

Abstract
The paucity of genetically informed, immune-competent tumor models impedes evaluation of conventional, targeted, and immune therapies. By engineering mouse fallopian tube epithelial organoids using lentiviral gene transduction and/or CRISPR/Cas9 mutagenesis, we generated multiple high grade serous tubo-ovarian carcinoma (HGSC) models exhibiting mutational combinations seen in HGSC patients. Detailed analysis of homologous recombination (HR)-proficient (Tp53−/−;Ccne1OE;Akt2OE ;KrasOE), HR-deficient (Tp53−/−;Brca1−/−;MycOE), and unclassified (Tp53−/−;Pten−/−;Nf1−/−) organoids revealed differences in in vitro properties (proliferation, differentiation, “secretome”), copy number aberrations, and tumorigenicity. Tumorigenic organoids had variable sensitivity to HGSC chemotherapeutics, evoked distinct immune microenvironments that could be modulated by neutralizing organoid-produced chemokines/cytokines. These findings enabled development of a chemotherapy/immunotherapy regimen that yielded durable, T-cell dependent responses in Tp53−/−;Ccne1OE;Akt2OE;Kras HGSC; by contrast, Tp53−/−;Pten−/−;Nf1−/− tumors failed to respond. Mouse and human HGSC models showed genotype-dependent similarities in chemosensitivity, secretome, and immune microenvironment. Genotype-informed, syngeneic organoid models could provide a platform for the rapid evaluation of tumor biology and therapeutics.
Keywords: Fallopian tube, organoids, HGSC, immune microenvironment, CCNE1
INTRODUCTION
The past 30 years of cancer research have yielded remarkable therapeutic advances along two main fronts (1,2). “Targeted therapies” were developed against oncogenic “driver” tyrosine and serine/threonine kinases or key downstream signaling components (3). Concomitantly, powerful “immune therapies” emerged, including “immune checkpoint inhibitors” (e.g., anti-CTLA4, anti-PD1, anti-PDL1) (4). These new modalities complement or replace conventional chemo-radiation therapy and are lifesaving for some patients. Nevertheless, most patients with metastatic solid tumors still succumb to their disease.
Targeted and immune therapies developed in parallel, usually using distinct experimental systems. Even today, targeted agents are typically tested against cancer cell lines/cell-derived xenografts (CDXs), patient-derived xenografts (PDXs), or more recently, human tumor spheroids/organoids. Such models are of human origin, have relevant mutational/epigenetic events, and sometimes retain a degree of tumor heterogeneity, but they do not allow evaluation of anti-tumor immune responses. PDXs can be established in “humanized” mice, but ~30% of human/mouse growth factors, cytokines, and chemokines fail to interact with the cognate receptor(s) in the other species, imposing intrinsic limits on “humanization” (5). Immune therapies, by contrast, are usually tested against syngeneic mouse tumors (6). These models (e.g., B16, CT26, MC38) are mainly carcinogen-induced, arise from unknown, irrelevant, or not the most relevant cell-of-origin, and often lack key causal mutations found in the cognate human disease. Some targeted agents/immune therapies have been evaluated in genetically engineered mouse models (GEMMs), which harbor disease-relevant genetic abnormalities and have intact immune systems (7). For most malignancies, however, only a few mutational combinations are generated, limiting the diversity of the human disease that can be analyzed. Most GEMMs introduce cancer-associated defects simultaneously into all epithelia in a target tissue; by contrast, real-world tumors initiate clonally and expand and progress in a sea of predominantly normal cells. Transplantable GEMM-derived models (e.g., Yum/Yummer melanoma cells (8)) have been generated, but they have the same truncal mutations and limited genetic diversity.
The tumor genotype, in the context of the cell-of-origin, determines its susceptibility to conventional and targeted therapies, intrinsic immunogenicity (e.g., by neoantigens, altered surface expression of MHC class I molecules and/or ligands for activating/inhibitory receptors on immune cells), and the spectrum of cytokines and chemokines (“secretome”) produced (9–11). Secretome components, in turn, recruit immune cells to the tumor microenvironment (TME). Save for mutation-selective agents (e.g., RASG12C inhibitors, osimertinib for mutant EGFR), targeted and conventional agents affect cells in the TME in addition to tumor cells (12,13). A suite of immune-competent mouse models bearing tumors with genetic defects seen in patient neoplasms might simulate tumor biology with greater fidelity and facilitate development of novel therapeutic agents or combinations of existing drugs.
We developed such a platform for the most common and deadly form of ovarian epithelial cancer, high grade serous tubo-ovarian cancer (HGSC) (14). HGSC patients usually present at an advanced stage with bulky metastatic spread throughout the peritoneum, although some have more discrete tumor deposits. Current treatment includes surgical “debulking” and platinum/taxane-based chemotherapy and often results in complete responses (CRs). Unfortunately, disease almost always recurs, eventually in drug-resistant form. Despite the recent addition of anti-angiogenics (Avastin) and PARP inhibitors (PARP-Is) to the HGSC armamentarium, survival has improved only marginally in the past 30 years, and most (70–90%) patients die from their disease (15). Clearly, better therapies are needed for this deadly malignancy.
Much is known about HGSC pathogenesis. Despite its appellation, HGSC most often initiates with mutation, deletion, or silencing of TP53 in fallopian tube epithelium (FTE), not the ovary. The Cancer Genome Atlas (TCGA) reveals additional pathogenic single nucleotide variants (SNVs), but HGSC is primarily a disease of copy number abnormalities (CNAs), including amplifications, deletions, and more complex chromosomal rearrangements, which affect multiple genes and pathways (16). The most clinically useful molecular classification groups HGSCs by homologous recombination (HR) status. Defects in known HR genes, including BRCA1, BRCA2, RAD51, or other Fanconi Anemia genes, occur in ~40% of cases; another ~15–20% have PTEN loss or EMSY amplification and are probably HR-deficient (17). Defective HR confers sensitivity to platinum agents (the mainstay of HGSC therapy), and some (but not all) of these defects confer PARP-I responsiveness (18,19). The remaining ~40% of tumors are HR-proficient, respond poorly to current therapy, and result in shorter survival (20). CCNE1 amplification, found in ~20% of HGSC, is notorious for causing chemo-resistance and poor outcome (21); hence, there is a particular need to develop new therapeutic strategies for these tumors. Despite this impressive progress in delineating the molecular anatomy of HGSC, how specific combinations of mutations determine the transformed phenotype, including the tumor transcriptome, secretome, anti-tumor immunity, and therapy response, remains poorly understood.
The paucity of genetically relevant, immune-competent models of HGSC poses a major barrier to addressing such issues. Many studies have used cancer cell lines, most of which (including the most frequently used) lack the characteristic genomic abnormalities of HGSC (22). Human HGSC organoids have been derived (23,24), but while organoids have been co-cultured with immune cells (25–27), such systems cannot fully simulate the anti-tumor response. ID8 cells have been the primary model for studying the host immune response to HGSC, but these cells originate from ovarian surface epithelium (OSE) and have wild type (WT) Tp53 (16,28). GEMMs that use FTE-selective promoter/enhancers to direct mutational events have been developed (29,30), but these involve artificial alterations (e.g., SV40 large T antigen expression) or realtively rare mutational combinations (e.g., Brca1/Pten/Tp53). Also, most are on mixed strain backgrounds, which impedes some tumor immunology studies. Notably, no immune competent models for CCNE1-amplified HGSC have been reported.
Exploiting our mouse FTE organoid culture system (31), combined with viral-based overexpression and CRISPR/Cas9 mutagenesis, we developed multiple new syngeneic models of HGSC. We demonstrate the utility of this platform for uncovering cellular genotype/phenotype relationships, complementation groups for tumorigenicity, the effect of tumor genotype on drug sensitivity, secretome, CNAs, tumor immune landscape, and metastatic spread and, most importantly, the rational development of a highly effective therapeutic combination for Ccne1-overexpressing HGSC using existing combinations of FDA-approved drugs.
RESULTS
FTE organoid-based platform for HGSC
Most HGSCs initiate from the distal fallopian tube (fimbria), which mainly comprises secretory (PAX8+) and ciliated (acetyl-α-tubulin+) epithelia (32,33). The initial event (except in germ line carriers of mutations in BRCA1/2 or other predisposing genes) is mutation of TP53 in a PAX8+ cell, which, together with other defects, evokes the precursor lesion serous tubal intraepithelial carcinoma (STIC). Additional SNVs/CNAs confer invasive potential and promote metastasis to the ovarian surface, peritoneum, and distal organs (34,35).
We used mouse FTE organoids (31) to model this complex biology. Briefly, fimbrial cells from Tp53f/f (or, where indicated, Brca1f/f:Tp53f/f mice) were seeded in Matrigel and cultured in defined media. Cyst-like organoids formed from single PAX8+ cells, a mixture of secretory and ciliated cells was seen after 6 days of culture, and tube-like epithelial folds developed by 10 days (Ref. 29, Supplementary Fig. S1). After expansion, floxed alleles were excised by infection with adenovirus-Cre (Ad-Cre), yielding parental Tp53−/− organoids or, where indicated, compound mutants (all in C57BL6/J background). Additional genetic changes were introduced by lenti- or retroviral gene transduction to model over-expression and/or CRISPR/Cas9 mutagenesis to model deletions or mutations (Supplementary Fig. S2). Models were tested in cellular assays or transferred to 2D cultures for larger scale studies. Tumorigenesis was assessed by orthotopic injection into the ovarian bursa (for details, see Methods). Our current collection of models is summarized in Supplementary Table S1. To evaluate the utility of this platform for simulating HGSC pathogenesis and therapeutics, we performed detailed studies on representative examples of HR-proficient, HR-deficient, and unclassified subgroups.
Tp53−/−; Brca1−/−;MycOE FTE organoids give rise to HGSC-like tumors
BRCA1/2 alterations are found in ~20% of HGSC (36), so we chose Tp53−/−/Brca1−/− models to represent the HR-deficient subgroup (Figs.1A and B; Supplementary Table S1). We infected Tp53 f/f;Brca1 f/f FTE with Ad-Cre, picked single organoids, and confirmed deletion of the relevant loci (Fig.1B and Supplementary Fig. S2A). Neither Tp53 nor Brca1 deletion alone or in combination altered organoid morphology or ciliated cell differentiation (Fig. 1C and Supplementary Fig. S2B and C), although Tp53−/−;Brca1−/− organoids were significantly larger than their parental counterparts, most likely due to enhanced proliferation (assessed by Ki67 staining). MYC is amplified in ~40% of HGSC and often co-occurs with Brca1 alterations (Fig. 1A). Over-expression of Myc in Tp53−/−/Brca1−/− organoids further increased proliferation and organoid size, while impeding ciliary differentiation (Figs. 1B and C and Supplementary Fig. S2A and B). Orthotopic injection of Tp53−/− or Tp53−/−/Brca1−/− FTE cells (2 × 106) did not result in tumors within the 6-month observation period. By contrast, Tp53−/−;Brca1−/−;MycOE organoid cells evoked ovarian masses and omental metastases, resulting in death of all injected mice within 4 months. These tumors expressed HSGC markers, including PAX8, Cytokeratin 7 (CK7), P16, and Wilms Tumor 1 (WT1), and were strongly Ki67+ (Figs. 1D–F). Hence, whereas compound BRCA1/TP53 deficiency is insufficient to cause HGSC, superimposing high MYC levels (or PTEN and/or NF1 deficiency; Supplementary Table S1) results in a highly invasive, metastatic, lethal malignancy.
Figure 1.
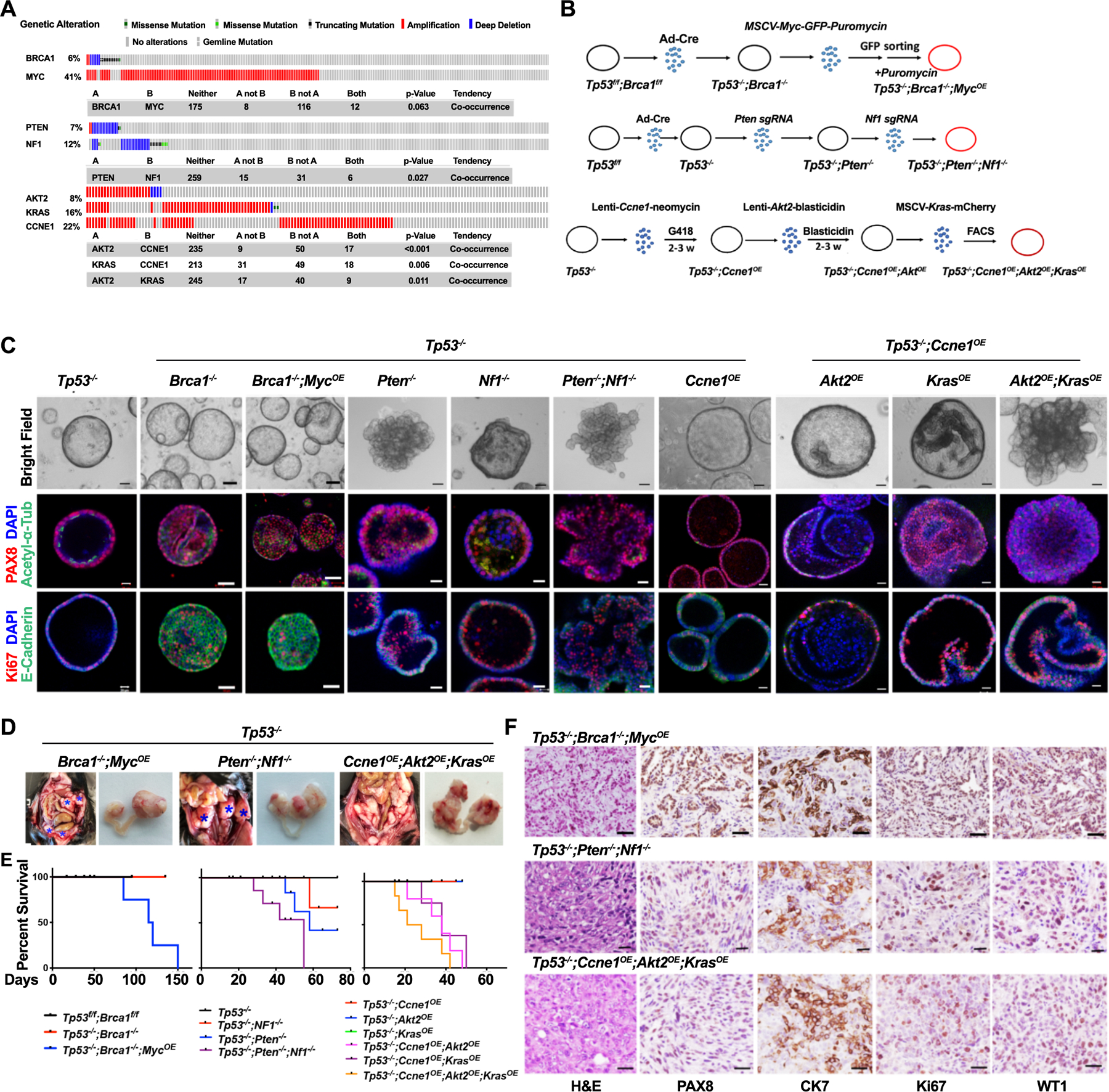
Generation of tumorigenic organoids. A, OncoPrint showing selected genetic alterations and co-occurrence of the indicated abnormalities in human HGSC (TCGA, Firehose Legacy). B, Schematic showing approach for generating tumorigenic organoids from parental Tp53f/f;Brca1f/f or Tp53f/f FTE organoids. C, Representative bright field images and immunofluorescence staining of organoids after 7 days in culture. Scale bars: 20 μm. D, Exposed abdomens (right panels) and dissected genital tracts (right panels) of mice bearing organoid-derived tumors of the indicated genotypes; asterisks indicate large metastatic deposits. E, K–M curves of mice following orthotopic injection of 2X106 organoid cells of the indicated genotypes, n=6/group. F, H&E-stained sections and immunohistochemical analysis for the HGSC markers PAX8, CK7 (Cytokeratin 7), Ki67, and WT1 (Wilms’ Tumor 1) in representative sections from the indicated ovarian tumors. Scale bars: 50 μm. See also Supplementary Figs. S1 and S2.
