

Abstract
The Anopheles gambiae complex consists of multiple morphologically indistinguishable mosquito species including the most important vectors of the malaria parasite Plasmodium falciparum in sub-Saharan Africa. Nine cryptic species have been described so far within the complex. The ecological, immunological, and reproductive differences among these species will critically impact population responses to disease control strategies and environmental changes. Here we examine whole-genome sequencing data from a longitudinal study of putative A. coluzzii in western Burkina Faso. Surprisingly, many specimens are genetically divergent from A. coluzzii and all other Anopheles species and represent a new taxon, here designated Anopheles TENGRELA (AT). Population genetic analysis suggests that the cryptic GOUNDRY subgroup, previously collected as larvae in central Burkina Faso, represents an admixed population descended from both A. coluzzii and AT. AT harbors low nucleotide diversity except for the 2La inversion polymorphism which is maintained by overdominance. It shows numerous fixed differences with A. coluzzii concentrated in several regions reflecting selective sweeps, but the two taxa are identical at standard diagnostic loci used for taxon identification and thus AT may often go unnoticed. We present an amplicon-based genotyping assay for identifying AT which could be usefully applied to numerous existing samples. Misidentified cryptic taxa could seriously confound ongoing studies of Anopheles ecology and evolution in western Africa, including phenotypic and genotypic surveys of insecticide resistance. Reproductive barriers between cryptic species may also complicate novel vector control efforts, for example gene drives, and hinder predictions about evolutionary dynamics of Anopheles and Plasmodium.
Keywords: Anopheles, vector, cryptic taxa, admixture, selective sweep, reproductive barrier
Introduction
Successfully controlling vector-borne diseases will require a comprehensive evolutionary genetic understanding of host species. For malaria, a major global infectious disease afflicting over 200 million people, the relevant vectors are Anopheles mosquitoes which transmit Plasmodium parasites (Miller, 2002; WHO, 2018). Mosquito-targeting interventions are by far the most effective at reducing malaria infection, substantially exceeding the impact of those that target the parasite directly, like artemisinin combination therapy (Bhatt et al., 2016). However, evolutionary processes such as selection for resistance to insecticides have repeatedly allowed mosquitoes to evade control efforts (Ranson & Lissenden, 2016). Similarly, adaptive resistance is likely to frustrate new control technologies such as gene drives, which involve manipulating mosquito evolution directly (Marshall et al., 2019). Control strategies will need to anticipate and foil these adaptive responses and thus can only succeed if Anopheles population genetics is thoroughly understood.
In sub-Saharan Africa, the most important definitive hosts for Plasmodium are mosquitoes of the Anopheles gambiae species complex. These mosquitoes are among the most genetically diverse animals on Earth (Leffler et al., 2012; Anopheles gambiae 1000 Genomes Consortium, 2017). The clade contains nine morphologically identical species, three of which were only described in the last decade (Coetzee et al., 2013; Barrón et al., 2019). The evolutionary relationships among these cryptic species are complicated due to incomplete lineage sorting and introgression facilitated by porous reproductive barriers (Fontaine et al., 2015). A majority of the genome can cross species boundaries, and this is a frequent and recent phenomenon in response to novel selective pressures such as insecticides (Clarkson et al., 2014; Main et al., 2015; Norris et al., 2015). Additionally, the taxonomic status of some rare yet distinct groups within this complex remains unclear. The subgroup GOUNDRY was identified in Burkina Faso and found to be genetically distinct from its closest known relative, A. coluzzii (Riehle et al., 2011; Crawford et al., 2015; Crawford et al., 2016). While GOUNDRY has not been formally described as a separate species, it is genetically distinct from any known species. Further characterization has been impeded because only larval stages have been collected in the field and collecting additional GOUNDRY individuals has proven difficult. There is a substantial need to better understand patterns of gene flow and partitioning of genetic diversity within the A. gambiae complex, in order to better predict and mitigate the inevitable evolutionary counterstrategies to vector control efforts.
In this paper, we use whole genome sequencing data to identify yet another new cryptic taxon within the A. gambiae complex, occurring in a country (Burkina Faso) where many previous surveys of anopheline mosquitoes have occurred. Though this taxon is closely related to A. coluzzii, it shows substantial yet incomplete reproductive incompatibility with it. This new taxon, Anopheles TENGRELA (AT), clarifies the origin of GOUNDRY and illuminates the complicated interplay between migration and isolation that characterizes these mosquitoes.
Materials and Methods
Samples and sequencing
We chose 287 specimens of putative A. coluzzii from larval collections in Tengrela, Burkina Faso (10.7° N, 4.8° W) (Table 1). All specimens were reared to adults and typed as A. coluzzii females based on morphology (Gillies & De Meillon, 1968) and standard molecular assays (Santolamazza et al, 2008). They comprised a longitudinal series across four years (2011, 2012, 2015, and 2016) which were examined as part of a study on insecticide resistance evolution. We extracted DNA from individual mosquitoes using a Qiagen DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s instructions. We sequenced whole genomic DNA with 151 bp paired-end reads on an Illumina HiSeq X instrument at the Broad Institute, using Nextera low-input sequencing libraries.
Table 1.
Samples examined.
| Data Source | Year | Sampling | Taxon | N |
|---|---|---|---|---|
| This study: whole genome sequencing | 2011 (N = 72) | Tengrela (southwestern Burkina Faso): puddles and/or rice paddies | AT | 51 |
| A. coluzzii | 20 | |||
| Unidentified | 1 | |||
| This study: whole genome sequencing | 2012 (N = 71), 2015 (N = 72), 2016 (N = 72) | Tengrela (southwestern Burkina Faso): rice paddies | AT | 0 |
| A. coluzzii | 208 | |||
| Unidentified | 7 | |||
| This study: amplicon genotyping | Various / unknown | Bounouna or Nafona (southwestern Burkina Faso) or unknown | AT | 0 |
| A. coluzzii | 79 | |||
| Unidentified | 3 | |||
| Crawford et al., 2016: whole genome sequencing | 2007–2008 | Central Burkina Faso | GOUNDRY | 12 |
| A. coluzzii | 10 |
Identification of AT
All reads were aligned to the Anopheles gambiae PEST reference genome (assembly AgamP4; Holt et al., 2002; Sharakhova et al., 2007) using bwamem v. 0.7.17 (Li & Durbin, 2009; command: bwa mem -M) and samtools v .1.8 (Li 2011; commands: samtools view -h -F 4 -b, samtools sort, samtools index) and variants were called using GATK v. 3.8–1 (McKenna et al., 2010; hard filtering of single nucleotide polymorphisms (SNPs) with QD < 5 and/or FS > 60, and indels with QD < 2 and/or FS > 200; --max-gaussians 4). Initial analysis was restricted to individuals with at least 8x median coverage, with variants filtered to have at least 8x coverage in all individuals, be at least 500 bp apart, and not show heterozygote excess in violation of Hardy-Weinberg expectations. With this dataset, AT was identified as distinct from A. coluzzii using principal component analysis (PCA) with the princomp function in R (v. 3.6.2; R Core Team, 2019). Lower-coverage individuals were subsequently designated as AT or A. coluzzii based on markers that differentiate these taxa in the high-coverage individuals; eight individuals (coverage 0–2x) could not be unambiguously assigned to taxon and were subsequently ignored, leaving 279 acceptable individuals.
Standard statistical tests and data visualization were performed in R (v. 3.6.2; R Core Team 2019). Phylogenetic analysis employed RAxML with -m GTRCAT (Stamatakis 2006). Anopheles genomes used in phylogenetic analysis (other than AT and A. coluzzii from Tengrela) were: A. coluzzii from Burkina Faso at locations (Bana, Pala, and Souroukoudinga) approximately intermediate between Tengrela and the GOUNDRY sites (ERS224009, ERS224023, ERS223804, ERS223963, ERS224782, ERS223946), A. gambiae (PEST reference genome, ERS223759, ERS224149, ERS223976, ERS224154, ERS224132, and ERS224151) A. arabiensis (SRR3715623 and SRR3715622), A. quadriannulatus (SRR1055286 and SRR1508190), A. bwambae (SRR1255391 and SRR1255390), A. melas (SRR561803 and SRR606147), and A. merus (SRR1055284). Annotation and analysis of key genomic regions and alleles was facilitated by VectorBase (Giraldo-Calderón et al., 2015) and the Ag1000G genomic resources (Anopheles gambiae 1000 Genomes Consortium, 2017).
Demographic analysis with GOUNDRY
In order to directly compare our specimens with GOUNDRY, we examined the previously published whole-genome sequences of GOUNDRY and A. coluzzii (Crawford et al., 2016; BioProject PRJNA273873). We then examined a representative dataset of 51 AT genomes, 51 A. coluzzii genomes from Tengrela, 12 GOUNDRY genomes, and 10 A. coluzzii genomes from GOUNDRY habitats (Kodougou and Goundry). To minimize artifacts due to read alignment, we trimmed our Tengrela reads to 100 bp to match the GOUNDRY data, and then aligned all reads to the PEST reference genome using bwamem and samtools as above and called genotypes jointly using bcftools v. 1.8 (Li 2011; commands: bcftools mpileup, bcftools call -m -Ov -v). For demographic analysis, we removed any variants with missing genotypes, as above we filtered variants to be at least 500 bp apart and to not show heterozygote excess in violation of Hardy-Weinberg expectations, we excluded sites in the 2La inversion, and we separated this jointly-called and filtered dataset into autosomes (84,550 sites) and X chromosome (9,212 sites).
We used this jointly called dataset for demographic analysis with dadi (Gutenkunsk, Hernandez, Williamson, & Bustamante, 2009). Our goal was to generate a plausible demographic model for these populations and in particular to test whether GOUNDRY derives from an admixture event between AT and A. coluzzii. We ran multiple models, including models without migration, without population size changes, or with only two periods of differing demographic parameters. Critically for this analysis, we ran models identical to the preferred model but with GOUNDRY originating entirely from either AT or A. coluzzii; rejecting these models thus demonstrates that GOUNDRY is admixed (Supp Table 1). These models estimate multiple parameters, the absolute values of which depend upon numerous assumptions that are challenging to validate. For example, heterozygosity per bp in our full dataset is 400x higher than in the filtered dataset because most variants are filtered, and we estimate that the full dataset represents 85% of the genome, with the rest occurring in long stretches (over 1kb) without called genotypes that may be inaccessible to our genotyping pipeline; thus we adjusted our estimate of nucleotide diversity upwards accordingly when inferring effective population size. However, this estimate is likely imprecise, as low-quality sites that were filtered away could overestimate heterozygosity if enriched for mismatched reads, or could underestimate heterozygosity if divergent reads failed to align to the reference genome, Similarly, we assumed a mutation rate of 3×10−9 (Keightley, Ness, Halligan, & Haddrill, 2014), and ten generations per year. In contrast to absolute values of these point estimates, the relative values among parameters (e.g. population size of AT versus A. coluzzii), and among model likelihoods, should be more robust. As above, we examined autosomal genotypes and X chromosome genotypes separately. To assess the robustness of our results, we ran dadi on datasets with different levels of filtration, such that the minimum distance among variants was 100, 200, 500, or 1000 bp; as with other analyses we present the 500 bp threshold as the default.
