

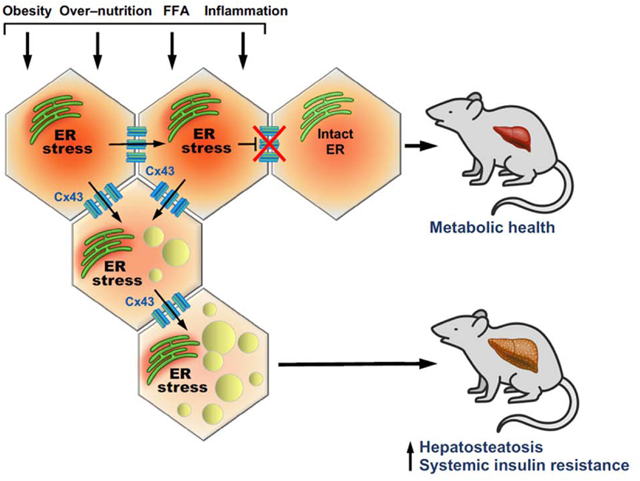
Summary
Endoplasmic reticulum stress (ERS) has a pathophysiological role in obesity-associated insulin resistance. Yet, the coordinated tissue response to ERS remains unclear. Increased connexin (Cx)43-mediated intercellular communication, has been implicated in tissue adaptive and maladaptive response to various chronic stresses. Here we demonstrate that in hepatocytes, ERS results in increased Cx43 expression and cell-cell coupling. Co-culture of ER stressed ‘donor’ cells resulted in inter-cellular transmission of ERS and dysfunction to ERS-naïve ‘recipient’ cells (‘bystander response’), which could be prevented by genetic or pharmacologic suppression of Cx43. Hepatocytes from obese mice were able to transmit ERS to hepatocytes from lean mice, and mice lacking liver Cx43 were protected from diet-induced ERS, insulin resistance, and hepatosteatosis. Taken together, our results indicate that in obesity, the increased Cx43-mediated cell-cell coupling allows inter-cellular propagation of ERS. This novel maladaptive response to over-nutrition exacerbates the tissue ERS burden, promoting hepatosteatosis and impairing whole-body glucose metabolism.
Keywords: Gap junctions, connexin 43, endoplasmic reticulum stress, intercellular communication, insulin resistance, diabetes, unfolded protein response
eTOC blurb:
Connexin-43 (Cx43) has a key role in cell-cell communication. While Cx43-mediated inter-cellular communication is an adaptive tissue response in acute stress, it may become deleterious under chronic, non-resolving stress conditions. Herein, Tirosh et al. discover that in obesity, Cx43 allows deleterious signals to disseminate between cells in the liver, leading to fatty-liver disease and metabolic alterations.
Graphical Abstract
Introduction
The acquisition of morphological and functional complexity in multicellular organisms was a major step in animal evolution, resulting in a fundamental need for the development of efficient routes for cell-cell communication (Rokas, 2008). Such interactions between the cellular constituents of an organ may be critical for many processes, from sharing molecular entities, to preemptive adaptations to an incoming insult, to propagation of stress. Gap junction (GJ) channels provide one of the most common forms of intercellular communication and fulfill similar functions in all multicellular animals (Herve and Derangeon, 2013). Multiple isoforms of GJ proteins form channels between the cytoplasm of adjacent cells, allowing the exchange of ions, signaling molecules and various metabolites between cells of most tissues. GJ channels are formed by paired connexons, each made up of six connexin (Cx) subunits. Cxs play pivotal roles in a wide range of physiological processes, such as differentiation and proliferation, electrical conductivity, neuronal signaling, hormone secretion, and immune functions (Bosco et al., 2011). Of the 21 human Cx genes (Koval et al., 2014), connexin 43 (Cx43), a 43-kDa protein, is the most ubiquitous and critical GJ protein, playing important developmental, regulatory and pathophysiological roles in many tissues.
While intercellular sharing of cytoplasmic content may be beneficial (the “Good Samaritan” effect), open GJ channels may also be hazardous (the “Bystander” effect) by providing a conduit through which toxins, inflammatory and other pathological alterations can spread cytotoxicity to adjacent cells (Lin et al., 1998; Perez Velazquez et al., 2003; Spray et al., 2013). The recognition of injuries that involve bystander cell damage has greatly expanded recently to include infectious agents and chemical compounds, radiation injury, and apoptosis. For example, blocking GJ channels was found to be hepatoprotective in toxic liver damage (Du et al., 2013; Patel et al., 2012), and ischemic injury in the brain (Rawanduzy et al., 1997) and heart (Garcia-Dorado et al., 1997) was shown to spread to unaffected cells via Cx43-mediated intercellular communication. In addition, some intracellular pathogens lead to pro-inflammatory pathway activation that spreads through GJs from infected to uninfected ‘bystander’ cells (Kasper et al., 2010), primarily through upregulation of Cx43 (Eugenin and Berman, 2007; Eugenin et al., 2011). While this mechanism to amplify the tissue inflammatory response may contribute to innate immunity, providing obvious advantages in tissue adaptation to acute infection or stress, it may also be deleterious to tissue homeostasis in the setting of chronic inflammatory or other stresses. Indeed, in several conditions of chronic inflammation, increased Cx expression and GJ activity were found to play an important pathological role. This includes Cx43-mediated increase in chemo-attraction of neutrophils and macrophages to atherosclerotic plaques (Morel et al., 2014), and an increase in neointima formation (Song et al., 2009). Cxs have been implicated in the adaptation to other chronic stresses including ER stress (Cheng et al., 2013), inflammation and fibrosis in chronic kidney disease (Abed et al., 2014), rheumatoid arthritis (Abed et al., 2014), and delayed wound healing (Qiu et al., 2003).
Obesity and type 2 diabetes have been well-characterized as conditions of chronic, low-level metabolic inflammation or metaflammation, and cellular stresses (Hotamisligil, 2017b; Wellen and Hotamisligil, 2005; Ye, 2013). In particular, hepatic ER stress and dysfunction play a key role in the network of stress signals orchestrating the development of insulin resistance and diabetes, thus promoting the disruption of systemic glucose homeostasis (Hotamisligil, 2010). While the mechanistic basis of ER dysfunction and the links with abnormal metabolic responses are intensely explored at the cellular and molecular level, the potential role of cell-cell communication in the coordination of the response at the tissue and organ level is not well understood.
Here we report that ER stress signals can spread in between cells and that Cx43 plays a critical role in this process. Under conditions of chronic ER stress, such as in the case of obesity, increased Cx43-mediated intercellular communication is a critical component of the maladaptive response to ER stress and dysfunction in the liver. We demonstrate that Cx43 expression is increased in the liver in response to obesity, and that ER stress leads to increased Cx43-mediated cell-cell coupling and intercellular transmission of ER stress, resulting in ER dysfunction in ‘bystander’ cells. Liver-specific deletion of Cx43 prevented the transmission of over nutrition-induced ER stress between hepatocytes, improved whole-body glucose metabolism, and attenuated hepatosteatosis.
Results
ER stress increases Cx43 expression and function in hepatocytes
Recently, increased intercellular communication by GJ primarily composed of Cx43 was implicated in the pathogenesis of atherosclerosis and neurodegenerative diseases, which are characterized by chronic low-grade inflammation and ER stress (Matus et al., 2011; Tabas, 2010). These observations raised the possibility that increased intercellular communication may be a common feature of conditions characterized by cellular stress. Obesity and diabetes are characterized by chronic ER stress in multiple organs including the liver, where maladaptive ER function contributes to metaflammation, impaired insulin signaling and abnormal glucose homeostasis (Hotamisligil, 2010). We asked whether such stress may also result in regulation of connexins in liver cells. In hepatocytes, the major Cx isoform is Cx32, and background expression of Cx43 in healthy animals is rather low but could be regulated under stress conditions (Vinken, 2012). We therefore assessed the direct effect of ER stress on Cx43 expression in a hepatocyte cell line and in isolated primary mouse hepatocytes. Chemical induction of ER stress in Hepa1–6 cells (Fig. 1A) or in primary isolated mouse hepatocytes (Fig. 1C) by tunicamycin (Tm), an inhibitor of N-linked glycosylation, or thapsigargin (Tg), an inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), resulted in increased levels of Cx43 protein, in parallel with induction of the ER stress marker 78 kDa glucose-regulated protein (GRP78) as well as phosphorylation of inositol-requiring enzyme (IRE1) and eukaryotic initiation factor 2 α (peIF2α) (Fig. S1A). Under these conditions, an increase in Cx43 gene expression was also noted (Fig. 1B and 1D, respectively). The induction of Cx43 expression by ER stress was similarly observed in a non-transformed hepatocyte cell line, AML12 cells (Fig. 1E and 1F). To assess whether the increase in Cx43 gene and protein expression is associated with increased intercellular communication, we subjected hepatocytes to the scrape-loading dye transfer assay using the fluorescent dye lucifer yellow (LY), which can only cross from one cell into adjacent cells via functional GJs. We found that both Tg and Tm treatment resulted in enhanced diffusion capacity of LY compared to control treatment, and this could be prevented by addition of the Cx43 selective blocking peptide GAP27 (Fig. 1G and 1H), indicating that ER stress increased GJ-mediated cell-cell coupling. Of note, treatment of cells with GAP27 did not significantly alter Cx43 gene or protein expression (Fig. S1E)
Figure 1: ER stress and obesity increases Cx43 expression and function in hepatocytes.
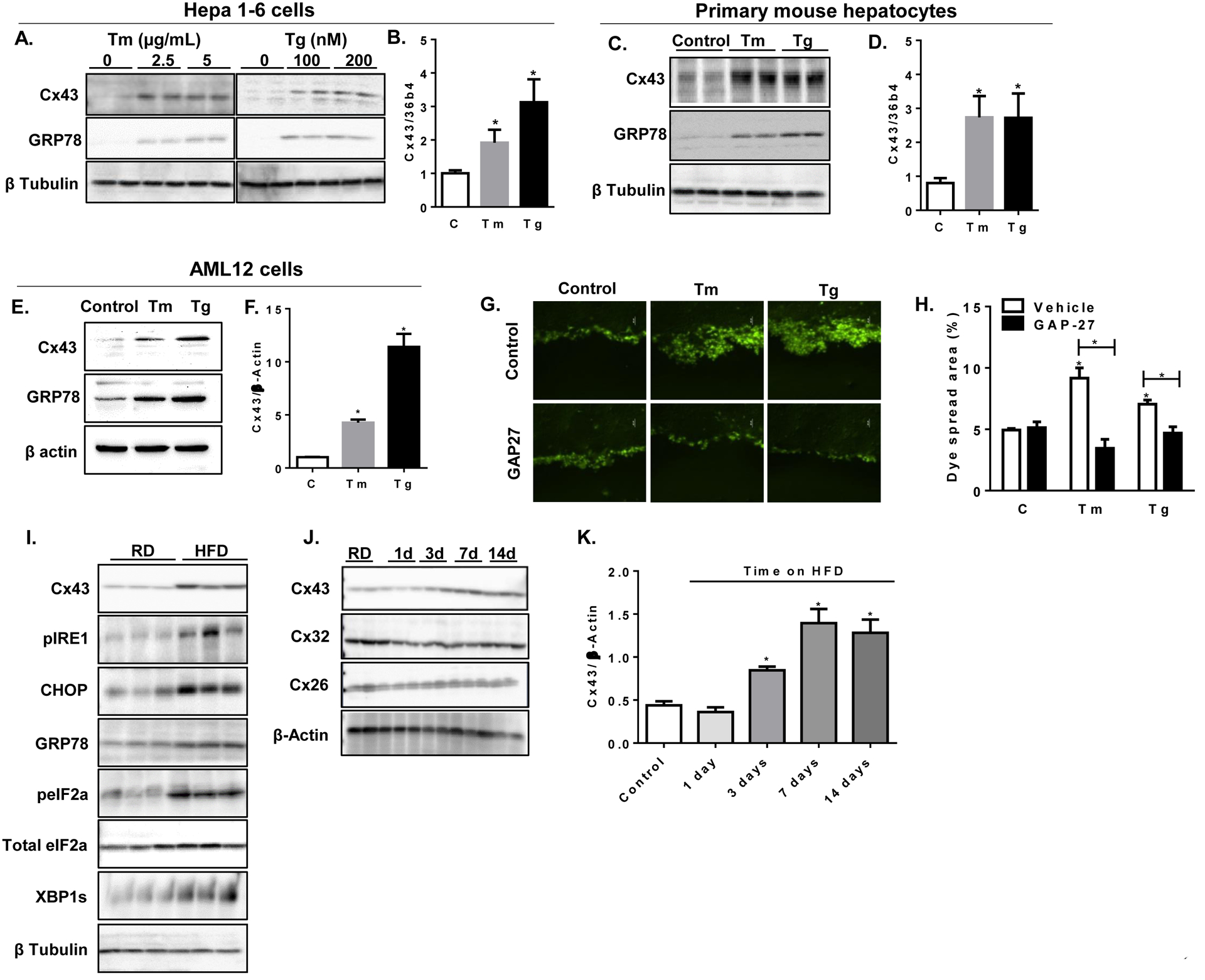
(A) Hepa1–6 cells were treated with either tunicamycin (Tm), thapsigargin (Tg), or with the vehicle DMSO (0) at the indicated concentrations for 6 hours. Cell lysates were subjected to western blot analysis and protein expression of Cx43, GRP78 and β tubulin was determined. (B) Cx43 gene expression was determined by quantitative, real-time PCR (qPCR) relative to 36b4 gene in Hep1–6 cells following 6-hour treatment with either Tm (5 μg/mL) or Tg (200 nM), C=control (vehicle treated cells). (C) Primary mouse hepatocytes were treated for 6 hours with either Tm (5 μg/mL) or Tg (200 nM) or vehicle and levels of Cx43, GRP78 and β tubulin were determined by western blot analysis. (D) Relative expression of Cx43 to 36b4 genes were determined by qPCR in DMSO- (C), Tm- or Tg-treated primary hepatocytes. (E) AML12 hepatocytes were treated for 6 hours with either Tm (5 μg/mL) or Tg (200 nM) or vehicle and levels of Cx43, GRP78 and β tubulin were determined by western blot, and quantified in (F). (G) Scrape loading dye transfer assay; Hepa1–6 cells were treated with DMSO (Vehicle), Tg or Tm in the presence or absence of the specific Cx43 inhibitory peptide GAP27 (150 nM) for 6 hours. (H) Quantification of the Lucifer Yellow diffusion by % area with the fluorescent dye. (I) C57BL/6 male mice were treated with regular chow diet (RD) or high fat diet (HFD) for 16 weeks. Liver homogenates were assayed for protein expression of Cx43, phosphorylated IRE1 (pIRE1), CHOP, GRP78, total and the phosphorylated form of eIF2alpha, spliced XBP1 and β tubulin. (J) C57BL/6 male mice were treated with regular chow diet (RD) or high fat diet (HFD) for the indicated times. Liver homogenates were assayed for protein expression of Cx43 and other liver connexin isoforms (Cx32 and Cx26). (K) Quantification of Cx43 protein expression by densitometry relative to β actin expression during a time course of HFD exposure. n=6 mice/group. *p<0.05 as compared to control (=RD).