Tp53−/−;Pten−/−;Nf1−/− FTE organoids also cause HGSC-like tumors
NF1 deficiency, due to NF1 mutation/deletion, is seen in ~12% of HGSC (37). PTEN loss also occurs fairly frequently (~7%) and is associated with poor prognosis (38) (Fig. 1A). We therefore assessed the effects of PTEN, NF1, or compound PTEN/NF1 deficiency on Tp53−/− FTE. Using lentiviral transduction, an sgRNA targeting Pten exon 2 was introduced into Tp53−/− organoids, three clones with bi-allelic deletion were identified and expanded, and PTEN deficiency was confirmed by immunoblotting. An analogous strategy was used to target Nf1 exon2 in Tp53−/− or Tp53−/−;Pten−/− organoids (Fig. 1B and Supplementary Fig. 2D). As expected, PTEN deficiency increased AKT (pAKT) and mTOR (pS6 and pS6K) activation, while loss of NF1 led to increased pERK1/2 (Supplementary Fig. 2D). Pten−/− organoids showed increased proliferation and organoid size, filled lumens, and decreased ciliary differentiation. Nf1 deletion decreased ciliary differentiation and altered organoid shape, but proliferation and luminal integrity were unaffected. Triple-deleted (Tp53−/−;Pten−/−;Nf1−/−) and Tp53−/−;Pten−/− organoids behaved similarly in these assays (Fig. 1C and Supplementary Fig. 2E).
We also tested the tumorigenicity of Tp53−/−;Pten−/−, Tp53−/−;Nf1-/−., and Tp53−/−;Pten−/−;Nf1−/− organoid cells (at least 2 clones each). Some double mutant-injected mice (8/20 Tp53−/−;Pten−/−, 8/24 Tp53−/−;Nf1−/−) developed tumors within 6 months, but Tp53−/−;Pten−/−;Nf1−/− cells showed more rapid and penetrant (28/30) tumorigenesis and also caused tumors more rapidly than did Tp53−/−;Brca1−/−;MycOE cells (Figs. 1D and E and Supplementary Table S1). Tp53−/−;Pten−/−;Nf1−/− tumors metastasized to the omentum and produced more ascites than did Tp53−/−;Pten−/−, Tp53−/−;Nf1−/−, Tp53−/−;Brca1−/−;MycOE, or Ccne1OE tumors (Fig. 1D). Tp53−/−;Pten−/−;Nf1−/− tumor-bearing mice expressed HGSC markers and had shorter life spans than double knockouts (Figs. 1E and F).
AKT2 and/or KRAS cooperate with CCNE1 to cause HGSC
Amplification or gain of CCNE1, encoding the cell-cycle regulator cyclin E1, is the best characterized driver of HR-proficient HGSC and accounts for ~20% of cases (17,20). AKT2 and KRAS amplification occur in 8% and 16%, respectively, of HGSC, and co-occur with CCNE1 amplification (Fig. 1A). To model CCNE1 amplification (CCNE1amp) alone or with KRASamp and/or AKT2amp, Ccne1, Akt2, and/or Kras were over-expressed (OE) sequentially in Tp53−/− FTE using lentiviruses harboring different selection markers (Fig. 1B). Over-expression/increased activation of each protein was confirmed by immunoblotting (Supplementary Fig. S2F). Organoid diameter/morphology were not affected significantly by CCNE1, AKT2, or KRAS over-expression alone (compared with parental Tp53−/− organoids). However, CCNE1, but not AKT2 or KRAS, overexpression significantly increased proliferation (Supplementary Fig. S2G). This increase was probably offset by a comparable increase in cell death, accounting for the lack of alteration of organoid size; notably, CCND1 over-expression has analogous effects on MCF10A mammary organoids (39). Superimposing Akt2OE or KrasOE on Tp53−/−;Ccne1OE organoids further enhanced proliferation, increased organoid diameter, lumen filling, and organoid disorganization, which was even more pronounced in quadruple mutants. Ccne1OE alone did not alter ciliary differentiation, but ciliated cells were virtually undetectable in triple and quadruple mutant organoids (Fig. 1C; Supplementary Fig. S2G). Tp53−/−;Ccne1OE cells did not give rise to tumors by 6 months after orthotopic injection. By contrast, Tp53−/−;Ccne1OE;Akt2OE, Tp53−/−;Ccne1OE;KrasOE and Tp53−/−;Ccne1OE;Akt2OE;KrasOE cells formed large, palpable ovarian tumor masses, massive omental metastasis and death within 2 months of injection (Figs. 1D and E). There was no apparent difference in tumor formation by each triple mutant, but quadruple mutants showed accelerated tumorigenesis and displayed histologic and immunohistochemical features of high grade, poorly differentiated, invasive carcinoma (Figs. 1E and F).
Hence several combinations of genetic abnormalities seen in human HGSC give rise to lethal ovarian cancers in immune-competent mice and can be used to assign complementation groups for in vitro properties (proliferation, differentiation, organoid morphology) and tumorigenic capacity. Other combinations of genetic abnormalities reported by TCGA also give rise to HGSC in mice (Supplementary Table S1).
Organoid genotype affects genome stability, drug sensitivity and secretome
HGSC is characterized by widespread CNAs/aneuploidy, which have been assigned to seven “copy number signatures” associated with distinct mutational processes and driver abnormalities (40). We used shallow (2X) whole genome sequencing (sWGS) to assess the CN status of our models. WT and Tp53−/− organoids (2 clones each) showed normal diploid profiles, whereas two independent Tp53−/−;Brca1−/− organoid clones exhibited gains of mouse chromosome 5 (Supplementary Fig. S3A). Tp53−/−;Brca1−/−;MycOE organoids (2 independent clones) showed additional, but distinct CNAs. Tp53−/−;Ccne1OE;Akt2OE;KrasOE (2 independent clones) and Tp53−/−;Pten−/−;Nf1−/− organoids (one clone assessed at different times) also had multiple CNAs. Notably, the two Tp53−/−;Ccne1OE;Akt2OE;KrasOE clones had some shared and other distinct CNAs, whereas a Tp53−/−;Pten−/−;Nf1−/− organoid clone assessed at different times had a stable, although markedly aneuploid genome (Fig. 2A).
Figure 2.
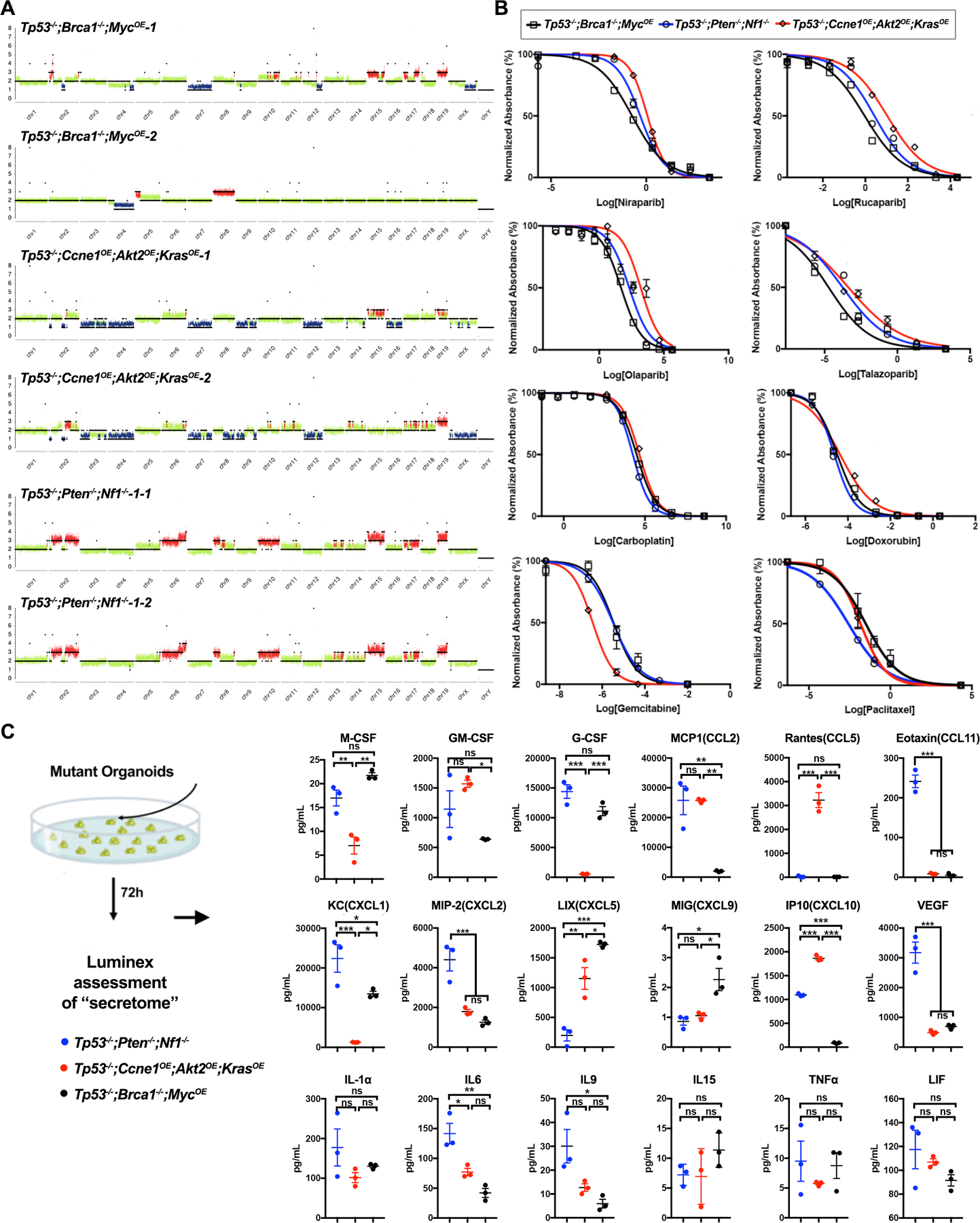
Genotype affects organoid copy number alterations, drug sensitivity, and secretome. A, Shallow WGS (sWGS) of the indicated tumorigenic organoids. Two independent clones are shown for Tp53−/−;Brca1−/−;MycOE and Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoids, respectively, the same Tp53−/−;Pten−/−;Nf1−/− organoid clone at two different times is shown. Copy number losses and gains are shown in blue and red, respectively. B, Dose-response curves for the indicated drugs in tumorigenic organoid lines of different genotypes. Cell viability was calculated relative to 0.01% DMSO-treated control cells, measured after 5 days of treatment. C, Levels of the indicated cytokines, chemokines, and growth factors from 72 hr-conditioned media from the indicated tumorigenic organoids. Only secreted factors that are detectable and differ between groups are shown. Error bars indicate ± SEM; **P<0.01, ***P<0.001, 2-way ANOVA. See also Supplementary Fig. S3.
Next, we tested these models for sensitivity to FDA-approved drugs and investigational/experimental agents for HGSC (Fig. 2B; Supplementary Fig. S3B). Organoids were titurated into small clumps, dissociated into single cells, and dispensed into 96-well Matrigel pre-coated plates (see Methods). Each agent was added at various doses, and cell viability was assessed 5 days later. As expected, Tp53−/−;Brca1−/−;MycOE cells showed increased sensitivity to PARP-Is (Fig. 2B), although differential sensitivity varied for individual PARP-Is and was less than seen in conventional ovarian cancer cell lines (41). Brca1-deleted cells showed slightly increased sensitivity to carboplatin, although there was substantial overlap with the other mutants. Comparison of Tp53−/−;Brca1−/− and Tp53−/−;Brca1−/−;MycOE organoids showed that MYC over-expression reduces PARP-I and/or platinum sensitivity (Supplementary Fig. S3C). Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoids were more sensitive to gemcitabine than the other models, consistent with the increased replication stress caused by CCNE1 overexpression (23). By contrast, and unexpectedly, Tp53−/−;Pten−/−;Nf1−/− cells showed enhanced susceptibility to paclitaxel, and comparisons Tp53−/−;Pten−/− and Tp53−/−;Nf1−/− cells attributed this difference to NF1 deficiency (Fig. 2B; Supplementary Fig. S3D). Tp53−/−;Brca1−/−;MycOE and Tp53−/−;Pten−/−;Nf1−/− organoids had increased sensitivity to the ATR inhibitor BAY1895344, whereas chloroquine, which inhibits endosomal acidification and is often used as an autophagy inhibitor, was differentially toxic for all genotypes (Tp53−/−;Pten−/−;Nf1−/−>Tp53−/−;Ccne1OE;Akt2OE;Kras>Tp53−/−;Brca1−/−;MycOE). All genotypes showed comparable sensitivity to the CDK7 inhibitor YKI-5–1241, the CDK7/9 inhibitor PHA767491, and the CDK2/7/9 inhibitor Seliciclib (Supplementary Fig. S3B).
We also used Luminex technology to assay organoid-conditioned media (Fig. 2C). Notably, engineered organoids secreted a complex mixture of chemokines, cytokines, and growth factors, and their secretome was genotype-dependent. As these factors could help initiate immune cell immigration and/or survival, these results raised the possibility that, as demonstrated below, tumor genotype instructs the TME.
Ovarian tumors with different genotypes have distinct transcriptomes
We used RNA sequencing to analyze the transcriptomes of tumors (4 each) of each genotype and normal FT. Unsupervised hierarchical clustering showed clear separation between tumor and normal samples and between each model, with Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors showing the greatest difference from the others (Fig. 3A). Pathway analysis revealed that, compared with normal FT, tumor transcriptomes were enriched primarily for KEGG gene sets associated with the immune response (e.g., cytokine/cytokine receptor interaction, chemokine signaling pathway, antigen processing and presentation, Leishmania infection, Toll like receptor signaling pathway, etc.), and, to a lesser extent, for processes related to proliferation (e.g., DNA replication, cell cycle, ribosome, etc.). Hallmark Gene Sets associated with inflammatory/immune (allograft rejection, TNFα signaling, interferon gamma response, interferon alpha response, complement signaling, etc.) and proliferative (G2/M checkpoint, MYC targets, KRAS signaling, mTORC signaling, etc.) processes and multiple Oncogenic Gene Sets also were enriched (Figs. 3B and C). Pairwise comparisons revealed significant differences between tumors, comporting with their distinct genotypes. For example, compared with Tp53−/−;Ccne1OE;Akt2OE;KrasOE models, Tp53−/−;Pten−/−;Nf1−/− tumors showed lower expression of “PTEN-down” and of “MEK-up,” “KRAS-up” and “EGFR-up” genes; these findings likely reflect stronger RAS/ERK pathway activation in KRAS over-expressing, compared with NF1-deficient, cells. By contrast, Tp53−/−;Pten−/−;Nf1−/− tumors showed enrichment for “KRAS-up” and AKT-up gene sets compared with their Tp53−/−;Brca1−/−;MycOE counterparts (Fig. 3C)
Figure 3.
