

Abstract
Objective:
The aim of this study was to characterize the fetal sonographic findings and the approach utilized to obtain a definitive diagnosis through molecular testing strategies.
Methods:
This is a retrospective case series of fetuses referred for consultation for prenatal findings suggestive of a skeletal dysplasia between March 1, 2014 and March 1, 2016. Ultrasound images, their timing in gestation and reported findings were reviewed and skeletal abnormalities were documented. Unique features were ascertained. The approach for molecular evaluation, and molecular results were extracted.
Results:
Nine cases were referred for evaluation secondary to prenatal sonographic features suggestive of a skeletal dysplasia. In 4 cases a skeletal dysplasia was suspected prior to 16 weeks gestation. Three of these, with mutations in CANT1, NEK1, and COL2A1 were considered lethal, while the fourth case had a non-lethal ALPL mutation. Similarly 2 of 3 cases diagnosed at 16–22 weeks gestation had lethal mutations in COL1A and DYNC2H1 while the fetus with Russell Silver survived. The final 2 cases diagnosed in the third trimester, both hypochondroplasia, were non-lethal dysplasias. A molecular diagnosis was obtained in 8/9 (88.9%) cases which encompassed eight different skeletal dysplasias. The final case declined molecular testing.
Conclusion:
Features of specific skeletal dysplasias can be visualized in utero and guide appropriate molecular testing. Sonographic details in addition to molecular genetic results aid in prognostic counseling.
Keywords: Prenatal ultrasound, Fetal, long bones, skeletal dysplasia, molecular diagnosis
Introduction
Skeletal dysplasias constitute the largest subgroup of the 461 genetic skeletal disorders outlined in the most recent nosology [1]. As early as 1954, fetal skeletal abnormalities were detected by x-ray [2]. In the 1970s ultrasonography became increasingly available. Sonographic technology improved dramatically over the next several decades leading to what are today detailed fetal anatomic surveys [3]. The accuracy of prenatal sonography is reported to be as high as 68% for skeletal dysplasias when compared to postnatal evaluation [4]. A clinical suspicion of a fetal skeletal dysplasia is often raised during the fetal anatomic survey ultrasound at 18–22 weeks gestation when short limbs are accompanied by other skeletal manifestations. Determining the specific diagnosis, however, can be challenging [5].
Perinatal and long-term outcomes of a fetus with a skeletal dysplasia is dependent on the severity of the skeletal anomalies, additional organ involvement, and the underlying genetic, environmental, or multifactorial etiology. Despite the advances in molecular laboratory diagnostics, the challenges engendered by the numerous types of osteochondrodysplasias, many of which have overlapping features, persist today in establishing a specific diagnosis. For example, in a fetus with severe micromelia, the differential diagnosis is extensive and includes mutations in multiple genes. The difficulty is compounded prenatally by the limitations due to variable gestational age at onset of certain features in many of these disorders (Figure 1).
Figure 1.
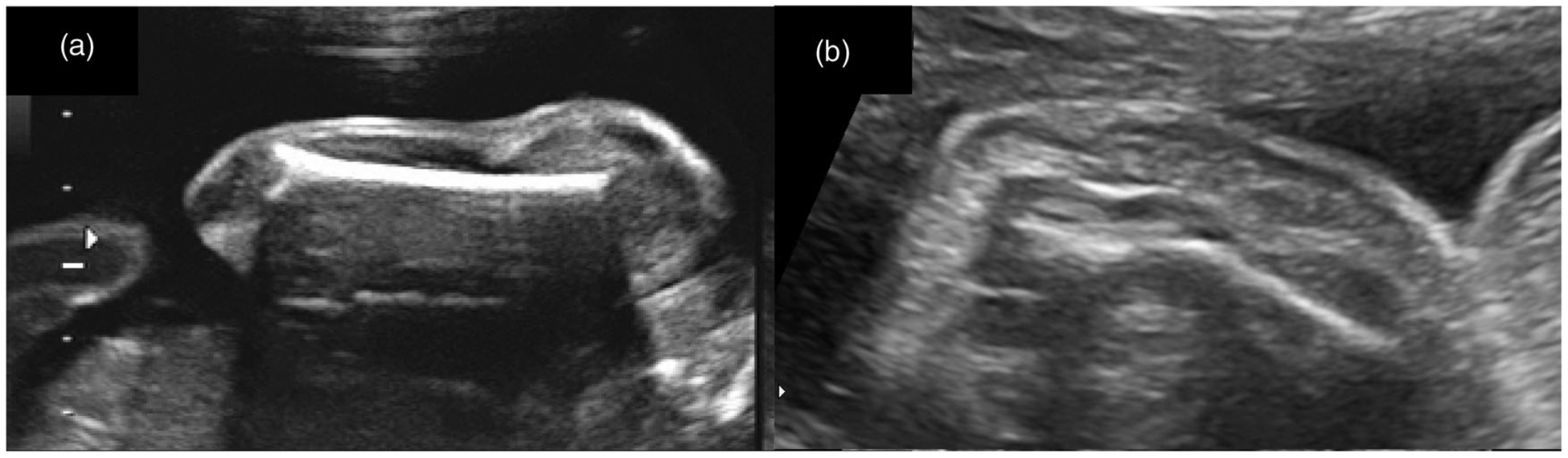
(a) Normal fetal humerus; (b) Bowed/fractured fetal humerus.