Obesity results in increased liver expression of Cx43
To test whether ER stress also induce an increase in Cx43 expression in vivo, we exposed C57BL/6 mice to high fat diet (HFD) for 16 weeks and assessed Cx43 expression in liver homogenates. As expected, HFD treatment resulted in increased hepatic ER stress, as indicated by the increased expression of the ER stress markers CCAAT-enhancer-binding protein homologous protein (CHOP), GRP78, and the spliced form of the X-box binding protein 1 (XBP-1s), and increased phosphorylation of IRE1 and eIF2α (Fig. 1I). Under these conditions, Cx43 expression was similarly elevated (Fig. 1I). Remarkably, the increase in Cx43 expression in the liver was an early event following exposure to over nutrition, observed following just 3 days on HFD (Fig. 1J, 1K and S1B). The expression of the two other liver connexin isoforms, Cx32 and Cx26, were not significantly altered by HFD at any time point examined (Fig. 1J).The marked increase in Cx43 expression following HFD exposure could be localized mainly to the plasma membrane (Fig. S1C).
The unfolded protein response (UPR) is transmittable via Cx43-mediated inter-cellular communication
We next explored whether the increased Cx43 expression and cell-cell coupling results in inter-cellular transmission of ER stress signals. For this, we designed a co-culture system in which ER stress generated in one population of cells (‘donors’) could be detected only if transmitted to ER-stress naïve cells (‘recipients’), as illustrated in figure 2A. HEK293 cells stably expressing the green fluorescent protein (GFP) were used as identifiable ‘donor’ cells, and pre-treatment with either Tg or Tm (for 4 hours) was used to induce ER stress. As recipients, we generated cells stably expressing either the UPR response element (UPRE) or the ER stress response element (ERSE) sub-cloned into the mCherry fluorescent protein construct, thus driving the expression of mCherry upon exposure to ER stress and/or activation of the UPR (UPRE-mCherry or ERSE-mCherry reporters, respectively, Fig. S2A). Following induction of ER stress, donor cells were washed 3 times, trypsinized, and re-plated into the recipient dish in a single location. Following 16 hours of co-culture, the mixed ‘donor’ and ‘recipient’ cell population was further subjected to direct imaging of fluorescent signals or to flow cytometry and sorting to quantify the extent of mCherry expression among the recipient cells. As demonstrated in figure 2B and quantified in figure 2C, pre-treatment of GFP donor cells with Tm or Tg resulted in increased number of both UPRE-mCherry and ERSE-mCherry recipient cells expressing mCherry. In order to better assess the extent of relative mCherry expression in recipient cells, we used fluorescent activated cell sorting analysis (FACS) of the mixed colonies and quantified the percent of mCherry expression among recipient cells. As indicated in figure 2D and S2B, co-culture with un-stressed GFP donors resulted in mCherry expression of 1.35% and 1.4% of UPRE-mCherry and ERSE-mCherry recipient cells, respectively. However, in co-cultures with ER stressed donors, we observed an approximately 10-fold increase in mCherry expression (11.4% and 14.4%, respectively), indicating an increased number of recipient cells with significant UPR activation.
Figure 2: UPR is transmitted between cells in a Cx43-dependent manner.
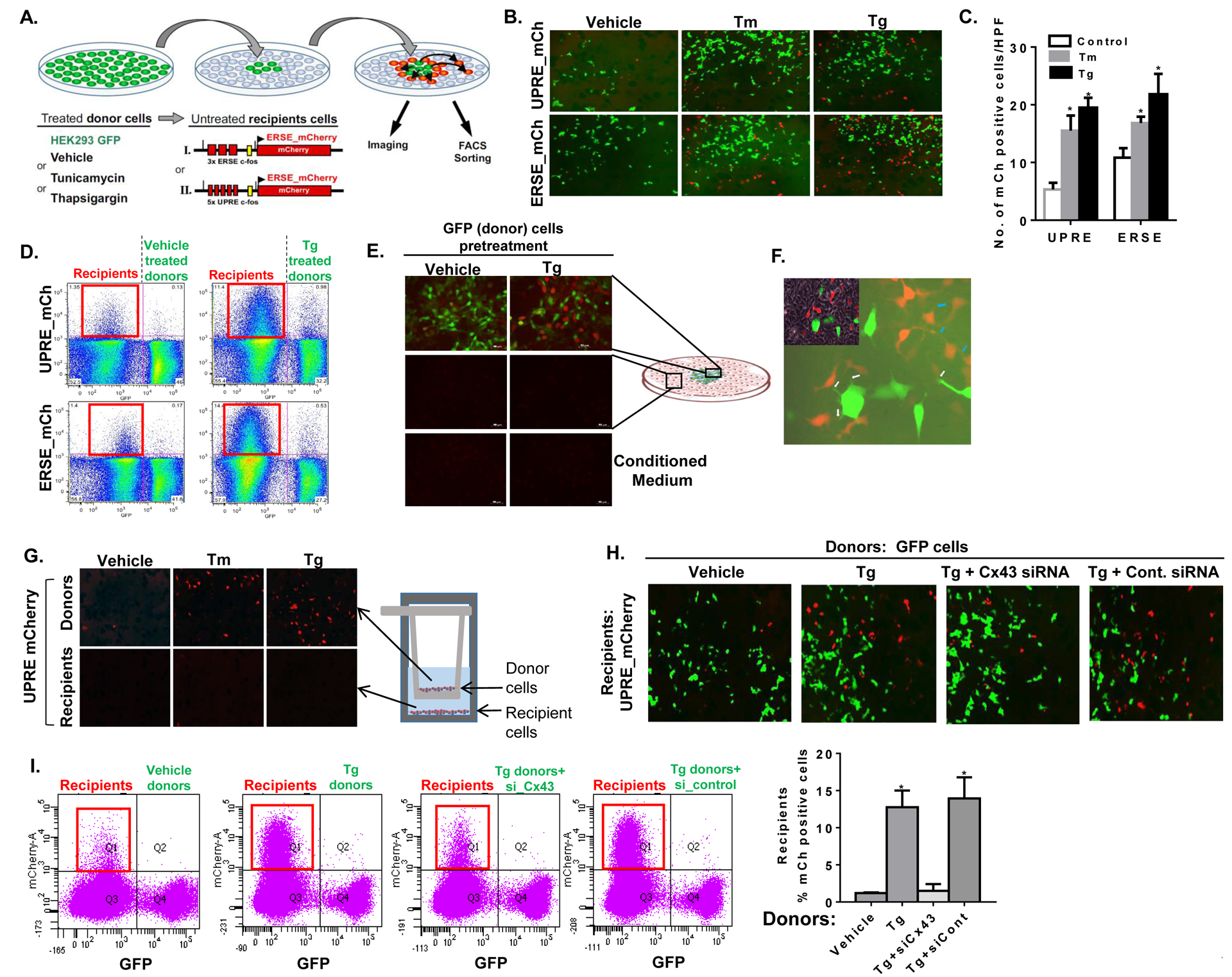
(A) Illustration of the cellular models used to assess the transfer of ER stress signals from donor cells to ER stress naïve recipient cells. HEK293 cells were stably transfected with the red fluorescent protein mCherry driven by either the ER stress or the UPR response element (ERSE- and UPRE-mCherry). These cells express mCherry following exposure to ER stress and were used as ‘recipients’. GFP-expressing HEK293 cells were used as ‘donor’ cells and were pre-treated with either DMSO (Vehicle), tunicamycin (Tm) or thapsgargin (Tg). Following several washes, donor cells were transferred to a near confluent dish of ‘recipient’ cells and mCherry activation was assessed by fluorescent microscopy and by flow cytometry. (B) Donor GFP cells were pre-treated with either vehicle (DMSO), Tm (5 μg/mL) or Tg (200 nM) for 6 hours, followed by co-culture with either UPRE-mCherry (UPRE-mCh) or ERSE-mCherry (ERSE-mCh) recipient cells. Images of the mixed population of donor (GFP) and ER-stressed recipient (mCherry positive) cells were captured following 16 hours of co-culture. (C) Mean ± SD of mCherry (mCh) positive cells in 10 high power field (HPF) images of the mixed donor-recipient cell population. *p<0.05 as compared to vehicle. (D) Fluorescence-activated cell sorting (FACS) analysis of the mixed cell populations of GFP donor cells, pre-treated with either vehicle (left panels) or Tg (right panels) with UPRE-mCh (upper panels) or ERSE-mCh (lower panels) recipient cells following an overnight co-culture. The red square (left upper quadrant in each panel) highlights the extent of mCherry expression among recipient cells. (E) mCherry expression among UPRE-mCherry recipient cells was assessed by fluorescent microscopy within and around the GFP donor colony (upper panels) and remotely, at the periphery of the dish (middle panels). The effect of conditioned media from vehicle or Tg-treated donor cells on mCherry expression in recipient cells was also tested (lower panels). (G) A x400 magnification demonstrating thin cellular extensions of donor (GFP positive) to recipient (mCherry positive) cells (white arrows) and of recipient-to-recipient cells (blue arrows). Trans-well co-culture system was used to prevent cell-cell contact between donor and recipient cells, while incubated in the same medium. The ability of donor cells to transmit ER stress to the recipients was tested under these conditions. (H, I) mCherry expression in UPRE-mCherry recipient cells co-cultured overnight with vehicle (DMSO), or Tg-treated GFP donor cells. Donor cells were either transfected with Cx43 siRNA (si-Cx43) or with control siRNA (si-control). Fluorescent images (G) and FACS analyses (H) are presented. Q1 (framed in red) represents the extent of recipient cells expressing mCherry in each condition. Quantification of two independent experiments is presented, p<0.05.
Cx-mediated inter-cellular communication requires donor-recipient proximity. We noted that mCherry expression was observed in recipients within or around the GFP donor colony (Fig. 2B and 2E), but not in recipient cells located remotely from donor cells (Fig. 2E, middle panels), indicating that the signal may be passed by direct cell-cell contact. Of note, it appeared that some of the mCherry positive recipient cells were not in close proximity to GFP donor cells. However, a x400 magnification revealed thin cellular extensions between donors and recipients (white arrows) that likely mediate cell-cell coupling (Fig. 2F). These cellular extensions are also identified between recipient only cells (blue arrows), which may enable second- and third-hand transmission of ER stress signals. Recent studies have also demonstrated that non-adjacent cells can communicate through tunneling nanotubes (TNTs), tubular extensions reaching 100 μm in length which contain Cx43 (Ribeiro-Rodrigues et al., 2017). In further support of the need for direct cell-cell contact to transmit ER stress, conditioned media collected from ER stressed donors was found to be insufficient to activate mCherry in recipient cells (Fig. 2E, lower panels). In addition, we tested the ability of stressed donor cells (similarly expressing the mCherry reporter) to transmit ER stress to recipient cells using a trans-well co-culture system. In these experiments, treatment with Tg or Tm induced ER stress in donor cells as expected (Fig. 2G upper panels and Fig S2C), but the signal was not detected in recipient cells (Fig. 2G lower panels and Fig. S2C), demonstrating that cell-cell contact is required. Finally, we found that the ability of donor cells to induce either UPRE-mCherry or ERSE-mCherry expression among recipients was significantly attenuated when expression of Cx43 was reduced in donor cells using Cx43 siRNA (Fig. 2H and Fig. S2D, respectively). FACS analysis of the mixed cell population demonstrated that silencing of Cx43 in donors (Fig. 2I and S2E) or in recipient cells (Fig. S2F) was able to prevent nearly 90% of recipient mCherry activation induced by ER stressed donors. An approximately 80% reduction in Cx43 protein expression was achieved by Cx43 siRNA treatment in this set of experiments (Fig. S2G) with no detectable effect on cell viability (Fig. S2H).
Assessment of Cx43-mediated inter-cellular transmission of endogenous ER stress
In order to study cell-cell transmission of ER stress in a chemical-free system (i.e. not induced by Tm or Tg), we generated two different donor cell lines that were selected to exhibit either high cell autonomous ER stress (highly stressed cells, HSC) or low expression of ER stress markers (unstressed cells, USC). These distinct cell populations were achieved by stable knock-down of endogenous genes using a genome-wide shRNA library in HEK293 cells expressing the UPRE-mCherry reporter as described above. Cells expressing both the ER stress reporter and the control vector were then fractionated by FACS into populations of HSCs or USCs by selecting the high (upper 20%) and low (lower 20%) spontaneous mCherry expression, respectively. Sorted cells were re-plated and sorted for 5 consecutive times to enrich the HSC (high mCherry expression) and USC (low mCherry expression) populations (Fig. 3A). As demonstrated in Fig. 3B, HSC donors had increased expression of the ER stress markers GRP78, phosphorylated protein kinase R (PKR)-like endoplasmic reticulum kinase (pPERK) and phosphorylated IRE1 compared to USC donors. Level of Cx43 protein was also increased in these cells. To test the ability of USCs and HSCs to activate the UPR in ‘bystanding’ recipient cells, we co-cultured them with recipient GFP HEK293 cells stably co-expressing UPRE-mCherry. Following an overnight co-culture, the mixed donor and recipient populations were subjected to FACS analysis. HSC donors could be easily detected by their extensive mCherry expression compared with USC donors (compare left upper quadrants of Figs. 3D and 3F with left upper quadrants of Figs. 3C and 3E). Similar to Tg-treated donors (Fig. 2D), untreated HSC donors induced a robust co-expression of mCherry among GFP+-recipients, which was ~3-fold greater than that observed in recipient cells cultured with USC donors (right upper quadrants in Figs. 3C and 3D, respectively, quantified in Fig. 3G). Silencing of Cx43 in donor cells significantly attenuated the ability of HSC cells to activate the UPRE in adjacent recipients (Figs. 3E–G). To further study UPR activation in recipient cells, GFP recipients were co-cultured with either USC or HSC donors, and sorted recipients were lysed and subjected to western blot analysis. As demonstrated in Fig. 3H, increased expression of GRP78, pIRE1 and pPERK was induced only by co-culture with HSC donors. Under these conditions, Cx43 expression was also induced only among recipient cells co-cultured with HSC donors (Fig. 3H). In addition, co-cultured with ER stressed HSC, but not with USC, also resulted in increased spliced XBP1 in recipient cells (Fig. 3I). In order to assess the consequences of activation of ER stress signals in recipient cells on insulin response, we treated sorted recipients, co-cultured with either USCs or HSCs, with insulin. As depicted in Fig 3J, the ability of acute insulin stimulation to increase Akt phosphorylation was markedly inhibited in recipients cells co-cultured with HSCs, but not with USCs, indicating that the co-culture with ER stressed cells can induce insulin resistance in ER stress naïve neighboring cells.
