

Abstract
Systemic chemotherapy is efficacious against triple-negative breast cancer (TNBC), but it is often associated with serious side effects. Here, we report a luteinizing hormone-releasing hormone (LHRH) receptor-targeted and tumor microenvironment-responsive nanoparticle system to selectively deliver chemotherapeutic drugs to TNBC cells. This delivery system (termed “LHRH-DCMs”) contains poly (ethylene glycol) and dendritic cholic acid as a micellar carrier, reversible intra-micellar disulfide bond as a redox-responsive crosslink, and synthetic high-affinity (D-Lys)-LHRH peptide as a targeting moiety. LHRH-DCMs exhibited high drug loading efficiency, optimal particle size, good colloidal stability, and glutathione-responsive drug release. As expected, LHRH-DCMs were more efficiently internalized into human TNBC cells through receptor-mediated endocytosis, resulting in stronger cytotoxicity against these cancer cells than nontargeted counterpart when encapsulated with paclitaxel (PTX). Furthermore, near-infrared fluorescence and magnetic resonance imaging demonstrated that LHRH-DCMs facilitated the tumor distribution and penetration of payloads in three different animal models of breast cancer, including cell line-derived xenograft (CDX), patient-derived xenograft (PDX), and transgenic mammary carcinoma. Finally, in vivo therapeutic studies showed that PTX-LHRH-DCMs outperformed both the corresponding nontargeted PTX-DCMs and the current clinical formulation (Taxol®) in an orthotopic TNBC model. These results provide new insights into approaches for precise drug delivery of TNBC.
Keywords: LHRH, micelles, disulfide crosslink, redox-responsive, targeted delivery, paclitaxel, breast cancer
1. Introduction
Breast cancer is the most commonly diagnosed cancer (24.2%) and the leading cause of cancer death (15%) among women worldwide in 2018.[1] Hormonal therapy and targeted therapy are effective for hormone receptor positive and human epidermal growth factor receptor 2 (HER2) positive breast cancer, respectively. Currently, chemotherapy is the only systemic treatment for triple-negative breast cancer (TNBC), which is characterized by the lack of expression of estrogen receptor (ER), progesterone receptor (PR), and HER2.[2] Although chemo- and adjuvant therapies are clinically efficacious, TNBC tends to recur after a short duration, and side effects associated with these therapies are serious and sometimes even life-threatening.[3] Therefore, the development of a new treatment strategy to selectively kill cancer cells is of great significance for the treatment of advanced breast cancer, especially TNBC.
Nanotechnology has recently shown great promise for the diagnosis and treatment of a wide range of diseases, including cancer.[4] It is well known that nanoparticles can be passively enriched at the tumor site through the enhanced permeability and retention (EPR) effects due to the leakage of tumor vasculature and defects in the lymphatic system.[5] Doxil® and Abraxane® are the first two anti-cancer nanodrugs approved by the US food and drug administration (FDA).[6] Although the toxicities of these nanodrugs were significantly lower than the parent drugs, their clinical efficacies are only marginally better, in part because of the relatively large particle size (130–140 nm), which limits their EPR effect and tissue penetration. Polymeric micelles may have some therapeutic advantages over liposomes and protein aggregates because they are significantly smaller (20–100 nm), and may have deeper tumor permeability.[7] We have developed a series of highly versatile and robust telodendrimer-based micellar nanoparticles comprising linear polyethylene glycol (PEG) and dendritic cholic acids (CA), which show multiple advantages in drug delivery, such as superior biocompatibility, narrow particle size (20–60 nm), and high drug loading capacity.[8, 9] In addition, in order to improve their colloidal stability in the blood circulation, we further developed two types of stimuli-responsive crosslinked micelles for programmable drug delivery. One of them, called the disulfide crosslinked micelles (DCMs), is obtained by introducing thiol groups into the telodendrimer to form intra-micellar disulfide crosslinks, which respond to the high level of glutathione (GSH) in tumor cells or exogenous N-acetylcysteine (NAC, Mucomyst®) given on demand.[10–12] The other one, called the boronate crosslinked micelles (BCMs), is responsive to the acidic pH in tumor microenvironment or exogenous mannitol (Osmitrol®) given on demand.[13]
It is generally believed that the modification of nanoparticles with cancer cell-targeting ligands will further promote the residence and penetration of delivered drugs in tumor tissue and cells.[14, 15] Luteinizing hormone-releasing hormone (LHRH) receptors (LHRH-R) are overexpressed in a variety of cancers such as breast cancer (64%),[16] prostate cancer (86%), ovarian cancer and endometrial cancer (80%),[17] but are rarely expressed in normal tissues,[18] making them suitable targets for cancer-specific delivery. Engel and co-workers reported that LHRH-R were expressed in 34 out of 69 human TNBC specimens (49%) by immunohistochemistry, and its mRNA was expressed in MDA-MB-231 and HCC1806 TNBC cell lines by real time RT-PCR.[19] Recent studies have shown that LHRH peptide or its analogs can be used to link theranostic agents, thus delivering them directly to cancer cells with high expression of LHRH-R, rather than normal cells with no or low expression.[20, 21] Peptides as targeting moieties may have certain advantages over conventional antibodies, including ease of preparation, low antigenicity, less nonspecific binding, and proteolytic stability (when using D- and unnatural amino acids).[22] In addition, LHRH agonist alone or in combination with tamoxifen have been used as adjuvant treatment in premenopausal women with advanced breast cancer.[23] (D-Lys)-LHRH is a synthetic degradation-resistant LHRH analog with a high binding affinity to LHRH-R. It has been used in several studies as a targeting strategy for LHRH-R positive tumors, but mainly focused on the delivery of small molecule anti-cancer drugs, such as doxorubicin (AEZS-108, AN-152) and Disorazol Z (AEZS-125).[19, 24] However, as far as we know, the high-specificity and high-affinity (D-Lys)-LHRH peptide has not yet been utilized to improve the targeted delivery of nanoparticle drugs in TNBC treatment.
In this study, (D-Lys)-LHRH peptide will be anchored onto our early developed redox-responsive DCMs as a targeting moiety to achieve more precise drug delivery to TNBC. It is expected that (D-Lys)-LHRH peptide will not only direct the nanoparticle drugs more specifically to breast tumor, but also promote their internalization into breast cancer cells through receptor-mediated endocytosis, thus greatly improving the anti-tumor efficacy of delivered drugs with reduced systemic toxicity. Firstly, the expression of LHRH-R, binding affinity and specificity of (D-Lys)-LHRH peptide were confirmed in human TNBC cells and tissues. Secondly, alkyne modifying (D-Lys)-LHRH peptide was covalently conjugated to the azide-functionalized distal terminal of PEG chain in the thiolated telodendrimer (PEG5k-Cys4-L8-CA8) via click chemistry. The physicochemical characteristics (e.g., morphology, particle size and drug release) of the resulting LHRH-targeted DCMs (LHRH-DCMs) were determined, and their uptake, cytotoxicity, and tumor-targeting capability were investigated in different TNBC cell lines and animal models, including cell line-derived xenograft (CDX), patient-derived xenograft (PDX), and transgenic mammary carcinoma. Finally, the therapeutic efficacy and safety profiles of paclitaxel (PTX)-encapsulated LHRH-DCMs were evaluated in an orthotopic model of TNBC.
2. Results and Discussion
2.1. (D-Lys)-LHRH Peptide Analog as a Specific Breast Cancer Targeting Ligand
Flow cytometric analysis demonstrated that LHRH-R was almost not detected in MCF10A normal human breast cell line (Figure 1A), but overexpressed on the cell surface of human TNBC cells (such as MDA-MB-231 cell line, Figure 1B). Tissue immunohistochemistry staining showed that LHRH-R was not expressed in normal human breast tissues (Figure 1C), but overexpressed in most human TNBC tissues (Figure 1D). The overexpression of LHRH-R in TNBC makes it an ideal target for cancer-specific drug delivery.
Figure 1.