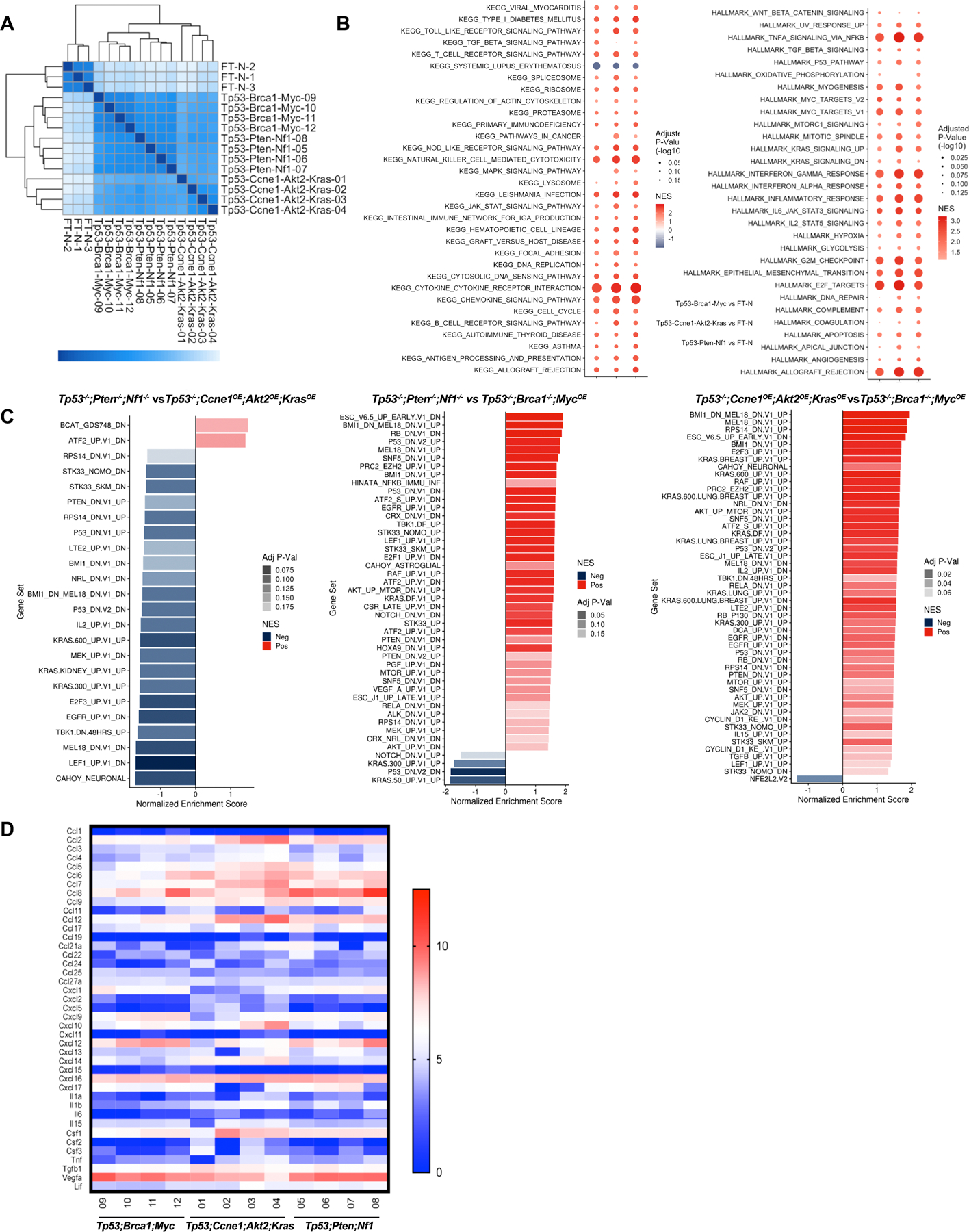
Tumors derived from organoids with different genotypes have distinct transcriptomes. A,Heat map showing sample distances by hierarchical clustering, based on variance stabilized expression levels of all genes in normal FT, Tp53−/−;Ccne1OE;Akt2OE;KrasOE, Tp53−/−;Pten−/−;Nf1−/− and Tp53−/−;Brca1−/−;MycOE tumors, respectively. Shading represents Euclidian distance for each sample pair. B, Enriched KEGG (left) and MSigDB Hallmark genes (right), ranked by fold-change between the indicated groups. Shading represents the FDR-adjusted p value within each category; color indicates direction of enrichment relative to the first group of the comparison. C, Pathway analysis comparing the indicated groups. color indicates direction of enrichment relative to the first group of the comparison. D, Heat map showing transcripts (log-transformed TPMs) of the indicated chemokines/cytokines/growth factors in representative tumors from each genotype. See also Supplementary Fig. S3.
We also examined chemokine, cytokine, and hematopoietic growth factor gene expression in each type of tumor (Fig. 3D). Most interleukins were expressed at low/undetectable levels in all models, as were many chemokines, whereas IL15, IL16, IL18, IL33, and IL34 were expressed significantly in all tumors (as in their cognate organoids; Fig. 2C). Lif, IL1b, Csf1 (MCSF) and to a lesser extent, Tnfα, were expressed at higher levels in Tp53−/−;Pten−/−;Nf1−/− and Tp53−/−;Ccne1OE;Akt2OE, compared with Tp53−/−;Brca1−/−;MycOE, tumors. Some chemokines (e.g., Cxcl12, Cxcl16) were expressed at similar, high levels in all models. Others showed genotype-specific differences: e.g., Ccl2 and Ccl5 were expressed most highly in Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors, at intermediate levels in Tp53−/−;Pten−/−;Nf1−/− tumors, and at lower levels in Tp53−/−;Brca1−/−;MycOE tumors. Cxcl1 levels were higher in Tp53−/−;Brca1−/−;MycOE and Tp53−/−;Pten−/−;Nf1−/− tumors. Ccl6–9 were expressed in most models, although at generally lower levels in Tp53−/−;Brca1−/−;MycOE tumors. By contrast, Cxcl9 was expressed at highest levels in Tp53−/−;Brca1−/−;MycOE tumors and at lowest levels in Tp53−/−;Ccne1OE; Akt2OE, KrasOE tumors, whereas Cxcl10 was expressed at higher levels in the latter. Vegfa and Tgfb1 transcripts were high in all of the models.
Comparing the organoid secretome with the tumor transcriptome suggested cytokines/chemokines that initiate and help to maintain the TME (e.g., CCL2, CCL5, CXCL10 for Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors; MCSF, CXCL1, and CXCL9 for Tp53−/−;Brca1−/−;MycOE tumors; MCSF, CXCL1, CCL2, and VEGF-A for Tp53−/−;Pten−/−;Nf1−/− tumors). CCL2, CCL5, and CXCL10 were also detected at high levels in the serum of tumor-bearing Tp53−/−;Ccne1OE;Akt2OE;KrasOE mice (213.6 ±56.39 pg/mL). Other factors might contribute to TME initiation but are no longer expressed at high levels in tumors themselves (e.g., G-CSF/Csf2 in Tp53−/−;Brca1−/−;MycOE and Tp53−/−;Pten−/−;Nf1−/− tumors). Some presumably emanate from primarily from tumor-infiltrating immune cells, rather than cancer cells themselves (e.g., GMSCF/Csf3 and CXCL5 in Tp53−/−;Brca1−/−;MycOE). We tested some of these predictions by perturbation experiments, as described below.
The HGSC microenvironment depends on tumor genotype
Given their markedly different secretomes, we suspected that organoids of different genotype might elicit distinct TMEs. To test this hypothesis, we assayed Tp53−/−;Brca1−/−;MycOE, Tp53−/−;Pten−/−;Nf1−/−, and Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors by flow cytometry using lymphoid and myeloid marker panels (Supplementary Figs. S4A and B). Levels of CD45+ immune cells (compared with CD45− tumor/stromal cells) were ~2-fold higher in Tp53−/−;Ccne1OE;Akt2OE;KrasOE and Tp53−/−;Brca1−/−;MycOE tumors than in Tp53−/−;Pten−/−;Nf1−/− tumors (Supplementary Fig. S4C). None of the models had many tumor-associated B (CD19+), NK (NK1.1+), or NKT (NK1.1+CD3+) cells (Fig. 4A; Supplementary Fig. S4C).
Figure 4.
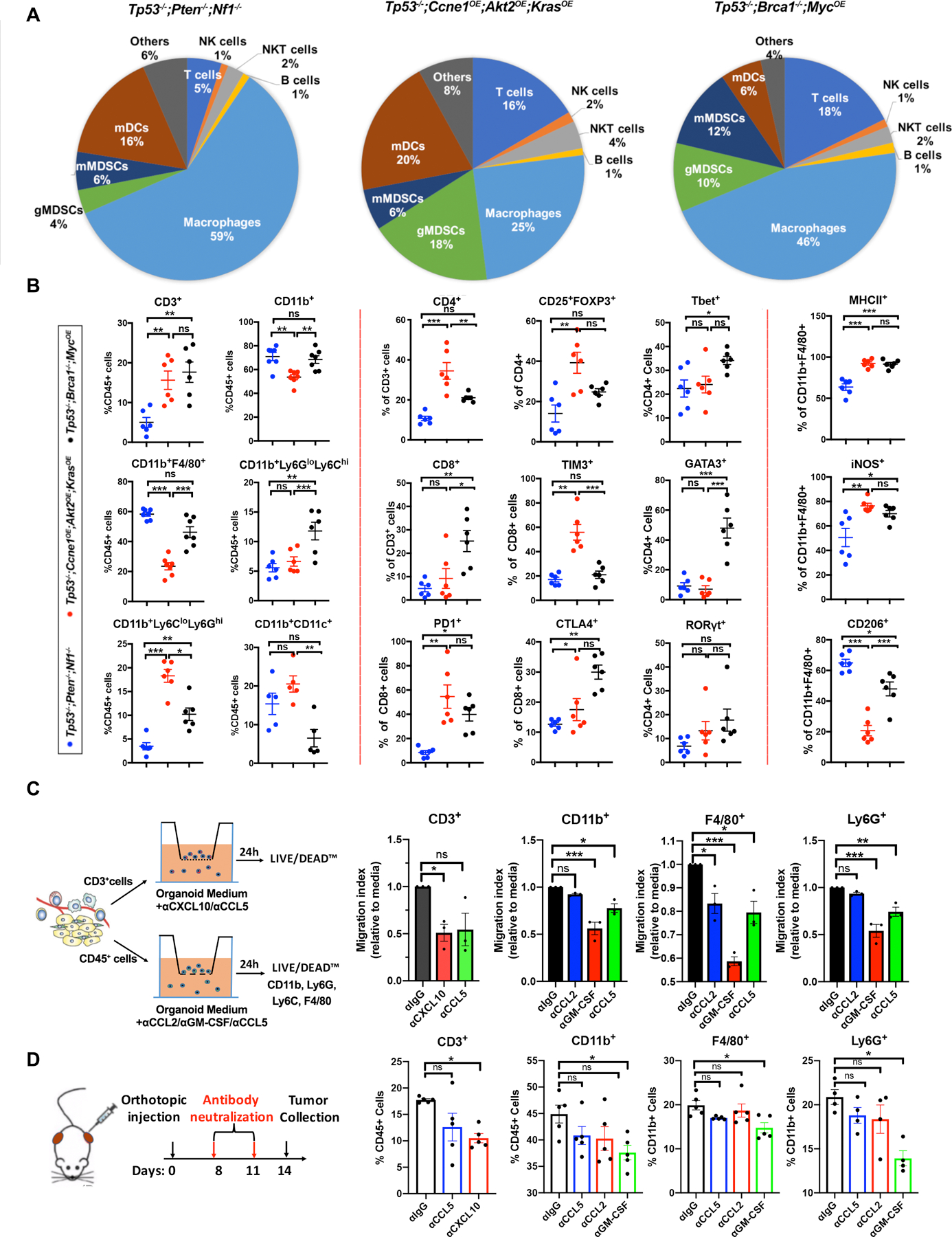
Tumor genotype determines immune landscape. A, Pie charts summarizing composition of immune cells (CD45+) in tumors with the indicated genotypes. Note that CD45+ cells (as % of total tumor cells) were significantly less in Tp53−/−;Pten−/−;Nf1−/− tumors, but similar in the other two genotypes (see Supplementary Fig. S4). B, Immune cell subtyping by flow cytometric analysis of representative tumors of the indicated genotypes. Each point represents a tumor from a different mouse. Data are presented as mean ± SEM, **P<0.01, ***P<0.001, 2-way ANOVA. C, Left panel: Diagram showing strategies for analyzing function of selected chemokines/cytokines in Tp53−/−;Ccne1OE;Akt2OE;KrasOE HGSC. Right panels: Effect of the indicated neutralizing antibodies on migration of T cells, CD11b+ cells, F4/80+ cells, and Ly6G+ cells in Transwell assays, quantified as Migration Index (migration with/without antibody) after 24 h co-culture of the indicated cell population with Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoid-conditioned medium. D, Left panel: Schematic showing in vivo antibody neutralization experiments. Right panels: Immune cell immigration into Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors, after injections with the indicated antibody. Each point represents a tumor from a different mouse. Data are presented as mean ± SEM, **P<0.01, ***P<0.001, 2-way ANOVA.
Nevertheless, the composition of the CD45+ population in tumors with different genotypes differed substantially (Figs. 4A and B). Tp53−/−;Pten−/−;Nf1−/− tumors had a predominant macrophage (CD11b+F4/80+) population, smaller numbers of myeloid dendritic cells (mDC, CD11b+CD11C+), granulocytic myeloid-derived suppressor cells (g-MDSC, CD11b+Ly6CloLy6Ghi), and monocytic myeloid-derived suppressor cells (m-MDSC, CD11b+Ly6GloLy6Chi), and sparse T lymphocytes (CD3+ cells). Given their lower fraction of total CD45+ cells (Supplementary Fig. S4C), absolute T cell number in Tp53−/−;Pten−/−;Nf1−/− tumors is even lower compared with the other models. The macrophages in Tp53−/−;Pten−/−;Nf1−/− tumors had greater “M2-like” character, with high percentages of CD11b+ F4/80+ cells expressing CD206 and a lower percentage of iNOS+ cells (Fig. 4B); most, however, co-expressed M1 and M2 markers, consistent with an “M0-like” state (42,43). Immunofluorescence (IF) staining provided direct confirmation of higher CD3+ cell infiltration into Tp53−/−;Brca1−/−;MycOE and Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors than into Tp53−/−;Pten−/−;Nf1−/− tumors, and highest levels of Ly6G+ cells in Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors (Supplementary Figs. S4D and S4E).
Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors were more inflamed, exhibiting infiltration with macrophages, mDCs, g-MDSCs, and T lymphocytes (Figs. 4A and B). Nearly half of the CD4+ T cells in these tumors were T regulatory cells (Tregs, CD25+FOXP3+), though, while most CD8+ cells showed “exhaustion” markers (TIM3+, PD1+). Tumor-associated macrophages (TAMs) expressed “M1-like” (MHCII+, iNOS+) and “M2-like” (CD206+) markers, although the former predominated.