The aim of this study was to examine the fetal sonographic findings, their gestational age at onset, and the strategies utilized to obtain the molecular diagnosis for fetuses with a skeletal dysplasia referred to a tertiary care center in 2014–2016 prior to the advent of the use of whole exome sequencing.
Materials and methods
Editorial policies and ethical considerations
This study was approved as a retrospective study by the Institutional Review Board (IRB) at Johns Hopkins Hospital. Informed consent was waived by the IRB. The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Cases
We conducted a retrospective case series at a tertiary university based setting involving patients with a fetal skeletal dysplasia at our prenatal diagnosis center between March 1, 2014 and March 1, 2016. An ultrasound database and prenatal genetic counseling records were first queried for cases referred due to a possible diagnosis of fetal skeletal dysplasia. The study group consisted of all cases in which a sonogram at our Center determined a certain or high likelihood of an osteochondrodysplasia. All ultrasounds were performed on a GE Voluson 730, E8, or E10 by a certified sonographer. An extensive, well developed, skeletal dysplasia sonographic evaluation was performed and interpreted by a sonologist, board-certified in Maternal-Fetal Medicine or in both Maternal-Fetal Medicine and Medical Genetics. In addition to diaphyseal, epiphyseal and metaphyseal characteristics of all the long bones, the evaluation includes images of structures that are not routinely evaluated such as the scapula, clavical and ilium. Additional clinical input through prenatal case review with a pediatric geneticist and specialist in postnatal congenital skeletal disorders was also sought. Her role was to review the ultrasound images in conjunction with the sonologist and to assist with genetic characterization of the features (Figure 2).
Figure 2.
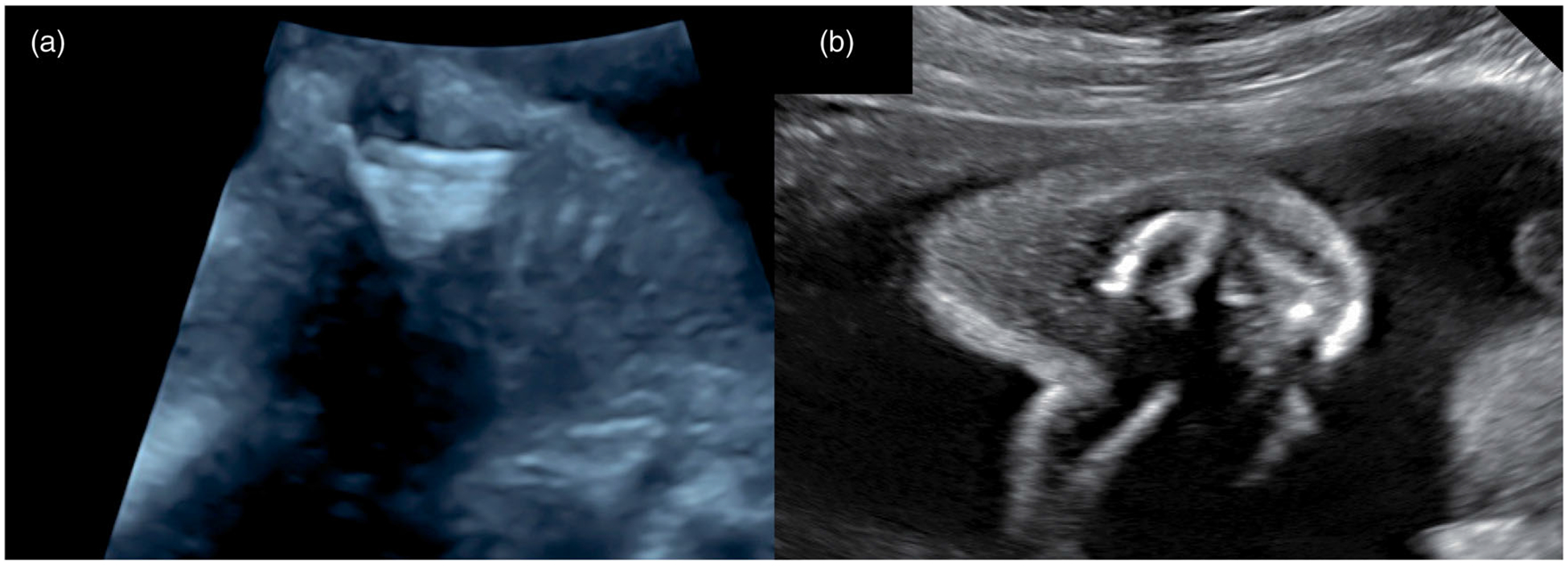
(a) Normal fetal scapula; (b) Hypoplastic and scaffolded fetal scapula.
