with scaled inputs due to different units of measurement between variables. Shape PCAs were conducted on morphometric residuals, and as such were not scaled. All phenotypic analyses, including ANOVAs and ANCOVAs and plotting were done in R version 3.4.3.
For ANCOVAs, we compared each phenotype (PC1 and PC2 for shape and armour, actual values for gill raker N and L) with each of our seven environmental variables. We explored five possible GLMs: 1) A null model; 2) phenotype varies by environment (linear slope); 3) phenotype varies by radiation (intercepts vary); 4) phenotype varies by environment and intercept varies by radiation (parallel slope, intercepts vary); 5) phenotype varies by environment in a radiation-specific manner (nonparallel slopes). The best model was chosen by backwards model selection using AIC, with simpler models (model 1 simplest) preferred if the change in AIC > −2. We then calculated F-statistics for the resulting GLM. For variable slopes, we used post-hoc Tukey tests to compare radiations.
Genotype-Environment/Phenotype Associations.
For each radiation separately (N=18 to 19 populations) we used Bayenv2 (46) to identify associations between genomic allele frequencies (N=10 to 21 individual fish, mean = 17. 8), the set of seven biotic and abiotic environmental variables (Ca, Na, pH, Zn, lake area, prevalence of Gyrodactylus spp. and Schistochephalus solidus) and the set of 12 phenotypic traits (ShapePC1, ShapePC2, ShapePC3, DS1, DS2, PS, LP, HP, BAP, Plate N, Gill Raker L and Gill Raker N) mentioned above. For each radiation, a matrix of genetic covariance was calculated using a subset of SNPs limited to a single SNP per RAD-locus and pruned for linkage disequilibrium (R2 < 0.4) in plink (89). This cut-off was selected to balance the trade-off between SNPs retained and minimising the effects of linkage. Covariance matrices were therefore calculated using 9619, 7983, 7300 and 5705 SNPs for Alaska, BC, Iceland and Scotland respectively. Covariance matrices were calculated across 100,000 iterations and averaged across 5 independent runs. Bayenv2 was run independently 8 times and final results were averaged across runs. The purpose of the covariance matrix is to rule out spurious associations between drifting allele frequencies associated with population structure and environmental/phenotypic variation. Some of these correlations did exist in our data, but poorly explain signals of genomic parallelism (Supplementary Fig. 7).
Environmentally and phenotypically associated SNPs were selected as having a log10-BayesFactor > 1.5 and an absolute Spearman’s rank coefficient above the 95th percentile. The combination of BayesFactor and non-parametric measure of correlation helps to avoid selecting SNPs with high BayesFactors due to single or few outlier populations with extreme allele frequencies and environmental/phenotypic variation (90). SNPs were grouped into 50kb, 75kb, 100kb, 200kb and 0.1 cM windows (Supplementary Table 14) to test the robustness of our results across different extents of linkage. Our 50kb dataset was composed of 4,868 windows with SNPs in all radiations, covering approximately 55% of the 447 Mb genome, with a further 1,940 windows sequenced in 2 or more radiations providing information on an additional 21.7% of the genome. To evaluate whether windows were environmentally or phenotypically associated, we adapted the methodology of (91). We calculated the upper 99% binomial expectation for the number of associated SNPs given the total number of SNPs in a specific window, and selected windows that had a greater number of associated SNPs than this expectation. We focused on repeated changes within the same genomic regions, rather than on reuse of the same mutations. This is because the causal mutations are unknown in most cases and may not be sequenced by reduced representation sequencing methods such as RAD-sequencing. This method also controls for variation in SNP density across windows and ensures that significant windows exhibit consistent allele frequency correlations across multiple SNPs. We visualised the genomic locations of associated windows using Manhattan plots (Supplementary Fig. 6) and plotted the residual number of outlier SNPs above the binomial expectation (Supplementary Fig. 8). Linkage groups I-XXI were visualised with the exception of XIX; windows on scaffolds were not visualised. Finally, we compared these associated windows across radiations to examine those that were parallel.