We confirmed the demographic results from dadi using the same jointly called dataset in other analyses. We conducted PCA as above. We also conducted an ADMIXTURE analysis (Alexander, Novembre, & Lange, 2009) with K ranging from 1 to 5, choosing the value of K with the lowest cross-validation error as recommended. Finally, we used TreeMix (Pickrell & Pritchard 2012) to generate a phylogeny among populations and test for migration. We first incorporated genotypes from 115 specimens of A. gambiae from Ag1000G, yielding a dataset with 49,645 autosomal and 997 X chromosome variants. We created a phylogeny with the possibility of migration (treemix function, -m 1 or -m 2, -root A. gambiae). We used the threepop and fourpop functions (-k 100) to calculate f3 and f4.
Genomic characterization of AT
Across the genome, population genetic statistics such as FST, π, Tajima’s D, and Dxy were calculated with Perl scripts incorporating the Perlymorphism scripts (Bio::PopGen, BioPerl version 1.007000; Stajich & Hahn, 2005). FST calculations followed the script FstPerSite.pl available at https://github.com/jacobtennessen/MalariaHallmarks/. Statistics were examined in overlapping sliding windows of 1 Mb or 100 kb. We identified putative selective sweep regions as those showing Tajima’s D under −2.5 and π under 0.0005. We considered “definitive differences” (i.e. fixed or nearly fixed alleles) between AT and A. coluzzii to be sites showing FST > 0.99. At several notable loci we directly compared genotypes in AT, GOUNDRY, and A. coluzzii (Table 2), including the intergenic region (IGS) of rRNA (Scott, Brogdon, & Collins, 1993; Fanello, Santolamazza, & della Torre, 2002), indels in SINE200 retrotransposon S200 X6.1 (Santolamazza et al., 2008), and “divergence island SNPs” showing nearly-fixed differences among previously described taxa (Lee et al., 2013). We identified numerous variants spanning over 20 Mb from 2L_20569357 to 2L_42087028 showing perfect linkage disequilibrium with each other in our data; we inferred that these variants represent the 2La inversion and used a set of a 70 such variants, all more than 1 kb apart, to genotype 2La in all individuals. Although all specimens were phenotyped as female, we examined coverage across the X and Y chromosomes in the reference genome in order to infer sex chromosome karyotype. X chromosome coverage approximately equal to autosomal coverage was interpreted as XX karyotype. Y chromosome coverage equal to or greater to autosomal coverage across the majority of the Y chromosome was interpreted to mean that sequence which is male-specific in PEST occurs in our female specimens. To infer whether such sequence could constitute a functional Y chromosome, we assessed coverage at sex-determining gene YG2.
Table 2.
Key genomic regions characterized in AT, GOUNDRY, and Tengrela A. coluzzii.
| Locus | Description | Chromosome | Position | Allele | AT Frequency | GOUNDRY Frequency | Tengrela Anopheles coluzzii Frequency |
|---|---|---|---|---|---|---|---|
| 2La | 22 Mb inversion | 2L | 20000000–42000000 | 2L+a (haplotype) | 48% | 70% | 3% |
| 2La2L+a (heterozygous genotype) | 73% (expect 50%; χ2 = 10.5; P = 0.001) | 31% (expect 42%; χ2 = 36.2; P < 0.0001) | 5% (expect 5%) | ||||
| Y-linked | Y-chromosome sequence in PEST, excluding sex-determining gene | Y | 50000–230000 | Y sequence present | 94% | 100% | 3% |
| Rdl | Insecticide resistance | 2L | 25429235 | SER (resistant) | 32% | 58% | 68% in 2011–2012; 38% in 2015–2016 |
| Kdr | Insecticide resistance | 2L | 2422652 | PHE (resistant) | 51% | 46% | 78% |
| CYP9K1 | Insecticide resistance | X | 15242000 | cyp-II (resistant?) | 100% | 67% | 11% |
| TEP1 | Plasmodium resistance | 3L | 11205000 | R (protective) | 0% | 12% | 98% |
| APL1A | Plasmodium resistance | 2L | 41271000 | APL1A2 (protective) | 15% | 17% | 80% |
| Xh | 1.5 Mb inversion | X | 8470000–10100000 (example SNP at 9361641) | Non-reference | 100% | 100% | 0% |
| Sweep region | Unknown phenotypic effect | 3R | 8900000–13300000 (example SNP at 13081325) | T (non-reference) | 100% | 88% | 0% |
| rDNA IGS | Intergenic region of ribosomal DNA (multiple copies) | UNK (sequence from Scott et al., 1993) | 580–581 | HhaI cut site (S-form specific; Fanello et al., 2002) | 0% | 8% | 0% |
| M/S diagnostic SNPs | Divergence island SNPs (Lee et al., 2013) | 2L | 209536, 1274353, 2430786, 2430915, 2431005 | M-form | 46–51% | 54–58% | 22–28% |
| 3L | 296897, 387877, 413944 | M-form | 99–100% | 100% | 100% | ||
| X | 20015634, 22105429, 22105860, 22497157,22750432, 22750572, 22944682 | M-form | 99–100% | 96% | 100% | ||
| S200 X6.1 | 230bp diagnostic indel | X | 22951000 | M-form insertion present | 100% | 100% | 100% |
| SNP within insertion | X | 22951586 | T (non-reference) | 100% | 100% | 1% |
We used several tactics to test for inbreeding and its potential effects. For all analyses including those mentioned above, we filtered out sites showing heterozygote excess in violation of Hardy-Weinberg equilibrium but not sites showing homozygote excess, since the former are more likely to be caused by alignment errors and the latter could be biologically real due to population structure. This filtering strategy could only bias the dataset toward excess homozygosity. To calculate the F coefficient, we used the -het function in PLINK (v1.90b3.32, Chang et al. 2015) treating AT, A. coluzzii, and GOUNDRY separately. A positive F coefficient is indicative of inbreeding. For all individuals, we calculated heterozygosity (H) in 1 Mb windows across the genome and looked for homozygosity tracts showing H ≤ 1e-06 (i.e. no more than one heterozygous polymorphism per Mb). To assess whether homozygosity tracts are genomic outliers or consistent with genome-wide levels of polymorphism, we examined the distribution of heterozygosity per genomic window per individual. We counted homozygous doubletons (i.e. homozygotes for an allele otherwise absent in the population) and tested whether these are enriched in homozygosity tracts. Finally, to test whether inbreeding would be likely to influence our results, we generated a perfectly homozygous set of autosomes in silico by selecting only a single allele per individual at all autosomal sites. Using this haploid dataset, we replicated our ADMIXTURE and dadi analyses.
Amplicon genotyping
We designed and tested an amplicon-based genotyping method to identify AT in additional samples. We selected 50 diagnostic SNPs and small indels, each with a non-reference allele that is fixed in all AT specimens but absent in Tengrela A. coluzzii and the Ag1000G data (Anopheles gambiae 1000 Genomes Consortium, 2017). For each of these, we designed a primer pair to amplify a PCR product 160 to 230 bp in size using BatchPrimer3 (You et al., 2008). We ensured that primers did not overlap common polymorphisms found in A. coluzzii or A. gambiae (Anopheles gambiae 1000 Genomes Consortium, 2017). We ordered primers with the Nextera Transposase Adapters sequences added to the 5′ end in order to eliminate the initial ligation step in library preparation (primer 1: 5′ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-[locus specific sequence]; primer 2: 5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-[locus specific sequence]).
Primers were tested individually and in various pools of primer pairs to amplify jointly in multiplex PCR using known samples of AT, A. coluzzii, and A. gambiae. We PCR-amplified 2ng of each DNA sample using the Veriti 96-well Fast Thermal Cycler (Applied Biosystems, Waltham, Massachusetts) with 12.5uL (62.5 U) of Multiplex PCR Master Mix (Qiagen, Hilden, Germany), 2.5 uL of the pre-mixed primer pool (200 nM), and 8 uL of nuclease-free water. The PCR conditions were as follows: 95 °C for 15 minutes; 30 cycles of 94 °C for 30 seconds and 60 °C for 90 seconds, 72 °C for 90 seconds; and 72 °C for 10 minutes. Initial confirmation of correctly sized amplicons was done by gel electrophoresis.
We performed a second round of PCR to attach i5 and i7 Illumina indices (Nextera XT Index Kit v2 set A; Illumina, San Diego, California) to the PCR amplicons. The second round PCR reaction included 5 uL of 1X Platinum SuperFi Library Amplification Master Mix (Thermo Fisher, Waltham, Massachusetts), 5 uL of the first round PCR product, and 1 uL of premixed i5/i7 index primers. The PCR conditions were as follows: 98 °C for 30 seconds; 10 cycles of 98 °C for 15 seconds, 60 °C for 30 seconds, and 72 °C for 30 seconds; and 72 °C for 1 minute. Confirmation of amplification was done by gel electrophoresis.
Amplicon libraries were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, California) at 1.8x with a final elution volume of 35 uL. Library concentrations were determined using Qubit HS DNA kit (Thermo Fisher, Waltham, Massachusetts). Library concentration and size was confirmed using the Agilent 2100 Bioanalyzer instrument. Libraries were pooled in equimolar concentration and diluted to 180 pmol. A 15% PhiX spike in was added to the diluted pool. We loaded 20uL of the final pool onto an Illumina iSeq 100 instrument at the Broad Institute per the manufacturer’s protocol and sequenced these with 151 bp paired-end reads.
Genotype could be inferred from the resultant reads without an alignment step, simply by counting reads containing diagnostic kmers (24–33bp) overlapping the target polymorphism. After determining that a pool of 5 primer pairs sequenced by iSeq was sufficient for taxon identification, we used it to amplify and sequence a novel set of 79 putative A. coluzzii larvae (from either near Tengrela in southwestern Burkina Faso or else of unknown origin; Table 1), alongside a known Tengrela A. coluzzii control and 16 empty well negative controls.
Results
A new cryptically isolated lineage
In whole-genome sequencing data of 279 putative A. coluzzii specimens from Tengrela, Burkina Faso (median coverage = 16x; dataset Tennessen et al., 2020), collected across four years, 51 specimens were unexpectedly distinct (Figure 1A). This signal is robust across both autosomes and the X chromosome, and for more stringent filtering approaches (Supp Figure 1). The divergent individuals, here designated Anopheles TENGRELA (AT), were all collected as larvae in 2011, the only year in which sample collection included temporary puddles in addition to rice paddies. Most (72%) specimens from 2011 were AT. AT is not a close match to any known sequenced samples of A. coluzzii or A. gambiae (Anopheles gambiae 1000 Genomes Consortium, 2017). It is similar but not identical to GOUNDRY, which was also described from temporary puddles in Burkina Faso, from sites 250–550 km from Tengrela (Riehle et al., 2011). The genetic divergence from sympatric A. coluzzii individuals collected simultaneously suggests a strong reproductive barrier.