Figure 3: Cx43-mediated intercellular transmission of UPR in a chemical-free system.
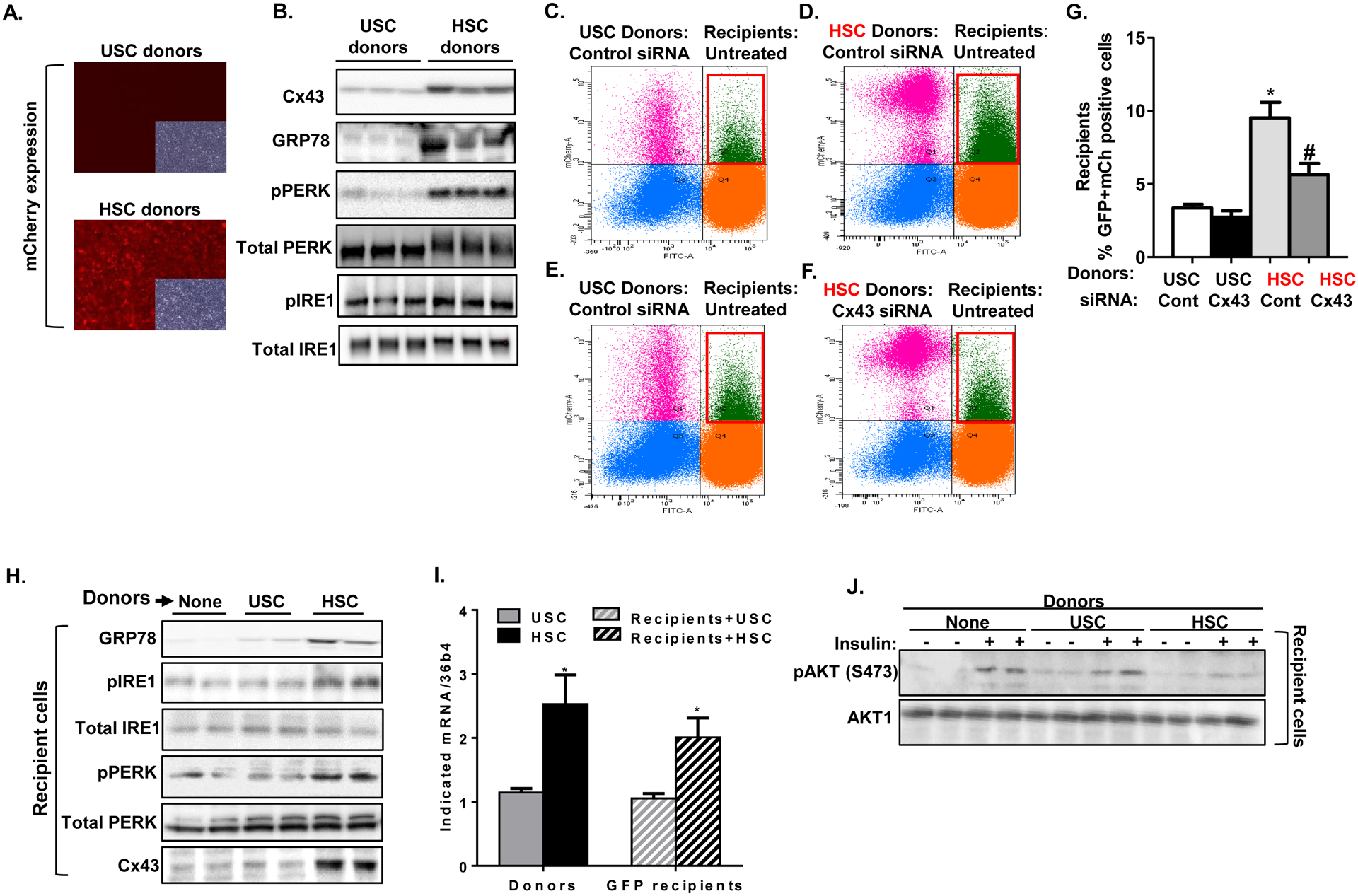
Intrinsically unstressed (USC) and highly stressed (HSC) HEK293 cells were generated by establishing stable knock-down of endogenous genes using genome wide shRNA library in ER stress reporter cells stably expressing the UPRE-mCherry reporter. (A) Baseline, unstimulated expression of mCherry in USC and HSC cells that were used as donors in these experiments. USC indicates unstressed donor cells. HSC represents donor cells with chemical-free high level of ER stress. (B) Western blot analysis assessing protein expression of Cx43, GRP78, phospho-PERK, phospho-IRE1, total IRE1 and β tubulin in USC and HSC donors. (C–F) Co-culture of USCs or HSCs (donors) with GFP cells stably co-expressing UPRE-mCherry (recipients) for 16 hours, followed by FACS analysis. Quadrant 2 (Q2), framed in red, represents the extent of GFP recipient cells co-expressing mCherry, indicative of ER stress activation in recipient cells. USC or HSC donors were either transfected with control siRNA (C and D, respectively) or with Cx43 siRNA (E and F, respectively). (G) Quantification of FACS analysis (mean ± SD) from 4 co-cultured dishes. *p<0.005 comparing mCherry response in recipients co-cultured with HSC vs. USC donors. #p<0.05 comparing mCherry response in recipients co-cultured with Cx43 knocked-down vs. control siRNA treated HSC donors. (H) GFP-expressing recipient cells co-cultured with either untreated USC or HSC donors were subjected to FACS and sorting. Protein expression of the ER stress markers GRP78, phospho-IRE1 and phospho-PERK, as well as Cx43 and β tubulin were assayed by western blot in lysed recipients. (I) mRNA levels of spliced XBP1 in sorted GFP expressing recipients co-cultured with either USC (striped grey) or HSC (striped black), and in their respective donors. (J) Sorted recipients co-cultured with either USC or HSC, were re-plated and then treated with insulin (100 nM) or vehicle (PBS) stimulation for 5 minutes. Treated cells were lysed and subjected to western blot analysis of phosphorylated AKT (Ser473) and total AKT.
Cx43-mediated intercellular transmission of ER stress results in decreased chaperone activity and decreased ER folding capacity in recipient cells
Transmission of ER stress signals to bystander cells could be either ‘adaptive’, resulting in increased ER folding capacity among recipients (i.e. pre-conditioning), or ‘maladaptive’, resulting in dysfunctional ER. To evaluate these possibilities, we next studied the effect of donor-to-recipient communication on ER function in recipient cells using two complementary independent cellular assays developed to quantitatively monitor ER function (Fu et al., 2015). In the first assay, the luminal domain of the Activating Transcription Factor 6 (ATF6) is fused to a reporter gene encoding luciferase (Cluc); high chaperone availability results in retention of the Cluc reporter in the ER, whereas chaperone consumption in a dysfunctional ER liberates the chimeric protein, resulting in increased luciferase secretion (illustrated in Fig. S3A). As an internal control, the reporter construct contains a second secreted Gaussia luciferase (Gluc), whose expression is not affected by ER stress. We expressed this novel functional ER reporter in recipient cells, co-cultured with donors that were pre-treated (or not) with Tg. Following 16 hours of co-culture, the mixed culture was acutely challenged with varying doses of Tg (experimental design is illustrated in Fig. 4A). ATF6 secretion (determined by luciferase activity in the media) was measured at baseline and following Tg challenge. As depicted in Fig. 4B, increasing concentrations of Tg decreased chaperone availability more rapidly in recipients co-cultured with stressed donors, compared to co-cultured with ER-stress naïve donors. Following Cx43 silencing, recipient cells became blind to the ER stress status of their neighboring donors, and were therefore no longer over sensitized to the acute treatment of Tg (Fig. 4C). A near complete inhibition of Cx43 expression was achieved in this set of experiments (Fig. S4A). In the second complementary functional assay, asialoglycoprotein receptor 1 (ASGR1) was similarly fused to Cluc protein to monitor ER folding capacity (Fig. S3B). In this system, compromising ER homeostasis leads to less efficient protein folding and, therefore, lower levels of ASGR-Cluc secretion (Fu et al., 2015). This construct also includes a Gluc reporter as an internal control. A significant impairment in the ability of recipient cells to properly fold and secrete ASGR-Cluc was observed following co-culture with ER stressed donors (Figure 4D). Preventing Cx43-mediated intercellular communication by Cx43 silencing was sufficient to completely protect recipients from the deleterious disruption of ER function induced by stressed donors, and to protect recipients from increased ER sensitivity to Tg, regardless of the ER status of their adjacent donors (Figure 4E). Similar results were obtained when donor cells were ER stressed by Tm (Figs. S4B–E), or when Tm was used as the ‘second hit’ to test ER function of recipient cells (Figs. S4F–I).
Figure 4: Cx43-mediated intercellular transmission of ER stress results in decreased chaperone activity and ER folding capacity in recipient cells.
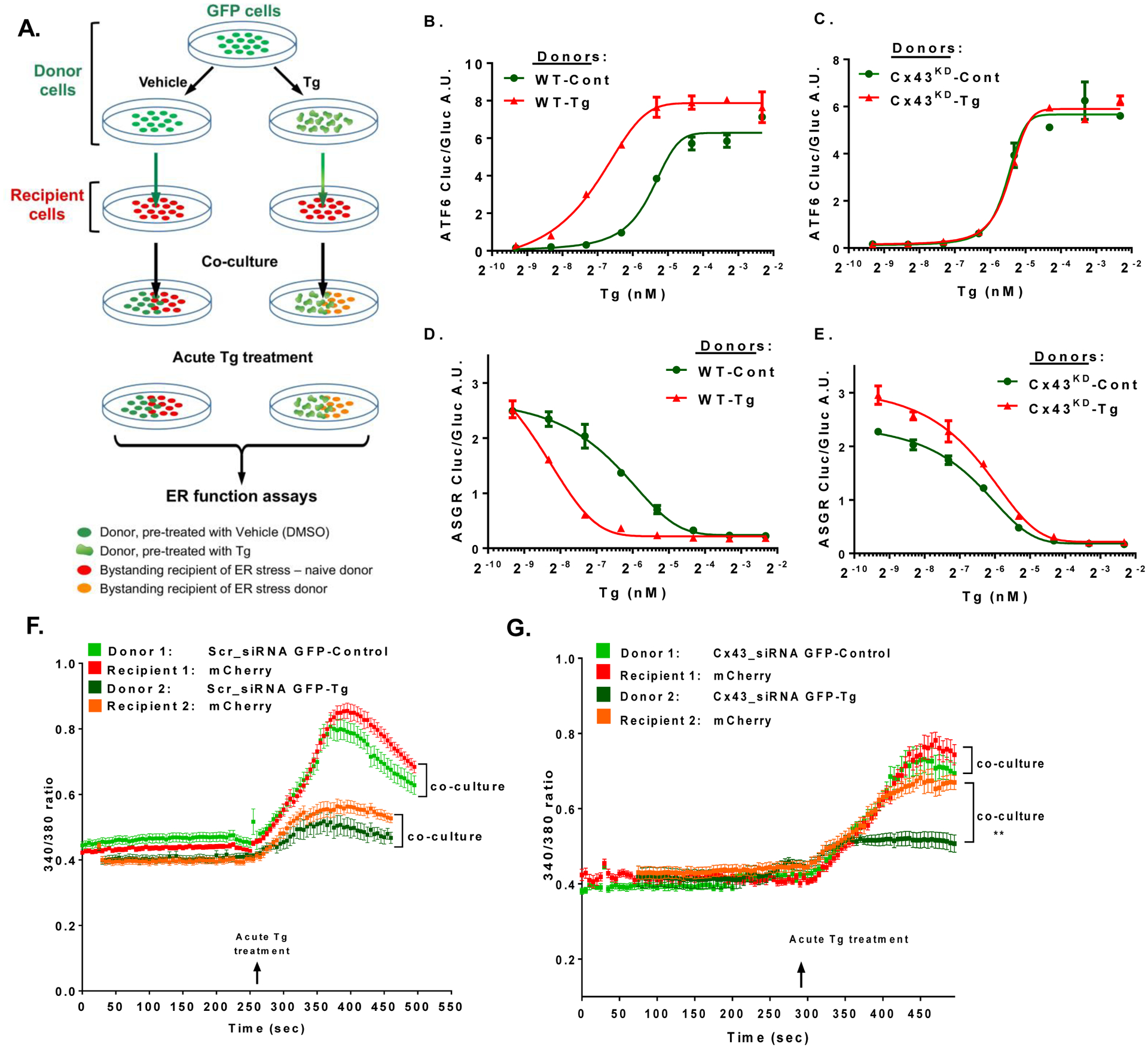
Two dual luciferase reporter constructs (detailed in Fig.S3) were expressed in recipient cells in the co-culture system illustrated in A. Following co-culture of donors and recipients overnight, the mixed cell population was stimulated acutely with Tg (200 nM) and ER function was assayed. Increased chaperone consumption following exposure to increasing concentrations of Tg as indicated (x-axis) results in release of ATF6-luciferase from the ER membrane and its increased activity in the media (panels B, C). In the ASGR reporter system (panels D, E), increased chaperone consumption with increasing concentrations of Tg (indicated in the x-axis) results in impaired ASGR folding, its retention in the ER, and decreased luciferase activity in the media. Donor cells were either transfected with control siRNA (WT; B and D) or with Cx43 siRNA (Cx43KD; C and E), and were pretreated with either DMSO (Cont.) or thapsigargin (Tg) for 6 hours. Following a co-culture for 16 hours, the mixed cell populations were acutely treated with Tg at increasing concentrations, and luciferase activity, indicative of ATF6 secretion (panels B, C), or of ASGR retention (panels D, E), was assayed. n=6 per group, graphs represent mean +/− SEM. Graphs are representative of two independent experiments. (F, G) Cytosolic Ca2+ measured with 4μM of Fura 2-AM in GFP labeled control donors (treated with Scr-siRNA, (F), and following knock down of Cx43 using Cx43 siRNA (Cx43-siRNA GFP; G). A total of 60–90 cells were counted per treatment in 3 experiments.