Finally, Tp53−/−;Brca1−/−;MycOE tumors had large percentages of macrophages and lower fractions of g-MDSCs, m-MDSCs, and mDCs. Unlike in the other models, CD4+ and CD8+ T cells in Tp53−/−;Brca1−/−;MycOE tumors were predominantly (>60%) CD44+ and strongly CTLA4+ and PD1+ (Fig. 4B; Supplementary Fig. S4C), suggesting activation. Compared with cognate cells in Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors, CD8+ cells in Tp53−/−;Brca1−/−;MycOE tumors showed less TIM3-positivity, suggestive of less exhaustion, and there were fewer Treg cells. Tp53−/−;Brca1−/−;MycOE tumors had more a balanced population of Th1 (Tbet+) and Th2 (GATA3+) cells (Th1/Th2: 0.7); the other models mostly had Th1 cells (Th1/Th2: 2.4 in Tp53−/−;Pten−/−;Nf1−/−; Th1/Th2: 3.4 in Tp53−/− in Ccne1OE;Akt2OE;KrasOE). Tp53−/−;Brca1−/−;MycOE macrophages also had more M1-like character (%CD206/%iNos:1.3) than did the other models (%CD206/iNOS: 0.2 in Ccne1OE;Akt2OE;KrasOE; %CD206/iNOS: 0.6 in Tp53−/−;Pten−/−;Nf1−/−).
PD-L1 expression also was genotype-dependent. In all models, ~40–45% of m-MDSCs were PD-L1+. In Ccne1OE;Akt2OE;KrasOE tumors, 45% of g-MDSCs also expressed PD-L1, whereas expression on g-MDSCs was lower in Tp53−/−;Brca1−/−;MycOE (25%) and Tp53−/−;Pten−/−;Nf1−/− (14%) tumors. By contrast, ~60% of Tp53−/−;Pten−/−;Nf1−/− TAMs were PD-L1+. Tp53−/−;Pten−/−;Nf1−/−, Ccne1OE;Akt2OE;KrasOE, and Tp53−/−;Brca1−/−;MycOE tumors showed PD-L1 expression on 2%, 8%, and 5% expression on CD45− cells (malignant cells, respectively).
To explore whether specific organoid-produced cytokines/chemokines elicit particular features of the TME, we focused on Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors and CCL2, CCL5, CXCL10, and GM-CSF; these proteins and their cognate transcripts were detected at high levels in organoid-conditioned media and tumors, respectively. First, we evaluated their effects on immune cell migration in vitro (Fig. 4C). Bulk CD45+ cells or CD3+ cells were purified from tumors and placed in the top well of a Transwell chamber. Conditioned media from Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoids was placed in the lower chamber with or without appropriate neutralizing antibodies. Anti-CXCL10 or -CCL5 blocked T cell migration, whereas anti-GM-CSF and to a lesser extent, anti-CCL5 blocked the migration of total CD11b+ cells. GM-CSF was the prime mediator of macrophage chemotaxis, whereas CCL5 and CCL2 were contributory. GM-CSF or CCL5 blockade impaired g-MDSC migration.
We also tested the effects of neutralizing these cytokines/chemokines on TME development. Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoid cells were injected orthotopically (Day 0), followed by neutralizing antibody injections at Days 8 and 11 (Fig. 4D). Consistent with the in vitro chemotaxis assays, CXCL10 blockade resulted in fewer T cells in the TME, while GM-CSF blockade resulted in decreases in TAMs and g-MDSCs, compared with isotype control-treatment. These results argue that CXCL10 and GM-CSF are important drivers of T cell, macrophage, and g-MDSC immigration into the Tp53−/−;Ccne1OE;Akt2OE;KrasOE TME, respectively. Although nominal decreases were observed, anti-CCL2 or -CCL5 did not significantly reduce tumor-associated T cell or myeloid cells infiltration compared to isotype control-injected mice (Fig. 4D). Combination effects are not, however, excluded. Indeed, a complex mix of immune modulatory factors, acting in concert, probably sculpt the microenvironment of these tumors.
Rationally derived combination therapy yields durable complete responses in Tp53−/−;Ccne1OE;AktOE;KrasOE HGSC
We next assessed the utility of our platform for developing HGSC therapies. To enable rapid clinical translation, we focused on CCNE1-overexpressing tumors, given their limited response to current therapies and poor prognosis, and on FDA-approved drugs. Consistent with our in vitro findings, gemcitabine administration to mice with Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors reduced, but it did not eliminate disease burden (Supplementary Figs. S5A and S5B). As in other tumor models (44–46), gemcitabine also decreased g-MDSCs (CD11b+Ly6CloLY6Ghi) in the TME, but other cell populations, most notably Tregs (CD24+CD25+FOXP3+) and T cells expressing exhaustion markers (TIM3/PD1), were unchanged (Supplementary Fig. S5C).
Given these data, we designed a regimen to attack tumor cells while normalizing the TME (Fig. 5A): gemcitabine to decrease tumor cells and g-MDSCs, anti-CTLA4 antibodies to target Tregs (47), and anti-PD-L1 antibodies to reactivate exhausted CD8 cells (48,49). This combination (GCP) produced complete responses (CRs) in 10/10 treated mice (Figs. 5B–E). Treatment was stopped after Day 35 (Fig. 5A), yet tumors failed to recur over a 60-day observation period (Fig. 5D; Supplementary Fig. S5C). Gemcitabine plus anti-PD-L1 (but not anti-CTLA4) evoked a greater decrease in tumor burden and ascites than gemcitabine alone, but no CRs. Gemcitabine/anti-CTLA4 reduced ascites but did not measurably diminish tumor burden (Fig. 5C; Supplementary Figs. S5D and S5E). Upon therapy cessation, tumors recurred in all mice in the 2-drug combination groups, leading to their rapid demise (Fig. 5D). Histological analysis of GCP-treated animals after 8 cycles revealed normal fat abutting minimal amounts of residual tumor in the injected bursae; by contrast, considerable tumor remained in mice treated with gemcitabine/anti-PD-L1 or gemcitabine/anti-CTLA4 (Fig. 5E). Multi-color IF confirmed that Ly6G+ cells were decreased in mice treated with gemcitabine, alone or in combination with anti-CTLA4 and/or anti-PD-L1. Only tumors from GCP-treated mice showed significantly increased T cell (CD3+) infiltration, which included CD4+ and CD8+ T cells (Fig. 5F). These mice also showed increases in granzyme B+ (cytolytic) cells, and decreased numbers of TAMs and Tregs (Supplementary Figs. S5F and S5G).
Figure 5.
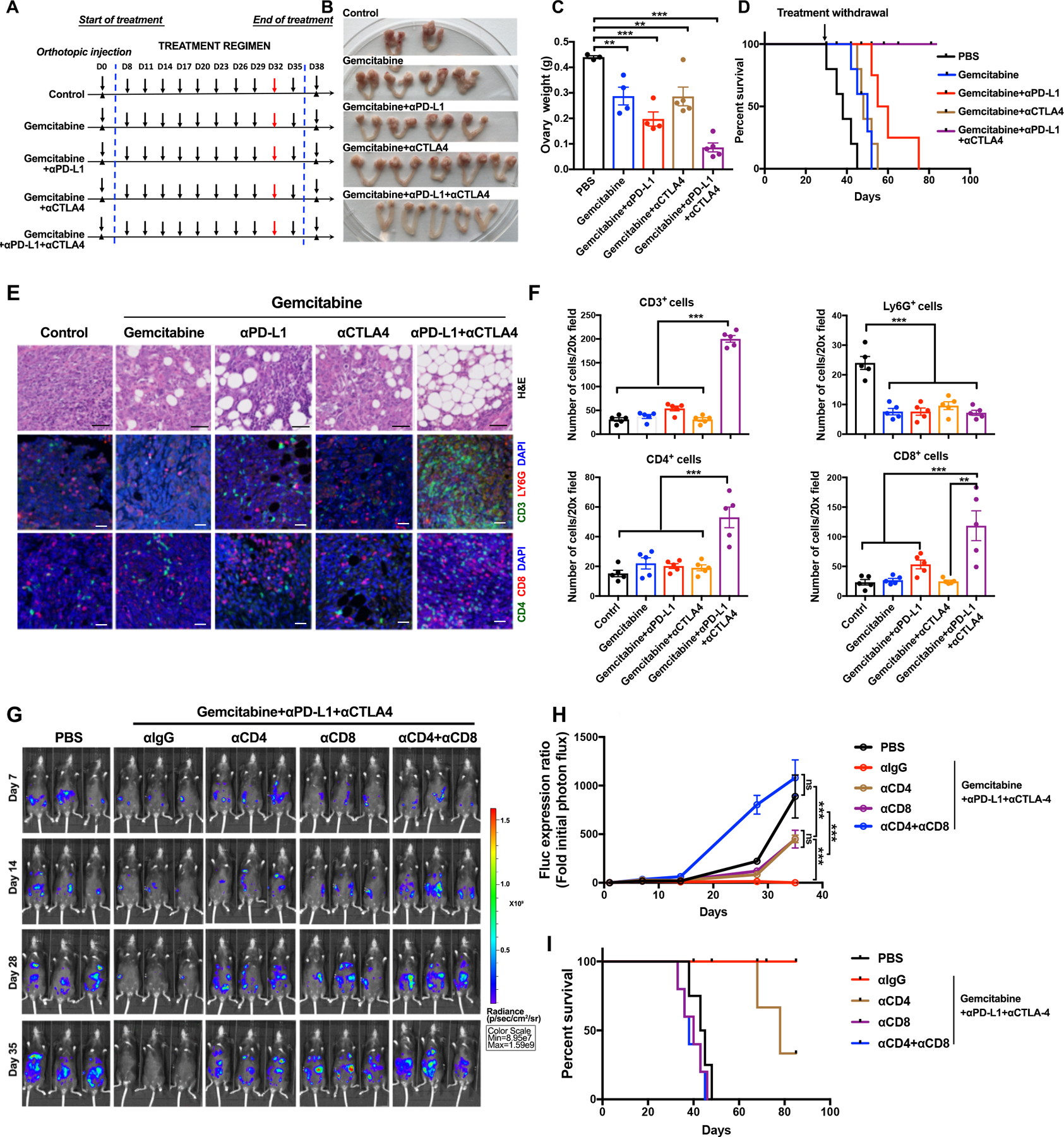
Rationally derived combination regimen results in complete responses in Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors. A, Schematic depicting treatment regimens (n=10 mice/group, 2 batches). For each set of experiments, five mice were sacrificed at Day 32 for histological analysis; the other 5 were continued on treatment until Day 35, then treatment was withdrawn and mice were followed thereafter for survival. B, Representative genital tracts from Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumor-bearing mice treated as indicated; mice were sacrificed at Day 32 of the scheme in (A). C, Ovary weights in mice from the indicated treatment groups. Each point represents one mouse. D, K-M curves of Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumor-bearing mice, treated as indicated in A until Day35 and then monitored for recurrence, n=5 mice/group. E, H&E and IF staining for the indicated immune markers and DAPI (nuclei) in ovarian sections from the indicated groups. Note that the ovarian fat pad has almost no tumor after Gemcitabine+αPD-L1+αCTLA4 treatment. Black scale bars: 50 μm, while scale bars: 20 μm. F, Quantification of the indicated immune cells from the sections in (E). Each point represents average cell number per 20X field from 5 independent sections of each mouse. Error bars indicate SEM; **P<0.01, ***P<0.001, 2-way ANOVA. G, Representative bioluminescence images of mice bearing orthotopic tumor allografts (expressing luciferase), treated as indicated, and measured at Days 7, 14, 28, and 35, respectively. H, Relative photon flux, quantified by the intensity of bioluminescence in the regions of interest (ROIs), determined at the indicated times in mice from each treatment group, n=5 mice/group. Error bars indicate SEM; ns, not significant, ***P<0.001, 2-way ANOVA. I, K-M curves for Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumor-bearing mice, treated as indicated. See also Supplementary Fig. S5 and Fig. S6.
The durability of the responses, and the attendant T cell influx, prompted us to ask if GCP responses were T cell-dependent. To this end, we depleted CD4+ and/or CD8+ T cells and re-assessed efficacy. To enhance our ability to monitor tumors, we transduced Tp53−/−;Ccne1OE;Akt2OE;KrasOE organoids with a luciferase-expressing lentivirus prior to implantation; control experiments showed that luciferase-expressing and parental tumors behaved similarly (Supplementary Fig. S6A). Depletion of the expected T cell population was confirmed by flow cytometry of peripheral blood (Supplementary Figs. S6B and S6C). Notably, CD4 or CD8 cell depletion impaired the response to GCP, whereas tumors from mice lacking CD4 and CD8 T cells actually grew faster in the presence of therapy than did tumors in PBS-treated mice with intact immune systems (Figs. 5G and 5H). GCP-treated, CD8- or CD4+CD8-depleted tumor-bearing mice had survival times similar to PBS-treated mice with intact immune systems. CD4-depletion impaired survival in the combination-treated group, but to a lesser extent (Fig. 5I).
Therapeutic efficacy is tumor genotype-specific
To ask if GCP efficacy was specific for Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumor-bearing mice, we tested the regimen in Tp53−/−;Pten−/−;Nf1−/− tumor-bearing mice. Remarkably, the latter were completely refractory to the GCP regimen, as measured by tumor burden and percentage of mice with ascites (Fig. 6A). We also tested the effects of single agent paclitaxel. Both models showed some response, but as predicted by our in vitro experiments, Tp53−/−;Pten−/−;Nf1−/− tumor-bearing mice experienced more regression than those with Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors (Figs. 6B–D). These differences in clinical parameters were accompanied by differences in survival. Although single agent paclitaxel did not result in CRs, it did evoke potentially beneficial changes in the TME, including an influx of CD44+ CD4 and CD8 T cells showing less evidence of exhaustion, and decreases in g-MDSCs and TAMs, with those remaining showing a more M1-like phenotype (Supplementary Figs. S7A–C).
Figure 6.