Data was extracted from electronic medical records for the cases. Ultrasonographic images, the gestational age and reported findings were determined and documented. Representative sonographic features that aided in a prenatal diagnosis were characterized. A list of distinctive features, which aided in clinical decision making in terms of the laboratory tests pursued for each of the cases, was then created.
Further medical chart review was performed to obtain demographics, any previous sonographic findings for each pregnancy, and all prenatal and neonatal testing results. Perinatal outcome data extracted from the electronic medical record included termination of pregnancy, intrauterine demise, gestational age at delivery, mode of delivery, postnatal clinical phenotype, and neonatal death or survival.
Results
A total of 4,943 fetuses were evaluated by prenatal ultrasonography in our Maternal-Fetal Prenatal Diagnostic Center over the study period. Nine (0.2%) cases were determined to have a certain or high likelihood for a fetal skeletal dysplasia. Demographics and perinatal outcomes of these 9 cases are provided in Table 1. Molecular testing was performed in 8/9(88.9%) of cases, and revealed a definitive diagnosis in all eight. A prenatal sample was obtained in 4 cases. In 4 cases the neonatal blood was sent for the genetic testing based on the prenatal clinical diagnosis. In one case (Case 5) all genetic testing was declined both prenatally and postnatally; with characteristic findings of hypochondroplasia, targeted FGFR3 sequencing was offered and declined.
Table 1.
Demographics of cases with fetal skeletal dysplasia.
| Category | Skeletal dysplasia N = 9 (%) |
|---|---|
| Maternal Age (years) | |
| <35 | 4 (44.4%) |
| ≥35 | 5 (55.6%) |
| Paternal Age (years) | |
| <35 | 4 (44.4) |
| 35–45 | 4 (44.4%) |
| >45 | 1 (11.1%) |
| Gravidity | |
| 1 | 3 (33.3%) |
| >1 | 6 (66.7%) |
| Parity | |
| Nulliparous | 5 (55.6%) |
| Multiparous | 4 (44.4%) |
| Fetal Gender | |
| Male | 5 (55.6) |
| Female | 4 (44.4) |
| Ethnicity | |
| Caucasian | 6 (66.7%) |
| African American | 0 |
| Asian | 1 (11.1%) |
| Pakistani | 1 (11.1%) |
| Iranian | 1 (11.1%) |
| History | |
| Prior affected/Prior SAB | 2 (22.2%) |
| Consanguinity | 1 (11.1%)* |
| Chromosomal microarray ordered | 3 (33.3%) |
| Abnormal ultrasound (GA) | |
| 11–14+1 weeks | 5/7 (71.4%) |
| < 21 weeks | 7/9 (77.8%) |
| Pregnancy Outcome | |
| Termination | 2 (22.2%) |
| Fetal Demise | 0 |
| Neonatal Demise | 3 (33.3%) |
| Preterm | 0 |
| Term | 7 (77.8%) |
| Survival >28 days | 4 (44.4%) |
Case #1.
Four cases were of autosomal recessive inheritance and 4 were autosomal dominant. Three out of 4 fetal skeletal dysplasias with advanced maternal age were de novo in genes with autosomal dominant inheritance patterns; although only 1 case was of advanced paternal age. The case with consanguinity had an autosomal recessive inheritance pattern. The genotype and phenotype of the 9 cases are documented in Table 2 and ordered based on the molecular strategies employed. Documented sonographic features represent composites of the first trimester, second trimester, and third trimester ultrasound findings.
Table 2.