To further demonstrate the role of Cx43 in mediating direct cell-cell coupling under ER stress conditions, we used quantitative, single-cell imaging of cytosolic Ca2+ levels, measured by Fura2-AM. For this set of experiments, we used GFP expressing cells as donors and constitutively expressing mCherry cells as recipients. As shown in Figure 4F, cytosolic Ca2+ concentration of recipient cells mirrored that of their coupled donors, with recipient cells exhibiting depleted ER Ca2+ stores to a similar extent as their ER-stressed donors. Silencing Cx43 in donor cells disrupted cell-cell coupling and maintained the ER Ca2+ storage capacity of the recipients, irrespective of intracellular Ca2+ homeostasis of their adjacent donors (Fig. 4G, and Figs. S4J–K).
Liver-specific deletion of Cx43 is protective against diet-induced ER stress, hepatosteatosis and systemic metabolic alterations
To assess the in vivo relevance of Cx43-mediated cell-cell coupling and ER stress transmission, we generated mice with liver-specific deletion of Cx43 (Cx43L-KO) by cross-breeding mice homozygous with loxP sites flanking exon 2 of Cx43 (Cx43flox/flox) with mice expressing Cre recombinase under the control of the albumin promoter. No significant changes in body weight (Fig. 5A), lean mass (Fig. 5B) or fat mass (Fig. 5C) were observed between Cx43L-KO and Cx43flox (WT) littermates following 12 weeks of exposure to high fat diet (HFD). However, liver-specific Cx43 deletion resulted in significantly improved insulin sensitivity (Fig. 4D) and glucose tolerance (Fig. 4E). As demonstrated in Figure 5F, Cx43L-KO mice demonstrated a marked improvement in non-alcoholic fatty liver disease (NAFLD) induced by HFD, with a significantly lower NAFLD Activity Score (NAS) as evaluated by independent pathologist blinded to genotype (Fig. 5G). In agreement with the marked attenuation observed in hepatosteatosis, unsupervised hierarchical clustering of key pathways-related genes demonstrated significant effect of hepatocyte Cx43 deletion on lipid metabolism transcriptome signature that promotes lipid degradation over lipid deposition. This includes a decrease in the expression of CD36, Pparg, Pklr and Plin4 with an increase in Acl1, ApoC3 and Plin5, respectively (detailed in figure S5A). The improvement in NAFLD was associated with a significant reduction in serum levels of alanine aminotransferase (ALT; Fig. 5H) and aspartate aminotransferase (AST; Fig. 5I). The hepatocyte-specific deletion of Cx43 also resulted in a decrease in total cholesterol and in LDL-cholesterol (Fig. 5J–K) with no significant changes in HDL-cholesterol or serum triglycerides (Fig. 5L–M). Of note, acute in vivo stimulation by insulin resulted in a marked increase in hepatic Akt phosphorylation in Cx43L-KO compared to control mice (Fig. 5N), indicating improved insulin sensitivity. In order to assess whether deletion of hepatocyte Cx43 had an effect on obesity-induced ER stress, we next used the ERAI (ER stress activated indicator)-Luciferase reporter mouse model (ERAI-Luc). In this mouse model (kindly provided by Dr. Iwawaki, Gunma University, Japan), the N-terminal domain and intron 1 of XBP1 is fused to luciferase cDNA (Iwawaki et al., 2004). Under ER stress conditions, the intron is removed and luciferase protein is translated. We crossed ERAI-Luc mice to mice with albumin Cre and either WT or Cx43flox/flox, to allow visualization of hepatic ER stress in vivo in the presence or absence of Cx43 expression in hepatocytes. As shown in Fig. 5O and quantified in Fig. 5P, deletion of liver Cx43 significantly attenuated luciferase activity in the liver, indicating that the improvement in insulin sensitivity and NAFLD observed in Cx43L-KO mice was associated with a marked reduction in hepatic ER stress. This was further validated in liver homogenates, demonstrating a decrease in ER stress markers (pIRE1 and GRP78) in Cx43L-KO livers as compared with WT mice (Fig. S5B). As expected, Cx43L-KO mice exhibited a near-complete reduction in Cx43 protein expression (Fig. S5B).
Figure 5: Deletion of Cx43 in hepatocytes improves, whereas overexpression impairs, ER function, systemic insulin sensitivity and whole body glucose metabolism in obesity.
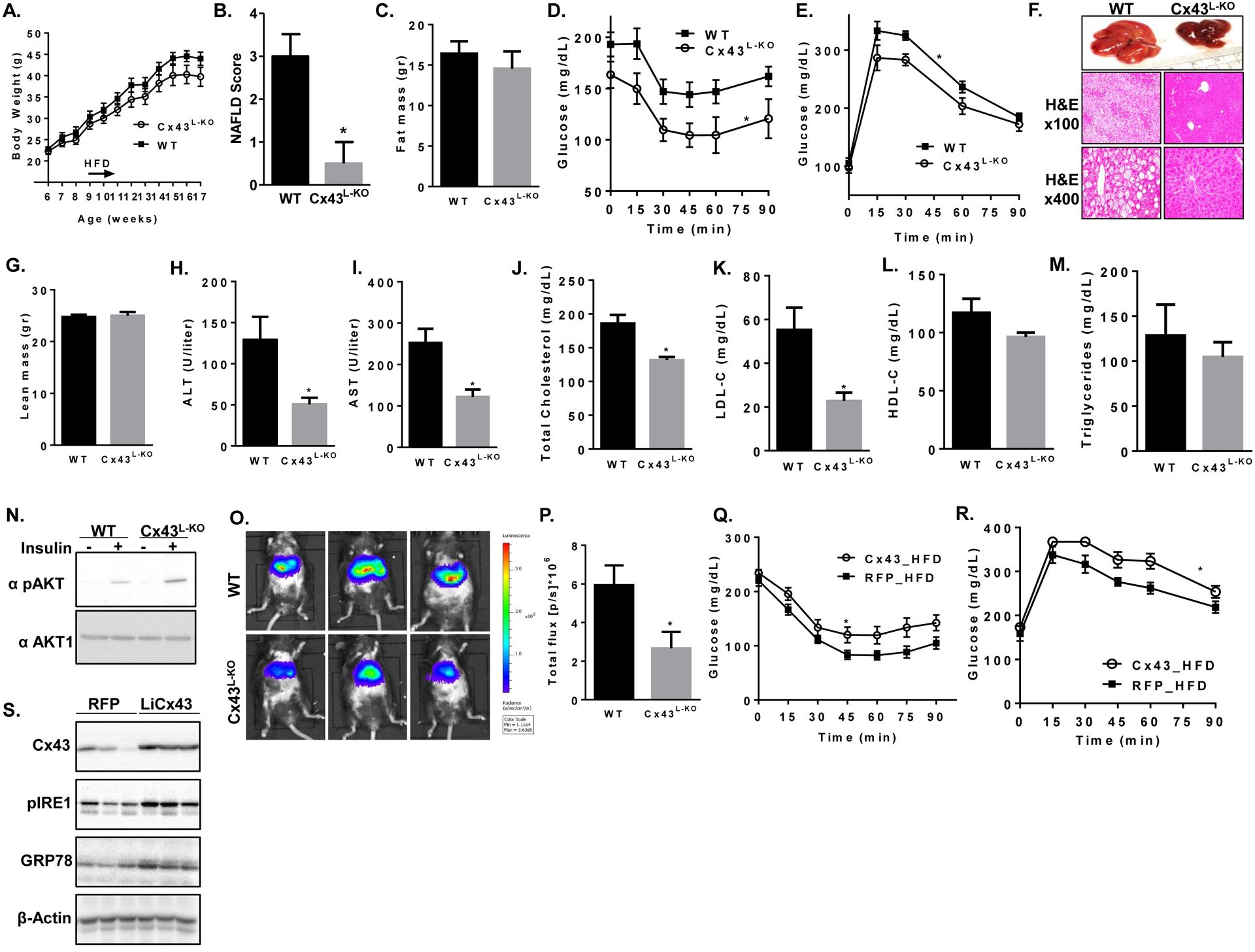
Cx43 liver-specific knockout (Cx43L-KO) and Cx43flox (WT) C57BL/6 mice were treated with high fat diet (HFD) for 12 weeks. n=9 WT and 8 Cx43L-KO mice (A) Weekly measurements of body weight. Measurement of lean (B) and fat (C) mass by dual-energy X-ray absorptiometry at 17 weeks of age. (D) Insulin tolerance test using regular human insulin (1 U/kg body weight, i.p. injection). (E) Glucose tolerance test (1 gr/kg glucose, i.p. injection). (F) Gross liver morphology and liver histology using hematoxylin and eosin (H&E) staining from a representative mouse from Cx43L-KO and WT groups are presented. (G) Non-alcoholic fatty liver disease (NAFLD) Activity Score (NAS) was determined by an independent pathologist blinded to genotype. *p<0.05. Serum ALT (H) and AST (I) as well as total cholesterol (J), LDL-Cholesterol (K), HDL-Cholesterol (L) and triglycerides (M) were measured at the end of the experiment. (N) WT and Cx43L-KO mice were stimulated acutely with intraperitoneal injection of either insulin (1unit/kg) or PBS. Livers were harvested 10 minutes following injection and subjected for western blot analysis for pAkt (Ser473) and total Akt. (O) WT mice expressing albumin promoter driven Cre (Alb-Cre) and Cx43L-KO were further crossed with the ER stress reporter mouse (ERAI-Luc). In vivo ER stress was measured by IVIS reading liver luciferase activity and quantified (P). In a separate cohort, WT mice were fed with HFD for 12 weeks, after which they received adenovirus containing either the red fluorescent protein (RFP, n=10) as control or the mouse GjA1 encoding Cx43 (Cx43, n=10). (Q) Insulin tolerance test (1 U/kg, i.p. injection) was performed on day 6 following infection with adenovirus and (R) glucose tolerance test (1 gr/kg, i.p. injection) on day 10. (S) Liver homogenates were subjected to western blot analysis and immunoblotting using the indicated antibodies.
Next, we studied the effect of intervening in Cx43 expression late in the development of diet-induced obesity and diabetes by injecting adenovirus containing either Cre or GFP into the tail vein of Cx43flox mice following 20 weeks of HFD, which resulted in deletion of ~25% of liver Cx43 (Fig. S5C). Similar to our observations with Cx43L-KO mice, acute reduction in liver Cx43 expression in obese and diabetic mice did not affect body weight (Fig. S5D) but was sufficient to improve blood glucose (Fig. S5E) and insulin sensitivity (Fig. S5F). Of note, this relatively mild decrease in liver Cx43 resulted in a marked improvement in NAFLD (Fig. S5G and S5H).
Lastly, in a complementary set of experiments, we overexpressed either the red fluorescent protein (RFP) or Cx43 in livers of HFD-fed mice through adenoviral gene delivery. No significant change in body weight was observed between the groups (Fig. S5I), but hepatic Cx43 overexpression in obese mice resulted in further deterioration of both insulin sensitivity (Fig. 5Q) and glucose tolerance (Fig. 5R), and was associated with increased expression of the ER stress markers GRP78 and pIRE1 (Fig. 5S).
Liver-specific deletion of Cx43 decreases hepatocyte-hepatocyte coupling and prevents inter-cellular transmission of ER stress
Reduced Cx43 expression in the liver resulted in a marked attenuation of HFD-induced ER stress, insulin resistance and hepatosteatosis. To assess whether the deletion of liver Cx43 indeed interfered with ER stress-induced hepatocyte-hepatocyte coupling, we used primary hepatocytes from WT or Cx43L-KO mice as donor cells, and assayed their capacity to induce ER stress in recipient hepatocytes isolated from ERAI-Luc mice, in which increased luciferase activity indicates ER stress (Fig. 6A). Donor cells for each genotype were either from HFD-fed mouse or from a regular diet (RD)-fed mouse. As depicted in figure 6B, a significant increase in luciferase activity among recipient ERAI-Luc hepatocytes was induced by donor hepatocytes from HFD-fed obese Cx43flox mice, but not by donor hepatocytes from Cx43flox lean mice. This obese-to-lean hepatocyte-hepatocyte transmission of ER stress could be completely prevented when isolated hepatocytes from Cx43L-KO mice were used as donors, regardless of their dietary intervention. As described above, Cx43L-KO mice had a marked improvement in ER stress (Figs. 5O, 5S) which may also explain the decreased capacity of Cx43L-KO hepatocytes to transmit ER stress to recipient ERAI-Luc hepatocytes. Therefore, we next used hepatocytes from ERAI-Luc crossed with either Cx43WT or Cx43L-KO mice to generate ER stress naïve recipients that either contain or are deleted for Cx43, respectively. Donor hepatocytes were isolated from WT mice kept for 16 weeks on HFD or RD. As demonstrated in Fig. 6C, the transmission of in vivo generated, obesity-induced hepatocyte ER stress to intact, ER stress naïve hepatocytes was markedly inhibited when recipient hepatocytes lacked Cx43. Taken together, these results indicate that deletion of hepatic Cx43 in either donors (Fig. 6B) or recipients (Fig. 6C) is sufficient to prevent cell-to-cell transmission of ER stress. Primary hepatocytes from Cx43L-KO mice were also incapable of activating cell-cell coupling when ER stress was induced acutely by Tm or Tg treatment in vitro, as assessed by scrape-loading dye transfer assay (Fig. S6A).
Figure 6: In vivo deletion of Cx43 in hepatocytes decreases cell-cell coupling and transmission of ER stress.