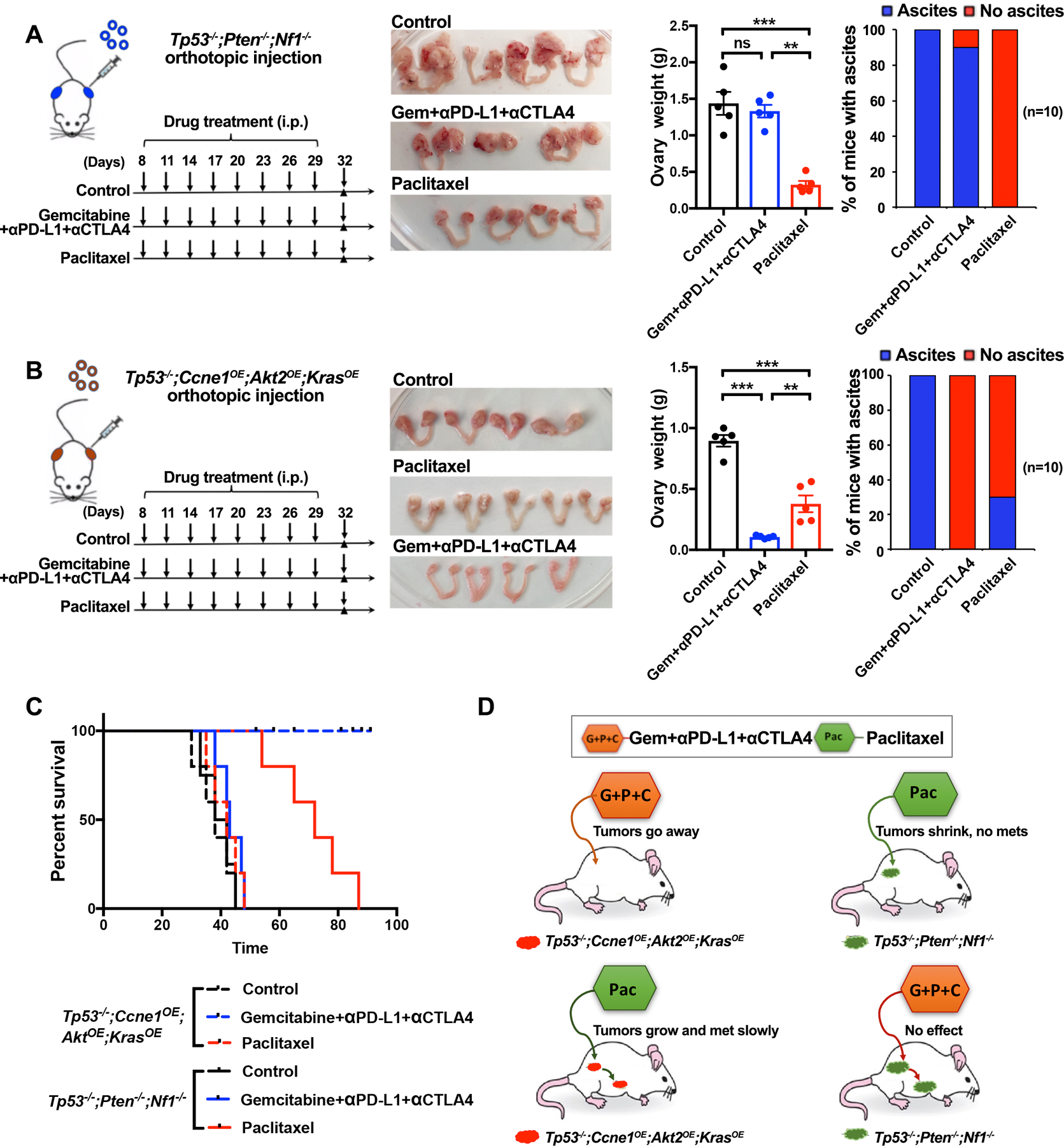
Treatment efficacy is tumor genotype-dependent. A, Left panel: Schematic showing treatment of Tp53−/−;Pten−/−;Nf1−/− tumor-bearing mice with Gemcitabine/α-PD-L1/α-CTLA-4 regimen (from Figure 6) or Paclitaxel. Second panel: Genital tracts from mice treated as indicated; Third panel: Ovary weights in treated mice. Right panel: % mice with ascites after indicated treatment. B, Left panel: Schematic showing treatment of Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumor-bearing mice with the indicated regimens. Second panel: Genital tracts from mice treated as indicated; Third panel: Ovary weights in treated mice. Right panel: % mice with ascites after indicated treatment. Data indicate means ± SEM, **p < 0.01, unpaired t test. C, K-M curves of tumor-bearing Tp53−/−;Pten−/−;Nf1−/− or Tp53−/−;Ccne1OE;Akt2OE;KrasOE mice, treated as indicated. Treatments were withdrawn at Day 32. D, Cartoon summarizing results, depicting tumor genotype-specificity of therapeutic efficacy. See also Supplementary Fig. S7.
Similarities between Ccne1OE models and human CCNE1amp HGSC
To further evaluate the translational potential of our mouse organoid platform, we established organoid lines from nine HGSC patients who had undergone genomic profiling. These included examples of the major alterations engineered into our mouse organoids (Supplemental Fig. S8A), including CCNE1Amp, BRCA1 or BRCA2 mutation (BRCAMut), and NF1 deletion (NF1Del). Different human organoids had distinct morphologies, but all expressed PAX8, were highly proliferative, and stained positively for the DNA damage marker γH2A.x (Fig. 7A). Although they showed a range of sensitivities, the CCNE1Amp lines were more gemcitabine-sensitive than the BRCAMut or NF1Del organoids. We noticed that the one CCNE1Amp organoid (HGS-3.1) tested by Kopper et al. also showed profound gemcitabine hypersensitivity (24). By contrast, NF1Del organoids were resistant to gemcitabine but more sensitive to paclitaxel, while, as expected, BRCAMut organoids were more sensitive to olaparib (Fig. 7B). Hence, human HGSC organoids showed a pattern of drug sensitivities similar to our mouse models.
Figure 7.
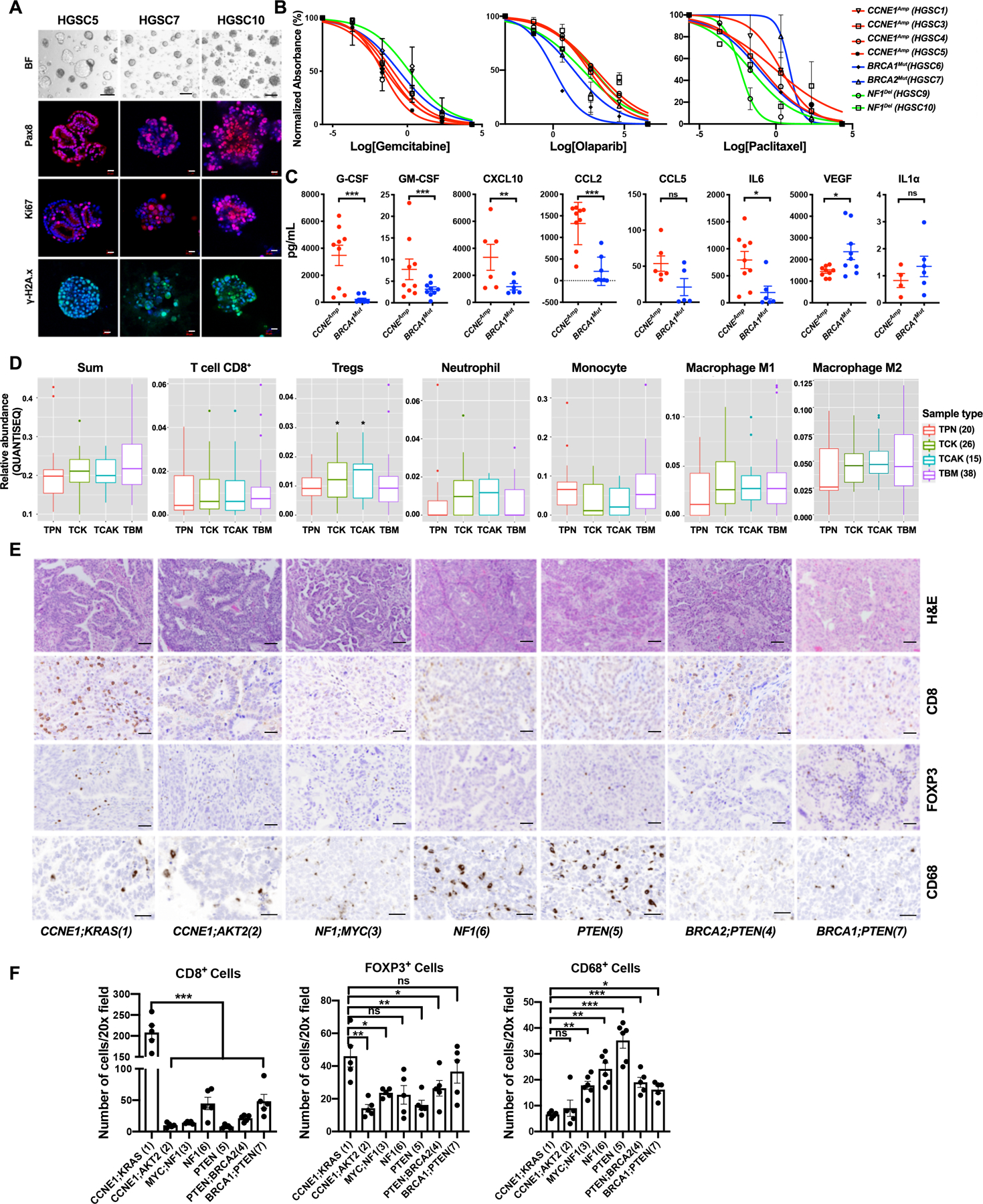
Similarities between human HGSC organoids and tumors and mouse models. A, Representative bright field images and IF staining of human HSGC organoids. Scale bars: bright field, 100 μm; IF: 20 μm. B, Dose-response curves for the indicated drugs in tumorigenic organoid lines of different genotypes. Cell viability was calculated relative to 0.01% DMSO-treated control cells, measured after 5 days of treatment. C, Levels of the indicated cytokines, chemokines, and growth factors in human HGSC organoid-conditioned media; error bars indicate ± SEM **P<0.01, ***P<0.001, 2-way ANOVA. D, Relative abundance of major immune cell subtypes in human HGSC samples with indicated genotypes from TCGA, as inferred by QUANTISEQ. TPN: TP53;PTEN;NF1, TCK: TP53;CCNE1;KRAS, TCAK: TP53;CCNE1;AKT2;KRAS, TBM: TP53;BRCA1;MYC. Numbers of samples per group are shown in parentheses. *P<0.05, t-test corrected for multiple comparisons by Benjamini-Hochberg method. E, H&E-stained sections and IHC analysis of the indicated markers in representative sections from human HGSC samples of the indicated tumor genotypes. Scale bars: 100 μm. F, Quantification of CD8+ cells, FOXP3+ (Treg) cells and CD68+ cells in tumors of the indicated genotypes; average cell numbers from five 20X fields were determined. Data represent mean ± SEM, **P<0.01, ***P<0.001, 2-way ANOVA. See also Supplementary Fig. S8.
We next compared the secretomes of CCNEAmp and BRCA1Mut organoids. Except for G-CSF, which was expressed at lower levels in Tp53−/−;Ccne1OE;Akt2OE;KrasOE than in Tp53−/−;Brca1−/−;MycOE organoids, the cytokines, chemokines, and growth factors detected in both species followed similar patterns in each (Fig. 7C). Of particular note, GM-CSF and CXCL10, which are functionally important for myeloid (g-MDSCs, macrophages), and T lymphocyte recruitment, respectively, to Tp53−/−;Ccne1OE;Akt2OE;KrasOE tumors (Fig. 4D), were significantly higher in CCNE1Amp organoids compared with their BRCA1Mut counterparts. Likewise, CCL2 and CCL5, which are required for chemotaxis in Transwell assays and had nominal, although not significant, effects in tumors, had a similar secretion pattern in mouse and human HGSC organoids, as did the angiogenic growth factor VEGF (Figs. 4C and D; Fig. 7C).
We did not have ready access to large numbers of HGSC cases for prospective characterization of their genomic abnormalities and TME. Instead, we inferred the immune landscape in tumors of different genotypes by applying the quanTIseq (50) and CIBERSORT (51) algorithms to TCGA data; CIBERSORT was implemented in “abs mode” to allow intra-sample comparison between cell types and inter-sample comparisons of the same cell type. HGSC cases were grouped as TP53−/−;PTEN−/−;NF1−/− (TPN), TP53−/−;CCNEamp/OE;KRASamp/OE (TPK), TP53−/−;CCNEamp/OE;AKT2OE;KRASamp/OE (TCAK) or TP53−/−;BRCA1−/−;MYCamp/OE (TBM) tumors, based on copy number and RNAseq profiles (for details, see Methods). The TCK group (which includes TCAK tumors) was included to increase sample size and because multiplex IHC showed that KRAS over-expression was primarily responsible for the major features of the Tp53−/−;Ccne1OE;Akt2OE;KrasOE TME (Supplementary Fig. S8B).
Consistent with the immune phenotypes observed in mouse organoid-derived tumors, quanTIseq revealed that CCNE1 (TCAK, TCK) and BRCA1 (TBM) tumors had nominally more total immune cells than the TPN tumors, although the differences did not reach statistical significance. There also was a trend towards increased CD8 cells and significantly higher levels of Tregs in TCAK and TCK tumors, as well as a trend towards more neutrophils (likely g-MDSCs) in TCK and TCAK tumors, compared with those of other genotypes. Also as in the mouse models, TBM tumors tended to have more monocytes, whereas all tumors tended to have a mixture of M1 and M2 macrophages (but predominantly the latter). Notably, TCK and TCAK tumors had similar inferred TMEs, comporting with the dominant effect of KRAS in the organoid-derived tumors (Fig. 7D; Supplementary Fig. S8B). The total immune cells and tumor-associated T cells predicted by CIBERSORT were similar to quanTIseq inferences, although the myeloid cell predictions differed from those inferred by quanTIseq and found in our mouse models (Supplementary Fig. S8C).
Finally, we performed IHC for CD8, FOXP3, and CD68 on seven primary HGSC samples for which we had genotype data (Supplementary Fig. S8D). Consistent with our mouse models and the quanTIseq/CIBERSORT analyses, the CCNE1Amp;KRASAmp tumor showed significantly more T cell infiltration that the CCNE1Amp;AKT2Amp tumor and the non-CCNE1amp samples (Figs. 7E and 7F). Tregs (FOXP3+) also were significantly higher in the CCNE1Amp;KRASAmp tumor. Although one of the two BRCAMut tumors (HGSC7) also showed a higher number of Tregs, we did not observe higher CD8+ T cell infiltration in BRCA1/2 tumors, which might reflect the co-occurring PTEN deletions in these two cases. Then BRCA1/2mut, PTENmut, and NF1mut tumors had more macrophages (CD68+) than the CCNE1Amp tumors, again consistent with the cognate mouse models. We could not obtain consistent, reliable staining for g-MDSC/neutrophils and therefore could not test whether CCNE1amp led to greater immigration of these cells in humans as in mice. Overall, although additional, more detailed analyses are clearly needed, these results indicate significant similarity between the phenotypes of our mouse models and human HGSC.
DISCUSSION
Like most solid tumors, HGSC is genetically complex and heterogeneous, yet with the exception of PARP-Is for BRCA-mutant tumors, current therapy for HGSC (as for most other neoplasms) is genotype-agnostic. Perhaps unsurprisingly, ~20% of HGSC patients experience minimal or no clinical benefit from this uniform approach, and even those who initially respond, nearly all relapse and die (9). Rational development of genotype-informed therapies is impeded by a paucity of relevant experimental systems. Our FTE organoid-based system remedies these deficiencies, enabling analysis of the effects of specific genetic aberrations on in vitro properties (proliferation, differentiation, morphology, genome stability, drug sensitivity, secretome), assignment of complementation groups for tumorigenicity, assessment and perturbation of the TME, and evaluation of drug therapies (Supplementary Fig. S1). Organoid-derived tumors derive from the “correct” cell-of-origin and form in the relevant anatomical location surrounded by normal host cells. We demonstrate the utility of this platform by developing a specific combination regimen that evokes durable complete responses in mice bearing Ccne1OE tumors but has no activity against Tp53−/−;Pten−/−;Nf1−/− tumors. The latter tumors, by contrast, are more sensitive to paclitaxel (Fig. 6D). Analysis of human HGSC organoids, primary tumors, and TCGA data reveal similarities between our mouse models and the human disease (Fig. 7). In concert, our results argue strongly against therapeutic approaches that treat HGSC as a single entity and support the development of new, genotype-informed strategies.
The HGSC cell-of-origin remains controversial. Transcriptomic (33,52,53), proteomic (54), epigenomic (52), and mouse modeling (31,55,56) data suggest that at least some cases initiate in OSE, but most often, HGSC initiates in FTE (33,57,58). Consequently, we focused our models on FTE organoids. Others have reported that orthotopic injection of 105 Tp53−/−;Brca1−/−;MycOE OSE cells also yields HGSC-like tumors, which kill recipients within 50 days (59). By contrast, mice injected with more (2X106) FTE cells of the same genotype survive for 70 days-150 days (Fig. 1E), consistent with our finding that the cell-of-origin influences HGSC biology (31). Our platform can be adapted easily to model OSE-derived HGSC, as well as other cancers for which mouse organoids can be cultured/engineered (60–62). Indeed, while our manuscript was in review, others reported that FTE (“oviductal” in their manuscript) and OSE organoids engineered with the same genetic abnormalities could give rise to HGSC, although OSE-derived tumors could only be established orthotopically after an initial sub-cutaneous passage. This study was restricted to Tp53−/−;Brca1−/+;Pten−/− and Tp53−/−;Brca1−/+;Nf1−/− combinations, which are not frequently seen in human HGSC, and used organoids from B6 × 129 mice, precluding transplantation into immune competent recipients and analysis of the TME (56).