Figure 1.
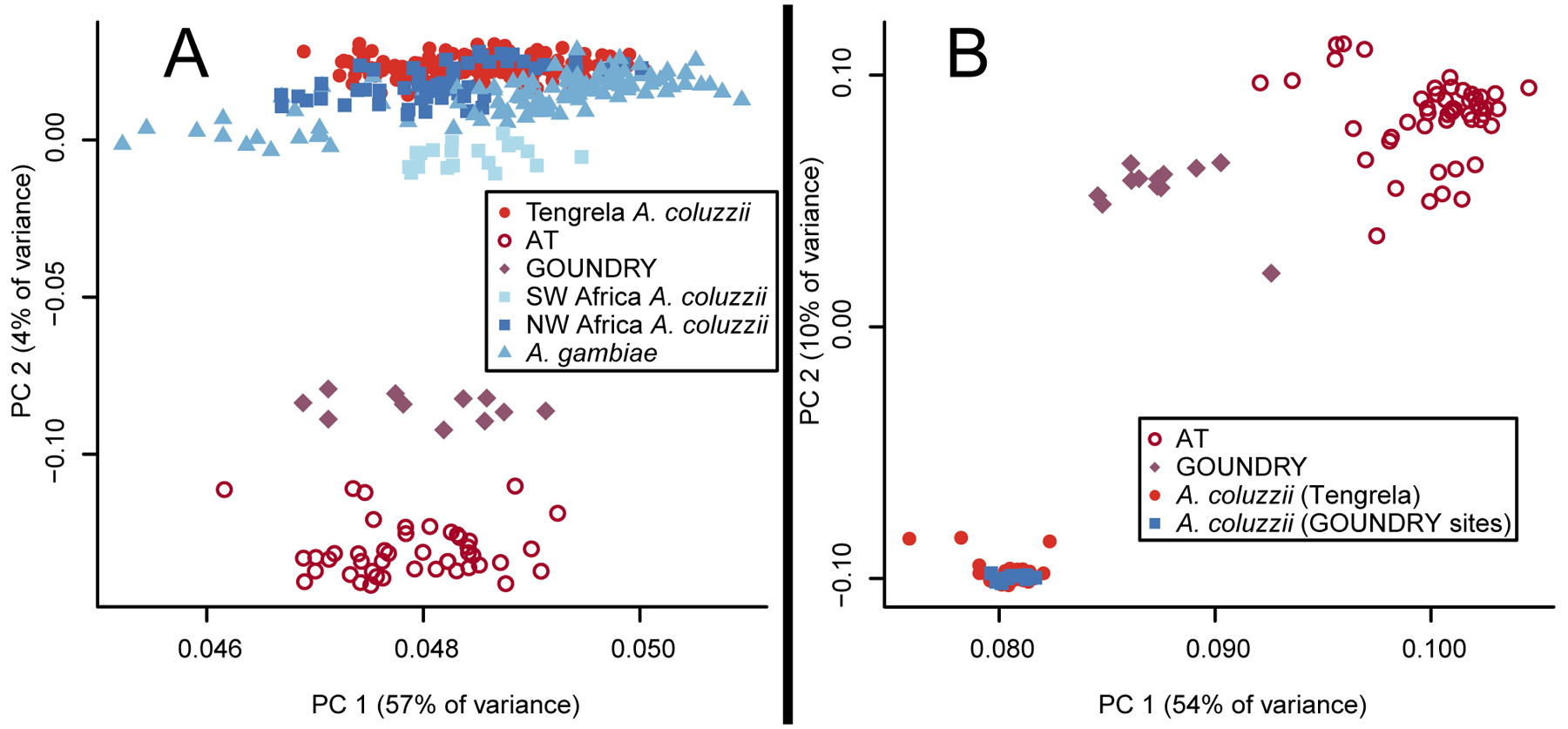
Genetic distinctiveness of AT. (A) In a PCA plot with Tengrela, GOUNDRY, and Ag1000G individuals, AT occurs as a distinct cluster close to GOUNDRY. Variants were chosen based on segregation in our data and may show ascertainment bias affecting the relationships of individuals within Ag1000G; the salient result is how AT and Tengrela A. coluzzii relate to these other individuals (B) AT remains distinct in a PCA after combining Tengrela samples with GOUNDRY and the A. coluzzii samples collected alongside GOUNDRY. To control for differences between studies, all reads were trimmed to the same length, and then alignment and genotyping were performed jointly.
To further investigate the distinction between AT and GOUNDRY, we jointly analyzed our Tengrela data alongside whole genome sequence data from GOUNDRY and sympatric A. coluzzii specimens (Crawford et al., 2016). To account for differences in sequencing platforms and genotyping algorithms, we trimmed all reads to 100 bp and jointly aligned them and called genotypes. While A. coluzzii from Tengrela and A. coluzzii from the GOUNDRY sites were genetically indistinguishable, AT and GOUNDRY remained distinct from each other (Figure 1B). Thus, the distinctiveness of AT is not owing to genotyping artifact, and the divergence between AT and GOUNDRY is greater than that seen for A. coluzzii populations over the same physical distance. We therefore infer that AT is not simply an additional population of GOUNDRY.
To examine the relationship between AT and the broader A. gambiae species complex, we divided the genome into 100 kb windows and ran a phylogenetic analysis with seven nominal species (Figure 2A). The AT genome is typically sister to A. coluzzii (42.1% of windows), A. gambiae (33.1% of windows), or to a clade containing only A. coluzzii and A. gambiae (20.4% of windows), with only 4.5% of windows displaying an alternate topology (Supp Figure 2). The relationship between AT and A. coluzzii is especially strong on the X chromosome (Figure 2A; Supp Figure 2), which in the A. gambiae complex is thought to reflect phylogenetic signal more accurately than the autosomes (Fontaine et al., 2015). As A. coluzzii is most often the closest species, and much of the similarity to A. gambiae is owing to the 2La inversion (Supp Figure 2), we consider A. coluzzii to be the sister taxon to AT for all subsequent analyses.
Figure 2.
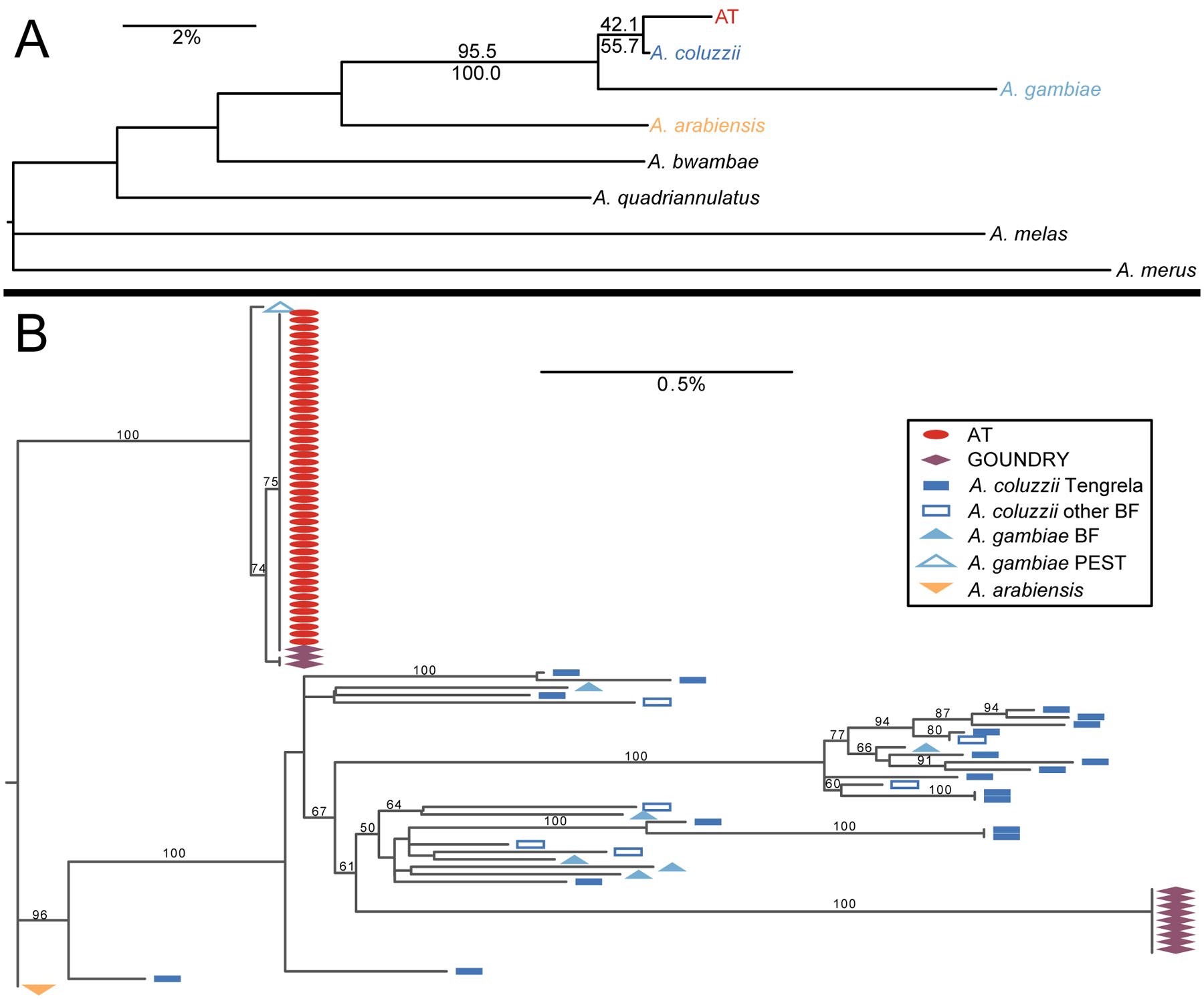
(A) Most common phylogenetic topology among sections of AT genome and nominal species of the A. gambiae complex. Numbers at branches are not bootstraps, but the percentage of 100 kb windows that support each clade (above branches: entire genome; below branches: X chromosome only). AT is sister to A. coluzzii across 42.1 % of the genome (55.7% of the X chromosome), more often than to any other species, and 95.5% of the genome (100.0% of the X chromosome) supports a clade with AT, A. coluzzii and A. gambiae to the exclusion of the other species. (B) The mitochondrial DNA phylogeny shows that AT shares a single haplotype that occupies a unique branch close to the A. gambiae PEST reference genome. GOUNDRY samples occur near AT or near A. coluzzii and A. gambiae samples from Tengrela and elsewhere in Burkina Faso (BF), consistent with an admixed origin.