As a positive control for the methods used we compared our results for parallelism across freshwater radiations with well-studied marine-freshwater parallelism in this species (19,29), and then examined genomic differentiation between all freshwater populations pooled within a radiation and four marine populations pooled together (one from each geographic region, Supplementary Table 1, Supplementary Information 2).
Parallelism statistics
We use the term genomic parallelism when referring to repeated changes within the same genomic regions, rather than the strict definition of genomic parallelism that refers to reuse of the same mutations. The use of ‘parallelism’ terminology is highly variable within the literature (12), but our usage is consistent with stickleback literature (19,47,77) and reflects parallelism of phenotypes. For all radiation groupings (11 combinations in total: one four radiation grouping, four three radiation groupings, and six two radiation groupings), we calculated the significance of parallel window counts using a permutation method. For each environmental or phenotypic variable, we randomly drew N windows from each radiation’s total pool where N was equivalent to the associated window count for each radiation. We then assessed the overlap of randomly associated windows across radiations and pooled the results over 10,000 iterations. The output from all permutations was used as a null distribution to infer p-values, which were then FDR-corrected using the R package qvalue (92).
Grouping of adjacent windows and expanding parallelism regions.
Windows of 50kb and above were based on a linkage assumption and to minimise non-independence between windows. There were, however, occasionally adjacent windows associated with the same variable across different groupings. Large regions of relatively strong linkage are plausible if recombination is reduced through processes such as genomic rearrangements. To investigate these, we grouped associated windows that were adjacent as well as those that were direct matches across radiations based on the likelihood of adjacent associated windows resulting independently being low, suggesting non-independence and probable linkage. We repeated the above permutations assuming adjacent windows to be single associated regions. These windows are available in Supplementary Table 13.
Multivariate vector comparison of environments and phenotypes.
Using the average trait values from marine and freshwater populations, we calculated the vectors of phenotypic change for armour, shape, and gill raker variables (3 vectors per population) separately between each freshwater population and the marine population from the same radiation. We then calculated the angles (θ) between all vectors from the same radiation (one distribution of θ values per radiation), and the angles between vectors from different radiations (one distribution of θ values per between-radiation comparison (N=6): for example, one distribution of θ values comprising of angles between each vector of Iceland vs each vector of Scotland). We then compared radiations pairwise, asking whether the distribution of angles between vectors from different radiations differed significantly from the distribution of angles calculated within a radiation (for example, comparing whether the distribution of θ values between Scottish vectors was significantly different from the distribution of θ values between Scottish and Icelandic vectors). We performed this analysis for all pairwise comparisons, comparing the “between-radiation” distribution to both “within-radiation” distributions separately (Fig. 2c, d). The analysis was performed separately for armour, shape, and gill raker variables. We lacked data for Scottish marine gill rakers, so these comparisons were not possible.
Comparing relative influences of environment, phenotype and genetics.
FST was calculated between each freshwater population and its relevant marine population (Alaska = MUD1, BC = LICA, Iceland = NYPS, Scotland = OBSM) in 50kb windows using the R package ‘PopGenome’ (93). For each MxF comparison, windows above the 95% quantile were classed as outliers. Outlier windows were compared across all pairwise freshwater comparisons (2,628 comparisons among 73 populations), with overlapping outliers representing MxF FST parallelism. Dissimilarity matrices of environment and phenotype were calculated as Euclidean distance in PCA space for the seven and 12 environmental and phenotypic variables respectively. The genetic dissimilarity matrix was composed of genome-wide pairwise FST estimates between freshwater populations. The matrix of MxF parallelism was associated to environmental, phenotypic and genetic dissimilarity matrices using Mantel tests (Spearman’s) with 9,999 permutations. Partial Mantel tests were performed with genetic distance as the conditional matrix for environmental and phenotypic effects on MxF parallelism, again with 9,999 permutations.
Supplementary Material
Acknowledgements
We thank Shaun Robertson, Rebecca Young, Abdul Rahman, Brian Santos, Sara Goodacre, Petur Halldorsson, Bjarni K. Kristjánsson, Dolph Schluter, Kieran Samuk, Diana Rennison and Sara Miller for help with the sampling and sampling permits. We are grateful to Ann Lowe and Laura Dean for help with the DNA extractions, to Cody Wiench, Amanda Stahlke and Sarah Hendricks for help making the RAD-libraries and to John Brookfield for discussion of probability calculations. This work was funded by a NERC grant (NE/J02239X/1 to A.D.C.M), and further support was provided by NIH grant P30GM103324.