Human HGSC is profoundly aneuploid, featuring amplifications, deletions, and complex rearrangements. Importantly, our engineered organoids also are aneuploid (Fig. 2A). Recent computational analyses identified recurrent patterns of abnormalities in human HGSC and defined seven specific CN “signatures” (40). That report noted correlations between specific driver genes/signaling aberrations and particular signatures but did not establish a causal relationship. Although we analyzed relatively few engineered organoids, our results suggest that different drivers cause distinct patterns of CNAs. Future, expanded studies will ask if mouse CN signatures also exist, potentially reflecting inter-species conservation of mutational processes, whether aneuploidy affects the anti-tumor immune response, and if the aneuploid genome is, at least in part, sculpted by the host TME.
Tumorigenic organoids showed several expected, but other unanticipated, sensitivities to small molecule inhibitors/drugs. In line with previous studies of human ovarian cancer cell lines, and Tp53−/−;Brca1−/−;MycOE OSE-derived cells, Brca1-mutant FTE-derived tumor organoids showed increased PARP-I sensitivity. Hypersensitivity was less in Tp53−/−;Brca1−/−;MycOE FTE organoids than in conventional Brca-mutant cell lines (63), however, and comparison of Tp53−/−;Brca1−/− and Tp53−/−;Brca1−/−;MycOE organoids shows that MYC confers relative PARP-I (and platinum) resistance (Supplementary Fig. S2C). Hence, MYC could be an important biomarker for PARP-I/platinum resistance in HGSC patients, as suggested previously (64). Although there was a class-specific increase in PARP-I sensitivity in Tp53−/−;Brca1−/−;MycOE organoids, the extent of hypersensitivity differed for individual PARP-Is. Our models could be used to elicit the mechanistic basis for such differences, as well as their respective effects on the TME. ATR inhibitors also showed increased efficacy against Tp53−/−;Brca1−/−;MycOE organoids, in accord with the HR-deficiency conferred by BRCA1 deficiency, whereas the sensitivity of Tp53−/−;Pten−/−;Nf1−/− cells to ATR inhibition comports with the reported role for nuclear PTEN in HR (65,66). The mechanisms underlying genotype-dependent differences in paclitaxel (for Tp53−/−;Pten−/−;Nf1−/− cells) and chloroquine (for Tp53−/−;Pten−/−;Nf1−/−>Tp53−/−;Ccne1OE;Akt2OE;Kras) sensitivity are less clear. Comparison of Tp53−/−;Pten−/− and Tp53/−−;Nf1−/− organoids implicate NF1 deficiency as the main cause increased paclitaxel sensitivity (Supplementary Fig. S3D); notably, NF1 associates with microtubules (28,67,68), the target of paclitaxel. KRAS-mutant cells require autophagy for survival (69), while PTEN deficiency or AKT2 over-expression, by increasing mTOR activity, should suppress autophagy. Conceivably, increased RAS activity, combined with lower basal autophagy due to increased mTOR, sensitizes FTE cells to further autophagy inhibition. Regardless, these differences emphasize the value of genotype-defined models for developing new therapies and identifying biomarkers and mechanisms of resistance. Although we tested a small number of agents, our models can be adapted to high throughput drug screens or genetic perturbations (e.g., CRISPR/Cas9 screens). Furthermore, the genotype-specific sensitivities that we observe suggest that only certain patient subsets will respond to standard-of-care single agents or combinations. For example, combining paclitaxel with platinum, a practice developed empirically (70,71), might only benefit patients with NF1-deficient tumors; others might simply incur taxane-based toxicity.
Human HGSC also has a complex TME, with differences in infiltrating immune cells and tumor-associated chemokines/cytokines associated with prognosis (72,73). As in many other malignancies, intratumor CD8+ cells and high CD8+/Treg ratio correlate with improved survival, whereas high levels of Tregs are a negative prognostic sign (74–77). Intratumor T cells have been associated with expression of CXCL9, CXCL10, CCL5, CCL21, and/or CCL22, whereas high VEGF levels inversely correlate with T cell infiltration (77–79). A large pan-cancer genomic analysis indicated that high levels of CCL5 RNA and protein (by IHC) correlate with intra-tumor CD8 cells in HGSC and other solid tumors (80). CCL5 and CXCL9 also correlated in this analysis, and dual expression of these chemokines was associated with better prognosis. Interestingly, ovarian cancers with high intra-tumor CD8+ cells but low CCL5 RNA had high levels of CXCL9 (81). By contrast, high levels of TAMs, particularly M2-like TAMs, and MDSCs correlate with poor outcome (82,83). Aside from describing greater T cell infiltration and better prognosis in BRCA-mutant tumors (72,84,85), examining the regulatory mechanism of specific immune regulatory molecules (e.g., silencing of CCL5) (81), and a very recent report correlating mutational signature 3 (which is associated with HRD) and immune score with response to combined PARP-I/anti-PD1 treatment, previous studies have been tumor genotype-agnostic. Yet the three mouse models that we examine in detail displayed major differences in TME, associated with major differences in chemokine/cytokine/growth factor expression (Figs. 2–4). Furthermore, perturbation experiments clearly identified specific secreted factors that influence TME (and likely tumor) development (Figs. 4C and 4D). As tumor genotype also affects response to targeted and conventional agents, understanding how genotypic differences direct host immune responses could aid in therapy development. Our ability to manipulate tumor (e.g., by further engineering of chemokine/cytokine genes) and host immune cells (e.g., by depletion studies, injection of tumorigenic organoid cells into various knockout backgrounds), and to study tumors over time, can provide insights into how the TME develops and responds to therapy.
Earlier reports noted differences in tumor immune infiltrates in other systems (10,86) and implicated MYC, KRAS, mTOR, YAP, and β-catenin signaling in cancer cells (11). Many of these studies used syngeneic tumor models, GEMMs, or GEMM-derived cell lines, and some pointed to specific cytokines/chemokines as the cause of differences in the TME. Nevertheless, the extent to which TME responses are “hard-wired” by specific oncogenic defects has remained unclear. For example, PTEN deficiency leads to impaired T cell infiltration owing to immunosuppressive myeloid cells in mouse prostate cancer (87) and melanoma (88) models. But whereas CXCL5 (mouse)/CXCL6 (human) are implicated in myeloid cell immigration in prostate cancer, CCL2 and VEGF are the apparent culprits in melanoma. We also observed increased myeloid cells in Tp53−/−;Pten−/−;Nf1−/− HGSC, along with increased levels of CCL2 and VEGF. However, MCSF1 and CXCL1 might also contribute to myeloid infiltration in this model, whereas CXCL5 is not elevated and is unlikely to play a role. These findings, and many others (10,11,89), argue that cellular context (e.g., cell-of-origin, cooperating mutations) might be as important as specific oncogenic abnormalities for determining the ultimate TME and anti-tumor immune response. Our ability to rapidly engineer FTE organoids with all major combinations of genetic defects seen in HGSC positions us to address this important issue.
Attempts to manage HGSC with immune therapy have not been very fruitful. Single-agent anti-CTLA-4 or anti-PD1/PD-L1 yield only modest results, with response rates of 10%−15% (90,91). Combining anti-PD1 and anti-CTLA4 increases response rate to 34%, but the clinical data are very immature (92). Our Brca1-mutant mouse model shows greater T cell infiltration, as does BRCA1-mutant HGSC (Fig. 7D); such tumors might show a better response to immune checkpoint inhibition, alone or in combination with PARP-Is (59,84,93). However, these responses are rarely durable, and whether other tumor genotypes confer more or less sensitivity is not clear. A major advantage of our organoid platform is its ability to rapidly suggest and credential potential therapies. Our chemo-immunotherapy combination of three approved drugs, gemcitabine, anti-PD-L1, and anti-CTLA4, led to durable, T-cell-dependent CRs in a highly aggressive, CCNE1-overexpressing HGSC model. It will be critical to test this combination in models in which Ccne1 over-expression is accompanied by other frequently co-occurring genetic defects (e.g., Mecom and/or Myc over-expression), as well as to develop and test combination immunotherapies with paclitaxel in PTEN/NF1-deficient HGSC; the changes in the TME that ensue following paclitaxel treatment already suggest several potential combination strategies.
A major consideration for any animal model is the extent to which it represents the cognate human disease. Although much more detailed studies are warranted, initial indications reveal similarities between our mouse models and human HGSC organoids in drug response (Fig. 7B), secretome (Fig. 7C), and TME (Figs. 7E and F; Supplementary Fig. S8). However, the latter analyses were limited by relatively small sample size, contradictory predictions of myeloid populations by quanTIseq and Cibersort, and lack of well-defined, consensus IHC/IF markers for identifying tumor-infiltrating myeloid cell subsets by IF/IHC (94,95).
In conclusion, our ability to rapidly generate multiple, genetically defined, complex HGSC organoid models should facilitate studies of the diversity and host response of this disease. Our models also suggest a genomics-informed, rationally based combination treatment for CCNE1-amplified HGSC, and suggest new interventional strategies for other genomic subgroups of this highly complex disease.
MATERIALS AND METHODS
Organoid Culture and Engineering
FTE organoids from Tp53f/f or Tp53f/f;Brca1f/f mice were established as described (31). Cultures were checked monthly for mycoplasma by PCR. Organoid cells were collected by using cold Cultrex® Organoid Harvesting Solution (Stem Cell Technologies) to dissolve Matrigel, following the manufacturer’s instructions. Tp53f/f female or Tp53f/f;Brca1f/f organoids were dissociated into single cells as described (96) and infected with 105 pfu Adenovirus-CMV-Cre (Vector Development Lab, Baylor College of Medicine) by “spinoculation” at 37 for 1h. Cell pellets were recovered and seeded into Matrigel in media containing nutlin-3 to enrich for Tp53−/− organoids. Organoids were released 7 days later, and multiple clones were picked and expanded. Deleted clones were identified by PCR (97). Primer sequences are provided in Supplementary Information.
Mouse Ccne1 and Akt2 were cloned into pLV-EF1a-IRES-Neo (Addgene#85139), with neomycin-resistance or pLV-EF1a-IRES-Blast (Addgene#64832) with blasticidin-resistance genes, respectively. For Myc-over-expression, we used the vector MSCV-transgene-PGK-Puro IRES-GFP, purchased from Addgene (#75124). Mouse Kras was cloned into pMSCV-IRES-mCherry (Addgene, #52114). Successful gene insertion was confirmed by Sanger sequencing. Pten (agatcgttagcagaaacaaa) or Nf1 (ctcgtcgaagcggctgacca) sgRNA sequences were designed with the CRISPR Design Tool (http://crispr.mit.edu/) and inserted into LentiCRISPR v2 (Addgene, #52961). For virus production, lentiviral vectors were co-transfected with psPAX2 and pMD2.G into HEK293T cells at a ratio of 10:7.5:2.5 or retroviral vectors were co-transfected with pVPack and VSV-G into HEK293T cells at a ratio of 10:6.5:3.5. All transfections were performed by using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific), according to the manufacturer’s instruction. Media were changed 8h after transfection, and viral supernatants were collected 48h later by passage through a 0.45-mm filter, aliquoted ,and stored at −80°C.
Organoids were dissociated into single cells and “spinoculated” with lentiviruses/retroviruses, also as described (96). Briefly, viral supernatants were added to cells in 48-well plates, centrifuged at 600g at 37℃ for 60 min, incubated at 37℃ for another 6–8 hours, collected, and re-seeded in Matrigel-containing media. Infected organoids were selected 72 hours after viral transduction with G418 (Thermo Fisher, 10131027) or blasticidin (Sigma, 15205), as indicated. Gene deletion and/or overexpression was assessed by PCR or immunoblotting. At least two independent clones of each genotype were used for experiments.
For human HGSC organoid cultures, samples were obtained from the University Health Network Tissue Bank (Toronto, ON, Canada) with written informed consent. All studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and Research Ethics Board approval (equivalent to Institutional Review Board (IRB) in U.S.). Tumor cells were isolated from fresh surgical material or ascites, as described previously (98,99). HGSC cells of the indicated genotypes were thawed, seeded in Matrigel, and cultured in human organoid growth medium, composed of: Ad+++ AdDMEM/F12 (Invitrogen); HEPES (Thermo Fisher Scientific, 100X diluted); penicillin/streptomycin and Glutamax, each 100X diluted (Life Technologies), supplemented with B27 (Invitrogen, 50X diluted), N2 supplement (Thermo Fisher Scientific, 100X diluted), 1.25 mM N-acetylcysteine (Sigma), 50 ng/ml EGF (Thermo Fisher Scientific), 500 ng/ml RSPO1 (Peprotech) or R-spondin-1-conditioned medium (25%−50%, v/v), WNT3a-conditioned medium (0–25%, v/v), 100 ng/ml Noggin (Peprotech), 10nM 17-β Estradiol (Sigma), 50 ng/ml EGF, 10 ng/ml FGF10, 0.5 μM A83–01 (Thermo Fisher Scientific), 50ng/ml human recombinant Heregulin-beta 1 and 10 μM Forskolin (both from STEMCELL Technologies). For the first 3 days after thawing, media were also supplemented with 10 μM Y-27632 (Sigma-Aldrich).
Cytokine Profiling
Cytokine, chemokine, and growth factor levels in 72 hr-conditioned media from organoid cultures were profiled using services at Eve Technologies (Calgary, Canada). The Mouse Cytokine Array/Chemokine Array 31-Plex (MD31) panel included: Eotaxin (CCL11), G-CSF, GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17A, IP-10, KC (CXCL1), LIF, LIX (CXCL5), MCP-1 (CCL2), M-CSF, MIG (CXCL9), MIP-1α (CCL3), MIP-1β (CCL4), MIP-2 (CXCL2), RANTES (CCL5), TNFα, and VEGF. The Human Cytokine Array/Chemokine Array 48-Plex (HD48) included: sCD40L, EGF, Eotaxin, FGF-2, Flt-3 ligand, Fractalkine, G-CSF, GM-CSF, GROα, IFNα2, IFNγ, IL-1α, IL-1β, IL-1ra, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17A, IL-17E/IL-25, IL-17F, IL-18, IL-22, IL-27, IP-10, MCP-1, MCP-3, M-CSF, MDC (CCL22), MIG, MIP-1α, MIP-1β, PDGF-AA, PDGF-AB/BB, RANTES, TGFα, TNFα, TNFβ, VEGF-A.
Drug Sensitivity Assays
Organoids were seeded into 96-well plates at 1,000 cells/well (Day 0). The indicated concentrations of Rucaparib (Selleckchem, S1098), Niraparib (MCE, HY-10619), Olaparib (Selleckchem, S1060), Gemcitabine (MCE, HY-B0003), Doxorubin (sigma, D1515), Paclitaxel (Selleckchem, S1150), Carboplatin (Sigma, 1096407), Seliciclib (MCE, HY-30237), PHA767491(Sigma, PZ0178), BAY1895344 (Selleckchem, S8666), Chloroquine (Selleckchem, S4157) and YKL-5–124 (a gift from Dr. Kwok-kin Wong) were added on the day following seeding (Day 1). Media were changed, and fresh drug was added on Day 3. Cell viability was assessed on Day 5 by adding10 μl PrestoBlue and incubating for 30 min in 37℃. Fluorescence was measured in a FlexStation® 3 Multi-Mode Microplate Reader (BOSTONind). Results were normalized to DMSO controls, and IC50 values were determined using Graphpad Prism 7.