We constructed a mitochondrial phylogeny of AT, GOUNDRY, and representative samples of A. coluzzii, A. gambiae, and A. arabiensis (Figure 2B). All AT individuals share the same haplotype, which does not occur in any other taxon except for a single GOUNDRY individual. GOUNDRY samples occur in two clades, the one containing AT and another one nested within A. coluzzii and A. gambiae, consistent with maternal ancestry from both lineages.
GOUNDRY derives from AT and A. coluzzii
GOUNDRY is closely related to AT, but genetically distinct (median FST = 0.11; mean FST = 0.12; Supp Figure 3). GOUNDRY also shows substantial recent ancestry from A. coluzzii.. Several independent analyses, described below, support the conclusion that GOUNDRY autosomes are admixed with 15–20% A. coluzzii ancestry, while GOUNDRY X chromosomes are almost entirely AT-like. There are relatively few fixed differences between GOUNDRY and AT, but several occur on all chromosomes including the X (Supp Figure 3).
Analysis of AT, A. coluzzii from Burkina Faso, and GOUNDRY autosomes with ADMIXTURE (Alexander et al., 2009) suggests two ancestral populations (K=2; Figure 3A). Populations 1 and 2 are closely approximated by the contemporary AT and A. coluzzii populations, respectively (AT: all individuals have 100.0% population 1 ancestry; A. coluzzii: mean population 1 ancestry = 0.1%, range = 0–7.4%). GOUNDRY is admixed from both populations, with a majority of ancestry from AT (mean population 1 ancestry = 84.6%, range = 76.6–88.7%). The X chromosome also suggests a two-population model differentiating contemporary AT and A. coluzzii, but it assigns GOUNDRY nearly complete population 1 (AT-like) ancestry (mean = 99.6%, range = 97.1–100%).
Figure 3:
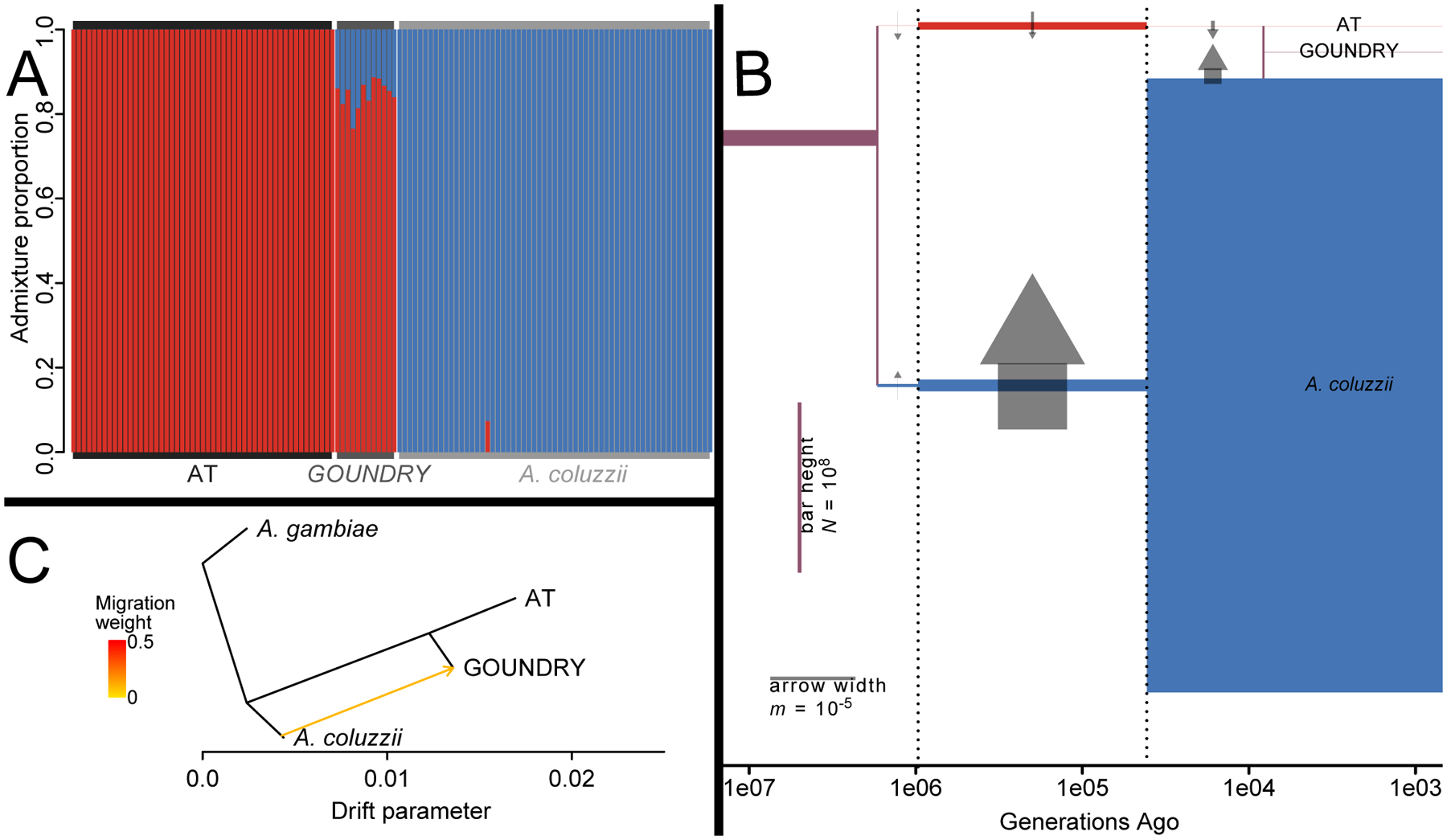
Relationships between AT, GOUNDRY, and A. coluzzii autosomes using jointly called genotypes. (A) Analysis with ADMIXTURE suggests two ancestral populations, closely approximated by contemporary AT and A. coluzzii, with GOUNDRY showing ancestry from both. (B) Analysis with dadi corroborates this model, with an AT/A. coluzzii split over one million generations ago, followed by ongoing gene flow and a recent admixed origin of GOUNDRY. Population sizes (heights of colored bars) and migration rates (widths of arrows) vary across three time periods (demarcated with dotted lines). (C) Analysis with TreeMix shows GOUNDRY as sister to AT but with in-migration from A. coluzzii.
We confirmed the signal of admixture by fitting demographic models to the two-dimensional site frequency spectrum with dadi (Gutenkunst et al., 2009). In our best-fitting autosomal model, the lineages of A. coluzzii and AT diverged 1.7 million generations ago, maintained a small degree of continuous gene flow, and then hybridized less than 10,000 generations ago to form GOUNDRY (Figure 3B; Supp Figure 4; Supp Table 1). Our model included three different time periods among which population sizes and migration rates were allowed to vary. Consistent with the relatively low nucleotide diversity, the effective population size (N) of AT was much smaller than A. coluzzii across all time periods. It was approximately 200-fold smaller in the earliest and longest period, expanded during the middle time period, and contracted again to be more than 10,000-fold smaller than A. coluzzii today (3.1×104 and 3.6×108, respectively). Migration rates (m) ranged from 10−8 to 10−5, such that the effective number of migrants per generation (m times N of the recipient population) has only sporadically been high enough to overcome the effects of drift (Nm > 1). In particular, gene flow from A. coluzzii into AT has only been non-negligible during the middle period 41,000–967,000 generations ago, when the AT population size was large and migration from A. coluzzii was substantial. GOUNDRY is admixed with 81.0% ancestry from AT and 19.0% ancestry from A. coluzzii, with N slightly larger than AT (4.1 × 104). All four filtration strategies for autosomal variants support the same overall scenario, with varying parameter estimates: the effective sizes of modern AT and GOUNDRY are 134 to 20,225 times smaller than A. coluzzii, AT has decreased in population size while A. coluzzii has increased, GOUNDRY is admixed with the proportion of A. coluzzii ancestry ranging from 13.7% to 30.7%, and the split between AT and A. coluzzii is 99 to 636 times older than the origin of GOUNDRY. In contrast, the best model for X chromosome data has GOUNDRY derived entirely from the AT branch without gene flow from A. coluzzii (Supp Table 1).
We further investigated the signal of admixture with TreeMix (Pickrell & Pritchard 2012), incorporating A. gambiae alongside A. coluzzii, AT, and GOUNDRY. Autosomal data suggest a tree with GOUNDRY sister to AT, but with relatively weak migration (weight = 0.10) from A. coluzzii to GOUNDRY (Figure 3C), and no support for any second migration event elsewhere in the tree. As corroboration, f4 is nonzero at 1.9e-04 (p < 0.05), indicating a significant but relatively weak signal of gene flow. However, f3 is positive for any of the four populations compared with any other two, meaning that this test provides no evidence for admixture. The X chromosome shows no evidence of GOUNDRY admixture, neither upon the phylogeny nor with f4 or f3.
Genomic characterization of AT
Mosquitoes in the A. gambiae complex are typically identified with established molecular markers. Analysis of these regions in AT reveals how this taxon easily goes undetected, both in our initial survey of these samples and possibly in other studies (Table 2). At IGS, AT reliably lacks the HhaI restriction site found in A. gambiae s. s., and instead harbors the AT dinucleotide typical of A. coluzzii (Fanello et al., 2002). At S200 X6.1, AT possesses the 230 bp insertion characteristic of A. coluzzii (Santolamazza et al., 2008). Interestingly, a SNP within this indel is nearly fixed between AT and A. coluzzii, suggesting that amplification of this region could still be diagnostic if it were sequenced and not merely assessed for band size. At divergence island SNPs, AT appears identical to Tengrela A. coluzzii, although such SNPs on chromosome 2L are polymorphic in both taxa.
In AT both haplotypes of the 2La inversion are common, unlike in A. coluzzii from Tengrela or elsewhere (Coluzzi et al., 2002; Neafsey et al., 2010), and thus this 22 Mb region represents one of its most striking differences from A. coluzzii (Figure 4A). The 2L+a haplotype occurs at a frequency of 48% and shows a remarkable heterozygote excess in violation of Hardy-Weinberg expectations (expected heterozygotes = 25.4, observed heterozygotes = 37; χ2 = 10.5; P = 0.001), suggesting at least half of homozygous genotypes are selected against. This observation is unexpected, since either the 2La or 2L+a haplotype can be the major allele across Anopheles populations, and thus the inversion is thought to be maintained by geographically-varying selection rather than heterozygote advantage (White et al., 2007). In contrast to AT, GOUNDRY 2La is in Hardy-Weinberg equilibrium (Riehle et al. 2011). Our results suggest that the genomic background of AT may facilitate overdominance at this inversion, and thus AT may be an important genetic reservoir which helps to maintain the polymorphism across the genus. Genes potentially under selection in 2La include the highly polymorphic APL1 gene complex, implicated in immunity against Plasmodium (Rottschaefer et al., 2011), and Rdl, at which a nonsynonymous Ala-Ser variant conveys resistance to the insecticide dieldrin (Du et sl. 2005). In APL1, the protective APL1A2 haplotype is rare in AT (15%) but is the major allele in Tengrela A. coluzzii (80%). At Rdl, resistant Ser occurs at 32% frequency in AT. In Tengrela A. coluzzii, this variant represents the largest shift in allele frequency across years (Supp Figure 5), decreasing from 68% in 2011–2012 to 38% in 2015–2016, consistent for selection against costly resistance as organochloride use has declined. While Rdl in A. coluzzii must occur on the (nearly fixed) 2La haplotype, in AT it is more often associated with 2L+a.