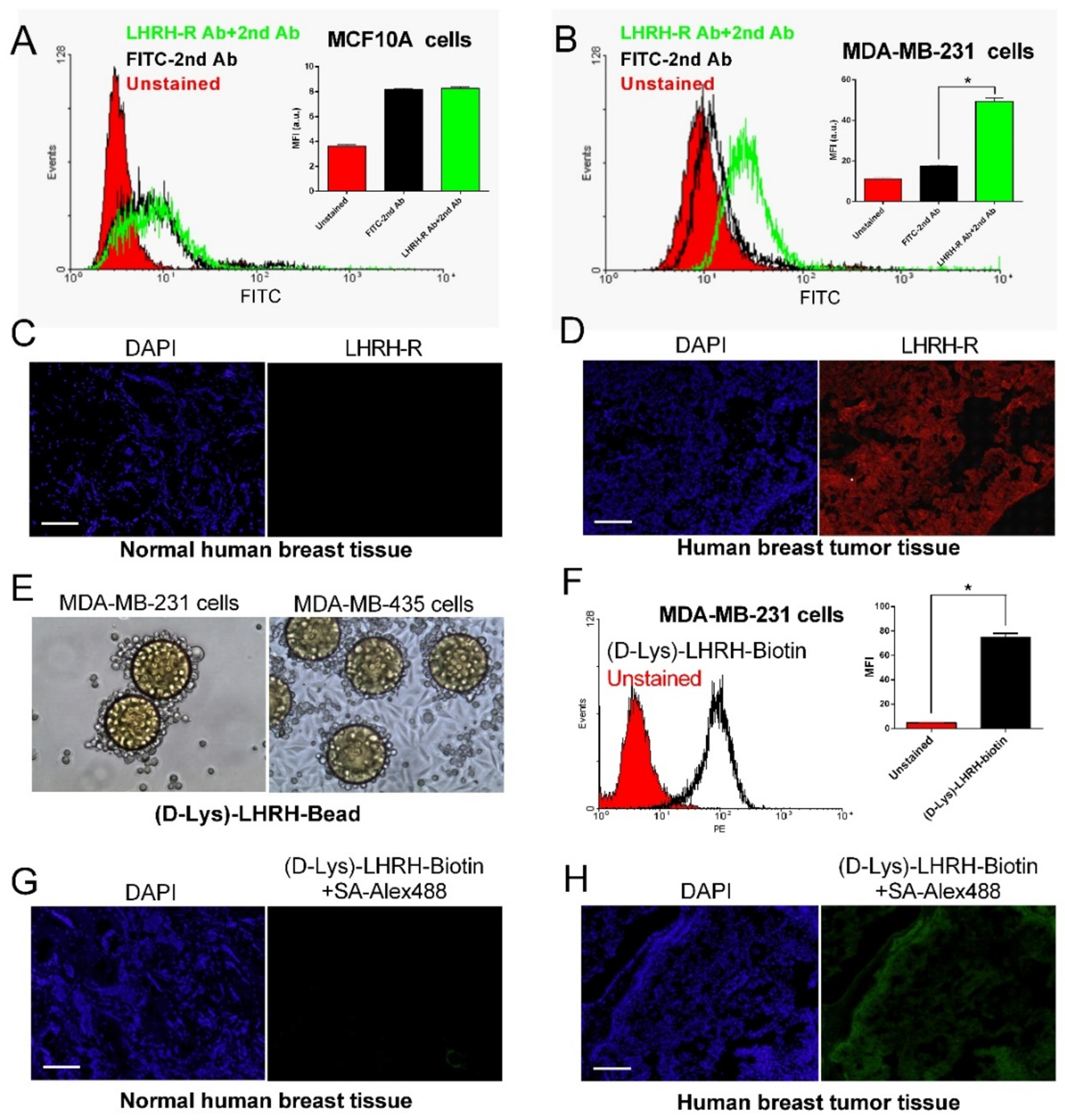
Overexpression of LHRH receptor (LHRH-R) in human breast cancer cells and tissue, and (D-Lys)-LHRH analogue showing high-specificity and high-affinity to LHRH-R overexpressed breast cancer. Flow cytometric analysis of LHRH-R expression on MCF10A normal human breast cells (A) and MDA-MB-231 human breast cancer cells (B) (n = 3). Immunohistochemical staining showing the expression of LHRH-R in normal human breast tissue (C) and human breast tumor tissue (D). (E) TentaGel resin beads displaying (D-Lys)-LHRH peptide showed strong binding with MDA-MB-231 and MDA-MB-435 cells after 2-hour incubation. (F) Flow cytometric analysis showed that biotinylated (D-Lys)-LHRH peptide had strong binding affinity to MDA-MB-231 cells (n = 3). Immunostaining results of biotinylated (D-Lys)-LHRH peptide were negative in normal mammary tissue (G), but were positive in human breast tumor tissue (H). Results are shown as mean ± SD. *p < 0.05, determined by one-way ANOVA with Tukey’s multiple comparisons. Scale bar: 100 μm.
(D-Lys)-LHRH peptide was coupled onto TentaGel resin beads by solid-phase synthesis for the cell binding experiment. As shown in Figure 1E, MDA-MB-231 and MDA-MB-435 breast cancer cells were able to strongly bind to the beads displaying (D-Lys)-LHRH peptide after 2-hour incubation. In contrast, no bead binding was observed for MCF10A normal human breast cells (data not shown). In addition, the binding capacity of biotinylated (D-Lys)-LHRH peptide to breast cancer cells was detected by streptavidin-PE (fluorochrome) staining. The results of flow cytometric analysis (Figure 1F) indicated that (D-Lys)-LHRH peptide had a strong binding affinity to MDA-MB-231 cells. Immunofluorescence staining further confirmed that (D-Lys) LHRH peptide did not bind to normal breast tissue (Figure 1G), but could specifically bind to human breast tumor tissue (Figure 1H) and transgenic murine mammary carcinoma tissue (Figure S-1). Therefore, as illustrated in Figure 2, (D-Lys)-LHRH peptide could be potentially used to direct nanoparticles to breast cancer cells and promote their cellular internalization through receptor-mediated endocytosis, thus ultimately improving the anti-cancer effect of nanoparticle drugs.
Figure 2.
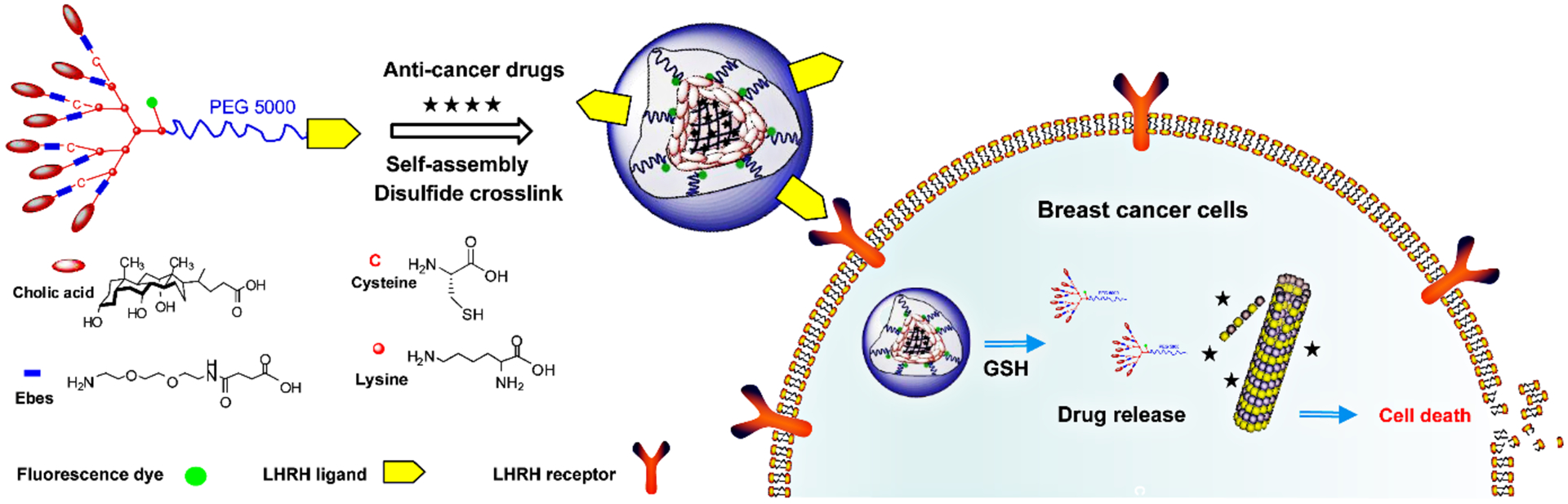
Schematic illustrating breast cancer-specific drug delivery of LHRH-DCMs. LHRH-DCMs are formed by the self-assembly and oxidation of (D-Lys)-LHRH conjugated PEG5k-Cys4-L8-CA8 telodendrimer, which will facilitate the precise delivery of therapeutic drugs into breast cancer cells through specific recognition and binding of over-expressed LHRH receptors on the cell membrane.
2.2. Synthesis of LHRH-PEG5k-Cys4-L8-CA8 Telodendrimer and Characterization of the Corresponding LHRH-DCMs
Click chemistry was used to covalently conjugate alkyne-containing (D-Lys)-LHRH peptide (MW = 1667.58, Figure S-2) onto the azide group at the distal terminus of PEG chain in PEG5k-Cys4-L8-CA8 telodendrimer, resulting in LHRH-PEG5k-Cys4-L8-CA8 telodendrimer (Figure 3A). The molecular weight of LHRH-PEG5k-Cys4-L8-CA8 telodendrimer was determined with MALDI-TOF mass spectrometry, which was mono-dispersed and almost identical to the theoretical value (data not shown). The chemical structure of LHRH-PEG5k-Cys4-L8-CA8 telodendrimer was also determined by 1H-NMR spectrometry. As shown in Figure S-3, the signals at 0.6–2.5 ppm and 3.5–3.7 ppm could be assigned to cholic acid and PEG chains, respectively, while the signals of D-Lys LHRH peptide were overlapped with the signals from the telodendrimer, with no distinguishable signals observed.
Figure 3.