Footnotes
Competing financial interests
The authors declare no competing financial interests.
Ethical compliance
Ethical approval for sampling in the UK was under Home Office licence 40/3486, in British Columbia under Professor Dolph Schluter’s UBC animal care certificate A11-0402, and in Alaska under University of Alaska Anchorage IACUC protocol 739596-1. No ethical approval was required in Iceland.
Data Accessibility
Bam files of aligned reads for each individual and corresponding sample information have been deposited in the European Nucleotide Archive database under the project PRJEB20851, with the sample accession numbers ERS1831811-ERS1833111, and run accession numbers ERR2055459-ERR2056759. Scripts used for all analyses are archived through Github/Zenodo (DOI:10.5281/zenodo.4024117).
References
- 1.Schluter D The ecology of adaptive radiations. (Oxford University Press, 2000). [Google Scholar]
- 2.Gavrilets S & Losos JB Adaptive radiation: Contrasting theory with data. Science 323, 732–737 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Arnold SJ, Bürger R, Hohenlohe PA, Ajie BC & Jones AG Understanding the evolution and stability of the G-matrix. Evolution 62, 2451–2461 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Losos JB Adaptive radiation, ecological opportunity, and evolutionary determinism : American society of naturalists E. O. Wilson award address. Am. Nat 175, 623–639 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Elmer KR et al. Parallel evolution of Nicaraguan crater lake cichlid fishes via non-parallel routes. Nat. Commun 5, 1–8 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Mahler DL, Ingram T, Revell LJ & Losos JB Exceptional convergence on the macroevolutionary landscape in island lizard radiations. Science 341, 292–295 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Lamichhaney S et al. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518, 371–375 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Gould SJ Wonderful life: The Burgess shale and the nature of history. (W.W. Norton and Company, 1989). [Google Scholar]
- 9.Schluter D Adaptive Radiation Along Genetic Lines of Least Resistance. Evolution 50, 1766 (1996). [DOI] [PubMed] [Google Scholar]
- 10.Roff D The evolution of the G matrix: Selection or drift? Heredity 84, 135–142 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Arendt J & Reznick D Convergence and parallelism reconsidered: what have we learned about the genetics of adaptation? Trends Ecol. Evol 23, 26–32 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Stuart YE Divergent uses of “parallel evolution” during the history of the American naturalist. Am. Nat 193, 11–19 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Oke KB, Rolshausen G, LeBlond C & Hendry AP How parallel is parallel evolution? A comparative analysis in fishes. Am. Nat 190, 1–16 (2017). [DOI] [PubMed] [Google Scholar]
- 14.McGee MD, Neches RY & Seehausen O Evaluating genomic divergence and parallelism in replicate ecomorphs from young and old cichlid adaptive radiations. Mol. Ecol 25, 260–268 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Soria-Carrasco V et al. Stick insect genomes reveal natural selection’s role in parallel speciation. Science 344, 738–742 (2014). [DOI] [PubMed] [Google Scholar]
- 16.MacColl ADC The ecological causes of evolution. Trends Ecol. Evol 26, 514–522 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Elmer KR & Meyer A Adaptation in the age of ecological genomics: Insights from parallelism and convergence. Trends Ecol. Evol 26, 298–306 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Conte GL, Arnegard ME, Peichel CL & Schluter D The probability of genetic parallelism and convergence in natural populations. Proc. R. Soc. B Biol. Sci 279, 5039–5047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones FC et al. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484, 55–61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stuart YE et al. Contrasting effects of environment and genetics generate a continuum of parallel evolution. Nat. Ecol. Evol 1, 1–7 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Jacobs A et al. Parallelism in eco-morphology and gene expression despite variable evolutionary and genomic backgrounds in a Holarctic fish. PLoS genetics 16, 1008658 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muschick M, Indermaur A & Salzburger W Convergent evolution within an adaptive radiation of cichlid fishes. Curr. Biol 22, 2362–2368 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Foster S & Bell M The evolutionary biology of the threespine stickleback. (Oxford Univ. Press, 1994). [Google Scholar]