Fetal sonographic findings and molecular diagnosis of fetal skeletal dysplasias.
| Prenatal sonographic findings | NT (mm) | GA at 1st Abnormal US | Sample type | Testing strategy/Genetic test result | Diagnosis | Inheritance pattern | Delivery | Vital status | |
|---|---|---|---|---|---|---|---|---|---|
| Long bones | Additional features | ||||||||
| 1. Micromelia | Small chest, small stomach, polyhydramnios, bilateral clubfeet, lethal | 6.6+ | 12 1/7 | Amniotic fluid | Skeletal Panel (76 genes) sequencing, dup/del: heterozygous c.1340G>A COL2A1 mutation | OMIM 151210 Platyspondylic skeletal dysplasia | De novo, AD | Term, 1LTCS | Neonatal demise |
| 2. Micromelia | Abnormal bowing, 90-degree angle in right mid-femur and mid-humerus, improvement with advancing gestational age, consistent with hypophosphastasia | 1.4 | 12 6/7 | Amniotic fluid | Karyotype: Normal Microarray: Normal ALPL sequencing: compound heterozygous c.648 + 1G>A and C.454C>T mutations | OMIM 241500 Hypophosphatasia | AR | Term, NSVD | Living |
| 3. Mesomelia | Hitchhiker thumbs, abnormal curvature of the spine (kyphosis/lordosis) bilateral syndactyly of hands, bilateral clubbed feet, abduction of knees | 4.3+ | 13 4/7 | Chorionic villi | Microarray: Regions of homozygosity CANT 1 sequencing: homozygous c.643G>T mutation | OMIM 251450 Desbuquois dysplasia | AR | TAB | - |
| 4. Micromelia | Hypoplastic left scapula, bell-shaped chest with short ribs, echogenic kidneys, bilateral clubbed feet possible cleft lip, lethal | 2.9 | 14 1/7 | Amniotic fluid | Karyotype: Normal Skeletal dysplasia ciliopathy panel (19 genes): homozygous C.3576G>A NEK1 mutation | OMIM 263520 Short-rib dysplasia 6 w/ or w/o polydactyly | AR | TAB | - |
| 5. Micromelia | Multiple fractures of the long bones consistent with Ol type III, lethal | - | 19 6/7 | Neonatal blood | COL1A1 sequencing: heterozygous c.1156G>A mutation | OMIM 120150 Ol type ll/lll | De novo, AD | Term, NSVD | Neonatal demise |
| 6. Micromelia | Frontal bossing, fetal growth restriction, hyperextended thumbs, 5th digit clinodactyly, duplicated renal artery | - | 20 1/7 | Neonatal blood | Karyotype: Normal Microarray: Normal FGRF3: Normal DMR1 (IGF2, H19): hypermethylation | OMIM 180860 Russell Silver | De novo, AD | Term, RLTCS | Living |
| 7. Micromelia | Handlebar scapula, bell-shaped chest with short ribs, jagged metaphyses, lethal | 1.1 | 20 4/7 | Neonatal blood | Skeletal dysplasia ciliopathy panel (19 genes): compound heterozygous c.4260+1G>A and c.7594C>T DYNC2H1 mutations | OMIM 613091 Short-rib dysplasia 3 w/ or w/o polydactyly | AR | Term, RLTCS | Neonatal demise |
| 8. Micromelia | Widened metaphyses at the femurs, foreshortened ribs, mild frontal bossing | 1.6 | 26 4/7* | NA | FGFR3 sequencing recommended, declined | OMIM 14600 Hypochondroplasia | - | Term, 1LTCS | Living |
| 9. Rhizomelia | Widened metaphyses at the femurs, foreshortened ribs, mildly-bell-shaped thorax, urinary tract dilation | 1.3 | 33 1/7* | Neonatal blood | FGFR3 sequencing: heterozygous c.1620C>G mutation | OMIM 14600 Hypochondroplasia | De novo, AD | Term, 1LTCS | Living |
NT: nuchal translucency; GA: gestational age; US: ultrasound; CVS: chorionic villus sampling; AR: autosomal recessive; AD: autosomal dominant; TAB : therapeutic abortion; term: full term delivery;1LTCS: primary low transverse cesarean section; RLTCS: repeat low transverse cesarean; section; NSVD: normal spontaneous vaginal delivery; OMIM: Online Mendelian Inheritance in Man; NA: not applicable.
Skeletal dysplasia diagnosed in the third trimester, not suspected on anatomy ultrasound,
NT >99%ile.