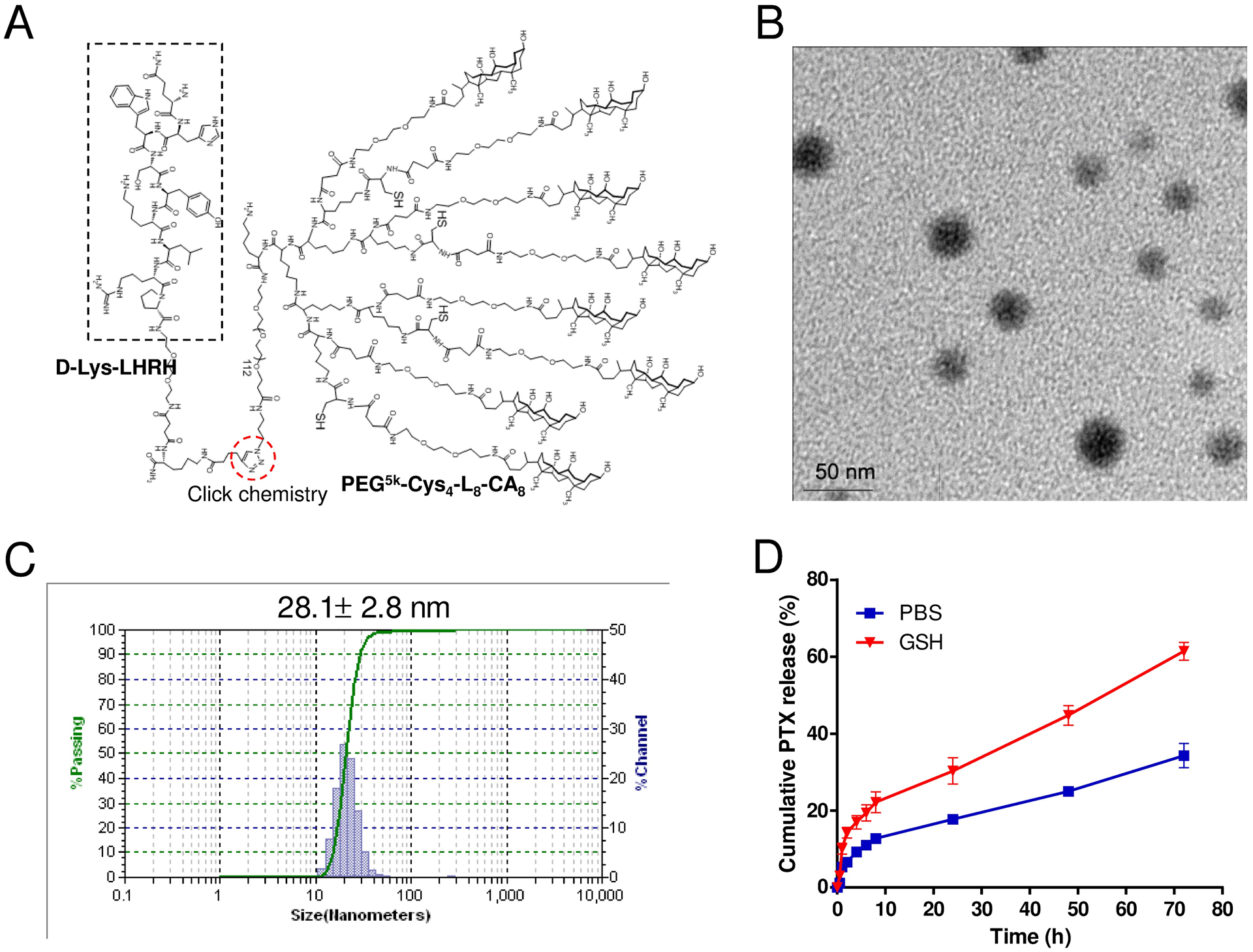
Physicochemical characterization of (D-Lys)-LHRH-PEG5k-Cys4-L8-CA8 telodendrimer and the resulting PTX-loaded LHRH-DCMs. (A) Chemical structure of (D-Lys)-LHRH-PEG5k-Cys4-L8-CA8 telodendrimer. Alkyne-containing (D-Lys)-LHRH peptide was conjugated to the azide group on the distal terminus of PEG chain in PEG5k-Cys4-L8-CA8 telodendrimer via “click chemistry”. The morphology (B) and particle size distribution (C) of PTX-loaded LHRH-DCMs (4 mg PTX in 20 mg/ml telodendrimer), measured by TEM and DLS, respectively. (D) Cumulative release of PTX from PTX-loaded LHRH-DCMs in PBS or GSH (10 mM) at 37 °C (n = 3). The concentration of PTX remained in the dialysis cartridge at various time points was measured by HPLC. Results are shown as mean ± SD.
The resulting LHRH-PEG5k-Cys4-L8-CA8 telodendrimer was able to self-assemble in aqueous solution to form spherical monodisperse core-shell micelles and can be further crosslinked by oxidation of free thiol groups in the telodendrimer to form disulfide bridges.[11] Hydrophobic drugs such as PTX could be efficiently encapsulated inside LHRH-DCMs via solvent evaporation method followed by H2O2-mediated oxidization. When the feeding ratio of PTX/telodendrimer (w/w) was 1:5, the encapsulation efficiency (EE) of LHRH-DCMs was approximately 93%, which was similar to that of nontargeted DCMs (Table 1, Figure S-4). TEM image (Figure 3B) demonstrated that PTX-loaded LHRH-DCMs (PTX-LHRH-DCMs) were spherical with uniform particle sizes of around 30 nm in diameter, which was consistent with the measurement results of DLS (Figure 3C). The zeta potential of PTX-LHRH-DCMs was almost neutral (0.44 mV, Table 1). The stability of PTX-LHRH-DCMs in terms of particle size was monitored in different conditions including FBS, and micelle-disrupting SDS with or without reducing agent GSH, respectively. The results (Figure S-5) demonstrated that the particle size of PTX-LHRH-DCMs in FBS and SDS were relatively stable after 24-hour incubation, suggesting that the crosslinked micelles could maintain their integrity in the blood circulation. However, in the presence of both SDS and GSH, the particle size of PTX-LHRH-DCMs disappeared completely, indicating the dissociation of micelles after reduction of the disulfide bonds. The PTX release profiles from PTX-LHRH-DCMs were measured by the dialysis method. As illustrated in Figure 3D, the drug release profile from PTX-LHRH-DCMs in PBS was biphasic, with a burst release in the first few hours followed by a slow linear release over the following days, resulting in PTX release less than 40% after 72 hours. However, the release rate of PTX from PTX-LHRH-DCMs in the presence of GSH was significantly faster than that in PBS, suggesting that the drug release from micelles can be accelerated by reducing agents. Thus, this two-stage drug release strategy of DCMs can be exploited to minimize the premature drug release during circulation, and trigger the rapid release of drugs by the high concentration of GSH in cancer cells, or by the administration of exogenous reducing agent such as NAC after the micelles have reached the tumor sites.[10]
Table 1.
The physicochemical characteristics of LHRH-DCMs
| Micelles | CMC (μM)a | Particle size (nm)b | PTX/Polymer ratio (w/w)c | PTX in micelles (mg/ml) | EE (%)d | Particle size after PTX loading (nm)b | Zeta potential (mV) |
|---|---|---|---|---|---|---|---|
| DCMs | 0.67 | 26.0 ± 1.6 | 1:5 | 3.8 ± 0.7 | 95 ± 2.0 | 29.2 ± 2.4 | 0.62 |
| LHRH-DCMs | 0.70 | 25.5 ± 1.9 | 1:5 | 3.7 ± 0.9 | 93 ± 2.5 | 28.1 ± 2.8 | 0.44 |
CMC was measured by fluorescence spectrometry using pyrene (2 μM) as a probe;
Measured by DLS;
The concentrations of telodendrimers were 20 mg/ml;
Encapsulation efficiency expressed as a percentage mean of three determinations ± standard deviation of PTX weight recovered in micelles compared to theoretical loaded weight.
2.3. Cellular Uptake Studies
The uptake of FITC-labeled LHRH-DCMs in human breast cancer cells was first qualitatively observed by confocal microscopy. After 2-hour incubation, nontargeted DCMs showed minimal nonspecific uptake in MDA-MB-231 and MDA-MB-435 cells, while the incorporation of (D-Lys) LHRH peptide significantly enhanced the uptake of DCMs in both cells (Figure 4A, B). In addition, most of the green fluorescence was distributed in the perinuclear region, suggesting that these micelles were internalized into the cytoplasm. More importantly, the addition of excess LHRH-R antibody dramatically inhibited the uptake of LHRH-DCMs in breast cancer cells, indicating that their internalization was mediated by the LHRH-R.
Figure 4.