Figure 4.
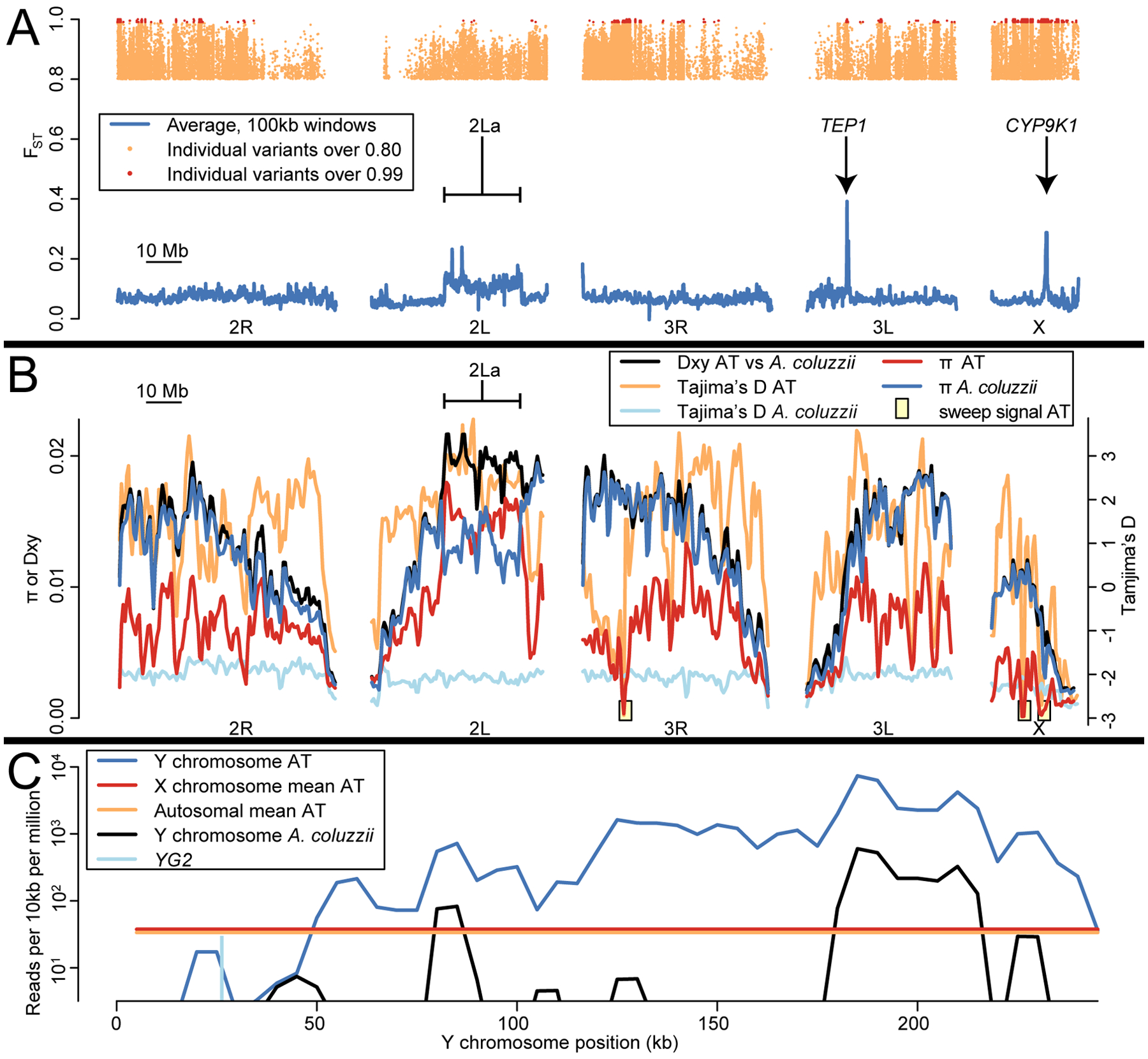
Unique genomic characteristics of AT. (A) FST across the genome between AT and Tengrela A. coluzzii. Tens of thousands of variants distributed across the genome are highly divergent between these taxa (FST > 0.8; orange dots at top), while over a thousand sites, concentrated in several clusters, are fixed or nearly so (“definitive differences”, FST > 0.99; red dots at top). Average FST in 100 kb windows is more modest (blue lines), but three regions stand out representing the 2La inversion, TEP1, and CYP9K1. (B) Nucleotide diversity (π), intertaxon divergence (Dxy), and site frequency spectra (Tajima’s D) in AT and A. coluzzii. Nucleotide diversity is low in AT except at the 2La inversion. Tajima’s D is mostly positive in AT, but three regions show low Tajima’s D, low π, and many high-FST sites (as shown in A), suggesting selective sweeps. (C) In most AT females, read coverage along most of the reference Y chromosome substantially exceeds the X/autosomal average. Coverage is negligible around sex-determining gene YG2. A. coluzzii females, in contrast, typically show negligible coverage across the entire Y except for a few repetitive sections.
Several other genomic regions are unusually divergent between AT and A. coluzzii (Table 2). Along with 2La, both TEP1 and CYP9K1 are outliers with respect to mean FST between these taxa (Figure 4A). TEP1 encodes a complement-like immunity protein that occurs in highly dissimilar allelic forms correlated with resistance to Plasmodium (White et al., 2011). All TEP1 alleles in AT are of the S (susceptible) type, while the R (resistant) type is nearly fixed in Tengrela A. coluzzii. CYP9K1 is a P450 gene associated with resistance to insecticide (Main et al., 2015). The cyp-II haplotype of the CYP9K1 region is fixed in AT and rare in Tengrela A. coluzzii; there is suggestive but inconclusive evidence that this allele conveys increased resistance (Main et al., 2015). In contrast to the pronounced divergence at CYP9K1, the well-known insecticide-resistance polymorphism Kdr (Donnelly et al., 2009) shows similar, intermediate frequencies across both AT and Tengrela A. coluzzii.
Nucleotide diversity is substantially lower in AT (π = 0.007) than in A. coluzzii (π = 0.012) (Figure 4B). This trend is consistent across the genome except within the 2La inversion (AT: π = 0.015 in 2La, π = 0.006 elsewhere; A. coluzzii: π = 0.013 in 2La, π = 0.012 elsewhere). This difference is also consistent among individuals, such that the range of heterozygosity per individual for AT does not overlap that of A. coluzzii (Supp Figure 6). Across the genome, divergence between these taxa as measured by Dxy closely matches π in A. coluzzii, as this taxon that contributes the majority of variation; the exception occurs in 2La, when both Dxy and AT π are maximized. The site frequency spectra of AT and A. coluzzii are also quite distinct. In A. coluzzii, Tajima’s D is very negative (D = −2.0) and fairly uniform across the genome (SD = 0.2), reflecting its recent population expansion (Figure 3B; Anopheles gambiae 1000 Genomes Consortium, 2017). In contrast, Tajima’s D in AT is positive on average (D = 1.3), consistent with a recent dramatic decrease in population size (Figure 3B), but it’s also quite variable across the genome (SD = 1.3). Three genomic regions in AT, comprising about 2% of the genome, show a combination of highly negative Tajima’s D and unusually low nucleotide diversity, and together they harbor a third of the “definitive differences” (defined here as FST > 0.99; Figure 4A) between AT and A. coluzzii. This suite of signals suggests positive selective sweeps in these three regions in AT (Figure 4B). One putative sweep is observed on 3R. Though the signal extends from approximately 8.9 Mb to 13.3 Mb, it is concentrated in a one-megabase section from 11.5 to 12.5 Mb which contains 125 definitive differences (7% of the genome-wide total) and also the lowest autosomal nucleotide diversity (π = 0.0003) and lowest Tajima’s D genome-wide (D = −2.9). Multiple genes occur in or near this region, with no obvious single selection target. Several of these genes have known phenotypic effects, including IR21a which encodes a thermoreceptor implicated in heat-mediated host-seeking (Greppi et al., 2020), and a cluster of cuticular proteins tied to insecticide resistance (Nkya et al., 2014; Huang et al., 2018). The other two selective sweep signals occur on the X chromosome, which even outside of these regions shows lower nucleotide diversity overall than the autosomes. The putative inversion Xh between 8.5 to 10.0 Mb, which also shows a sweep signal in GOUNDRY (Crawford et al., 2016), contains 15% of all definitive differences with A. coluzzii and has the lowest nucleotide diversity in the AT genome (π =0.0001). The third signal overlaps CYP9K1 on the X between 13.5 to 16.0 Mb. It accounts for 10% of all definitive differences and also shows low nucleotide diversity (π = 0.0004). These two sweep regions on X show the lowest Tajima’s D values in the genome outside of the 3R sweep region (Xh vicinity D = −2.6; CYP9K1 vicinity D = −2.7).
A final major genomic feature of AT concerns the Y chromosome. All AT individuals were morphologically typed as female and confirmed as such due to coverage similarity between the X chromosome and autosomes. However, 94% of them showed high coverage across the majority of the Y chromosome, except for the first 50 kb which includes the sex-determining region and sex-determining gene YG2 (Figure 4C; Table 2). For most of the Y chromosome after 50 kb, coverage exceeded the autosomal and X-chromosome averages by over an order of magnitude, implying that this sequence occurs repeatedly. This pattern is also observed in GOUNDRY, but not in A. coluzzii from Tengrela (Table 2). The most likely explanation is that multiple copies of most of the Y chromosome have merged with an autosome or the X chromosome in AT, consistent with the highly repetitive and dynamic nature of the Anopheles Y (Hall et al., 2016).