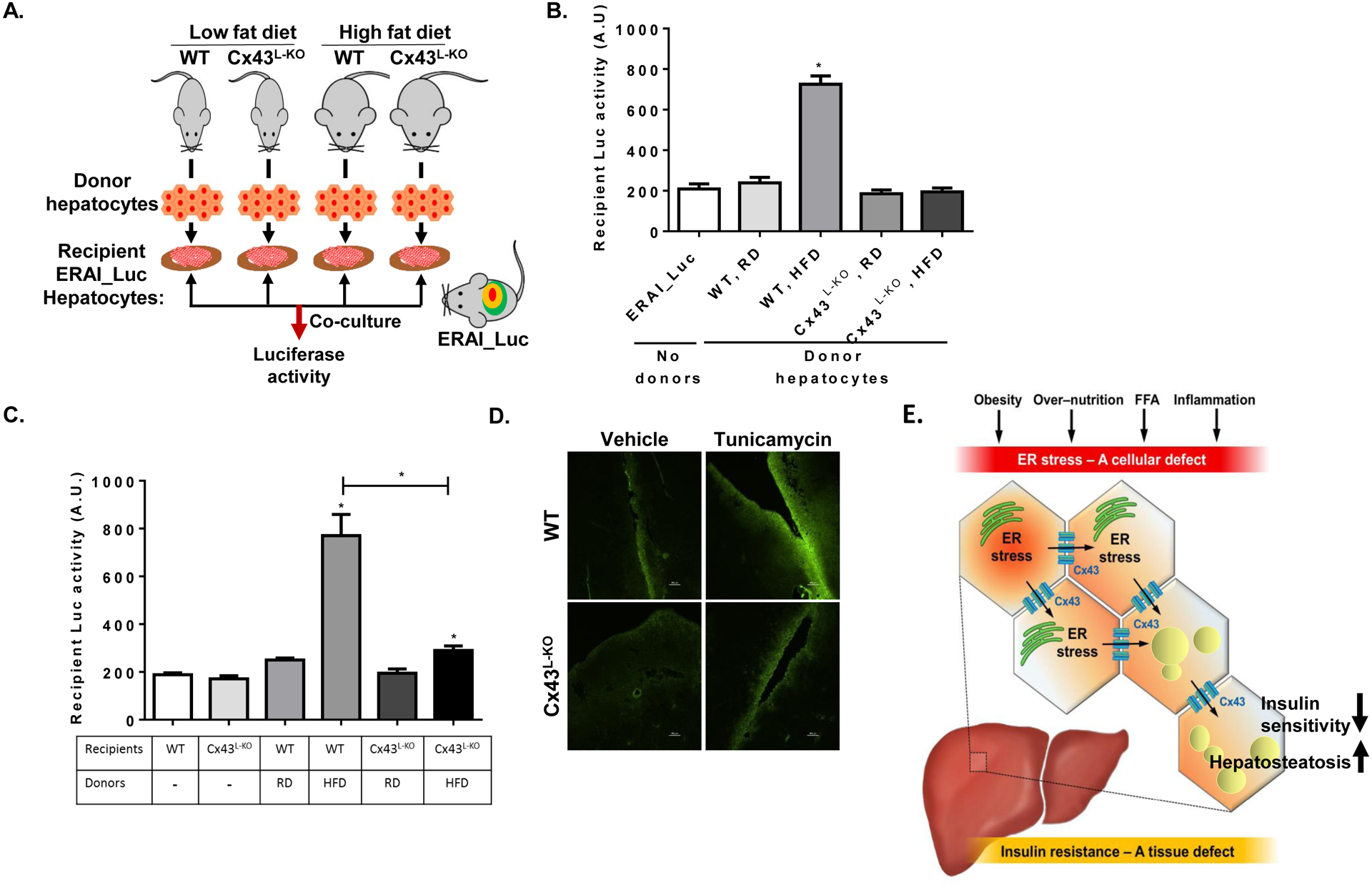
(A) Illustration of the experimental model for co-culture of primary isolated mouse hepatocytes from WT or Cx43L-KO on regular or high fat diet (donor hepatocytes) with recipient hepatocytes isolated from ERAI-Luciferase (ERAI-Luc) mouse kept on a regular diet. (B) Following isolation, donor and recipient isolated hepatocytes were co-cultured overnight and luciferase activity, indicative of the recipients’ degree of ER stress exposure, was determined. n=6 in each treatment. (C) Co-culture of primary mouse hepatocytes. Donor cells were isolated from WT mice on either regular chow diet (RD) or following 12 weeks of high fat diet (HFD). Recipient cells were isolated from ERAI-Luc mice that were either wild type for Cx43 (WT) or deficient in hepatocyte Cx43 (Cx43L-KO). All recipient mice were kept on a regular chow diet. n=3–5 for each treatment. (D) Diffusion of the fluorescent dye Lucifer Yellow away from a scalpel cut made in liver parenchyma. Livers were obtained from either wild type (upper panels) or Cx43L-KO (lower panels), treated the prior day with an intraperitoneal injection of either DMSO (Vehicle) or Tunicamycin (2 mg/kg). (E) Illustration summarizing the suggested role of Cx43-mediated inter-cellular communication in liver in the propagation of ER stress signals under obese conditions.
As obesity is involved in the activation of multiple inflammatory and stress pathways, we next assessed the effect of acute intraperitoneal tunicamycin injection, as a model for in vivo ER stress that is independent of obesity, to induce hepatocyte-hepatocyte coupling. Indeed, the acute induction of ER stress by tunicamycin injection resulted in an increase in functional GJ-mediated intercellular communication in the liver as assessed using a tissue version of the scrape-and-load test (Patel et al., 2012) (Fig. 6D, right upper panel). The ER stress-induced hepatocyte-hepatocyte coupling was abolished in livers of Cx43L-KO mice (Fig, 6D right lower panel). Of interest, the increase in hepatic Cx43-mediated intercellular communication induced by acute induction of ER stress was apparently an adaptive response, as blocking this effect in Cx43L-KO mice resulted in exaggerated hepatocellular injury (Fig. S6B and S6C), and enhanced induction of liver ER stress (Fig. S6D). Indeed, as opposed to the deleterious effects of a forced increase in hepatic Cx43 expression (by adeno_Cx43) in the setting of chronic over nutrition (Fig. 5Q–S), following acute ER stress, adenoviral delivery of Cx43 was hepatoprotective (Fig. S6E–F).
Discussion
This study demonstrates that regulation of intercellular communication is a critical component of tissue adaptation to metabolic stress. In the liver tissue of obese mice, Cx43 is upregulated under ER stress conditions in hepatocytes, leading to increased Cx43-mediated intercellular communication. The increased cell-cell coupling allows transmission of ER stress signals from ‘stressed’ to neighboring, ER-stress naïve ‘bystander’ cells, resulting in impaired ER function and insulin resistance. Disruption of Cx43 in hepatocytes in vivo prevents this transmission and protects the liver from over-nutrition-induced ER stress and hepatosteatosis, leading to improved systemic glucose metabolism (illustrated in Fig. 6E).
Intercellular communication is supposedly a ubiquitous tool for tissue adaptation to various stresses. However, our results indicate that in obesity, the sustained elevation in Cx43-mediated cell-cell coupling may become maladaptive by mediating tissue propagation of ER stress and dysfunction, thus promoting insulin resistance. This apparent discrepancy highlights cell-cell coupling as a ‘double-edged sword’ in normal physiology and in pathophysiological processes. In general, increased Cx-mediated intercellular communication is fundamental in adaptation of tissues to acute environmental stresses, allowing modulation of inflammatory responses and enhancement of tissue repair (Chanson et al., 2005; De Maio et al., 2002). More specifically, Cxs have been implicated in delaying the initiation of acute lung inflammation (Johnson and Koval, 2009), decreasing neuronal vulnerability to acute ischemia and oxidative stress (Blanc et al., 1998) or acute glutamic acid toxicity (Ozog et al., 2002), and improving liver adaptation to acute hepatotoxic insult (Maes et al., 2016). In addition, inhibiting Cx43 following bile duct ligation produces detrimental effects with markedly greater hepatocellular necrosis (Balasubramaniyan et al., 2013). Liver injury following ischemia and reperfusion was worse in Cx43 heterozygous mice, resulting in increased liver necrosis (Trevisan et al., 2019), and Cx43 haploinsufficiency resulted in excessive liver fibrosis by CCl4 -induced hepatic injury (Cogliati et al., 2011), highlighting the adaptive role of Cx43 and cell-cell communication in the response of the liver to acute insults. Similarly, when wild type and Cx43L-KO mice were exposed to tunicamycin, we observed a key adaptive role for Cx43-mediated hepatocyte coupling in the tissue response to this acute injury. In addition, our results with both Cx43 deletion and over-expression in mouse models in vivo indicate that similarly, Cx43 is hepato-protective in the adaptive response to acute induction of ER stress (Fig. S6B–C and S6E–F). However, in the setting of chronic, non-resolving tissue stress, increased inter-cellular communication may become maladaptive and facilitate the spread of tissue injury and dysmetabolism. To the best of our knowledge, this is the first time that connexin-mediated intercellular communication has been demonstrated to play a role in the pathophysiology of metabolic stress-induced ER dysfunction, leading to impaired tissue and systemic metabolic homeostasis. Our results demonstrate that following chronic over-nutrition, the increased Cx43-mediated cell-cell coupling becomes maladaptive by mediating tissue propagation of ER dysfunction, and consequently, aggravating hepatic dysfunction. Thus, upregulation of Cx43 and enhanced intercellular communication in response to acute environmental stresses may turn an adaptive response to acute stress into a maladaptive response to chronic stress.
This effect seems to be a common feature of evolutionarily conserved short-term adaptive response systems that play roles in the pathophysiology of obesity mediated metabolic alterations. More specifically, key mediators of obesity-induced insulin resistance, such as activation of the innate and adaptive inflammatory pathways and the unfolded protein response, are adaptive traits that become maladaptive in the setting of chronic metabolic stress (Hotamisligil, 2017a) (Hotamisligil, 2017b) (Saltiel and Olefsky, 2017). A positive energy balance is likely the initial trigger for activation of these ubiquitous adaptive responses to restore metabolic homeostasis and to relieve the anabolic pressure. However, when metabolic resolution cannot be achieved, the sustained engagement of these homeostatic mechanisms becomes maladaptive, and contributes to chronic tissue dysfunction (Hotamisligil, 2017b). Our findings support this concept that a well-characterized connexin-mediated intercellular communication in the setting of chronic over-nutrition results in a maladaptive response that contributes to hepatic ER dysfunction. In this regard, deletion of Cx43 prevented ER stress-induced coupling of hepatocytes and transmission of ER stress, which was sufficient to provide robust protection from HFD-induced liver ER stress and hepatosteatosis. This further underscores the importance of liver ER integrity for maintaining metabolic homeostasis, as previous studies have demonstrated that interventions that alleviate hepatic ER stress have substantial effects on systemic metabolism (Fu et al., 2011; Ozcan et al., 2006; Yang et al., 2015). Of note, inflammatory signals, oxidative stress and glucotoxicity are all important players in the pathophysiology of insulin resistance and have also been shown to increase Cx43 expression and cell-cell coupling (Le et al., 2014; Riquelme and Jiang, 2013; Willebrords et al., 2016). Thus, inhibition of Cx43 mediated inter-cellular communication may be beneficial in preventing tissue dissemination of multiple harmful signals, in addition to ER stress.
Other than Cx43, the connexin isoforms (21 in humans and 20 in mice) have distinct patterns of tissue expression and participate in key biological activities that, for the most part, are tissue-specific. Cx43, however, is expressed in essentially all tissues and organs at various levels, and has been shown to be relevant in the adaptive response to multiple stress signals and tissue insults. For example, Cx43 is upregulated in the setting of chronic inflammation, hypoxia-reperfusion injuries, oxidative stress, and in response to mechanical signals, changes in intra- and extracellular osmolarity and Ca2+ concentration, and ER stress (Wang and Mehta, 1995; Wang et al., 1995). Moreover, in hepatocytes, which exhibit low baseline expression of Cx43, the adaptation to hepatotoxic insults has been demonstrated to be dependent on Cx43 (Cogliati et al., 2011) and Fig. S6). Therefore, it is likely that Cx43 expression is suppressed under normal conditions, but upregulated as part of a ubiquitous adaptive response to maintain tissue homeostasis upon exposure to stress.
Intercellular transmission of ER stress has been previously demonstrated between various cancer cells and myeloid or dendritic cells (Cullen et al., 2013; Mahadevan et al., 2012; Mahadevan et al., 2011; Rodvold et al., 2017) via an as yet unidentified secreted factor. Secretion of an ER stress-inducing factor was suggested as a mechanism through which cells can facilitate tumor progression, induce clonal fitness, and improve survival in the setting of micro-environmental and exogenous stressors (Rodvold et al., 2017). While we cannot exclude an additional paracrine mode of ER transmission by secreted factor(s) in hepatocytes, our results suggest that cell-cell contact, or at least cell-cell proximity is required for intercellular transmission of ER stress. Of note, Cx43 can also mediate the release of intracellular signals to the extracellular matrix. Recent data suggest that Cx43 hemichannels, in addition of being structural precursors for the formation of intact GJs, may form a direct connection between the cell cytoplasm and extra-cellular matrix (Belousov et al., 2017; D’Hondt et al., 2014). This additional route of utilizing Cx43 for intercellular communication has recently attracted attention (Belousov et al., 2017; Willebrords et al., 2017), although the co-culture and trans-well experiments presented in this work suggest that this is unlikely to be the leading mechanism mediating intercellular transmission of ER stress by Cx43.
Despite the importance of Cx-mediated intercellular communication in physiology and pathology of multiple tissues, the potential roles in regulating systemic metabolic homeostasis have not been well understood. In vitro studies suggest that a transient increase in Cx43 expression may be important for adipocyte differentiation (Yeganeh et al., 2012), while evidence for an in vivo role for adipose tissue Cx43-mediated inter-cellular communication has only recently become available. Zhu et al. reported that Cx43-mediated adipocyte-adipocyte communication in white adipose tissue is necessary for propagation of sympathetic signals in response to cold exposure, leading to ‘beiging’ of adipocyte clusters (Zhu et al., 2016). Cx43 was also suggested to be required to maintain mitochondrial integrity in brown adipose tissue, affecting systemic thermogenesis and energy expenditure (Kim et al., 2017), thus highlighting an additional potential role for this ubiquitous Cx isoform.
An important question that remains unanswered regards the exact mediator that signals ER stress between coupled cells. A variety of approaches have been used to investigate permeability properties of connexin channels. Assessments of unitary conductance, ion selectivity, permeability to various sizes, charges, and chemistries of Cx43 GJs concluded that no atomic ions are large enough to be excluded, and most molecules up to 1kDa in size may cross between coupled cells (Bosco et al., 2011). Indeed, Cx43 GJs have been demonstrated to facilitate the exchange of ions, small metabolites, second messengers, microRNAs and linear peptides between the cytoplasm of connected cells (Ribeiro-Rodrigues et al., 2017). Yet, Cx43 channels have demonstrated a high cation selectivity compared to other connexins (Ribeiro-Rodrigues et al., 2017). Although the literature on permeation of biological molecules is limited, Cx43 mediated transfer of Ca2+ between adjacent cells has been demonstrated in many cell types including astrocytes (Rakers et al., 2017), cardiomyocytes (Li et al., 2012), bone cells (Loiselle et al., 2013), cancer cells (Paemeleire et al., 2000) (Toyofuku et al., 1998), and others (Ribeiro-Rodrigues et al., 2017). In this study, we demonstrate that ER stress results in comparable intracellular Ca2+ dynamics between neighboring cells (Fig. 4F and 4G), and that this is dependent on Cx43-mediated intercellular coupling. Given the increase in cytoplasmic Ca2+ in the setting of ER stress, one can speculate that under these conditions Cx43 GJs mediate the transmission of Ca2+ ions from an ER stressed cell an ER stress naïve coupled cell. Yet, we cannot conclude whether such potential ion flux is mediating cell-cell transmission of ER stress.