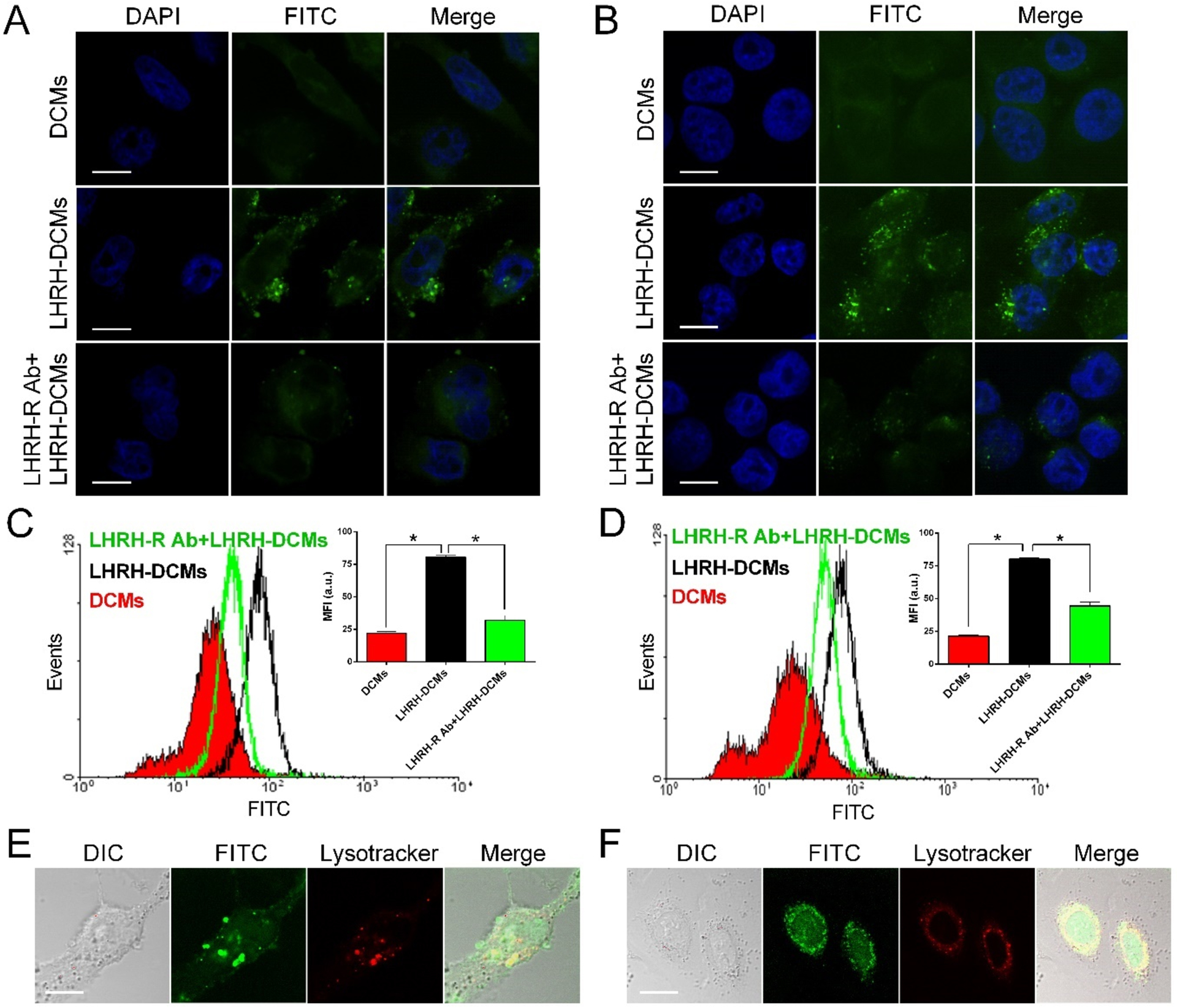
The uptake and intracellular fate of LHRH-DCMs in breast cancer cells. Confocal microscopic images of MDA-MB-231 cells (A) and MDA-MB-435 cells (B) after 2-hour incubation with FITC-labeled DCMs or LHRH-DCMs (green). To confirm that the uptake of LHRH-DCMs in breast cancer cells was mediated by LHRH-R, excess amount of LHRH-R antibody (200 μM) was added prior to the incubation. Quantitative uptake of FITC-labeled LHRH-DCMs in MDA-MB-231 cells (C) and MDA-MB-435 cells (D) were detected by flow cytometry. Cells were incubated with FITC-labeled DCMs and LHRH-DCMs with or without the prior addition of LHRH-R antibody. The mean fluorescence intensity (MFI) of cells is shown as insets (n = 3). Intracellular tracking of FITC-labeled LHRH-DCMs in live MDA-MB-231 cells (E) and MDA-MB-435 cells (F). Cells were incubated with FITC-labeled LHRH-DCMs for 2 hours, stained with lysosome tracker (red), and then subjected to the live-cell imaging by confocal microscopy. Results are shown as mean ± SD. *p < 0.05, determined by one-way ANOVA with Tukey’s multiple comparisons. Scale bar: 10 μm.
The uptake efficiencies of LHRH-DCMs in breast cancer cells were further quantitatively analyzed by flow cytometry. As shown in Figure S-6, LHRH-DCMs exhibited low non-specific uptake similar to nontargeted DCMs in MCF10A normal human breast cells. However, the uptake of LHRH-DCMs in both MDA-MB-231 (Figure 4C) and MDA-MB-435 (Figure 4D) breast cancer cells was significantly higher than that of nontargeted DCMs (P < 0.05). For instance, the uptake levels of LHRH-DCMs in MDA-MB-231 and MDA-MB-435 cells were 3.7-fold, and 3.8-fold higher than that of nontargeted DCMs, respectively. Meanwhile, the addition of excess LHRH-R antibody significantly inhibited the uptake of LHRH-DCMs in both breast cancer cells, but not MCF10A normal human breast cells (Figure S-6). In addition, a significant decrease in the uptake of LHRH-DCMs in MDA-MB-231 and MDA-MB-435 breast cancer cells was observed when incubated at 4 °C (Figure S-7), suggesting an energy-dependent active process.[25] These data indicated that the enhanced uptake of LHRH-DCMs in breast cancer cells was mainly due to receptor-mediated endocytosis.
The intracellular fate of LHRH-DCMs in live breast cancer cells was further investigated by co-localization method. As shown in the confocal images (Figure 4E and 4F), dot-shape fluorescent foci (green) were formed in the cytoplasm of MDA-MB-231 and MDA-MB-435 cells after 2-hour incubation with FITC-labeled LHRH-DCMs, and were co-localized with the lysosomal compartment (red), suggesting that LHRH-DCMs were mainly trapped in the endocytic vesicles of breast cancer cells upon their uptake. Similar observations have been previously reported by us and others.[14, 21]
2.4. In Vitro Cytotoxicity
The cytotoxicity of blank and PTX-loaded LHRH-DCMs against normal human breast cells and human breast cancer cells were evaluated by MTT assay. Blank LHRH-DCMs did not show detectable cytotoxicity against both MCF10A (Figure 5A) and MDA-MB-231 cells (Figure 5B) at all concentrations up to 2 mg/mL. When loaded with PTX, LHRH-DCMs exhibited dose-dependent cytotoxicity against both MCF10A cells and MDA-MB-231 cells. Compared with the equivalent concentration of PTX-DCMs, the cytotoxicity of PTX-LHRH-DCMs in MCF10A normal breast cells was similar (Figure 5C), but it was significantly higher in MDA-MB-231 breast cancer cells (Figure 5D, P < 0.05). The stronger cytotoxicity of PTX-LHRH-DCMs in breast cancer cells may be associated with increased intracellular PTX concentration due to higher cell uptake.[14]
Figure 5.
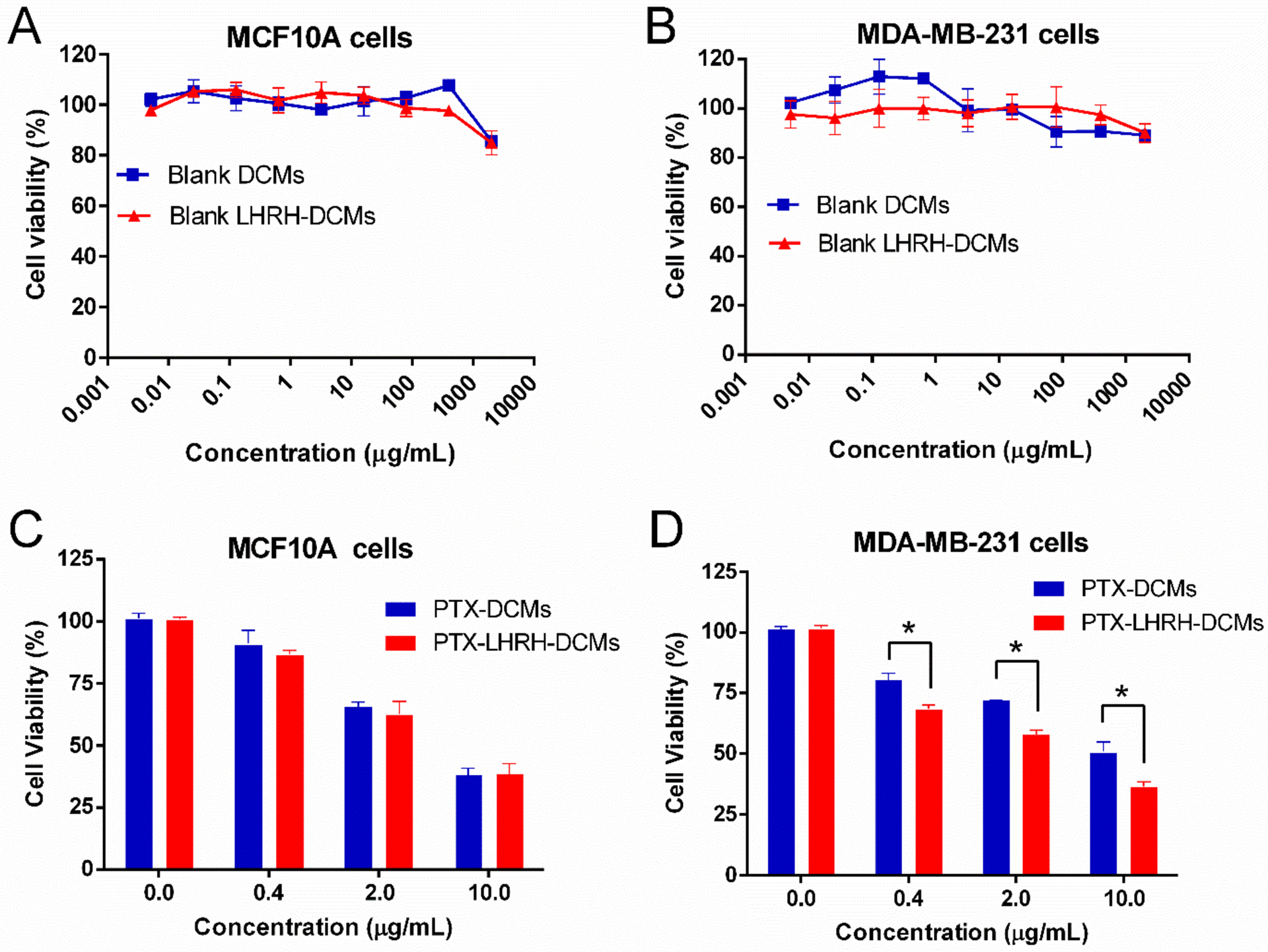
Cytotoxicity of blank and PTX-loaded LHRH-DCMs in normal human mammary epithelial cells (MCF10A) and human breast cancer cells (MDA-MB-231). The cytotoxicity of blank DCMs and LHRH-DCMs in MCF10A (A) and MDA-MB-231 cells (B) (n = 3). Cells were incubated with different concentration of blank DCMs and LHRH-DCMs for 72 hours, and the cell viability was measured by MTS assay. The cytotoxicity of PTX-DCMs and PTX-LHRH-DCMs in MCF-10A (C) and MDA-MB-231 cells (D) (n = 3). Cells were incubated with equivalent concentration of PTX-DCMs and PTX-LHRH-DCMs for 2 hours, then washed and incubated in fresh media for a total of 72 hours before the cell viability measurement. Results are shown as mean ± SD. *p < 0.05, determined by one-way ANOVA with Tukey’s multiple comparisons.