Given the signal of inbreeding reported in GOUNDRY (Crawford et al. 2016), we examined AT extensively for inbred individuals. There is no overall homozygote excess in AT. The F coefficient of inbreeding is close to zero and slightly negative on average (mean = −0.035, median = −0.027). As in GOUNDRY, we observe large tracts of homozygosity in most individuals (Figure 5A). However, regions of low heterozygosity are to be expected when overall nucleotide diversity in the population is low (Figure 4B), so this is not necessarily a signal of inbreeding. Heterozygosity does not show a bimodal distribution (i.e. distinct regions of high or low heterozygosity), but rather most low heterozygosity regions appear to be the tail end of a continuous distribution (Figure 5B). Furthermore, fewer than 0.01% of variants are homozygous doubletons, and these are not enriched in homozygosity tracts (5 doubletons are in tracts with mean heterozygosity ≤ 1e-06; expect 7.7; p > 0.1). Finally, our haploid dataset demonstrated that our demographic conclusions are robust to inbreeding. For example, in ADMIXTURE a two-population model is still favored, separating AT from A. coluzzii, with GOUNDRY showing 78.5–91.3% AT-like ancestry. Similarly, in dadi an admixed model is favored with GOUNDRY showing 85.4% ancestry from AT (Supp Table 1).
Figure 5.
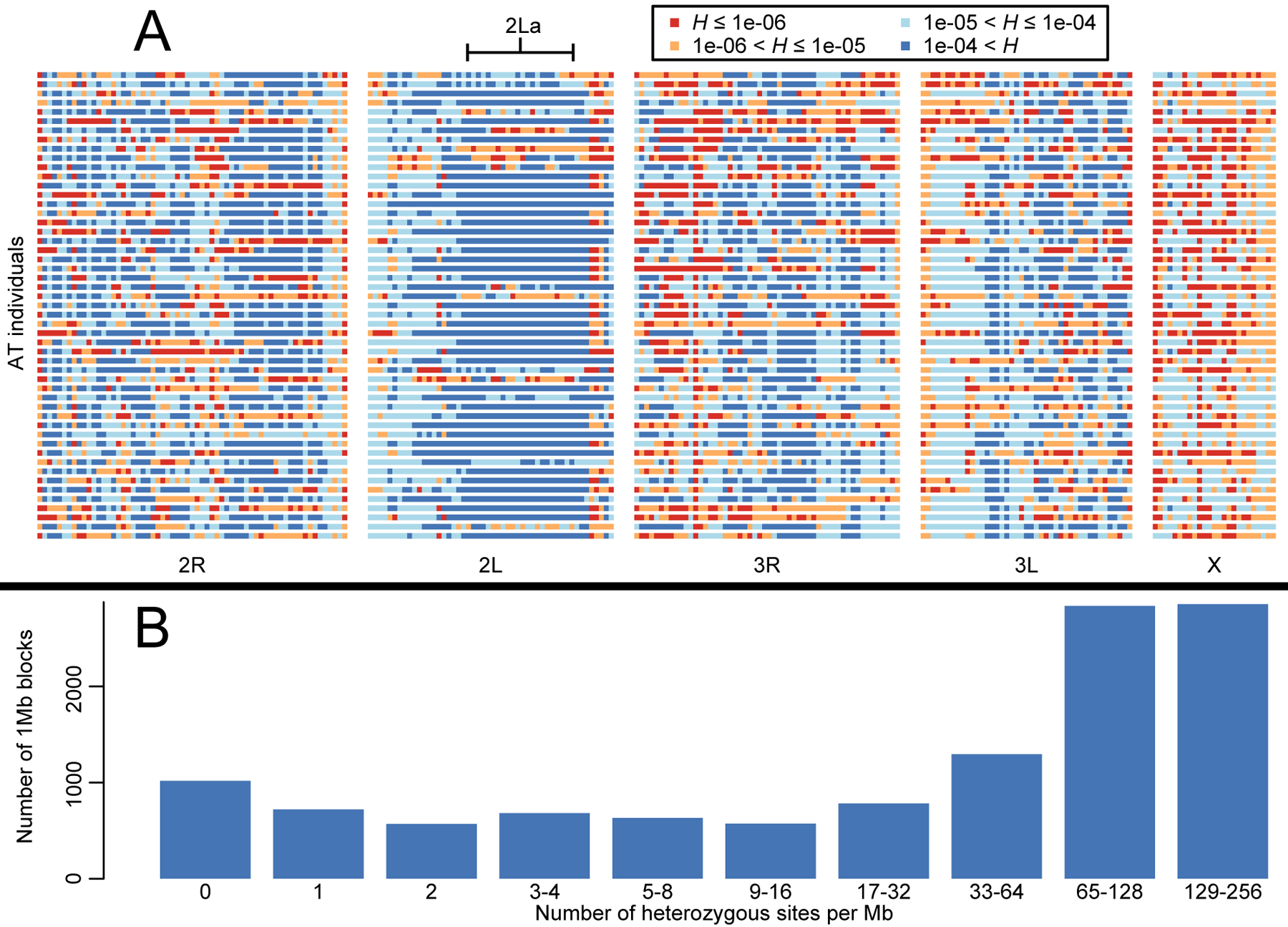
Regions of low heterozygosity in AT. (A) Heterozygosity (H) in blocks of 1 Mb across the genome for all 51 AT individuals. There are many long homozygosity tracts (red) in most individuals, but these are consistent with the relatively low genetic diversity observed across the population, except in the 2La inversion. (B) Histogram of the heterozygosity blocks depicted in A, based on number of heterozygous sites. Heterozygosity shows a relatively smooth distribution with only a slight uptick for homozygous blocks (0 or 1 heterozygous sites), indicating little evidence for inbreeding driving homozygosity.
A diagnostic protocol
In order to search for AT among additional samples, we developed a diagnostic protocol based on amplicon sequencing. We first identified 50 diagnostic SNPs and small indels that distinguish AT from Tengrela A. coluzzii and the Ag1000G samples (Supp Table 2; Supp Figure 7). We tested several pools of primer pairs and found that a pool of five primer pairs could be jointly amplified and sequenced to sufficient coverage on an Illumina iSeq 100. At these loci, the diagnostic allele is fixed in AT, polymorphic in GOUNDRY (33–63% frequency), and absent in A. coluzzii and A. gambiae. We genotyped each locus by searching the reads for kmers overlapping the target polymorphism. In a set of 10 AT specimens and 10 Tengrela A. coluzzii specimens, each locus yielded an unambiguous genotype in every specimen (median number of read pairs per locus per specimen = 7905; mean = 9047; range = 954 to 25,180; Figure 6). Specifically, for all loci at all AT specimens, more than 90% of read pairs had the AT allele, and for all loci at all A. coluzzii specimens, more than 90% of read pairs had the A. coluzzii allele. The small numbers of incorrect reads could be due to index hopping among these jointly-sequenced samples (van der Valk, Vezzi, Ormestad, Dalén, & Guschanski, 2020) or low-level contamination. Coverage at the negative control was lower but non-negligible (76 to 1460 read pairs per locus, mean = 411.6), presumably due to similar errors. However, for four out of five negative control loci, both alleles occurred in more than 20% of read pairs. These results suggest that taxonomy can be confidently assigned and false positives avoided if two filters are applied. First, specimens should be excluded if they have fewer than 10% of the expected number of reads pairs (i.e. fewer than about 4000 read pairs for a typical iSeq lane with 96 individuals). Second, for each specimen, a majority of loci should implicate the same taxon with at least 90% of reads. Either one of these filters would exclude our negative control.
Figure 6.
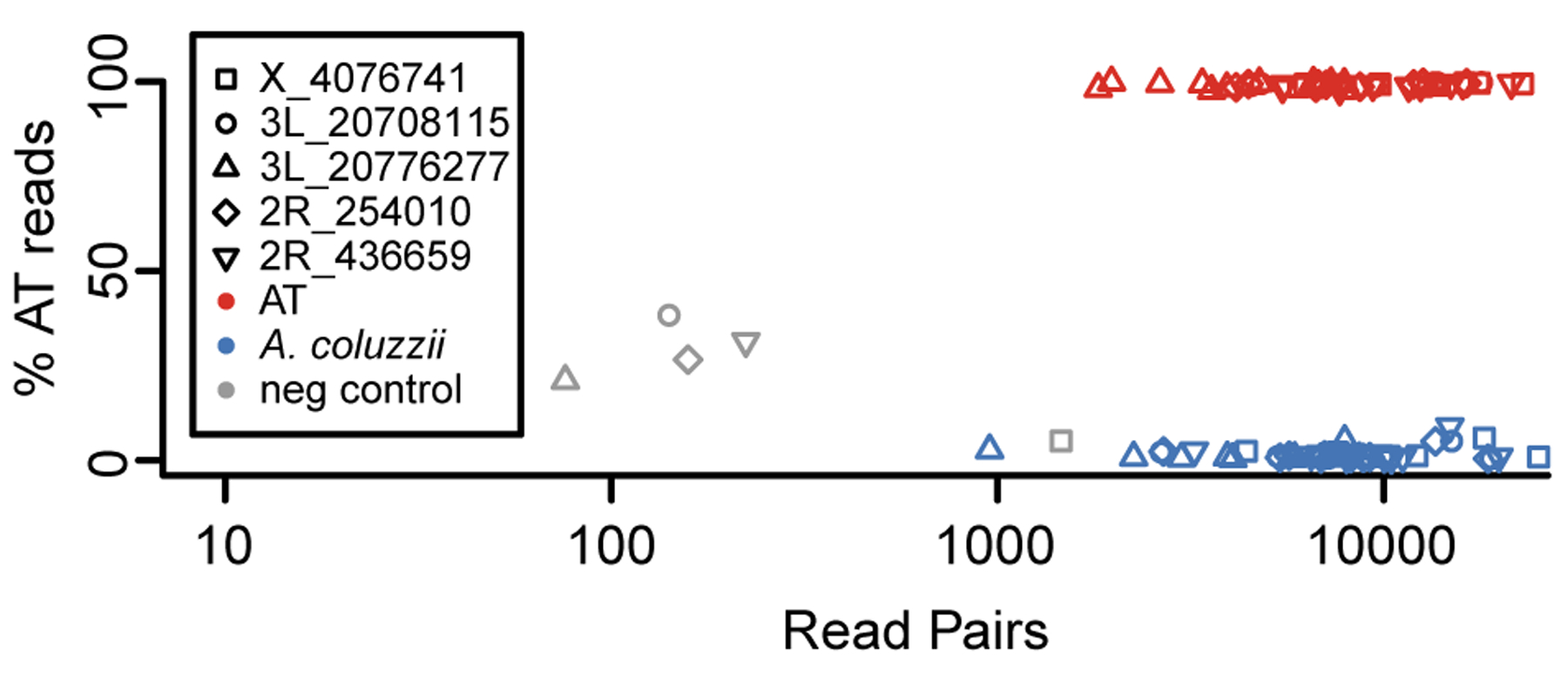
Distinguishing AT from A. coluzzii using amplicon genotyping. A pool of five primer pairs were selected that amplify five polymorphisms diagnostic for AT, and jointly amplified PCR products were sequenced (10 AT, 10 A. coluzzii, and one negative control). For each marker, the vast majority of read pairs are consistent with the known taxonomic category as inferred from whole genome sequencing, allowing for unambiguous identification. A small number of read pairs were erroneously assigned to the negative control (“neg”), indicating that a conservative test should exclude any individual with unusually low coverage and/or intermediate frequencies of both alleles.