- 24.Taylor EB & McPhail JD Historical contingency and ecological determinism interact to prime speciation in sticklebacks, Gasterosteus. Proc. R. Soc. B Biol. Sci 267, 2375–2384 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaeuffer R, Peichel CL, Bolnick DI & Hendry AP Parallel and nonparallel aspects of ecological, phenotypic, and genetic divergence across replicate population pairs of lake and stream stickleback. Evolution 66, 402–418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravinet M, Prodöhl PA & Harrod C Parallel and nonparallel ecological, morphological and genetic divergence in lake-stream stickleback from a single catchment. J. Evol. Biol 26, 186–204 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Magalhaes IS, D’Agostino D, Hohenlohe PA & MacColl ADC The ecology of an adaptive radiation of three-spined stickleback from North Uist, Scotland. Mol. Ecol 25, 4319–4336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colosimo PF et al. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307, 1928–1933 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Jones FC et al. A genome-wide SNP genotyping array reveals patterns of global and repeated species-pair divergence in sticklebacks. Curr. Biol 22, 83–90 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raeymaekers JAM et al. Adaptive and non-adaptive divergence in a common landscape. Nat. Commun 8, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rennison DJ, Stuart YE, Bolnick DI & Peichel CL Ecological factors and morphological traits are associated with repeated genomic differentiation between lake and stream stickleback. Philos. Trans. R. Soc. B Biol. Sci 374, 20180241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacColl ADC & Aucott B Inappropriate analysis does not reveal the ecological causes of evolution of stickleback armour: A critique of Spence et al. 2013. Ecol. Evol 4, 3509–3513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spoljaric MA & Reimchen TE 10 000 Years later: Evolution of body shape in Haida Gwaii three-spined stickleback. J. Fish Biol 70, 1484–1503 (2007). [Google Scholar]
- 34.De Schamphelaere KAC et al. Reproductive toxicity of dietary zinc to Daphnia magna. Aquat. Toxicol 70, 233–244 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Martins C, Jesus FT & Nogueira AJA The effects of Copper and Zinc on survival, growth and reproduction of the cladoceran Daphnia longispina: introducing new data in an “old” issue. Ecotoxicology 26, 1157–1169 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Miller CT et al. Modular skeletal evolution in sticklebacks is controlled by additive and clustered quantitative trait loci. Genetics 197, 405–420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan YF et al. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a pitxl enhancer. Science 327, 302–305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thompson KA, Osmond MM & Schluter D Parallel genetic evolution and speciation from standing variation. Evol. Lett 3, 129–141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson TC & Cresko WA Ancient genomic variation underlies repeated ecological adaptation in young stickleback populations. Evol. Lett 2, 9–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paccard A et al. Repeatability of Adaptive Radiation Depends on Spatial Scale: Regional Versus Global Replicates of Stickleback in Lake Versus Stream Habitats. J. Hered 111, 43–56 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Baldo L, Riera JL, Salzburger W & Barluenga M Phylogeography and ecological niche shape the cichlid fish gut microbiota in Central American and African lakes. Frontiers in Microbiology 10: 2372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang B, Kemppainen P, Momigliano P & Merilä J On the causes of geographically heterogeneous parallel evolution in sticklebacks. Nature Ecol. Evol 1–11 (2020). [DOI] [PubMed] [Google Scholar]