2.5. Biodistribution and Tumor Targeting of LHRH-DCMs in Various Breast Cancer Models
The in vivo biodistribution and tumor targeting capability of LHRH-DCMs were visualized by the NIRF optical imaging approach, which has the characteristics of deep penetration, low auto-fluorescence, less tissue absorption, and scattering.[22] DiD dye as a drug surrogate was efficiently encapsulated into DCMs/LHRH-DCMs for NIRF optical imaging. After intravenous injection in orthotopic MDA-MB-231 xenograft bearing mice, both DCMs and LHRH-DCMs were immediately distributed throughout the whole body and gradually accumulated at the tumor site through the EPR effect (Figure 6A). However, LHRH-DCMs demonstrated higher tumor uptake than nontargeted DCMs at 48 hours post-injection. This is probably because the active targeting of LHRH-DCMs enhances the retention of micelles at the tumor site by reducing the passive transport away from the tumor.[26] Ex vivo images (Figure 6B) further confirmed the preferential uptake of LHRH-DCMs in the tumor. Relatively high uptake of micelles was also observed in the liver, which was likely due to non-specific clearance of macrophages (Kupffer cells).[27] Furthermore, the histological distribution of micelles in the tumor tissue was observed under the confocal microscopy. As shown in Figure 6C, most DiD-labeled DCMs (red) mainly distributed in the perivascular region (CD31, green), which is consistent with previous histological observations of passive accumulation of liposomes and micelles in tumor tissue.[28] On the contrary, LHRH-DCMs were able to extravasate from the tumor vasculature, penetrate deeply into tumor stroma, bind to LHRH-R on the breast cancer cells, and eventually become internalized.
Figure 6.
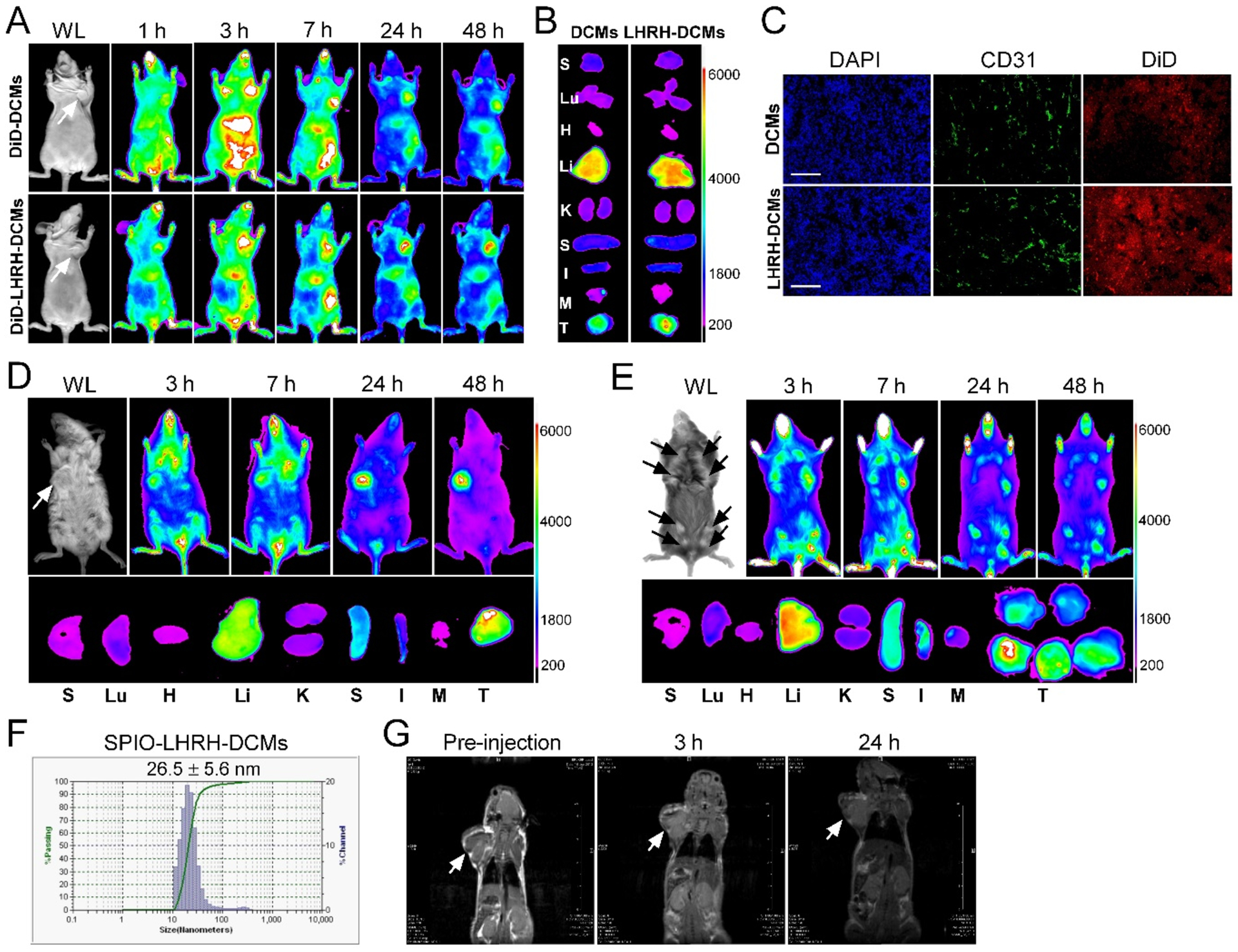
Biodistribution and tumor targeting of LHRH-DCMs in cell line-, patient-derived xenograft and transgenic models of breast cancer via NIRF optical imaging and MRI. In vivo (A), ex vivo (B), and microscopic (C) tracking of DiD fluorescently labeled LHRH-DCMs in orthotopic MDA-MB-231 xenograft model. Tumor bearing mice were injected intravenously with DiD-labeled DCMs and LHRH-DCMs, respectively, and scanned with Kodak multimodal imaging system IS2000MM at different time point. At 48 hours post-injection, major organs and tumor tissue were dissected for ex vivo imaging. The microscopic distribution of LHRH-DCMs (red) in the tumor section was observed under confocal microscopy, with the nuclear staining with DAPI (blue) and vascular endothelial cell staining with CD31 antibody (green). Scale bar: 100 μm. In vivo and ex vivo NIRF optical imaging of DiD-labeled LHRH-DCMs distribution in patient-derived xenograft (D) and transgenic (E, MMTV-PyMT) mouse model of breast cancer, respectively. (F) Particle size of superparamagnetic iron oxide nanoparticles (SPIO)-loaded LHRH-DCMs. (G) Representative MRI images of orthotopic MDA-MB-231 tumor bearing mice after intravenous injection of SPIO-loaded LHRH-DCMs.
In addition to CDX model, the biodistribution of LHRH-DCMs was also investigated in PDX model of breast cancer with high expression of LHRH-R. In vivo and ex vivo NIRF optical imaging in orthotopic breast cancer PDX bearing mice (Figure 6D) demonstrated that LHRH-DCMs were more likely to accumulate in breast tumors than other normal organs. In addition, confocal images of tumor sections showed that LHRH-DCMs can penetrate the entire tumor tissue (Figure S-8).