We then used this pool of five primer pairs to amplify and sequence a novel set of 79 putative A. coluzzii specimens. All negative controls, and three real specimens, had fewer than 4000 read pairs each and were discarded by the criteria outlined above. The remaining 76 specimens had acceptable coverage (median number of read pairs per locus per specimen = 1799; mean = 3466; range = 12 to 25,357) and were all genotyped as unambiguously A. coluzzii (maximum observed proportion of read pairs with AT allele: 2.6%). Thus, we failed to detect AT in this novel sample set.
Discussion
We present a novel taxon within the Anopheles gambiae species complex, Anopheles TENGRELA (AT). AT is related to but genetically distinct from sympatric A. coluzzii and shows numerous fixed or nearly-fixed differences across the genome, as well as large difference in genomic architecture such as the 2La inversion polymorphism, the fixed Xh inversion, and the presence of Y-chromosome-associated sequence in females. These differences are presumably maintained by strong reproductive barriers. However, reproductive isolation is not complete, as our demographic analysis indicated ongoing gene flow and a hybridization event within the last thousand years (Figure 3). Such occasional gene flow is typical among nominal species of the A. gambiae complex (Fontaine et al., 2015; Main et al., 2015). Lacking adequate phenotypic and ecological data, we refrain from formally describing AT. However, it represents a unique lineage and is not nested within A. coluzzii nor any other described species.
AT is most similar to GOUNDRY, another aberrant Anopheles population from Burkina Faso. Our results indicate that GOUNDRY is an admixed population with most of its ancestry originating from an AT-like lineage, but with substantial A. coluzzii ancestry as well. GOUNDRY autosomes show 15–20% A. coluzzii ancestry, while there is no evidence of A. coluzzii ancestry on the X chromosome, which in the A. gambiae complex is typically more impervious to introgression (Fontaine et al. 2015). Some GOUNDRY mitochondrial haplotypes are AT-like but most occur in a distinct clade closer to A. coluzzii and A. gambiae (Figure 2B). There is no evidence for reproductive barriers between AT and GOUNDRY; rather, GOUNDRY appears to be a relatively new population recently diverged from the AT lineage via considerable introgression from A. coluzzii. GOUNDRY is known to segregate for both S-form and M-form markers (Riehle et al. 2011), but the GOUNDRY genomes examined here resemble A. coluzzii (M-form) at diagnostic markers (Table 2). Thus, it is possible that GOUNDRY has a more complex history involving other ancestral lineages, beyond our ability to assess with the available data. The taxonomic status of AT and GOUNDRY remains unresolved, but these two populations are not synonymous, and AT appears to be more representative of the ancestral phylogenetic lineage.
Our demographic model suggests that AT and A. coluzzii diverged 1.7 million generations ago, or 170,000 years ago assuming ten generations per year. This result depends on numerous estimates, including mutation rate and the proportion of the genome genotyped accurately, and is therefore imprecise. In general, we endeavored to be conservative in our estimate of differentiation between AT and A. coluzzii. For example, we assumed no effect of purifying selection on the variants used in demographic analysis, but if purifying selection has acted it would mean the true time since divergence of AT and A. coluzzii is even greater than estimated here. Regardless, our results suggest that cladogenesis predates the rise of agriculture in sub-Saharan Africa and was not driven by adaptation to anthropogenically-disturbed habitats. The population sizes of both lineages increased following this split, bolstered by cross-lineage migration. Then approximately 4,000 years ago AT decreased dramatically in population size while A. coluzzii increased further as is well documented (Anopheles gambiae 1000 Genomes Consortium, 2017), leading to a modern A. coluzzii population thousands of times larger than AT. If effective population size today approximates census size, the relative rarity of AT could partially explain why it has not been detected previously. Interestingly, the AT population crash occurred around 2000 BCE during the advent of African agriculture (Shaw, 1972), which is hypothesized to have fostered the diversification of the A. gambiae complex (Coluzzi et al., 2002; Crawford et al., 2016). Thus, the decline of AT leading to its present-day rarity may have been driven by anthropogenic modifications to habitat, which perhaps favored A. coluzzii instead. Our demographic model is similar to the one previously suggested for comparing GOUNDRY and A. coluzzii (Crawford et al., 2016). Relative to that model, our estimate of the split with A. coluzzii is slightly older, and there are minor differences in population size changes and migration rates. By incorporating AT, we reveal GOUNDRY’s surprisingly recent origin as a distinct lineage.
We know little about the ecology of AT. While GOUNDRY larvae have been reported in several sites across the hot, arid Sudano-Sahelian region of central Burkina Faso, AT in Tengrela occurs in the cooler, wetter, tropical savannah Sudanese climactic zone of southwestern Burkina Faso. If GOUNDRY is derived from a local AT population, it would suggest that AT possesses a broad tolerance for variable tropical climate. Alternatively, the A. coluzzii ancestry of GOUNDRY may have permitted it to colonize more arid climates than AT can tolerate. We only observed AT in 2011, the sole year when samples were collected in temporary puddles and not just rice paddies, which suggests an ecological specificity. Although we cannot rule out immediate local decline or extinction of AT between 2011 and 2012, such a dramatic change seems implausible, especially when the absence of AT can be explained by our relatively small and ecologically limited sampling scheme. Puddle-specificity of AT is also consistent with the known habitat of GOUNDRY (Riehle et al., 2011). The putative selective sweep regions in AT may contain genes that convey unique adaptive features to AT, but these remain to be characterized. Insecticide resistance alleles are present in AT (Table 2), including at CYP9K1 which occurs within a putative selective sweep region. Selection pressure from regular exposure to insecticide would imply that AT may commonly occur in human-dominated habitats like the Tengrela village. We do not know if adults are anthropophagous, or if they carry Plasmodium. Vectorial capacity is highly plausible for AT given its close relationship to important malaria-transmitting taxa, so it will be critical to identify and study AT adults to understand their potential impact on human health.
Regardless of any direct vectorial capacity, cryptic Anopheles taxa have the potential to stymie malaria control efforts in at least three ways. First, reproductive barriers can thwart efforts to eliminate or modify the A. gambiae complex via gene drive (Marshall et al., 2019). A gene drive targeting A. coluzzii will not necessarily spread to sympatric AT, which might even expand its population size in response to a population crash of A. coluzzii. Second, because reproductive barriers are porous, adaptive genetic diversity maintained within AT can be shared with congeners via introgression, enhancing their capacity to survive and evolve. Rare, semi-isolated taxa like AT can thus serve as allelic reservoirs, facilitating adaptation to insecticides, gene drives, or even climate change. For example, the 2La inversion is associated with adaptation to climate (Cheng et al., 2012), and it shows heterozygote advantage in AT. Overdominance in AT could explain the persistence of this polymorphism in a rare taxon with otherwise low nucleotide diversity. In contrast, selection tends to eliminate one other the other 2La haplotype in A. coluzzii and A. gambiae populations, so AT could be an important genetic reserve for these species if a previously disfavored and purged allele becomes once again beneficial. Finally, misidentified cryptic taxa can confound scientific studies and lead to incorrect inferences about Anopheles biology and inaccurate predictions about disease epidemiology and the outcomes of vector control. For example, the A. gambiae species complex first came to light following confusing discordances in insecticide resistance phenotype between field and captive mosquito populations (Davidson 1956). Captive mating experiments subsequently demonstrated the discordance was due to the inability to discern A. gambiae from another morphologically identical member of the complex, A. arabiensis (Davidson 1956, Davidson & Jackson, 1962). More recently, Gildenhard et al. (2019) noted a striking difference in TEP1 allele frequencies between two populations of A. coluzzii in Burkina Faso, which was attributed to ecologically-varying selection. While this is a very plausible and potentially accurate explanation, the results could also be explained by misidentified AT within the samples, as AT has a very different TEP1 profile from A. coluzzii (Figure 4A, Table 2). Since AT and A. coluzzii appear similar at standard diagnostic loci (Table 2), this is just one of many studies on A. coluzzii that could be potentially confounded by AT.
It will be crucial to correctly identify Anopheles taxa in order to draw accurate conclusions that can inform disease control policy. Our amplicon-based diagnostic protocol (Figure 6; Supp Table 2) provides a clear methodology for identifying AT. The pool of five primer pairs can be amplified and sequenced jointly, yielding high accuracy. For further genotyping as needed, we provide primer pair sequences for a total of 50 diagnostic polymorphisms (Supp Table 2; Supp Figure 7), though not all primers have been empirically validated. Future work could improve upon this methodology, perhaps via a PCR and electrophoresis method to target these polymorphisms without sequencing. An important lesson from AT is that critical diversity can be missed if only a small number of diagnostic markers are assessed, so we do not advocate a protocol based on any one locus alone. We encourage Anopheles biologists to genotype existing DNA samples, especially of putative A. coluzzii from Burkina Faso and adjacent countries, and to seek AT in future surveys. As AT has only been found as larvae from a single site in a single year, we cannot fully characterize it biologically without additional observations. AT will probably not be the last cryptic taxon discovered within this extraordinarily diverse clade. Mapping these intertwined lineages across Africa will be an essential ongoing task with an inestimable impact on human health.
Supplementary Material
Acknowledgements
Our thanks to the residents of Tengrela where this study was conducted, and especially volunteer field workers from this village. We are grateful to the field team from CNRFP for helping with mosquito collection. We also thank Sanjay Nagi, Patricia Pignatelli, Natalie Lissenden, Nithya Swaminathan, Akanksha Khorgade, and Tim Farrell for assistance with sample collection, molecular preparation, and/or bioinformatics. Data interpretation was aided by helpful commentary from Angela Early, Stephen Schaffner, Aimee Taylor, Seth Redmond, and others. Mosquito collections in Burkina Faso were supported by EC FP7 Project grant no: 265660 “AvecNet” and Wellcome Trust Collaborative Award (200222/Z/15/Z). This project has been funded in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Grant Number U19AI110818 to the Broad Institute.
Footnotes
Data Accessibility
Raw Illumina reads from whole-genome sequencing have been deposited in NCBI SRA, Bioproject ID PRJNA639055, at https://www.ncbi.nlm.nih.gov/sra.