The marked metabolic phenotype we observed upon manipulating Cx43 expression in hepatocytes was perhaps surprising, as Cx32 is the predominant Cx isoform in hepatocytes. Previous reports have demonstrated that interruptions of Cx32 expression or function promote hepatic steatosis in various animal models (Sagawa et al., 2015; Tiburcio et al., 2017). In addition, a recent publication similarly suggested a protective role for Cx32 against nonalcoholic steatohepatitis (Willebrords et al., 2017). While in humans, Cx32 expression associates with steatohepatitis (Luther et al., 2018), we did not detect any changes in Cx32 expression during HFD in mice. As for Cx43, although most hepatic Cx43 expression is attributed to the non-parenchymal cell population (Vinken, 2012; Willebrords et al., 2015) and intact hepatocytes exhibit low expression of Cx43, a significant increase in its expression has been demonstrated in various liver pathologies (Vinken et al., 2013). Here, we show that Cx43 is upregulated very early in the course of diet-induced obesity. Thus, the inducible nature of hepatic Cx43 under pathological conditions may provide an attractive opportunity for its inhibition, potentially with minimal interference of other hepatic functions.
Limitations of Study –
This study highlights the importance of Cx43-mediated intercellular communication as an important conduit by which deleterious signals promote hepatic ER stress and dysfunction. However, a limitation of the study is that we do not provide evidence as for the identity of the potential ‘messenger(s)’ responsible for this cell-cell transmission of ER stress. As discussed above, additional questions emerged from our work that remains to be fully answered, including the differentiation between gap junctions and hemichannels as the main mode of transmission, and the potential for cellular signals, other than ER stress, to similarly utilize Cx43-dependent intercellular communication to promote tissue stress and systemic insulin resistance. Finally, the potential translation of our findings to other key metabolic tissues, such as the adipose tissue, is yet to be determined.
In summary, given the strong evidence that increased ER stress in obesity plays a key role in the pathogenesis of metabolic dysfunction, interfering with Cx43-mediated cell-cell coupling in hepatocytes may block tissue propagation of ER dysfunction and thus represent a novel approach to alleviate fatty liver disease, systemic insulin resistance and abnormal glucose homeostasis.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
This is a collaborative study conducted in two independent laboratories. Further information and requests for resources and reagents should be directed to and will be fulfilled by either one of the corresponding authors; The Lead Contact is Dr. Amir Tirosh (Amir.Tirosh@Sheba.Health.gov.il).
Materials Availability
All cellular models for assessment of ER stress and ER function as listed above are available from the lead contact upon request (Dr. Amir Tirosh, Amir.Tirosh@Sheba.Health.gov.il).
Data and Code Availability
The RNA sequencing data reported in this paper was deposited in ArrayExpress (Accession Number: E-MTAB-9687).
Experimental Models and Subject Details
Animal care and use
Mice were bred and housed at both Harvard School of Public Health and Sheba Medical Center animal facilities on a 12 hr light/dark cycle. Mice had ad libitum access to standard chow diet (PicoLab Mouse Diet 20 from LabDiet) or a high fat diet (60% calories from fat, Research Diets). C57BL/6J mice were purchased from Jackson laboratory. To produce mice deficient in liver Cx43 (Cx43L-KO) we bred homozygous B6.129S7-Gja1tm1Dlg/J mice with loxP sites flanking exon 2 of the Gja1 gene with B6.Cg-Speer6-ps1Tg(Alb-cre)21Mgn/J mice expressing Cre recombinase under the control of the albumin promoter (both from The Jackson Laboratory). Cross breeding of Cx43flox and Cx43L-KO with ERAI (ER stress activated indicator)-Luc reporter mice; (ERAI-Luc, Dr. Iwawaki, Gunma University, Japan) generated mice expressing the ERAI-Luc transgene with intact or deficient expression of liver Cx43. For HFD studies, pups were placed on standard chow diet for 3 week at weaning, and at 6 weeks of age male mice were switched to a high fat diet at the indicated times. All experimental procedures were performed on male mice. Unless otherwise indicated, all experiments were performed with mice aged 12–16 weeks. All experimental procedures were approved by the Harvard T.H. Chan School of Public Health and by Sheba Medical Center Institutional Animal Care and Use Committees. All mice, in both animal facilities, underwent routine health status check by a certified veterinarian. Randomly assigned littermates were used when comparing Cx43flox to Cx43L-KO mice.
Generation of ER stress reporter cell lines, unstressed cells (USC) and highly ER stressed (HSC) HEK293 cells
Female HEK293 ER stress reporter cells were generated by stable expression of UPRE-mCherry or ERSE-mCherry reporters as described previously (Oh et al., 2012). Specifically, UPRE-mCherry and ERSE-mCherry reporters were constructed respectively as follows: tandem repeats of 5 mammalian UPRE cis-response element (CCAATCGGAGGCCTCCACG) and 3 tandem repeats of mammalian ERSE cis-response element (CTCGAGACAGGT-GCTGACGTGGCGATTC) were subcloned into the mCherry fluorescent protein construct (pmCherry-1, Clontech) using standard PCR protocols, replacing the standard promoter sequence controlling the mCherry protein expression. HEK293 cells were then transfected with the reporter constructs using Lipofectamine 2000 (ThermoFisher Scientific), according to the manufacturer’s instructions. Cells with the stable reporter expression were obtained using Blasticidin (Invivogen) as selection agent. These cells were used as recipient cells.
Stable High- and Low-UPR activity HEK293 cells were generated by establishing stable knock-down of endogenous genes using genome wide shRNA library in ER stress reporter cells that stably express UPRE-mCherry. Monoclonaly selected reporter cells were then used for genome wide RNAi knock-down using a pooled genome-wide shRNA library (GIPZ Lentiviral shRNAmir, V2L, Open Biosystems). To generate pooled viral lentiviral particles, HEK293T cells were transfected with shRNA library pools. Approximately 2 × 107 stable ER stress reporter HEK293 cells were transduced with viral shRNA library and 6 × 106 cells were stably transfected. MOI (multiplicity of infection) of viral particles were tittered to avoid multiple transduction of single cells by viral particles. Cells stably expressing the shRNA hairpins were obtained by using Puromycin (Invivogen) as selection agent. The pools of stable cells that express both ER stress reporters (UPRE-mCherry) and shRNA vectors were then fractionated by using FACS (FACSAria II, BD) respectively as UPRE High activity cells by selecting the high spontaneous mCherry expression (top 20–30% of the cell population) or UPRE Low activity cells by selecting low mCherry expression (bottom 20–30% of the cell population) followed by 24 hours of 10 nM Tunicamycin treatment (Sigma). Sorted cells were replated and after 7 days of growth at normal conditions the FACS sorting cycles were repeated. FACS sorting cycles were repeated 5 consecutive times until an enriched low UPR activity (unstressed cells, USC) and high UPR activity (highly stressed cells, HSC) cell populations were obtained.
ER function reporter cell lines
The generation of the ER function reporter cell lines expressing either the pGluc/ATF6-Cluc or the pGluc/ASGR-Cluc in female HEK293 cells is detailed in a previous publication (Fu et al., 2015). Briefly, full length coding sequence of Cypridina luciferase (Cluc) protein was cloned in pCDNA3.1(−) cloning plasmid downstream of CMV promoter. Then, the Gaussia luciferase (Gluc) coding sequence controlled by CMV promoter was cloned. ATF6LD, luminal domain was cloned inside Cypridina luciferase gene after signal peptide to generate ATF6LD-Cluc expressing plasmid. ASGR coding sequence of mouse Asialoglycoprotein receptor 1 protein (ASGR) was cloned inside Cypridina luciferase gene after signal peptide sequence to generate ASGR-Cluc expressing plasmid. Reporter-expressing HEK293 cells co-cultured with HEK293A ‘donors’ experiments were performed in 96-well plates. At the end of the co-culture and following the acute treatment with either tunicamycin or thapsigargin as indicated in the figure legends, 10 μl medium was transferred into two 96-well white plates for luciferase assays following the manufacturer’s protocol. Briefly, 50 μl of luciferase substrate (1μM Cypridina or 10 mM CTZ in 100 mM Tris buffer, pH=7.5) was added to the 10 μl medium and incubated in the dark for 5–10 minutes. The luminescence was read on EnVision plate reader (Perkin Elmer).
Primary hepatocyte isolation
Male mice were anesthetized using 2mg/ml xylazine combined with 2mg/ml ketamine in PBS and the liver perfused through the portal vein with 10–15mL warm (37°C) HBSS buffer (Invitrogen, CA) supplemented with 10mM Hepes, 1 mM EGTA and 5mM of glucose, pH 7.4. Next, the liver were perfused with 20mL of HBSS buffer supplemented with 10mM Hepes, 1.2mM CaCl2 and 5 mM glucose, in the presence of 0.3mg/mL Collagenase X (WAKO, Japan) in tissue digestion. The buffers were perfused at a speed of 3mL/min. After digestion, the hepatocytes were carefully released and sedimented at 500 rpm for 2 minutes and washed two times with medium 199 (Invitrogen, CA). The hepatocytes were then homogenized in Williams E medium in the presence of 5% CCS. To separate live from dead cells, the hepatocyte suspension was layered on a 30% Percoll gradient and centrifuged 1500 rpm for 10 minutes. The healthy cells were recovered at the bottom of the tube and plated in Williams E medium supplemented by 5% CCS and 1mM glutamine for experimentation. All experiments in primary hepatocytes were conducted within 24 hours of isolation and plating in order to maintain the Cx expression pattern as closed as possible to the in vivo setting.
METHODS DETAILS
Adenovirus-mediated overexpression of Cx43
For exogenous Cx43 expression, purified, de-salted adenovirus preparations expressing the mouse GJA1 gene or RFP containing adenovirus as control were purchased from Abm. The adenovirus was administered to 18 week old C57BL/6J male mice being on 12 weeks on a high fat diet (D12492i; Research Diets) at a titer of 1 × 109 IFU/mouse by a tail vein injection. Metabolic studies were performed between day 7–12 after infection. The animals were sacrificed after 14 days of infection for ex vivo experiments.
Adenovirus-mediated knock-down of Cx43
For acute deletion of liver Cx43, adenovirus containing either the Cre construct or GFP as control were purchased from UI Viral Vector Core Web, University of Iowa. The adenovirus was administered to 33-week-old C57BL/6J male Cx43flox mice being on a high fat diet for 25 weeks (D12492i; Research Diets) at a titer of 1 × 109 IFU/mouse by a tail vein injection. Metabolic studies were performed at least 7 days after infection.
Glucose and insulin tolerance tests
Animals were subjected to an intraperitoneal (i.p.) glucose injection (1.0 g⋅kg−1) after overnight fasting, and blood glucose levels were measured throughout the first 120 min of the metabolic response. Insulin tolerance tests were performed by intraperitoneal injection of human regular insulin (1.0 IU·kg−1) following 6hrs of day time food withdrawal.
Histology
Livers were fixed in 10% zinc formalin overnight and then transferred to 70% ethanol. Tissue processing, sectioning, and staining with hematoxylin and eosin was performed by the Harvard Medical School rodent histopathology core. Paraffin-embedded specimens were stained with H&E. Olympus CX41 microscope linked to a charge-coupled device (CCD) camera was used to image H&E-stained slides
Cell Culture
Hepa 1–6 cells and HEK293A cells were cultured in DMEM with 10% FCS. AML-12 cells were cultured in DMEM:F12 Medium supplemented with10% FCS supplemented with 1 μg/ml insulin, 0.55 μg/ml transferrin, 0.5 ng/ml selenium and 40 ng/ml dexamethasone. All cells were cultured at 37°C in a humidified incubator that was maintained at a CO2 level of 5%.
ER stress induction in cell lines
Hepa1–6 cells and primary hepatocytes were treated with either 2.5 and or 5 μg/ml tunicamycin (Tm), 100 and or 200 nM thapsigargin (Tg), or with the vehicle (DMSO) for 6 hours. AML12 cells were treated with either 5 μg/ml tunicamycin (Tm), 200 nM thapsigargin (Tg), or with the vehicle (DMSO) for an overnight. Cell lysates were subjected to Western blot analysis and protein expression of Cx43, GRP78 and β tubulin was determined
Immunoblots
Tissues and cells used for immunoblot analysis were lysed in buffer containing 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 0.1% w/v SDS, 0.5% w/v Na-Deoxycholate, 1% v/v Nonidet P-40, 1 mM EDTA, 1 mM EGTA, 2.5 mM Na-pyrophosphate, 1 mM NaVO4, 10 mM NaF and fresh protease inhibitors from Sigma. Lysates were centrifuged at 4°C for 15 min at 13,000 g and the supernatant was transferred to a fresh tube. Protein concentrations were determined using the Pierce BCA Protein Assay kit from ThermoFisher Scientific, and samples were stored in Laemmli buffer at ≤ −20°C until further use. Electrophoresis of protein was run on SDS-PAGE. Protein in gels were transferred to polyvinylidene fluoride (PVDF) membranes, blocked in TBST containing 5% milk, and then incubated overnight with primary antibodies in TBST containing 0.5% bovine serum albumin (BSA, Sigma) at 4°C on a rotating table. After washing three times for 5 min with TBST containing 0.5% milk, anti-rabbit or anti-mouse HRP-conjugated antibody from Santa Cruz Biotechnologies were applied for 1–2 hours in TBST containing 0.5% milk at room temperature. After washing for 5 min with TBST containing 0.5% BSA three times, membranes were incubated with Super Signal West Femto Maximum Sensitivity Substrate from ThermoFisher Scientific to generate a chemiluminescent signal detected with a FluorChem M imager from ProteinSimple. Digital images were analyzed with AlphaView software or ImageJ.
Fluorescence-activated cell sorting (FACS) for determination of mCherry expression
Mixed donor and recipient cell populations in the various co-culture protocols were washed 3 times with PBS and were dissociated by incubation in 0.25% trypsin/EDTA for 2 min complemented by gentle trituration with a p200 pipette, filtered, and immediately processed on the cytometer. Conventional FACS separation was conducted based on red (mCherry) and green (FIT-C) fluorescence. Sorted cells were collected directly in lysis buffer for protein determination and Western blot analyses. Experiments were performed at the Beth Israel Deaconess Medical Center Flow Cytometry Core facility, Boston, MA.
Cytosolic Ca2+ assessment
Cytosolic Ca2+ levels were determined using Fura-2 AM (Invitrogen). Briefly, cells were loaded with 4μM Fura-2AM and 1μM Pluronic F-127 in HBSS for 60 min at room temperature and then washed. Fluorescence images were obtained using an Olympus IX70 with 40x objective and alternatively illuminated with 340 and 380 nm light for 250ms (Lambda DG-4; Sutter Instrument Co.). Cytosolic Ca2+ measurements were obtained in the presence of calcium free medium. Emission light > 510 nm was captured using a CCD camera (Orca-ER; Hamamatsu Photonics). Images were collected every 5 seconds, background corrected and analyzed with Slidebook.