As the tumor xenografts (human cancer cell line- or patient-derived xenograft) are derived from an exogenous source, tumor vascularization (mouse endothelial cells) in the xenograft may not present the same way as it occurs in human. Tumor-bearing transgenic mice might provide more clinically relevant information about the passive and active targeting of nanoparticle drugs in tumors, as the tumor formation in the transgenic model is “spontaneous”, and tumor cells and endothelial cells derive from the same host (mouse), excluding the tumor vasculature artifacts that may occur in tumor xenograft models.[29] Therefore, the tumor-targeting capacity of LHRH-DCMs was also validated in transgenic murine mammary carcinoma mouse model (MMTV-PyMT, LHRH-R positive). In vivo optical imaging (Figure 6E) demonstrated that LHRH-DCMs were able to gradually accumulate and maintain in different sizes of spontaneous breast tumors through EPR effect and active targeting. Ex vivo images at 48 hours post-injection further confirmed the high uptake of LHRH-DCMs in the tumor tissue.
In addition, MRI was also used to monitor the tumor targeting and biodistribution of LHRH-DCMs in orthotopic breast cancer xenograft models. SPIO as an MRI imaging contrast agent was physically encapsulated into LHRH-DCMs, with the average particle size of 26 nm in diameter (Figure 6F). SPIO-loaded LHRH-DCMs were intravenously injected into orthotopic MDA-MB-231 xenograft bearing mice, and MRI images were obtained at different time points post-injection. As shown in Figure 6G, SPIO-loaded LHRH-DCMs distributed throughout the whole body after injection and were able to gradually accumulate into tumor site over time.
2.6. Therapeutic Efficacy of PTX-LHRH-DCMs in Orthotopic Breast Cancer Model
The anti-tumor activity of PTX-LHRH-DCMs was evaluated in an orthotopic MDA-MB-231 xenograft mouse model. Tumor bearing mice (n = 6~7) were intravenously administrated with PBS, Taxol® (10 mg/kg), PTX-DCMs (10 and 25 mg PTX/kg PTX) and PTX-LHRH-DCMs (10 and 25 mg PTX/kg) every four days for a total of 6 doses, respectively. In another group of 25 mg/kg PTX-LHRH-DCMs, NAC (100 mg/kg) was injected intraperitoneally 24 hours after each administration to trigger the drug release when the micelles accumulated at tumor sites. Compared to PBS Control group, all PTX preparations were able to significantly inhibit tumor growth (P < 0.05, Figure 7A). However, both micellar formulations of PTX showed better inhibition of tumor growth than free PTX at the equivalent dose, which may be due to the EPR effect allowing more drugs to enter the tumor site. It is worth noting that the tumor growth rate of mice in the PTX-LHRH-DCMs group was significantly slower than that in the PTX-DCMs group (P < 0.05). The survival rate of mice in each group was expressed by Kaplan-Meier survival curve (Figure 7B). Overall, compared with PBS control group, all PTX preparations significantly prolonged the survival of tumor bearing mice. However, at the equivalent dose, mice treated with PTX-LHRH-DCMs survived longer than those treated with Taxol® and PTX-DCMs. The median survival time of PBS control, Taxol®, PTX-DCMs (10, 25 mg/kg), PTX-LHRH-DCMs (10, 25 mg/kg), and PTX-LHRH-DCMs + NAC groups were 32, 42, 50, 82, 55, 90, and >90 days, respectively. It should be noted that PTX-LHRH-DCMs + NAC was the most efficacious in all groups. No palpable tumors were detected in 5 of the 6 mice in this group by day 90 (Figure 7C). NAC is a reducing agent, which is commonly used for the treatment of acetaminophen overdose,[30] and detoxification when combined with chemotherapeutic agents (e.g. cisplatin, carboplatin).[31] However, in some cases, the administration of NAC may also reduce the therapeutic efficacy of these chemotherapeutic drugs. In this study, PTX-LHRH-DCMs combined with NAC achieved better anti-tumor effects than PTX-LHRH-DCMs alone, indicating that NAC plays an important role in reducing intra-micellar disulfide bonds and releasing drugs on demand.
Figure 7.
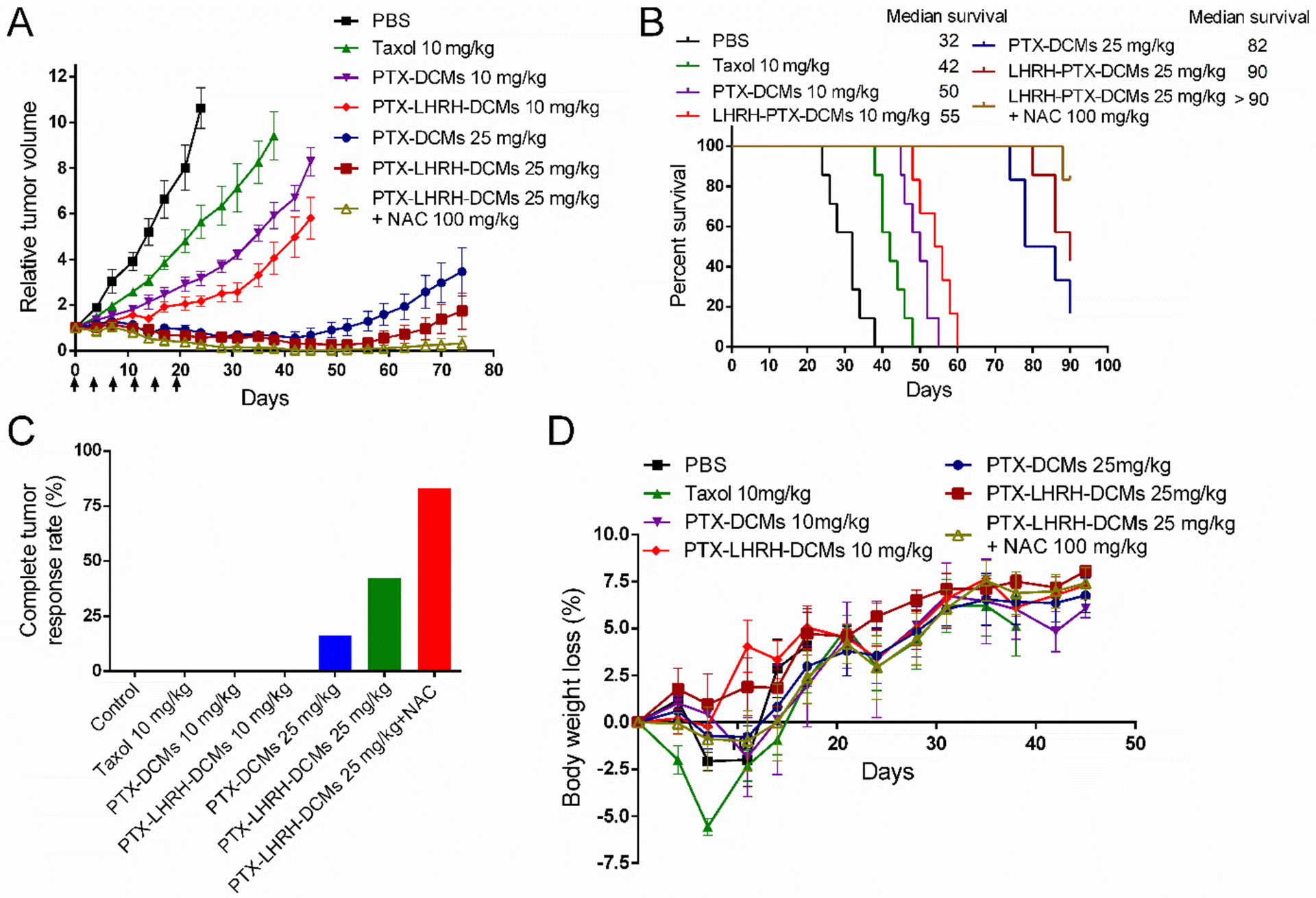
Anti-tumor efficacy and toxicity profiles of PTX-LHRH-DCMs in an orthotopic breast cancer model. In vivo tumor growth inhibition (A), Kaplan-Meier survival curves (B), complete tumor response rate (C) and body weight changes (D) of orthotopic MDA-MB-231 tumor bearing mice after intravenous administration of various PTX formulations. Tumor bearing mice (n = 6~7) were intravenously injected with PBS, Taxol® (10 mg/kg), PTX-DCMs (10 and 25 mg/kg), and PTX-LHRH-DCMs (10 and 25 mg/kg), respectively. The dosage was given every four day for a total of 6 doses. In addition, N-acetylcysteine (NAC, 100 mg/kg) was given in another group of 25 mg/kg PTX-LHRH-DCMs 24 hours after each administration so as to trigger the release of drugs when the targeted DCMs was enriched at the tumor site. Results are shown as mean ± SD. *p < 0.05, determined by one-way ANOVA with Tukey’s multiple comparisons.
Toxicities were evaluated by animal behaviors, body weight changes, CBC, and serum chemistry. The mice in the 10 mg/kg Taxol® group tended to show decreased activity within 10 minutes after injection. This may be due to the use of Cremophor EL and ethanol as vehicles, as well as the rapidly increasing peak level of PTX in the blood.[32] In addition, initial body weight loss was observed in mice treated with Taxol®. However, there was no significant change in overall activity and weight loss of mice after treatment with PTX-DCMs and PTX-LHRH-DCMs compared to the control group (Figure 7D). CBC (Table S-1) and serum chemistry results (Table S-2) demonstrated that all the parameters including WBC, RBC, ALT, AST, TBIL, CREA, and BUN, were all within the normal range, excluding the potential hematologic, hepatic and renal toxicities.