References
- Alexander DH, Novembre J, & Lange K (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655–1664. doi: 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anopheles gambiae 1000 Genomes Consortium. (2017). Genetic diversity of the African malaria vector Anopheles gambiae. Nature, 552, 96–100. doi: 10.1038/nature24995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrón MG, Paupy C, Rahola N, Akone-Ella O, Ngangue MF, Wilson-Bahun TA, … Ayala D (2019) A new species in the major malaria vector complex sheds light on reticulated species evolution. Scientific Reports, 9, 14753. doi: 10.1038/s41598-019-49065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Weiss DJ, Cameron E, Bisanzio D, Mappin B, Dalrymple U, … Gething PW (2015). The effect of malaria control on Plasmodium falciparum in Africa between 2000 and 2015. Nature, 526, 207–211. doi: 10.1038/nature15535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. doi: 10.1186/s13742-015-0047-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, White BJ, Kamdem C, Mockaitis K, Costantini C, Hahn MW, & Besansky NJ (2012). Ecological genomics of Anopheles gambiae along a latitudinal cline: a population-resequencing approach. Genetics, 190, 1417–1432. doi: 10.1534/genetics.111.137794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson CS, Weetman D, Essandoh J, Yawson AE, Maslen G, Manske M, … Donnelly MJ (2014). Adaptive introgression between Anopheles sibling species eliminates a major genomic island but not reproductive isolation. Nature Communications, 5, 4248. doi: 10.1038/ncomms5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee M, Hunt RH, Wilkerson R, della Torre A, Coulibaly MB, & Besansky NJ (2013). Anopheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex. Zootaxa, 3619, 246–274. [PubMed] [Google Scholar]
- Coluzzi M, Sabatini A, della Torre A, Di Deco MA, & Petrarca V (2002). A polytene chromosome analysis of the Anopheles gambiae species complex. Science, 298, 1415–1418. [DOI] [PubMed] [Google Scholar]
- Crawford JE, Riehle MM, Guelbeogo WM, Gneme A, Sagnon N, Vernick KD, … Lazzaro BP (2015). Reticulate speciation and barriers to introgression in the Anopheles gambiae species complex. Genome Biology and Evolution, 7, 3116–3131. doi: 10.1093/gbe/evv203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JE, Riehle MM, Markianos K, Bischoff E, Guelbeogo WM, Gneme A, … Lazzaro BP (2016). Evolution of GOUNDRY, a cryptic subgroup of Anopheles gambiae s.l., and its impact on susceptibility to Plasmodium infection. Molecular Ecology, 25, 1494–1510. doi: 10.1111/mec.13572 [DOI] [PubMed] [Google Scholar]
- Davidson G (1956). Insecticide resistance in Anopheles gambiae Giles. Nature, 178, 705–706. doi: 10.1038/178705a0 [DOI] [PubMed] [Google Scholar]
- Davidson G, & Jackson CE (1962). Incipient speciation in Anopheles gambiae Giles. Bulletin of the World Health Organization, 27: 303–305 [PMC free article] [PubMed] [Google Scholar]
- Donnelly MJ, Corbel V, Weetman D, Wilding CS, Williamson MS, & Black WC (2009). Does kdr genotype predict insecticide-resistance phenotype in mosquitoes? Trends in Parasitology, 25: 213–219. doi: 10.1016/j.pt.2009.02.007 [DOI] [PubMed] [Google Scholar]
- Du W, Awolola TS, Howell P, Koekemoer LL, Brooke BD, Benedict MQ, … Zheng L (2005). Independent mutations in the Rdl locus confer dieldrin resistance to Anopheles gambiae and An. arabiensis. Insect Molecular Biology, 14, 179–183. [DOI] [PubMed] [Google Scholar]
- Fanello C, Santolamazza F, & della Torre A (2002). Simultaneous identification of species and molecular forms of the Anopheles gambiae complex by PCR-RFLP. Medical and Veterinary Entomology, 16, 461–464. [DOI] [PubMed] [Google Scholar]
- Fontaine MC, Pease JB, Steele A, Waterhouse RM, Neafsey DE, Sharakhov IV, … Besansky NJ (2015). Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science, 347, 1258524. doi: 10.1126/science.1258524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildenhard M, Rono EK, Diarra A, Boissière A, Bascunan P, Carrillo-Bustamante P, … Levashina EA (2019). Mosquito microevolution drives Plasmodium falciparum dynamics. Nature Microbiology, 4, 941–947. doi: 10.1038/s41564-019-0414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies MT, & De Meillon B. (1968). The Anophelinae of Africa South of the Sahara (Ethiopian Zoogeographical Region) (2nd ed.). Johannesburg: South African Institute for Medical Research. [Google Scholar]
- Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas E, Topalis P, … Lawson D (2015). VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Research, 43, D707–13. doi: 10.1093/nar/gku1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Hernandez RD, Williamson SH, & Bustamante CD (2009). Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genetics, 5, e1000695. doi: 10.1371/journal.pgen.1000695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greppi C, Laursen WJ, Budelli G, Chang EC, Daniels AM, van Giesen L, … Garrity PA (2020). Mosquito heat seeking is driven by an ancestral cooling receptor. Science, 367, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AB, Papathanos PA, Sharma A, Cheng C, Akbari OS, Assour L, … Besansky NJ (2016). Radical remodeling of the Y chromosome in a recent radiation of malaria mosquitoes. Proceedings of the National Academy of Sciences of the United States of America, 113, E2114–2123. doi: 10.1073/pnas.1525164113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Guo Q, Sun X, Zhang C, Xu N, Xu Y, … Shen B (2018). Culex pipiens pallens cuticular protein CPLCG5 participates in pyrethroid resistance by forming a rigid matrix. Parasites & Vectors, 11, 6. doi: 10.1186/s13071-017-2567-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, & Haddrill PR (2014). Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics, 196, 313–320. doi: 10.1534/genetics.113.158758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, … Hoffman SL (2002). The genome sequence of the malaria mosquito Anopheles gambiae. Science, 298, 129–149. [DOI] [PubMed] [Google Scholar]
- Lee Y, Marsden CD, Norris LC, Collier TC, Main BJ, Fofana A, … Lanzaro GC (2013). Spatiotemporal dynamics of gene flow and hybrid fitness between the M and S forms of the malaria mosquito, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America, 110, 19854–19859. doi: 10.1073/pnas.1316851110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, Bullaughey K, Matute DR, Meyer WK, Ségurel L, Venkat A, … Przeworski M (2012). Revisiting an old riddle: what determines genetic diversity levels within species? PLoS Biology, 10, e1001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics, 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main BJ, Lee Y, Collier TC, Norris LC, Brisco K, Fofana A, … Lanzaro GC (2015) Complex genome evolution in Anopheles coluzzii associated with increased insecticide usage in Mali. Molecular Ecology, 24, 5145–5157. doi: 10.1111/mec.13382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM, Raban RR, Kandul NP, Edula JR, León TM, & Akbari OS (2019). Winning the tug-of-war between effector gene design and pathogen evolution in vector population replacement strategies. Frontiers in Genetics, 10, 1072. doi: 10.3389/fgene.2019.01072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, … DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20, 1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Lawniczak MKN, Park DJ, Redmond SN, Coulibaly MB, Traoré SF, … Muskavitch MAT (2010). SNP genotyping defines complex gene-flow boundaries among African malaria vector mosquitoes. Science, 330, 514–517. doi: 10.1126/science.1193036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris LC, Main BJ, Lee Y, Collier TC, Fofana A, Cornel AJ, & Lanzaro GC (2015). Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proceedings of the National Academy of Sciences of the United States of America, 112, 815–820. doi: 10.1073/pnas.1418892112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nkya TE, Poupardin R, Laporte F, Akhouayri I, Mosha F, Magesa S, … David JP (2014). Impact of agriculture on the selection of insecticide resistance in the malaria vector Anopheles gambiae: a multigenerational study in controlled conditions. Parasites & Vectors, 7, 480. doi: 10.1186/s13071-014-0480-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, & Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genetics, 8, e1002967. doi: 10.1371/journal.pgen.1002967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2019). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/ [Google Scholar]
- Ranson H, & Lissenden N (2016). Insecticide resistance in African Anopheles mosquitoes: a worsening situation that needs urgent action to maintain malaria control. Trends in Parasitology, 32, 187–196. doi: 10.1016/j.pt.2015.11.010 [DOI] [PubMed] [Google Scholar]
- Riehle MM, Guelbeogo WM, Gneme A, Eiglmeier K, Holm I, Bischoff E, … Vernick KD (2011). A cryptic subgroup of Anopheles gambiae is highly susceptible to human malaria parasites. Science, 331, 596–598. doi: 10.1126/science.1196759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottschaefer SM, Riehle MM, Coulibaly B, Sacko M, Niaré O, Morlais I, … Lazzaro BP (2011). Exceptional diversity, maintenance of polymorphism, and recent directional selection on the APL1 malaria resistance genes of Anopheles gambiae. PLoS Biology, 9, e1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santolamazza F, Mancini E, Simard F, Qi Y, Tu Z, & della Torre A (2008). Insertion polymorphisms of SINE200 retrotransposons within speciation islands of Anopheles gambiae molecular forms. Malaria Journal, 7, 163. doi: 10.1186/1475-2875-7-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JA, Brogdon WG, & Collins FH (1993). Identification of single specimens of the Anopheles gambiae complex by the polymerase chain reaction. The American Journal of Tropical Medicine and Hygiene, 49, 520–529. [DOI] [PubMed] [Google Scholar]
- Sharakhova MV, Hammond MP, Lobo NF, Krzywinski J, Unger MF, Hillenmeyer ME, … Collins FH (2007). Update of the Anopheles gambiae PEST genome assembly. Genome Biology, 8, R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw T (1972). Early agriculture in Africa. Journal of the Historical Society of Nigeria, 6, 143–191. [Google Scholar]
- Stajich JE, Hahn MW (2005). Disentangling the effects of demography and selection in human history. Molecular Biology and Evolution, 22, 63–73. [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. [DOI] [PubMed] [Google Scholar]
- [dataset] Tennessen JA, Ingham VA, Toé KH, Guelbéogo WM, Sagnon N, Kuzma R, … Neafsey DE; 2020; Whole-genome sequencing of Anopheles mosquitoes from Tengrela, Burkina Faso; NCBI SRA; BioProject ID PRJNA639055 [Google Scholar]
- van der Valk T, Vezzi F, Ormestad M, Dalén L, & Guschanski K (2020). Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Molecular Ecology Resources, 20, 1171–1181. doi: 10.1111/1755-0998.13009 [DOI] [PubMed] [Google Scholar]
- White BJ, Hahn MW, Pombi M, Cassone BJ, Lobo NF, Simard F, Besansky NJ (2007). Localization of candidate regions maintaining a common polymorphic inversion (2La) in Anopheles gambiae. PLoS Genetics, 3, e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BJ, Lawniczak MK, Cheng C, Coulibaly MB, Wilson MD, Sagnon N, … Besansky NJ (2011). Adaptive divergence between incipient species of Anopheles gambiae increases resistance to Plasmodium. Proceedings of the National Academy of Sciences of the United States of America, 108, 244–249. doi: 10.1073/pnas.1013648108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You FM, Huo N, Gu YQ, Luo MC, Ma Y, Hane D, … Anderson OD (2008). BatchPrimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics, 9, 253. doi: 10.1186/1471-2105-9-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.