Gene expression analysis
Total RNA was isolated using Trizol Reagent (Invitrogen) and cDNA was synthesized with iScript Reverse Transcription Supermix (Bio-Rad). Real-time qPCR analysis was performed using Power SYBR Green Master Mix (Thermo Fisher Scientific) in ViiA 7 Real-Time PCR systems (Applied Biosystems). Data were normalized to acidic ribosomal phosphoprotein P0 (Rplp0, 36b4) expression. The primer sequences used:
Cx43 Forward: CTT TGA CTC TGA TTA CAG AGC TTA A
Cx43 Reverse: GTC TCA CTG TTA CTT AAC AGC TTG A
36b4 Forward: AGA TTC GGG ATA TGC TGT TGG C
36b4 Reverse: TCG GGT CCT AGA CCA GTG TTC
Incision loading/dye transfer experiments and image analysis
Hepa 1–6 cells were seeded in 24 well plate 1×105 cells/well in complete growth medium. Thirty hours later, cells were treated with tunicamycin (Tm) (5 μg/ml) or thapsigargin (Tg) (200 nM) or vehicle (DMSO) for an overnight. Cells were further treated with 0.5 mM Gap27 or Vehicle (PBS) for 3 hours. Gap junction-permeable Lucifer yellow dye solution (5 mg/ml in medium) or vehicle was added to the cells and a scratch was made in the confluent monolayer with a sterile pipette tip. Cells were incubated for 5 min at 37°C, the dye was removed, and cells were washed and fixed in a 4% paraformaldehyde solution for 10 min. The cells were visualized using a fluorescence microscope (Nikon Eclipse TE 200). Five fields of view were imaged. The % of Lucifer yellow dye travel distance was quantified using ImageJ software (NIH).
The detection of gap junction intercellular communication ex vivo was performed as previously described (Igarashi et al., 2014). Briefly, liver samples were excised and placed on a plastic plate. Using a sharp blade, two incisions (7–8 mm long and 1–2 mm depth) were made on the surface of each specimen. The sample was covered with 0.5% Lucifer Yellow in PBS and kept for 3 min at room temperature. The tissue was washed with PBS and fixed in formalin solution overnight at room temperature. Five μm sections were prepared by cutting the paraffin block perpendicular to the incision line. Lucifer Yellow staining was detected with a fluorescence microscope (Nikon Eclipse TE 200).
In-Vivo Imaging System
Bioluminescence analysis were performed using the In-Vivo Imaging System (IVIS Lumina LT series III, Caliper life sciences). For in vivo analysis, mice were intraperitoneally administered with D-Luciferin dissolved in PBS w/o Mg2+ and Ca2+ (150 mg/kg). After 10 min, isoflurane anaesthetized animal was put into the IVIS system, and signal was acquired. Signal analysis was performed using Living Image Software (PerkinElmer, Waltham, MA, USA). Data were expressed as photons per second per square centimeter per steradian (p/s/cm2/sr). To allow detection of the emitted signal, prior to imaging hair was removed from the abdomen and chest of the mice. During the imaging period mice were anaesthetized with isoflurane. For ex vivo analysis, mice were injected with 150mg/kg of luciferin just prior to euthanasia. Immediately after necropsy, livers were placed individually into a 24mm plate and covered with 300 μg/ml luciferin.
In vivo metabolic studies, liver enzymes and lipid profile
Glucose tolerance test (GTT) and insulin tolerance test (ITT) assays were performed according to standard published protocols (Charles et al., 2017). For GTT assays, mice were fasted for 16 hours before injected with indicated doses of glucose. For ITT assays, mice were fasted for 6 hours before injected with indicated doses of insulin. In both assays, blood glucose was monitored at the indicated time points using glucometer. For assessment of hepatic insulin signaling, WT and Cx43L-KO mice on HFD were stimulated acutely with intraperitoneal injection of insulin (1unit/kg body, following 16h fast). Livers were harvested 10 minutes following injection. Protein extracts were subjected for western blot analysis as described above with indicated antibodies (adopted from (Titchenell et al., 2015)). Serum levels of total cholesterol, triglycerides, HDL, LDL, VLDL, ALT and AST were measured by AU5800 (Beckman Coulter Diagnostics).
RNA sequencing
Liver tissue samples from WT and Cx43L-KO mice (two samples each) were processed for RNA sequencing. Paired-end RNA sequencing was performed on Illumina platforms, yielding 44M reads (average, minimum 39.2M), with a threshold quality score of Q30 in >93% of reads. Raw data quality was further validated using FastQC and trimmomatic (Bolger et al., 2014). Reads alignment was executed by Star (Dobin et al., 2013) using mm10 as reference genome. Differential gene expression analysis was quantified using HTSeq-counts (Anders et al., 2015) and finalized through the DESeq2 pipeline (Love et al., 2014) for normalization and final downstream analysis. To assess the impact of transcriptome changes of Cx43-deficient liver tissue, we performed targeted analysis of the key pathways involved. The genes were defined according to the KEGG database and included the following gene-sets: mmu00010 Glycolysis / Gluconeogenesis, mmu00061 Fatty acid biosynthesis, mmu00071 Fatty acid degradation, mmu03320 PPAR signaling pathway, and mmu04923 Regulation of lipolysis in adipocytes. Heatmap was produced on R studio platform.
Quantification and Statistical Analysis
Data were expressed as Mean ± Standard Error of the Mean (SEM). Significance was defined as p < 0.05 determined using the two-tailed, unpaired Student’s t test, one-way analysis of variance (ANOVA) or two-way ANOVA as indicated in the figure legends. Following initial characterization of Cx43WT and Cx43L-KO mice, the number of mice per group was calculated to detect 50% difference in hepatosteatosis using a power of 0.8 and an alpha of 0.5 (https://clincalc.com/stats/samplesize.aspx). Data of the final calculated cohort met the assumption with a power >0.9 given a greater than 80% difference in NAFLD score between groups. Data analysis was performed using GraphPad Prism software version 7.0.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Antibodies | ||||
| Anti Cx43 | Sigma Aldrich | C6219 | ||
| Anti Cx43 | Abcam | ab113770 | ||
| Anti GRP78 | Santa Cruz Biotechnology |
sc-376768 | ||
| Anti β Tubulin | Santa Cruz Biotechnology |
sc-9104 | ||
| Anti β Actin | MP Biomedicals | 691001 | ||
| Anti- CHOP | Cell Signaling | # 2895 | ||
| Anti phospho-IRE1 S-724 | Abcam | ab48187 | ||
| Anti Cx32 | Abcam | ab66613 | ||
| Anti Cx26 | Thermo Fisher Scientific |
71-0500 | ||
| Anti phospho-PERK -Thr 981 | Santa Cruz Biotechnology |
sc-32577 | ||
| Anti phospho eIF2α | Cell Signaling | 3597S | ||
| Anti eIF2α | Cell Signaling | 9722S | ||
| Anti phospho AKT (S473) | Santa Cruz Biotechnology |
|||
| Anti AKT | Santa Cruz Biotechnology |
sc-8312 | ||
| Anti XBP-1 (M-186) | Santa Cruz Biotechnology |
Cat#: sc-7160 | ||
| Anti-rabbit HRP conjugate | Santa Cruz Biotechnology |
Cat# sc-2004 | ||
| Anti-mouse HRP conjugate | Santa Cruz Biotechnology |
Cat# sc-2005 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||||
| Tunicamycin | Sigma Aldrich | T7765; CAS # 11089-65-9 | ||
| Thapsigargin | Sigma Aldrich | T9033; CAS # 67526-95-8 | ||
| Lucifer Yellow | Thermo Fisher Scientific |
L453 | ||
| Lucifer Yellow VS dilithium salt | Sigma Aldrich | L3510 | ||
| Carbenoxole | Sigma Aldrich | C4790 | ||
| TRIzol | ThermoFisher Scientific |
Cat# 15596018 | ||
| Nitrocellulose membrane | Bio-Rad | Cat# 162-0115 | ||
| Dulbecco’s modified medium (DMEM) | GIBCO | Cat# 11965 | ||
| Cosmic Calf Serum (CCS) | Hyclone | Cat# SH30087.03 | ||
| Fetal Bovine Calf Serum (FBS) | Atlanta Biologicals | Cat# S11550 | ||
| Antibiotics | Lonza | Cat# 17-603E | ||
| L-Glutamine | GIBCO | Cat# 25030-081 | ||
| D-Luciferin, Firefly, potassium salt, 1.0 g/vial XenoLight D-luciferin K+ Salt Bioluminescent substrate, |
Caliper PerkinElmer |
(Part #: 122796) | ||
| DPBS x10, w/o Mg2+ and Ca2+ | Biological Industries |
02-023-5A | ||
| Critical Commercial Assays | ||||
| Connexin 43 siRNA (h) | Santa Cruz Biotechnology |
sc-29276 | ||
| Connexin 43 human siRNA sequences | Dharmacon ON-TARGETplus | LU-002000-00-0008 | ||
| Non targeting siRNA | Dharmacon ON-TARGETplus Non-targeting Pool |
D-001810-10-05 | ||
| GJA1 Adenovirus (Mouse) | ABM (Applied Biomedical Materials) | 221788A | ||
| Ad5CMVCre-eGFP - HT | UI Viral Vector Core Web, University of Iowa |
VVC-U of Iowa-1174-HT | ||
| Ad5CMVeGFP- HT | VVC-U of Iowa-4-HT | |||
| Luciferase Assay System | Promega | Cat# E1501 | ||
| GAP-27 | Sigma Aldrich | G1794-2MG | ||
| Lucifer yellow VS dilithium | Sigma Aldrich | L3510-25MG | ||
| MTT assay | Abcam | ab211091 | ||
| Lipofectamin 3000 Reagent | Thermo Fischer | L3000001 | ||
| Experimental Models: Organisms/Strains | ||||
| Mouse: C57BL/6J | The Jackson Laboratory | Jax: 000664 | ||
| Mouse: B6.129S7-Gja1tm1Dlg/J, Cx43flox; (Cx43flox) | The Jackson Laboratory | Jax:008039 | ||
| Mouse: B6.Cg-Speer6-ps1Tg(Alb-cre)21 Mgn /J Albumin promoter driven Cre recombinase expressing transgenic |
The Jackson Laboratory | Jax:003574 | ||
| Mouse: ERAI (ER stress activated indicator)-Luc reporter mice; (ERAI-Luc) | Dr. Iwawaki, Gunma University, Japan | (Iwawaki et al., 2004) | ||
| Experimental Models: Cell Lines | ||||
| Hepa 1-6 cells | ATCC | CRL-1830 | ||
| Primary murine hepatocytes | This paper | N/A | ||
| HEK 293A cells | Thermo Fisher Scientific |
Cat# R70507 | ||
| AML12 | ATCC | CRL-2254 | ||
| GFP tagged, HEK 293A cells | This paper | N/A | ||
| mCherry tagged, HEK 293A cells | This paper | N/A | ||
| mCherry following the UPR response element (UPRE); UPRE_mCherry | This paper | N/A | ||
| mCherry following the ER stress response element (ERSE); ERSE_mCherry | This paper | N/A | ||
| GFP tagged, HEK293A co-expressing mCherry following the UPR response element (UPRE); GFP_UPRE_mCherry | This paper | N/A | ||
| ATF6a luminal domain fused to luciferase (Cluc) in HEK293 cells; (ATF6-Cluc) | (Fu et al., 2015) | N/A | ||
| Asialoglycoprotein receptor 1 (ASGR1 fused to Cluc in HEK293 cells; (ASGR-Cluc) | (Fu et al., 2015) | N/A | ||
| Software and Algorithms | ||||
| ImageJ | NIH | https://imagej.nih.gov/ij/ | ||
| FACSDiva | BD Biosciences | Version 6.1.3 | ||
| Graphpad prism | Graphpad | Version 7 | ||
| Alphaview | Proteinsimple | https://www.proteinsimple.com/software_alphaview.html | ||
| Illustrations images | Dreamstime | Dreamstime.com | ||
| Living Image ® 4.7.2 (64-bit) | PerkinElmer | |||
| Others | ||||
| Standard chow PicoLab Mouse Diet 20 | LabDiet | Cat# 5058 | ||
| Rodent Diet With 60 kcal% Fat | Research Diets | Cat# D12492i; http://www.researchdiets.com/ |
||
Highlights:
Obesity leads to hepatic ER stress and increased connexin 43 (Cx43) expression
Cx43-mediated cell-cell coupling allows ER stress to disseminate between adjacent cells
Propagation of ER stress between cells induces ER dysfunction and insulin resistance
Inhibition of Cx43 improves ER function and attenuates NAFLD and insulin resistance
Acknowledgments
We are grateful to Kathryn Claiborn and Martin McGrath for supporting this project. We thank the Sabri Ülker Center/Hotamisligil lab members for discussions and express our gratitude for use of resources, advice and expertise and for Mr. Nissim Oz (A.T. lab) for his assistance with the in-vivo experiments. We would also like to thank Dr. Iwawaki (Gunma University, Japan) for providing us with the ERAI-Luc mouse model. This work was supported by the National Institute of Health (NIH)/National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) Career Development Award to A.T. (K08 DK097145-01). This research was also supported by the Israel Science Foundation to A.T. (grant No. 922/17). A.T. was also supported by an NIH T32 Training Grant T32DK007529.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: Dr. Gokhan Hotamisligil is an Advisory Board member of Cell Metabolism.