Chemotaxis assays
To assess tumor-infiltrating myeloid cell migration, CD45+ cells were isolated from Tp53−/−;Ccne1OE;Akt2OE;Kras tumors by using CD45 MicroBeads (Miltenyi Biotec, 130–052-301). Cell culture inserts (8 μm pore size) were placed into 24-well plates, and 5X105 CD45+ cells were added into each insert. Tp53−/−;Ccne1OE;Akt2OE;Kras organoid-conditioned medium (500 μl) with or without anti-CCL5 (1μg/mL), -CCL2 (1μg/mL) or -GM-CSF (1μg/mL) was added to the bottom chamber. For T cell migration assays, tumor-infiltrating T cells were purified by using the EasySep™ Mouse T Cell Isolation Kit (STEMCELL Technologies, Catalog # 19851), 5X105 purified cells were added into inserts (pore size=3 μm), and conditioned medium with or without anti-CXCL10 (1μg/mL) or -CCL5 (1μg/mL) was added to the bottom chamber. After incubation for 24h at 37C, inserts were removed, and cells that had migrated to each bottom well were collected, stained with the LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit (Thermo Fisher, L23105) and the indicated cell surface markers, and quantified by flow cytometry. Each antibody was tested in triplicate.
Animal Experiments
Tp53f/f female mice were from Dr. Kwok-kin Wong and Tp53f/f;Brca1f/f mice (97) were provided by Dr. Richard Possemato (both NYUGSoM). Female C57BL/6 mice (6–8 weeks old) were purchased from Charles River Laboratories. All animal experiments were approved by, and conducted in accordance with the procedures of, the IACUC at NYUGSoM (Protocol no.170602).
For orthotopic tumorigenicity assays, organoid cell pellets were collected and injected into ovarian bursae, as described (31). Briefly, mice were anesthetized by IP injection of Xylazine (10 mg/kg) and Ketamine (50 mg/kg), shaved, and cleaned with betadine. A dorsal incision above the ovary was made, followed by incision of the peritoneal cavity. The ovary was externalized and, using an insulin syringe with a 31G needle, 2 X 106 cells in 50 μl PBS/Matrigel (1:1 v/v) were injected through the ovarian fat pad into the bursa. Injected ovaries were returned to the peritoneal cavity, and incisions were sealed with wound clips. Mice that developed tumors were euthanized at the indicated time(s), or for survival experiments, they were monitored until death or upon veterinary recommendation. Where indicated, mice received IP injections of gemcitabine (50 mg/kg), paclitaxel (40 mg/kg), anti-CTLA4 (50 μg, clone 9H10, BioXcell), and/or anti-PD-L1 (50 μg, clone 4H2, BioXcell), beginning 8 days after cell implantation. Dosing was repeated every three days, as indicated. Control mice were injected with PBS or isotype control antibody (clone LTF-2, BioXcell).
CD4+ and/or CD8+ T cells were depleted by IP injection of 200 μg of InVivoMAb anti-mouse CD4 (clone GK1.5, BioXcell) and/or InVivoMAb anti-mouse CD8α (clone 2.43, BioXcell) respectively, one week after cell implantation. Injections were repeated every 3 days (100). Other mice received isotype control antibody (clone LTF-2, BioXcell). Depletion of the appropriate lymphoid population was confirmed by flow cytometry of peripheral blood and reassessed every 2 weeks for the duration of the study. Peripheral blood was collected from tail veins into heparinized microhematocrit capillary tubes, centrifuged, and prepared for flow cytometry by lysing RBCs in ACK buffer, followed by serial washes in RPMI. For cytokine neutralizations, mice were injected IP with 50 μg anti-CXCL10 (clone 134013, Thermo Fisher), 50 μg anti-CCL5 (clone 53405, Thermo Fisher), 100 μg anti-CCL2 (clone 2H5, BioXcell), 100 μg anti-GM-CSF (clone MP1–22E9, BioXcell), or isotype control IgG (100 μg), as indicated, one-week after implantation of Tp53−/−;Ccne1OE;Akt2OE;Kras organoid cells as above. Antibody injections were repeated every 3 days, and tumors were collected 2 days after the final injection and analyzed by flow cytometry.
Bioluminescence Imaging
Mice were injected with 150 mg/kg D-luciferin Firefly (PerkinElmer Part Number #122799), and luminescence was assessed 15 min. later by using a PerkinElmer IVIS Lumina III imaging system. Images were analyzed with Living Image Software 4.7.3.
Flow Cytometry
Tumors were minced, chopped, and digested with Gentle Collagenase, 0.012% Dispase (w/v) and DNaseI (STEMCELL Technologies) at 37℃ for 1 hour. Single cell suspensions were obtained by passage through a strainer (70 μm), washed in FACS buffer (PBS with 5% FBS), incubated with LIVE/DEAD Fixable Zombie Yellow Fixable Viability Kit (Biolegend, 423104) for 30 min., and blocked with anti-CD16/32 (Biolegend, clone 93) for 5 min. on ice. Primary fluorophore-conjugated antibodies were added, and samples were incubated on ice for 45 min. FOXP3 Fixation/Permeabilization Buffer Set (BioLegend) was used for intracellular markers, according to the manufacturer’s instructions. Antibodies for flow cytometry are listed in Supplementary Table S2. Flow cytometry was performed on an LSR II flow cytometer at the Flow Cytometry Core of the PCC Precision Immunology Shared Resource and analyzed with FlowJo software. Organoids cultured 6 days after infection with MSCV-Kras-mCherry were collected and digested as above, passed through a strainer (70 μm) to obtain single-cell suspensions, centrifuged at 1000×g for 5 min, and resuspended in PBS containing 2% FBS, 10 μM Y-27632, (STEMCELL Technologies Inc.), and DAPI (1 μg/ml). FACS was performed immediately on a MoFloTM XDP, and mCherryhi and mCherryneg cells were seeded at 5,000/well.
Histology, Immunofluorescence, and Immunohistochemistry
Mouse tumor tissues were fixed in 4% PFA for 48 hours, processed, and embedded for standard histology, IHC, and IF. Clinical molecular profiling results were used to identify appropriate HGSC cases. Formalin-fixed/paraffin-embedded tissue blocks were retrieved from institutional archives under IRB approval (study # i16–01086). Sections (5 μm) were de-paraffinized, rehydrated, stained with hematoxylin and eosin (H&E) or subjected to antigen retrieval (citrate) at 120℃ in a pressure cooker for 15 minutes for 5 minutes. For IHC, endogenous peroxidase activity was quenched in 3% H2O2 in methanol for 15 min, and sections were blocked in 0.5% BSA in PBS for 1h. Primary antibodies were added overnight at 4℃, then slides were washed in PBS three times for 10 min, incubated with secondary antibodies for 1 h at room temperature, and washed again. Antigens were detected by using the HRP Polymer Detection Kit and DAB peroxidase (HRP) substrate (34002, Life Technologies). For IF, after antigen retrieval, slides were washed in PBS three times for 10 min and then blocked in 0.5% BSA in PBS for 1h. Primary antibodies were incubated at 4°C overnight, and sections were washed in PBS (3 times, 10 min each), followed by incubation with Alexa Fluor secondary antibodies, as indicated. Washed slides were mounted with Prolong™ Gold Antifade Mountant (Thermo Fisher, P36930). For IF, organoids were released from Matrigel (as above), transferred to a µ-Slide 8 Well Glass Bottom (Ibidi), fixed in 4% PFA (pH 7.4) for 20 min., permeabilized in 1% Triton X-100 in PBS, and blocked in PBS, 1% BSA, 3% normal goat serum, 0.2% Triton X-100. After overnight incubation with primary antibody at 4℃, organoids were washed three times for 10 min. in PBS and incubated at room temperature with the appropriate Alexa Fluor secondary antibody (1:200). Organoids were washed with PBS and mounted using ProLong gold antifade (Molecular Probes, Invitrogen). Antibodies for IHC/IF are described in Supplementary Table S3. IHC slides were scanned by using a Leica SCN400 F whole-slide scanner. IF images were taken with a ZEISS LSM 700 confocal microscope.
Immunoblotting
Cell pellets were resuspended in SDS lysis buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 1% SDS, 2 mM Na3VO4), supplemented with protease (40 µg/ml PMSF, 2 µg/ml antipain, 2 µg/ml pepstatin A, 20 µg/ml leupeptin, and 20 µg/ml aprotinin) and phosphatase (10 mM NaF, 1mM Na3VO4, 10 mM β-glycerophosphate, and 10 mM sodium pyrophosphate) inhibitors. Total lysate was resolved by SDS–PAGE, followed by transfer to Immobilon-FL PVDF membranes (Millipore). Membranes were blocked in 1% BSA/TBS containing 0.1% Tween20 for 30 min. and incubated for 1h in blocking buffer containing the indicated antibodies (Supplemental Table S3), followed by IRDye-conjugated secondary antibodies (LI-COR). Images were obtained by using a LI-COR ODYSSEY CLx quantitative IR fluorescent detection system.
RNA Extraction and Sequencing
Tumors were lysed in Trizol, and RNA was extracted using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA sequencing was performed by the PCC Genome Technology Center Shared Resource (GTC). Libraries were prepared by using the Illumina TruSeq Stranded Total RNA Sample Preparation Kit and sequenced on an Illumina NovaSeq 6000 using 150 bp paired-end reads. Sequencing results were de-multiplexed and converted to FASTQ format using Illumina bcl2fastq software. Average read pairs/sample were 35.4 million. Data were processed by the PCC Applied Bioinformatics Laboratories shared resource (ABL). Briefly, reads were adapter- and quality-trimmed with Trimmomatic(101) and then aligned to the mouse genome (build mm10/GRCm38) using the splice-aware STAR aligner(102). The featureCounts program(103) was utilized to generate counts for each gene, based on how many aligned reads overlap its exons. Counts were normalized and tested for differential expression, using negative binomial generalized linear models implemented by the DESeq2 R package (104). For pairwise differential expression analysis between tumor groups, normal FT samples were not included in the model. Statistical analysis and visualization of gene sets were performed using the fgsea(105) and clusterProfiler R packages(106).
Shallow Whole Genome Sequencing (sWGS)
Organoid DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen), according to the manufacturer’s instructions. Libraries were prepared using the Nextera DNA Flex Library Kit (Illumina, 96rxn kit, cat# 20025520). To save costs, the manufacturer’s protocol was miniaturized by reducing reactions to one fourth of the recommended volumes. Following PCR amplification (5 cycles total), water (38 ul) was added to the amplified material (12.5 ul) to increase the volume to 50ul for the final 1x Ampure XP bead cleanup (Beckman Coulter, #A63882). Library DNA was evaluated on an Agilent Tapestation 2200 with high sensitivity DNA screen tape to verify library size of ~50 bp, and each library was quantified by qPCR using the Kapa-Roche Library Quant kit (Illumina, cat# KK4824) and a Bio-Rad CFX384 real time PCR system. Libraries were run on half of an SP300 flow cell (paired end 150 dual indexing run) using the Illumina Novaseq 6000 system. Sequencing reads were adapter and quality trimmed with Trimmomatic (101) and then aligned to the mouse reference genome (build mm10/GRCm38) using the Burrows-Wheeler Aligner with the BWA-MEM algorithm (107). Low confidence mappings (mapping quality <10) and duplicate reads were removed using Sambamba (108). Further local indel realignment and base-quality score recalibration was performed using the Genome Analysis Toolkit (GATK) (109). The average coverage ranged from 1.5 to 2.2X. Copy number profiles were calculated using Control-FREEC (110) with a fixed window size of 50kb.
Cibersort and quanTIseq Analyses
The immune cell constitution of TCGA samples was inferred by downloading TCGA-OV RNA-seq and CNV data (HTSeq - Counts) from the GDC data portal, and normalizing RNA-seq reads to transcripts per million (TPM). Samples with a CNV score of −2 or with a score of −1 and a TPM value within the bottom 33% of all samples were defined as having PTEN-, NF1-, or BRCA1-loss, respectively. Samples with a CNV score of 2 or with a score of 1 and TPM value within the top 33% of all samples were defined as having CCNE1-, AKT2-, KRAS-, or MYC- gain, respectively. Samples were then identified as TPN, TCK/TCAK, or TBM based on the gain/loss status of each gene. To avoid ambiguity, we excluded samples belonging to more than one genotype group. Tumor infiltrating immune cells were inferred using quanTIseq (50) and CIBERSORT (51) in abs. mode. For deconvolution, we used TIMER2.0 (73) with TPM data as input. To compare groups of samples, we first performed t-tests of the abundance of each cell population, and then adjusted P values for multiple comparisons by the Benjamini-Hochberg procedure.
Quantification and Statistical Analysis
Bioinformatic analyses were performed in R (version 3.5.1). All other statistical analyses were performed using Graphpad Prism, San Diego, CA. Statistical tests used, sample sizes (n) and P values are displayed in the figures and figure legends. P< 0.05 was considered significant.
Supplementary Material
SIGNIFICANCE:
The lack of genetically informed, diverse, immune-competent models poses a major barrier to therapeutic development for many malignancies. Using engineered fallopian tube organoids to study the cell-autonomous and -non-autonomous effects of specific combinations of mutations found in HGSC, we suggest an effective combination treatment for the currently intractable CCNE1-amplified subgroup.
Acknowledgements
We thank the PCC Experimental Pathology, Precision Immunology, Microscopy, GTC, Preclinical Imaging Laboratory, and ABL shared resources for technical support, and Drs. Justin Mastroianni (PCC) for assistance with IVIS imaging. We thank Drs. Kwan Ho Tang, Mitchell Geer, and Carmine Fedele (Neel lab) and Drs. Myles Brown and Xiaole Shirley Liu (Dana Farber Cancer Institute) for helpful advice and discussions. Work on this project was supported by grants MOP-191992 from the Canadian Institutes for Health Research and 02–20 from the Mary Kay Foundation to B.G.N. S.Z. was supported by a post-doctoral fellowship from the Ovarian Cancer Research Fund Alliance. S.I. was supported by the Ludwig Fund for Cancer Research. D.A.L and F.D. were supported by The Honorable Tina Brozman Foundation, DOD CDMRP award W81XWH-19–1-0232PCC. D.A.L and N.O. were supported by The V Foundation for Cancer Research, DOD CDMRP award W81XWH-15–1-0429. Shared resources are supported by P30 CA01687. R.A.W. was funded by grants from the National Institutes of Health (R01 CA0784561 and P01 CA080111), Samuel Waxman Cancer Research Foundation, Breast Cancer Research Foundation, and Ludwig Fund for Cancer Research.
Footnotes
Competing interests: B.G.N. is a co-founder, holds equity in, and received consulting fees from Navire Pharmaceuticals and Northern Biologics, Inc. He also is a member of the Scientific Advisory Board and receives consulting fees and equity from Avrinas, Inc, and was an expert witness for the Johnson and Johnson ovarian cancer talc litigation in US Federal Court. His spouse has or held equity in Amgen, Inc., Regeneron, Moderna, Inc., Gilead Sciences, Inc., and Arvinas, Inc. D.A.L has consulting/advisory role for Tesaro/GSK, Merck, receives research funding to institution from Merck, Tesaro, Clovis Oncology, Regeneron, Agenus, Takeda, Immunogen, VBL Therapeutics, Genentech, Celsion, Ambry, Splash Pharmaceuticals. He also is a founder of Nirova BioSense, Inc. The other authors declare no competing interests.
Data Availability
RNA sequence data have been deposited in the GEO database under the accession code GSE147276. sWGS data were deposited in SRA under BioProject accession number PRJNA613661. All other data supporting the findings of this study are available within the article, the Supplemental information files, or the corresponding author upon request.