- 43.Mäkinen HS & Merilä J Mitochondrial DNA phylogeography of the three-spined stickleback (Gasterosteus aculeatus) in Europe-Evidence for multiple glacial refugia. Mol. Phylogenet. Evol 46, 167–182 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Liu S, Hansen MM & Jacobsen MW Region-wide and ecotype-specific differences in demographic histories of threespine stickleback populations, estimated from whole genome sequences. Mol. Ecol 25, 5187–5202 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Fang B, Merilä J, Ribeiro F, Alexandre CM & Momigliano P Worldwide phylogeny of three-spined sticklebacks. Mol. Phylogenet. Evol 127, 613–625 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Coop G, Witonsky D, Di Rienzo A & Pritchard JK Using environmental correlations to identify loci underlying local adaptation. Genetics 185, 1411–1423 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hohenlohe PA et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo B, DeFaveri J, Sotelo G, Nair A & Merilä J Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks. BMC Biol. 13, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glazer AM, Cleves PA, Erickson PA, Lam AY & Miller CT Parallel developmental genetic features underlie stickleback gill raker evolution. Evodevo 5, 19 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Day T, Pritchard J & Schluter D A comparison of two sticklebacks. Evolution 48, 1723–1734 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Franchini P et al. Genomic architecture of ecologically divergent body shape in a pair of sympatric crater lake cichlid fishes. Mol. Ecol 23, 1828–1845 (2014). [DOI] [PubMed] [Google Scholar]
- 52.Garduno-Paz MV, Couderc S & Adams CE Habitat complexity modulates phenotype expression through developmental plasticity in the threespine stickleback. Biol. J. Linn. Soc 100, 407–413 (2010). [Google Scholar]
- 53.McCairns RJS & Bernatchez L Plasticity and heritability of morphological variation within and between parapatric stickleback demes. J. Evol. Biol 25, 1097–1112 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Peichel CL & Marques DA The genetic and molecular architecture of phenotypic diversity in sticklebacks. Philos. Trans. R. Soc. B Biol. Sci 372, 20150486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marques DA et al. Genomics of Rapid Incipient Speciation in Sympatric Threespine Stickleback. PLoS Genet. 12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burke MK, Liti G & Long AD Standing genetic variation drives repeatable experimental evolution in outcrossing populations of Saccharomyces cerevisiae. Mol. Biol. and Evol 31, 3228–3239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang L, Aggarwal DD, Rashkovetsky E, Korol AB and Michalak P Rapid genomic changes in Drosophila melanogaster adapting to desiccation stress in an experimental evolution system. BMC Genomics 17, 233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gompert Z & Messina FJ Genomic evidence that resource‐based trade‐offs limit host‐range expansion in a seed beetle. Evolution 70, 1249–1264 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Berner D, Moser D, Roesti M, Buescher H & Salzburger W Genetic architecture of skeletal evolution in european lake and stream stickleback. Evolution 68, 1792–1805 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Pease JB, Haak DC, Hahn MW & Moyle LC Phylogenomics Reveals Three Sources of Adaptive Variation during a Rapid Radiation. PLoS Biol. 14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lowry DB et al. Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol. Ecol. Resour 17, 142–152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKinney GJ, Larson WA, Seeb LW & Seeb JE RADseq provides unprecedented insights into molecular ecology and evolutionary genetics: comment on Breaking RAD by Lowry et al. (2016). Mol. Ecol. Resour 17, 356–361 (2017). [DOI] [PubMed] [Google Scholar]
- 63.Roesti M, Kueng B, Moser D & Berner D The genomics of ecological vicariance in threespine stickleback fish. Nat. Commun 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Catchen JM et al. Unbroken: RADseq remains a powerful tool for understanding the genetics of adaptation in natural populations. Mol. Ecol. Resour 17, 362–365 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Roesti M, Moser D & Berner D Recombination in the threespine stickleback genome - Patterns and consequences. Mol. Ecol 22, 3014–3027 (2013). [DOI] [PubMed] [Google Scholar]
- 66.Samuk K et al. Gene flow and selection interact to promote adaptive divergence in regions of low recombination. Mol. Ecol 26, 4378–4390 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Meier JI, Marques DA, Wagner CE, Excoffier L & Seehausen O Genomics of parallel ecological speciation in Lake Victoria cichlids. Mol. Biol. Evol 35, 1489–1506 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Cruickshank TE & Hahn MW Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol 23, 3133–3157 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Terekhanova NV et al. Fast Evolution from Precast Bricks: Genomics of Young Freshwater Populations of Threespine Stickleback Gasterosteus aculeatus. PLoS Genet. 