References
- Abed A, Toubas J, Kavvadas P, Authier F, Cathelin D, Alfieri C, Boffa JJ, Dussaule JC, Chatziantoniou C, and Chadjichristos CE (2014). Targeting connexin 43 protects against the progression of experimental chronic kidney disease in mice. Kidney international 86, 768–779. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramaniyan V, Dhar DK, Warner AE, Vivien Li WY, Amiri AF, Bright B, Mookerjee RP, Davies NA, Becker DL, and Jalan R (2013). Importance of Connexin-43 based gap junction in cirrhosis and acute-on-chronic liver failure. Journal of hepatology 58, 1194–1200. [DOI] [PubMed] [Google Scholar]
- Belousov AB, Fontes JD, Freitas-Andrade M, and Naus CC (2017). Gap junctions and hemichannels: communicating cell death in neurodevelopment and disease. BMC cell biology 18, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc EM, Bruce-Keller AJ, and Mattson MP (1998). Astrocytic gap junctional communication decreases neuronal vulnerability to oxidative stress-induced disruption of Ca2+ homeostasis and cell death. Journal of neurochemistry 70, 958–970. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D, Haefliger JA, and Meda P (2011). Connexins: key mediators of endocrine function. Physiological reviews 91, 1393–1445. [DOI] [PubMed] [Google Scholar]
- Chanson M, Derouette JP, Roth I, Foglia B, Scerri I, Dudez T, and Kwak BR (2005). Gap junctional communication in tissue inflammation and repair. Biochimica et biophysica acta 1711, 197–207. [DOI] [PubMed] [Google Scholar]
- Charles KN, Li MD, Engin F, Arruda AP, Inouye K, and Hotamisligil GS (2017). Uncoupling of Metabolic Health from Longevity through Genetic Alteration of Adipose Tissue Lipid-Binding Proteins. Cell reports 21, 393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YS, Dai DZ, and Dai Y (2013). AQP4 KO exacerbating renal dysfunction is mediated by endoplasmic reticulum stress and p66Shc and is attenuated by apocynin and endothelin antagonist CPU0213. European journal of pharmacology 721, 249–258. [DOI] [PubMed] [Google Scholar]
- Cogliati B, Da Silva TC, Aloia TP, Chaible LM, Real-Lima MA, Sanches DS, Matsuzaki P, Hernandez-Blazquez FJ, and Dagli ML (2011). Morphological and molecular pathology of CCL4-induced hepatic fibrosis in connexin43-deficient mice. Microscopy research and technique 74, 421–429. [DOI] [PubMed] [Google Scholar]
- Cullen SJ, Fatemie S, and Ladiges W (2013). Breast tumor cells primed by endoplasmic reticulum stress remodel macrophage phenotype. American journal of cancer research 3, 196–210. [PMC free article] [PubMed] [Google Scholar]
- D’Hondt C, Iyyathurai J, Himpens B, Leybaert L, and Bultynck G (2014). Cx43-hemichannel function and regulation in physiology and pathophysiology: insights from the bovine corneal endothelial cell system and beyond. Frontiers in physiology 5, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maio A, Vega VL, and Contreras JE (2002). Gap junctions, homeostasis, and injury. Journal of cellular physiology 191, 269–282. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Williams CD, McGill MR, Xie Y, Farhood A, Vinken M, and Jaeschke H (2013). The gap junction inhibitor 2-aminoethoxy-diphenyl-borate protects against acetaminophen hepatotoxicity by inhibiting cytochrome P450 enzymes and c-jun N-terminal kinase activation. Toxicology and applied pharmacology 273, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, and Berman JW (2007). Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 12844–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, Clements JE, Zink MC, and Berman JW (2011). Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. The Journal of neuroscience : the official journal of the Society for Neuroscience 31, 9456–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yalcin A, Lee GY, Li P, Fan J, Arruda AP, Pers BM, Yilmaz M, Eguchi K, and Hotamisligil GS (2015). Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Science translational medicine 7, 292ra298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dorado D, Inserte J, Ruiz-Meana M, Gonzalez MA, Solares J, Julia M, Barrabes JA, and Soler-Soler J (1997). Gap junction uncoupler heptanol prevents cell-to-cell progression of hypercontracture and limits necrosis during myocardial reperfusion. Circulation 96, 3579–3586. [DOI] [PubMed] [Google Scholar]
- Herve JC, and Derangeon M (2013). Gap-junction-mediated cell-to-cell communication. Cell and tissue research 352, 21–31. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2017a). Foundations of Immunometabolism and Implications for Metabolic Health and Disease. Immunity 47, 406–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS (2017b). Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185. [DOI] [PubMed] [Google Scholar]
- Igarashi I, Maejima T, Kai K, Arakawa S, Teranishi M, and Sanbuissho A (2014). Role of connexin 32 in acetaminophen toxicity in a knockout mice model. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie 66, 103–110. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, and Miura M (2004). A transgenic mouse model for monitoring endoplasmic reticulum stress. Nature medicine 10, 98–102. [DOI] [PubMed] [Google Scholar]
- Johnson LN, and Koval M (2009). Cross-talk between pulmonary injury, oxidant stress, and gap junctional communication. Antioxidants & redox signaling 11, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper CA, Sorg I, Schmutz C, Tschon T, Wischnewski H, Kim ML, and Arrieumerlou C (2010). Cell-cell propagation of NF-kappaB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 33, 804–816. [DOI] [PubMed] [Google Scholar]
- Kim SN, Kwon HJ, Im SW, Son YH, Akindehin S, Jung YS, Lee SJ, Rhyu IJ, Kim IY, Seong, et al. (2017). Connexin 43 is required for the maintenance of mitochondrial integrity in brown adipose tissue. Scientific reports 7, 7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koval M, Molina SA, and Burt JM (2014). Mix and match: investigating heteromeric and heterotypic gap junction channels in model systems and native tissues. FEBS letters 588, 1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le HT, Sin WC, Lozinsky S, Bechberger J, Vega JL, Guo XQ, Saez JC, and Naus CC (2014). Gap junction intercellular communication mediated by connexin43 in astrocytes is essential for their resistance to oxidative stress. The Journal of biological chemistry 289, 1345–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Meng Q, Yu X, Jing X, Xu P, and Luo D (2012). Regulatory effect of connexin 43 on basal Ca2+ signaling in rat ventricular myocytes. PloS one 7, e36165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, and Nedergaard M (1998). Gap-junction-mediated propagation and amplification of cell injury. Nature neuroscience 1, 494–500. [DOI] [PubMed] [Google Scholar]
- Loiselle AE, Jiang JX, and Donahue HJ (2013). Gap junction and hemichannel functions in osteocytes. Bone 54, 205–212. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther J, Gala MK, Borren N, Masia R, Goodman RP, Moeller IH, DiGiacomo E, Ehrlich A, Warren A, Yarmush, et al. (2018). Hepatic connexin 32 associates with nonalcoholic fatty liver disease severity. Hepatol Commun 2, 786–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, McGill MR, da Silva TC, Abels C, Lebofsky M, Maria Monteiro de Araujo C, Tiburcio T, Veloso Alves Pereira I, Willebrords J, Crespo Yanguas S, Farhood A, et al. (2016). Involvement of connexin43 in acetaminophen-induced liver injury. Biochimica et biophysica acta 1862, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, and Zanetti M (2012). Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming. PloS one 7, e51845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, and Zanetti M (2011). Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proceedings of the National Academy of Sciences of the United States of America 108, 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus S, Glimcher LH, and Hetz C (2011). Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Current opinion in cell biology 23, 239–252. [DOI] [PubMed] [Google Scholar]
- Morel S, Chanson M, Nguyen TD, Glass AM, Richani Sarieddine MZ, Meens MJ, Burnier L, Kwak BR, and Taffet SM (2014). Titration of the gap junction protein Connexin43 reduces atherogenesis. Thrombosis and haemostasis 112, 390–401. [DOI] [PubMed] [Google Scholar]
- Oh RS, Pan WC, Yalcin A, Zhang H, Guilarte TR, Hotamisligil GS, Christiani DC, and Lu Q (2012). Functional RNA interference (RNAi) screen identifies system A neutral amino acid transporter 2 (SNAT2) as a mediator of arsenic-induced endoplasmic reticulum stress. The Journal of biological chemistry 287, 6025–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, and Hotamisligil GS (2006). Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozog MA, Siushansian R, and Naus CC (2002). Blocked gap junctional coupling increases glutamate-induced neurotoxicity in neuron-astrocyte co-cultures. Journal of neuropathology and experimental neurology 61, 132–141. [DOI] [PubMed] [Google Scholar]
- Paemeleire K, Martin PE, Coleman SL, Fogarty KE, Carrington WA, Leybaert L, Tuft RA, Evans WH, and Sanderson MJ (2000). Intercellular calcium waves in HeLa cells expressing GFP-labeled connexin 43, 32, or 26. Molecular biology of the cell 11, 1815–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SJ, Milwid JM, King KR, Bohr S, Iracheta-Vellve A, Li M, Vitalo A, Parekkadan B, Jindal R, and Yarmush ML (2012). Gap junction inhibition prevents drug-induced liver toxicity and fulminant hepatic failure. Nature biotechnology 30, 179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, and Naus CC (2003). Gap junctions and neuronal injury: protectants or executioners? The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 9, 5–9. [DOI] [PubMed] [Google Scholar]
- Qiu C, Coutinho P, Frank S, Franke S, Law LY, Martin P, Green CR, and Becker DL (2003). Targeting connexin43 expression accelerates the rate of wound repair. Current biology : CB 13, 1697–1703. [DOI] [PubMed] [Google Scholar]
- Rakers C, Schmid M, and Petzold GC (2017). TRPV4 channels contribute to calcium transients in astrocytes and neurons during peri-infarct depolarizations in a stroke model. Glia 65, 1550–1561. [DOI] [PubMed] [Google Scholar]
- Rawanduzy A, Hansen A, Hansen TW, and Nedergaard M (1997). Effective reduction of infarct volume by gap junction blockade in a rodent model of stroke. Journal of neurosurgery 87, 916–920. [DOI] [PubMed] [Google Scholar]
- Ribeiro-Rodrigues TM, Martins-Marques T, Morel S, Kwak BR, and Girao H (2017). Role of connexin 43 in different forms of intercellular communication - gap junctions, extracellular vesicles and tunnelling nanotubes. Journal of cell science 130, 3619–3630. [DOI] [PubMed] [Google Scholar]
- Riquelme MA, and Jiang JX (2013). Elevated Intracellular Ca(2+) Signals by Oxidative Stress Activate Connexin 43 Hemichannels in Osteocytes. Bone research 1, 355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, Willert K, Lin JH, and Zanetti M (2017). Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Science signaling 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokas A (2008). The origins of multicellularity and the early history of the genetic toolkit for animal development. Annual review of genetics 42, 235–251. [DOI] [PubMed] [Google Scholar]
- Sagawa H, Naiki-Ito A, Kato H, Naiki T, Yamashita Y, Suzuki S, Sato S, Shiomi K, Kato A, Kuno T, et al. (2015). Connexin 32 and luteolin play protective roles in non-alcoholic steatohepatitis development and its related hepatocarcinogenesis in rats. Carcinogenesis 36, 1539–1549. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, and Olefsky JM (2017). Inflammatory mechanisms linking obesity and metabolic disease. The Journal of clinical investigation 127, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Yu X, Cui X, Zhu G, Zhao G, Chen J, and Huang L (2009). Blockade of connexin 43 hemichannels reduces neointima formation after vascular injury by inhibiting proliferation and phenotypic modulation of smooth muscle cells. Exp Biol Med (Maywood) 234, 1192–1200. [DOI] [PubMed] [Google Scholar]
- Spray DC, Hanstein R, Lopez-Quintero SV, Stout RF Jr., Suadicani SO, and Thi MM (2013). Gap junctions and Bystander Effects: Good Samaritans and executioners. Wiley interdisciplinary reviews. Membrane transport and signaling 2, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I (2010). The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circulation research 107, 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiburcio TC, Willebrords J, da Silva TC, Pereira IV, Nogueira MS, Crespo Yanguas S, Maes M, Silva ED, Dagli ML, de Castro, et al. (2017). Connexin32 deficiency is associated with liver injury, inflammation and oxidative stress in experimental non-alcoholic steatohepatitis. Clinical and experimental pharmacology & physiology 44, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titchenell PM, Chu Q, Monks BR, and Birnbaum MJ (2015). Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nature communications 6, 7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, and Tada M (1998). Intercellular calcium signaling via gap junction in connexin-43-transfected cells. The Journal of biological chemistry 273, 1519–1528. [DOI] [PubMed] [Google Scholar]
- Trevisan AM, Cogliati B, Homem AR, Aloiav TPA, Aquino Neto N, Moreira JM, Reno LDC, Naumann AM, Galvao FHF, Andraus W, et al. (2019). The liver injury following ischemia and reperfusion is worse in experimental knockout heterozygote mouse model for expression of connexin 431. Acta cirurgica brasileira 34, e201901003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinken M (2012). Gap junctions and non-neoplastic liver disease. Journal of hepatology 57, 655–662. [DOI] [PubMed] [Google Scholar]
- Vinken M, Maes M, Cavill R, Valkenborg D, Ellis JK, Decrock E, Leybaert L, Staes A, Gevaert K, Oliveira AG, et al. (2013). Proteomic and metabolomic responses to connexin43 silencing in primary hepatocyte cultures. Archives of toxicology 87, 883–894. [DOI] [PubMed] [Google Scholar]
- Wang Y, and Mehta PP (1995). Facilitation of gap-junctional communication and gap-junction formation in mammalian cells by inhibition of glycosylation. European journal of cell biology 67, 285–296. [PubMed] [Google Scholar]
- Wang Y, Mehta PP, and Rose B (1995). Inhibition of glycosylation induces formation of open connexin-43 cell-to-cell channels and phosphorylation and triton X-100 insolubility of connexin-43. The Journal of biological chemistry 270, 26581–26585. [DOI] [PubMed] [Google Scholar]
- Wellen KE, and Hotamisligil GS (2005). Inflammation, stress, and diabetes. The Journal of clinical investigation 115, 1111–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willebrords J, Cogliati B, Pereira IVA, da Silva TC, Crespo Yanguas S, Maes M, Govoni VM, Lima A, Felisbino DA, Decrock E, et al. (2017). Inhibition of connexin hemichannels alleviates non-alcoholic steatohepatitis in mice. Scientific reports 7, 8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willebrords J, Crespo Yanguas S, Maes M, Decrock E, Wang N, Leybaert L, da Silva TC, Veloso Alves Pereira I, Jaeschke H, et al. (2015). Structure, Regulation and Function of Gap Junctions in Liver. Cell communication & adhesion 22, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willebrords J, Crespo Yanguas S, Maes M, Decrock E, Wang N, Leybaert L, Kwak BR, Green CR, Cogliati B, and Vinken M (2016). Connexins and their channels in inflammation. Critical reviews in biochemistry and molecular biology 51, 413–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP, Yalcin A, Fu S, and Hotamisligil GS (2015). METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J (2013). Mechanisms of insulin resistance in obesity. Frontiers of medicine 7, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeganeh A, Stelmack GL, Fandrich RR, Halayko AJ, Kardami E, and Zahradka P (2012). Connexin 43 phosphorylation and degradation are required for adipogenesis. Biochimica et biophysica acta 1823, 1731–1744. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Gao Y, Tao C, Shao M, Zhao S, Huang W, Yao T, Johnson JA, Liu T, Cypess AM, et al. (2016). Connexin 43 Mediates White Adipose Tissue Beiging by Facilitating the Propagation of Sympathetic Neuronal Signals. Cell metabolism 24, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data reported in this paper was deposited in ArrayExpress (Accession Number: E-MTAB-9687).