REFERENCES
- 1.Robert L, Ribas A, Hu-Lieskovan S. Combining targeted therapy with immunotherapy. Can 1+1 equal more than 2? Semin Immunol 2016;28(1):73–80 doi 10.1016/j.smim.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol 2019;20(3):e175–e86 doi 10.1016/S1470-2045(19)30026-9. [DOI] [PubMed] [Google Scholar]
- 3.Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer 2017;17(2):93–115 doi 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12(4):252–64 doi 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh NC, Kenney LL, Jangalwe S, Aryee KE, Greiner DL, Brehm MA, et al. Humanized Mouse Models of Clinical Disease. Annu Rev Pathol 2017;12:187–215 doi 10.1146/annurev-pathol-052016-100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, et al. Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery. Cancer Immunol Res 2017;5(1):29–41 doi 10.1158/2326-6066.CIR-16-0114. [DOI] [PubMed] [Google Scholar]
- 7.Kersten K, de Visser KE, van Miltenburg MH, Jonkers J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol Med 2017;9(2):137–53 doi 10.15252/emmm.201606857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW. The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res 2016;29(5):590–7 doi 10.1111/pcmr.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24(5):541–50 doi 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li J, Stanger BZ. The tumor as organizer model. Science 2019;363(6431):1038–9 doi 10.1126/science.aau9861. [DOI] [PubMed] [Google Scholar]
- 11.Wellenstein MD, de Visser KE. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018;48(3):399–416 doi 10.1016/j.immuni.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 12.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 2020;10(1):54–71 doi 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575(7781):217–23 doi 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 14.Narod S Can advanced-stage ovarian cancer be cured? Nat Rev Clin Oncol 2016;13(4):255–61 doi 10.1038/nrclinonc.2015.224. [DOI] [PubMed] [Google Scholar]
- 15.Bowtell DD, Bohm S, Ahmed AA, Aspuria PJ, Bast RC Jr., Beral V, et al. Rethinking ovarian cancer II: reducing mortality from high-grade serous ovarian cancer. Nat Rev Cancer 2015;15(11):668–79 doi 10.1038/nrc4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474(7353):609–15 doi 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov 2015;5(11):1137–54 doi 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashworth A A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 2008;26(22):3785–90 doi 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 19.Franzese E, Centonze S, Diana A, Carlino F, Guerrera LP, Di Napoli M, et al. PARP inhibitors in ovarian cancer. Cancer Treat Rev 2019;73:1–9 doi 10.1016/j.ctrv.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 20.Nakayama N, Nakayama K, Shamima Y, Ishikawa M, Katagiri A, Iida K, et al. Gene amplification CCNE1 is related to poor survival and potential therapeutic target in ovarian cancer. Cancer 2010;116(11):2621–34 doi 10.1002/cncr.24987. [DOI] [PubMed] [Google Scholar]
- 21.Au-Yeung G, Lang F, Azar WJ, Mitchell C, Jarman KE, Lackovic K, et al. Selective Targeting of Cyclin E1-Amplified High-Grade Serous Ovarian Cancer by Cyclin-Dependent Kinase 2 and AKT Inhibition. Clin Cancer Res 2017;23(7):1862–74 doi 10.1158/1078-0432.CCR-16-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 2013;4:2126 doi 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ Jr., et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov 2018;8(11):1404–21 doi 10.1158/2159-8290.CD-18-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopper O, de Witte CJ, Lohmussaar K, Valle-Inclan JE, Hami N, Kester L, et al. An organoid platform for ovarian cancer captures intra- and interpatient heterogeneity. Nat Med 2019;25(5):838–49 doi 10.1038/s41591-019-0422-6. [DOI] [PubMed] [Google Scholar]
- 25.Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018;174(6):1586–98 e12 doi 10.1016/j.cell.2018.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018;175(7):1972–88 e16 doi 10.1016/j.cell.2018.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnalzger TE, de Groot MH, Zhang C, Mosa MH, Michels BE, Roder J, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J 2019;38(12) doi 10.15252/embj.2018100928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O, et al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000;21(4):585–91 doi 10.1093/carcin/21.4.585. [DOI] [PubMed] [Google Scholar]
- 29.Maniati E, Berlato C, Gopinathan G, Heath O, Kotantaki P, Lakhani A, et al. Mouse Ovarian Cancer Models Recapitulate the Human Tumor Microenvironment and Patient Response to Treatment. Cell Rep 2020;30(2):525–40 e7 doi 10.1016/j.celrep.2019.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell 2013;24(6):751–65 doi 10.1016/j.ccr.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S, Dolgalev I, Zhang T, Ran H, Levine DA, Neel BG. Both fallopian tube and ovarian surface epithelium are cells-of-origin for high-grade serous ovarian carcinoma. Nat Commun 2019;10(1):5367 doi 10.1038/s41467-019-13116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun 2017;8(1):1093 doi 10.1038/s41467-017-00962-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun 2017;8(1):990 doi 10.1038/s41467-017-01217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’t Veer L, Garber JE, et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002;346(21):1616–22 doi 10.1056/NEJMoa012158. [DOI] [PubMed] [Google Scholar]
- 35.Perets R, Drapkin R. It’s Totally Tubular….Riding The New Wave of Ovarian Cancer Research. Cancer Res 2016;76(1):10–7 doi 10.1158/0008-5472.CAN-15-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuchenbaecker KB, Ramus SJ, Tyrer J, Lee A, Shen HC, Beesley J, et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat Genet 2015;47(2):164–71 doi 10.1038/ng.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollis RL, Gourley C. Genetic and molecular changes in ovarian cancer. Cancer Biol Med 2016;13(2):236–47 doi 10.20892/j.issn.2095-3941.2016.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martins FC, Santiago I, Trinh A, Xian J, Guo A, Sayal K, et al. Combined image and genomic analysis of high-grade serous ovarian cancer reveals PTEN loss as a common driver event and prognostic classifier. Genome Biol 2014;15(12):526 doi 10.1186/s13059-014-0526-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002;111(1):29–40 doi 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 40.Macintyre G, Goranova TE, De Silva D, Ennis D, Piskorz AM, Eldridge M, et al. Copy number signatures and mutational processes in ovarian carcinoma. Nat Genet 2018;50(9):1262–70 doi 10.1038/s41588-018-0179-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baloch T, Lopez-Ozuna VM, Wang Q, Matanis E, Kessous R, Kogan L, et al. Sequential therapeutic targeting of ovarian Cancer harboring dysfunctional BRCA1. BMC Cancer 2019;19(1):44 doi 10.1186/s12885-018-5250-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 2014;41(1):14–20 doi 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reinartz S, Schumann T, Finkernagel F, Wortmann A, Jansen JM, Meissner W, et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int J Cancer 2014;134(1):32–42 doi 10.1002/ijc.28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res 2005;11(18):6713–21 doi 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 45.Deshmukh SK, Tyagi N, Khan MA, Srivastava SK, Al-Ghadhban A, Dugger K, et al. Gemcitabine treatment promotes immunosuppressive microenvironment in pancreatic tumors by supporting the infiltration, growth, and polarization of macrophages. Sci Rep 2018;8(1):12000 doi 10.1038/s41598-018-30437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol 2015;6:187 doi 10.3389/fimmu.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun 2013;45:49–57 doi 10.1016/j.jaut.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017;170(6):1120–33 e17 doi 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017;545(7652):60–5 doi 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019;11(1):34 doi 10.1186/s13073-019-0638-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12(5):453–7 doi 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawrenson K, Fonseca MAS, Liu AY, Segato Dezem F, Lee JM, Lin X, et al. A Study of High-Grade Serous Ovarian Cancer Origins Implicates the SOX18 Transcription Factor in Tumor Development. Cell Rep 2019;29(11):3726–35 e4 doi 10.1016/j.celrep.2019.10.122. [DOI] [PubMed] [Google Scholar]
- 53.Hao D, Li J, Jia S, Meng Y, Zhang C, Wang L, et al. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin Cancer Res 2017;23(23):7400–11 doi 10.1158/1078-0432.CCR-17-0638. [DOI] [PubMed] [Google Scholar]
- 54.Coscia F, Watters KM, Curtis M, Eckert MA, Chiang CY, Tyanova S, et al. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat Commun 2016;7:12645 doi 10.1038/ncomms12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szabova L, Yin C, Bupp S, Guerin TM, Schlomer JJ, Householder DB, et al. Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res 2012;72(16):4141–53 doi 10.1158/0008-5472.CAN-11-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lohmussaar K, Kopper O, Korving J, Begthel H, Vreuls CPH, van Es JH, et al. Assessing the origin of high-grade serous ovarian cancer using CRISPR-modification of mouse organoids. Nat Commun 2020;11(1):2660 doi 10.1038/s41467-020-16432-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karnezis AN, Cho KR. Of mice and women - Non-ovarian origins of “ovarian” cancer. Gynecol Oncol 2017;144(1):5–7 doi 10.1016/j.ygyno.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 58.Karnezis AN, Cho KR, Gilks CB, Pearce CL, Huntsman DG. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer 2017;17(1):65–74 doi 10.1038/nrc.2016.113. [DOI] [PubMed] [Google Scholar]
- 59.Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep 2018;25(11):2972–80 e5 doi 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clevers H Modeling Development and Disease with Organoids. Cell 2016;165(7):1586–97 doi 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- 61.Neal JT, Kuo CJ. Organoids as Models for Neoplastic Transformation. Annu Rev Pathol 2016;11:199–220 doi 10.1146/annurev-pathol-012615-044249. [DOI] [PubMed] [Google Scholar]
- 62.Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer 2018;18(7):407–18 doi 10.1038/s41568-018-0007-6. [DOI] [PubMed] [Google Scholar]
- 63.Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin Cancer Res 2018;24(13):3163–75 doi 10.1158/1078-0432.CCR-18-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yi J, Liu C, Tao Z, Wang M, Jia Y, Sang X, et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019;43:225–37 doi 10.1016/j.ebiom.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013;341(6144):395–9 doi 10.1126/science.1236188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007;128(1):157–70 doi 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 67.Gregory PE, Gutmann DH, Mitchell A, Park S, Boguski M, Jacks T, et al. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat Cell Mol Genet 1993;19(3):265–74 doi 10.1007/bf01233074. [DOI] [PubMed] [Google Scholar]
- 68.Xu H, Gutmann DH. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain Res 1997;759(1):149–52 doi 10.1016/s0006-8993(97)00328-4. [DOI] [PubMed] [Google Scholar]
- 69.Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013;504(7479):296–300 doi 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 70.Kampan NC, Madondo MT, McNally OM, Quinn M, Plebanski M. Paclitaxel and Its Evolving Role in the Management of Ovarian Cancer. Biomed Res Int 2015;2015:413076 doi 10.1155/2015/413076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boyd LR, Muggia FM. Carboplatin/Paclitaxel Induction in Ovarian Cancer: The Finer Points. Oncology (Williston Park) 2018;32(8):418–20, 22–4. [PubMed] [Google Scholar]
- 72.Rodriguez GM, Galpin KJC, McCloskey CW, Vanderhyden BC. The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers (Basel) 2018;10(8):E242 doi 10.3390/cancers10080242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farkkila A, Gulhan DC, Casado J, Jacobson CA, Nguyen H, Kochupurakkal B, et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat Commun 2020;11(1):1459 doi 10.1038/s41467-020-15315-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10(9):942–9 doi 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 75.Stumpf M, Hasenburg A, Riener MO, Jutting U, Wang C, Shen Y, et al. Intraepithelial CD8-positive T lymphocytes predict survival for patients with serous stage III ovarian carcinomas: relevance of clonal selection of T lymphocytes. Br J Cancer 2009;101(9):1513–21 doi 10.1038/sj.bjc.6605274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol 2012;124(2):192–8 doi 10.1016/j.ygyno.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 2003;348(3):203–13 doi 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- 78.Bronger H, Singer J, Windmuller C, Reuning U, Zech D, Delbridge C, et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer 2016;115(5):553–63 doi 10.1038/bjc.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol 2017;17(9):559–72 doi 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Negus RP, Stamp GW, Hadley J, Balkwill FR. Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol 1997;150(5):1723–34. [PMC free article] [PubMed] [Google Scholar]
- 81.Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 2019;35(6):885–900 e10 doi 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Drakes ML, Stiff PJ. Regulation of Ovarian Cancer Prognosis by Immune Cells in the Tumor Microenvironment. Cancers (Basel) 2018;10(9):E302 doi 10.3390/cancers10090302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015;27(4):462–72 doi 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016;7(12):13587–98 doi 10.18632/oncotarget.7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Clarke B, Tinker AV, Lee CH, Subramanian S, van de Rijn M, Turbin D, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol 2009;22(3):393–402 doi 10.1038/modpathol.2008.191. [DOI] [PubMed] [Google Scholar]
- 86.Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan B, Miura Y, et al. Differences in Tumor Microenvironment Dictate T Helper Lineage Polarization and Response to Immune Checkpoint Therapy. Cell 2019;179(5):1177–90 e13 doi 10.1016/j.cell.2019.10.029. [DOI] [PubMed] [Google Scholar]
- 87.Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov 2016;6(1):80–95 doi 10.1158/2159-8290.CD-15-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer 2018;18(3):139–47 doi 10.1038/nrc.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu JF, Gordon M, Veneris J, Braiteh F, Balmanoukian A, Eder JP, et al. Safety, clinical activity and biomarker assessments of atezolizumab from a Phase I study in advanced/recurrent ovarian and uterine cancers. Gynecol Oncol 2019;154(2):314–22 doi 10.1016/j.ygyno.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 91.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol 2015;33(34):4015–22 doi 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 92.Ghisoni E, Imbimbo M, Zimmermann S, Valabrega G. Ovarian Cancer Immunotherapy: Turning up the Heat. Int J Mol Sci 2019;20(12):E2927 doi 10.3390/ijms20122927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, et al. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 2019;79(2):311–9 doi 10.1158/0008-5472.CAN-18-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elliott LA, Doherty GA, Sheahan K, Ryan EJ. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front Immunol 2017;8:86 doi 10.3389/fimmu.2017.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering myeloid-derived suppressor cells: isolation and markers in humans, mice and non-human primates. Cancer Immunol Immunother 2019;68(4):687–97 doi 10.1007/s00262-019-02302-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koo BK, Stange DE, Sato T, Karthaus W, Farin HF, Huch M, et al. Controlled gene expression in primary Lgr5 organoid cultures. Nat Methods 2011;9(1):81–3 doi 10.1038/nmeth.1802. [DOI] [PubMed] [Google Scholar]
- 97.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A 2007;104(29):12111–6 doi 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci U S A 2011;108(16):6468–73 doi 10.1073/pnas.1005529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cybulska P, Stewart JM, Sayad A, Virtanen C, Shaw PA, Clarke B, et al. A Genomically Characterized Collection of High-Grade Serous Ovarian Cancer Xenografts for Preclinical Testing. Am J Pathol 2018;188(5):1120–31 doi 10.1016/j.ajpath.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 100.Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018;49(1):178–93 e7 doi 10.1016/j.immuni.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014;30(15):2114–20 doi 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29(1):15–21 doi 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014;30(7):923–30 doi 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 104.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korotkevich GSV, and Sergushichev A. Fast gene set enrichment analysis. bioRxiv 2019; 10.1101/060012. [DOI]
- 106.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16(5):284–7 doi 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60 doi 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015;31(12):2032–4 doi 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303 doi 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 2012;28(3):423–5 doi 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequence data have been deposited in the GEO database under the accession code GSE147276. sWGS data were deposited in SRA under BioProject accession number PRJNA613661. All other data supporting the findings of this study are available within the article, the Supplemental information files, or the corresponding author upon request.