10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Westram AM et al. Clines on the seashore: The genomic architecture underlying rapid divergence in the face of gene flow. Evol. Lett 2, 297–309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shimada Y, Shikano T & Merilä J A high incidence of selection on physiologically important genes in the three-spined stickleback, gasterosteus aculeatus. Mol. Biol. Evol 28, 181–193 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Xie KT et al. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science 363, 81–84 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Henning F & Meyer A The Evolutionary Genomics of Cichlid Fishes: Explosive Speciation and Adaptation in the Postgenomic Era. Annu. Rev. Genomics Hum. Genet 15, 417–441 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Kess T, Galindo J & Boulding EG Genomic divergence between Spanish Littorina saxatilis ecotypes unravels limited admixture and extensive parallelism associated with population history. Int. J. Bus. Innov. Res 17, 8311–8327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bohutínská M et al. Genomic basis of parallel adaptation varies with divergence in Arabidopsis and its relatives. Preprint at https://www.biorxiv.org/content/10.1101/2020.03.24.005397v1.full (2020). [DOI] [PMC free article] [PubMed]
- 76.Rennison DJ, Samuk K, Owens GL & Miller SE Shared patterns of genome-wide differentiation are more strongly predicted by geography than by ecology. Am. Nat 195, 192–200 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Lucek K, Sivasundar A, Roy D & Seehausen O Repeated and predictable patterns of ecotypic differentiation during a biological invasion: lake–stream divergence in parapatric Swiss stickleback. J. of Evol. Biol 26, 2691–2709 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Berner D, Roesti M, Hendry AP and Salzburger W Constraints on speciation suggested by comparing lake‐stream stickleback divergence across two continents. Mol. Ecol 19, 4963–4978 (2010). [DOI] [PubMed] [Google Scholar]
- 79.Giles N Behavioural effects of the parasite Schistocephalus solidus (Cestoda) on an intermediate host, the three-spined stickleback, Gasterosteus aculeatus L. Anim. Behav 31, 1192–1194 (1983). [Google Scholar]
- 80.Spence R, Wootton RJ, Barber I, Przybylski M & Smith C Ecological causes of morphological evolution in the three-spined stickleback. Ecol. Evol 3, 1717–1726 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reimchen TE Incidence and intensity of Cyathocephalus truncatus and Schistocephalus solidus infection in Gasterosteus aculeatus. Can. J. Zool 60, 1091–1095 (1982). [Google Scholar]
- 82.MacColl ADC Parasite burdens differ between sympatric three-spined stickleback species. Ecography 32, 153–160 (2009). [Google Scholar]
- 83.Stutz WE, Lau OL & Bolnick DI Contrasting patterns of phenotype-dependent parasitism within and among populations of threespine stickleback. Am. Nat 183, 810–825 (2014). [DOI] [PubMed] [Google Scholar]
- 84.Bassham S, Catchen J, Lescak E, von Hippel FA & Cresko WA Repeated selection of alternatively adapted haplotypes creates sweeping genomic remodeling in stickleback. Genetics 209, 921–939 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Etter PD, Preston JL, Bassham S, Cresko WA & Johnson EA Local de novo assembly of rad paired-end contigs using short sequencing reads. PLoS One 6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ali OA et al. Rad capture (Rapture): Flexible and efficient sequence-based genotyping. Genetics 202, 389–400 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Catchen J, Hohenlohe PA, Bassham S, Amores A & Cresko WA Stacks: An analysis tool set for population genomics. Mol. Ecol 22, 3124–3140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Paradis E, Claude J & Strimmer K APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Chang CC et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Günther T & Coop G Robust identification of local adaptation from allele frequencies. Genetics 195, 205–220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yeaman S et al. Convergent local adaptation to climate in distantly related conifers. Science 353, 1431–1433 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Storey J qvalue: Q-Value estimation for false discovery rate control. R package version 2.0.0. (2015). [Google Scholar]
- 93.Pfeifer B, Wittelsbürger U, Ramos-Onsins SE & Lercher MJ PopGenome: An efficient swiss army knife for population genomic analyses in R. Mol. Biol. Evol 31, 1929–1936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.