3. Conclusion
We have developed LHRH-targeted DCMs with several unique features, including high PTX loading efficiency, optimal particle size, redox-responsive drug release, and breast cancer-specific targeting. As expected, PTX-loaded LHRH-DCMs demonstrated enhanced uptake and cytotoxicity in LHRH-R overexpressed TNBC cells via receptor-mediated endocytosis, but not in normal human breast cells. Furthermore, NIRF and MRI imaging in various TNBC animal models showed that the decoration of (D-Lys)-LHRH peptide promoted the accumulation and internalization of DCMs in breast cancer tissue and cells, thus greatly improving the anti-tumor efficacy of delivered PTX and reducing the systemic toxicity. Therefore, LHRH-DCMs have great translational potential in the targeted therapy of breast cancer.
4. Experimental Section
Materials:
NH2-PEG-NH2 was purchased from Rapp Polymere (Tübingen, Germany). Paclitaxel was purchased from AK Scientific Inc. (Mountain View, CA). DiD (D-307), DAPI, and LysoTracker® Red were purchased from Thermo Fisher Scientific (Waltham, MA). CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit (MTS) was purchased from Promega (Madison, WI, USA). Cholic acid, fluorescein isothiocyanate (FITC), glutathione (GSH), and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). Organic super-paramagnetic iron oxide (SPIO, 5 nm) nanoparticles in Chloroform were purchased from Ocean Nanotech (San Diego, California).
Cell Culture and Animals:
MCF-10A (human mammary epithelial cells), MDA-MB-231 and MDA-MB-435 (human TNBC cells) were purchased from American Type Culture Collection (ATCC, Manassas, VA). MCF-10A cells were cultured in MEBM medium (Lonza/Clonetics Corporation) supplemented with 100 ng/mL cholera toxin. MDA-MB-231 and MDA-MB-435 cells were cultured in Leibovitz’s L-15 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C without CO2.
8-week-old female nude mice were obtained from Harlan Laboratories (Indianapolis, Indiana). Transgenic murine mammary carcinoma (MMTV-PyMT) mice and NSG mice were obtained from the Jackson Laboratory (Sacramento, CA). All mice were kept in specific pathogen-free (SPF) conditions, and all animal experiments were conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, Davis (No. 09–15584). Orthotopic mouse models of breast cancer CDX and PDX were established by the injection of MDA-MB-231 cells (1 × 107) and TNBC PDX specimen (BR0620F, obtained from the Jackson Laboratory) suspended in PBS/Matrigel (100 μL) in the mammary fat pad of nude mice and NSG mice, respectively.
LHRH-R Expression in Breast Cancer Cells/Tissue:
MCF10A, MDA-MB-231, and MDA-MB-435 cells were incubated with monoclonal anti-human LHRH-R antibody (Santa Cruz Biotechnology), washed with PBS, then incubated with FITC-labeled secondary antibody, washed with PBS, and analyzed by flow cytometry. The expression of LHRH-R in normal human breast tissue and human breast tumor tissue was examined by immunohistochemistry. Briefly, the tissue cryo-section was stained with primary LHRH-R antibody for 2 hours, then with FITC-labeled secondary antibody for 30 minutes, and washed three times with cold PBS. The nuclei were stained with DAPI, and the slides were observed under a fluorescence microscope.
Binding Affinity of (D-Lys6)-LHRH to Breast Cancer Cells/Tissue:
TentaGel resin beads displaying (D-Lys6)-LHRH peptide were incubated with MDA-MB-231 or MDA-MB-435 cells for 2 hours, washed with PBS, and observed under optical microscopy. Next, the binding affinity of (D-Lys6)-LHRH peptide to breast cancer cells was determined. In brief, MDA-MB-231 cells were incubated with biotinylated (D-Lys6)-LHRH peptide for 2 hours, and with streptavidin-PE for 30 minutes, then washed with PBS, and submitted to flow cytometric analysis. To confirm the binding capacity and specificity of (D-Lys6)-LHRH peptide to breast cancer tissue, the sections of human normal mammary gland and breast tumor tissue were stained with biotinylated (D-Lys6)-LHRH peptide for 2 hours, then with streptavidin-Alex488 for 30 minutes, and washed three times with cold PBS. The nuclei were stained with DAPI, and the slides were observed under a fluorescence microscope.
Synthesis of LHRH-PEG5k-Cys4-L8-CA8 Telodendrimer:
N3-PEG5k-Cys4-L8-CA8 telodendrimer was synthesized as described previously.[10, 14] (D-Lys)-LHRH analog, Gln-His-Trp-Ser-Tyr-(D-Lys)-Leu-Arg-Pro-Ebes-Lys, was synthesized by solid-phase reaction using the standard Fmoc chemistry. The ε-amino group of lysine at the C-terminal of LHRH analog was coupled with 5-hexenoic acid to realize alkynyl functionalization, which was then conjugated to the N3-PEG5k-Cys4-L8-CA8 telodendrimer via CuI catalyzed cycloaddition to obtain LHRH-PEG5k-Cys4-L8-CA8 telodendrimer.[33] Its molecular structure was measured by 1H-NMR. The amino group of the proximal lysine in the backbone of telodendrimer was coupled with FITC-NHS ester to obtain FITC-labeled telodendrimer.
Preparation and Characterization of PTX-loaded LHRH-DCMs:
PTX-loaded LHRH-DCMs (PTX-LHRH-DCMs) were prepared by the solvent evaporation method as described previously.[8] Briefly, PTX and PEG5k-Cys4-L8-CA8/LHRH-PEG5k-Cys4-L8-CA8 telodendrimer (1:1) were dissolved in chloroform, followed by the solvent-evaporation, hydration with PBS, and sonication. The resulting drug-loaded micelles were oxidized with hydrogen peroxide (H2O2) to form disulfide cross-linkage.
The morphology and particle size of PTX-LHRH-DCMs were measured by transmission electron microscopy (TEM), and dynamic light scattering (DLS), respectively. The stability of PTX-LHRH-DCMs in FBS (50%, v/v), sodium dodecyl sulfate (SDS, 2.5 mg/mL) with or without glutathione (GSH, 10 mM) was monitored by the change of particle size using DLS. The drug release of PTX-LHRH-DCMs was determined by dialysis. In brief, PTX-LHRH-DCMs in dialysis cartridges (3.5 kDa molecular-weight cutoff) were dialyzed against PBS or GSH (10 mM) at 37 °C, and the residual concentration of PTX in dialysis cartridge at different timepoints was determined by HPLC.
Cellular Uptake:
MDA-MB-231 and MDA-MB-435 cells incubated with 2 μM FITC-labeled DCMs and LHRH-DCMs with or without the addition of 200 μM LHRH-R antibody for 2 hours at 4 °C or 37 °C, respectively. Then, the cells were washed with PBS and submitted to confocal microscope observation and flow cytometry analysis, respectively. The cell nuclei were stained with DAPI.
To investigate the intracellular tracking of LHRH-DCMs upon cellular uptake, MDA-MB-231 and MDA-MB-435 cells seeded in 8-well chamber slides were incubated with FITC-labeled LHRH-DCMs for 2 hours, and the lysosomes were stained with LysoTracker Red (50 nM). Live cell imaging was acquired with a confocal microscope.
Cytotoxicity:
The in vitro cytotoxicity of blank/PTX-loaded DCMs and LHRH-DCMs against MCF10A and MDA-MB-231 cells was evaluated by MTT assay. The cells were incubated with blank DCMs and blank LHRH DCMs for 72 hours, or PTX-DCMs and PTX-LHRH-DCMs for 2 hours followed by PBS wash, fresh medium replacement and additional 70-hour culture. Finally, MTT reagent was added and the cell viability was detected by the absorbance (570 nm, 660 nm) using a microplate reader.
NIRF Optical Imaging:
DiD-loaded DCMs and LHRH-DCMs were intravenously injected into orthotopic MDA-MB-231 xenograft or breast cancer PDX (BR0620F) bearing mice, and MMTV-PyMT transgenic mammary carcinoma mice, respectively. These mice were scanned with Kodak imaging system (IS2000MM) at different time points post-injection. At 48 hours, tumor tissue and major organs were collected for ex vivo imaging. To determine the micro-distribution of LHRH-DCMs in the tumor section, tumor tissue was sliced with a microtome, stained with CD31 antibody (Millipore), and observed under a confocal microscope.
MRI Imaging:
SPIO (5 nm) were encapsulated into LHRH-DCMs to obtain SPIO-LHRH-DCMs for MRI imaging. Orthotopic MDA-MB-231 xenograft bearing mice were intravenously injected with SPIO-LHRH-DCMs and imaged with Bruker Biospec 7T MRI scanner at different time point. T2-weight MRI images (coronal section) were captured with the following parameters: repetition time 1000 ms, echo time 10 ms, field of view 8.00/6.00 cm, and slice thickness 2.0 mm.
Therapeutic Study:
orthotopic MDA-MB-231 xenograft bearing mice (n = 6~7) were intravenously administered with PBS, 10 mg/kg Taxol®, 10 and 25 mg/kg PTX-DCMs, 10 and 25 mg/kg PTX-LHRH-DCMs once every four days for a total of 6 doses, respectively. Additionally, N-acetylcysteine (NAC, 100 mg/kg) was injected intravenously 24 hours after each administration of 25 mg/kg PTX-LHRH-DCMs in another group. Tumor sizes were measured twice a week, and the relative tumor volume (RTV) was equal to the ratio of tumor volume at a given time point to that before initial treatment. When the tumor volume reached 1500 mm3, the mice were euthanized, which was considered as the end point of survival data. The weight of mice was measured twice per week. One the 7th day after the last administration, blood samples were collected for complete blood count (CBC) and serum chemistry measurement, including alanine aminotransferase (ALT), aspartate transaminase (AST), total bilirubin (TBIL), creatinine (CREA), and blood urea nitrogen (BUN).
Statistical Analysis:
All quantitative data were expressed as the mean ± standard deviation (SD). One-way analysis of variance (ANOVA) with Tukey’s post hoc test was performed in comparisons between more than two groups. A p-value of <0.05 was considered as statistical significance. In vitro experiments were performed in triplicates for each experimental group, unless otherwise specified. In vivo therapeutic animal experiments were performed with a sample size of 6–7 for each experimental group. Data were analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA) software.
Supplementary Material
Acknowledgements
The authors thank the financial support from NIH/NCI (R01CA115483), NIH/NIBIB (5R01EB012569), DoD BCRP Award (W81XWH-10-1-0817), National Natural Science Foundation of China (81572617 & 81630101), and Sichuan Science and Technology Program (2019JDRC0019).
Footnotes
Conflict of Interest
K.S.L. is the founding scientist of LamnoTherapeutics Inc. which plans to develop the nanotherapeutics described in the manuscript. All other authors declare no conflict of interest.
Contributor Information
Nell Suby, Department of Obstetrics and Gynecology, School of Medicine, University of California Davis, CA 95817, USA.
Wenwu Xiao, Department of Biochemistry & Molecular Medicine, UC Davis Cancer Center, University of California Davis, Sacramento, CA 95817, USA.
Rinki Agrawal, Department of Obstetrics and Gynecology, School of Medicine, University of California Davis, CA 95817, USA.
Michael Vu, Department of Biochemistry & Molecular Medicine, UC Davis Cancer Center, University of California Davis, Sacramento, CA 95817, USA.
Hongyong Zhang, Division of Hematology & Oncology, Department of Internal Medicine, School of Medicine, University of California Davis, CA 95817, USA.
Kit S. Lam, Department of Biochemistry & Molecular Medicine, UC Davis Cancer Center, University of California Davis, Sacramento, CA 95817, USA Division of Hematology & Oncology, Department of Internal Medicine, School of Medicine, University of California Davis, CA 95817, USA.
References
- [1].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, CA: a cancer journal for clinicians 2018, 68, 394. [DOI] [PubMed] [Google Scholar]
- [2].Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. , Science 1989, 244, 707. [DOI] [PubMed] [Google Scholar]
- [3].O’Reilly EA, Gubbins L, Sharma S, Tully R, Guang MH, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell M, McCann A, BBA clinical 2015, 3, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Zhang J, Chen J, Ren J, Guo W, Li X, Chen R, Chelora J, Cui X, Wan Y, Liang XJ, Hao Y, Lee CS, Biomaterials 2018, 181, 92; [DOI] [PubMed] [Google Scholar]; b) Zhang J, Nie W, Chen R, Chelora J, Wan Y, Cui X, Zhang X, Zhang W, Chen X, Xie HY, Lee CS, Nano letters 2019, 19, 658. [DOI] [PubMed] [Google Scholar]
- [5].Matsumura Y, Maeda H, Cancer research 1986, 46, 6387. [PubMed] [Google Scholar]
- [6].Barenholz Y, Journal of controlled release : official journal of the Controlled Release Society 2012, 160, 117. [DOI] [PubMed] [Google Scholar]
- [7].Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K, Bioconjugate chemistry 2005, 16, 122. [DOI] [PubMed] [Google Scholar]
- [8].Xiao K, Luo J, Fowler WL, Li Y, Lee JS, Xing L, Cheng RH, Wang L, Lam KS, Biomaterials 2009, 30, 6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Li Y, Xiao K, Luo J, Lee J, Pan S, Lam KS, Journal of controlled release : official journal of the Controlled Release Society 2010, 144, 314; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, Biomaterials 2011, 32, 3435; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Xiao K, Luo J, Li Y, Lee JS, Fung G, Lam KS, Journal of controlled release : official journal of the Controlled Release Society 2011, 155, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li Y, Xiao K, Luo J, Xiao W, Lee JS, Gonik AM, Kato J, Dong TA, Lam KS, Biomaterials 2011, 32, 6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xiao K, Li YP, Wang C, Ahmad S, Vu M, Kuma K, Cheng YQ, Lam KS, Biomaterials 2015, 67, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Xiao K, Lin TY, Lam KS, Li Y, Nanoscale 2017, 9, 7765; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiao K, Liu Q, Al Awwad N, Zhang H, Lai L, Luo Y, Lee JS, Li Y, Lam KS, Nanoscale 2018, 10, 8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li Y, Xiao W, Xiao K, Berti L, Luo J, Tseng HP, Fung G, Lam KS, Angewandte Chemie 2012, 51, 2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Xiao K, Li Y, Lee JS, Gonik AM, Dong T, Fung G, Sanchez E, Xing L, Cheng HR, Luo J, Lam KS, Cancer research 2012, 72, 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Peiris PM, He F, Covarrubias G, Raghunathan S, Turan O, Lorkowski M, Gnanasambandam B, Wu C, Schiemann WP, Karathanasis E, Nanoscale 2018, 10, 6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Moriya T, Suzuki T, Pilichowska M, Ariga N, Kimura N, Ouchi N, Nagura H, Sasano H, Pathology international 2001, 51, 333. [DOI] [PubMed] [Google Scholar]
- [17].Li X, Taratula O, Taratula O, Schumann C, Minko T, Mini reviews in medicinal chemistry 2017, 17, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tieva A, Stattin P, Wikstrom P, Bergh A, Damber JE, The Prostate 2001, 47, 276. [DOI] [PubMed] [Google Scholar]
- [19].Seitz S, Buchholz S, Schally AV, Weber F, Klinkhammer-Schalke M, Inwald EC, Perez R, Rick FG, Szalontay L, Hohla F, Segerer S, Kwok CW, Ortmann O, Engel JB, BMC cancer 2014, 14, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Westphalen S, Kotulla G, Kaiser F, Krauss W, Werning G, Elsasser HP, Nagy A, Schulz KD, Grundker C, Schally AV, Emons G, International journal of oncology 2000, 17, 1063; [DOI] [PubMed] [Google Scholar]; b) Leuschner C, Kumar CS, Hansel W, Soboyejo W, Zhou J, Hormes J, Breast cancer research and treatment 2006, 99, 163; [DOI] [PubMed] [Google Scholar]; c) Li M, Tang Z, Zhang Y, Lv S, Li Q, Chen X, Acta biomaterialia 2015, 18, 132. [DOI] [PubMed] [Google Scholar]
- [21].Dharap SS, Wang Y, Chandna P, Khandare JJ, Qiu B, Gunaseelan S, Sinko PJ, Stein S, Farmanfarmaian A, Minko T, Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS, Nat Chem Biol 2006, 2, 381. [DOI] [PubMed] [Google Scholar]
- [23].a) L. H.-a. i. E. B. C. O. group, Cuzick J, Ambroisine L, Davidson N, Jakesz R, Kaufmann M, Regan M, Sainsbury R, Lancet 2007, 369, 1711; [DOI] [PubMed] [Google Scholar]; b) Klijn JG, Blamey RW, Boccardo F, Tominaga T, Duchateau L, Sylvester R, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2001, 19, 343. [DOI] [PubMed] [Google Scholar]
- [24].a) Grundker C, Volker P, Griesinger F, Ramaswamy A, Nagy A, Schally AV, Emons G, American journal of obstetrics and gynecology 2002, 187, 528; [DOI] [PubMed] [Google Scholar]; b) Yu SS, Athreya K, Liu SV, Schally AV, Tsao-Wei D, Groshen S, Quinn DI, Dorff TB, Xiong S, Engel J, Pinski J, Clinical genitourinary cancer 2017, 15, 742. [DOI] [PubMed] [Google Scholar]
- [25].Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC, Journal of controlled release : official journal of the Controlled Release Society 2014, 190, 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Danhier F, Feron O, Preat V, Journal of controlled release : official journal of the Controlled Release Society 2010, 148, 135. [DOI] [PubMed] [Google Scholar]
- [27].Sadauskas E, Wallin H, Stoltenberg M, Vogel U, Doering P, Larsen A, Danscher G, Particle and fibre toxicology 2007, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].von Maltzahn G, Ren Y, Park JH, Min DH, Kotamraju VR, Jayakumar J, Fogal V, Sailor MJ, Ruoslahti E, Bhatia SN, Bioconjug Chem 2008, 19, 1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nielsen CH, Kimura RH, Withofs N, Tran PT, Miao Z, Cochran JR, Cheng Z, Felsher D, Kjaer A, Willmann JK, Gambhir SS, Cancer research 2010, 70, 9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mahmoudi GA, Astaraki P, Mohtashami AZ, Ahadi M, International medical case reports journal 2015, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Salamon S, Kramar B, Marolt TP, Poljsak B, Milisav I, Antioxidants 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rowinsky EK, Chaudhry V, Cornblath DR, Donehower RC, Journal of the National Cancer Institute. Monographs 1993, 107. [PubMed] [Google Scholar]
- [33].Lu J, Shi M, Shoichet MS, Bioconjug Chem 2009, 20, 87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.