Molecular testing results
Previous to referral, testing with either a karyotype and/or chromosomal microarray (CMA) was performed in 4 cases. A karyotype had been initially performed prior to referral in 3 of these four cases, 2 of which had abnormal ultrasound sonographic findings at less than 16 weeks gestation. All karyotypes were normal. Three of the four cases had had CMAs, including 2 of the cases with normal karyotypes; the CMA was also normal in these 2 cases. The abnormalities detected by a CMA are limited to duplications and deletions. Skeletal dysplasias generally occur secondary to a nucleotide change and thus, require testing that identifies genetic changes at this level, such as panel testing or whole exome sequencing. In case 3, as expected by family pedigree, the CMA detected areas of homozygosity. These regions assisted in focusing the molecular diagnostic workup [6].
In 5 cases, initial molecular diagnostic evaluation included sequencing of a single targeted gene based on fetal phenotype. Based on prenatal findings, a pathogenic variant was identified in the initial gene sequenced in 4/5 of these cases leading to the diagnoses of perinatal hypophosphatasia (case 2), Desbuquious dysplasia (case 3), osteogenesis imperfecta (case 5), and hypochondroplasia (case 9). The targeted gene testing in case 3 was prescribed through the finding of CANT1 in an area of homozygosity identified by CMA as previously mentioned, as well as the prenatal phenotype by ultrasound which fit well with the CANT1-associated disorder, Desbuquois dysplasia. In case 6 FGFR3 testing had been submitted prior to referral which was negative, and Russell-Silver syndrome was suspected prenatally at our evaluation at 28+3 weeks gestation due to frontal bossing and uniform growth lag of the abdomen and long bones. DMR1 hypermethylation studies were performed postnatally which confirmed the diagnosis of Russell-Silver syndrome.
Skeletal gene panel testing was performed in the remaining 3 cases. Two of these cases were suspected to have a short-rib polydactyly syndrome based on the finding of short ribs and abnormal scapulae, thus in these two cases the diagnoses of short-rib dysplasia 6 (case 4) and short-rib dysplasia 3 (case 7) were made with a 19 gene ciliopathy panel. The final case involved a broader differential diagnosis and a 76 gene panel led to a diagnosis of platyspondylic skeletal dysplasia (case 1).
Sonographic findings
Results of an ultrasound performed at 11–14+1 weeks for nuchal translucency screening were available in 7 cases. Abnormalities were visualized at this time in 5/7(71.4%) of these early ultrasounds, however, only 2/7(28.6%) of these cases had increased nuchal translucencies (NT). The fetus with platyspondylic skeletal dysplasia (case 1) and the fetus with Desbuquois dysplasia (case 3) had an increased NTs. The case of perinatal hypophosphatasia was ascertained as concern for phocomelia in the first trimester (case 2). A non-lethal form of hypophosphatasia was later suspected on the fetal anatomic survey at 23+1 weeks due to the appearance of bowed limbs without evidence of hypomineralization. An ultrasound at 14 weeks for the case of short-rib thoracic dysplasia 6 revealed a somewhat prominent NT (2.9 mm), short long bones, and clubbed feet, yet, the finding of an abnormal, hypoplastic left scapula with very short ribs later at 17 weeks led to the final diagnosis of a short-rib dysplasia (case 4). The first trimester ultrasound was normal for the case of short-rib dysplasia 3, however short long bones were noted at 20+4weeks (case 7); similar to case 4, the short ribs and abnormal scapula visualized on the 22+3 weeks fetal anatomy sonogram guided appropriate multi-gene panel testing. The only cases with an early ultrasound that had normal findings were the 2 cases with hypochondroplasia (cases 8 and 9). First trimester ultrasound screenings were either not performed or not available for 2 cases.
A skeletal dysplasia was suspected in an additional 2 cases at the time of their second trimester fetal anatomic survey for a total of 7/9 (77.8%) cases with a suspicion of skeletal dysplasia prior to 21 weeks gestation. These 2 cases included a classic presentation of severe osteogenesis imperfecta (case 6) with multiple long bone fractures that had not had an early ultrasound, and the case of Russell-Silver syndrome (case 7). The latter case of Russell Silver syndrome was referred for concern for a skeletal dysplasia after FGFR sequencing had been performed which was negative. Our differential diagnosis included osteochondrodysplasia, however, given evocative features of fetal growth restriction and frontal bossing, Russell-Silver syndrome was the leading diagnosis at the time of our prenatal consultation. Prenatal findings were confirmed postnatally with the addition of a triangular face. This diagnosis was confirmed molecularly by methylation studies after delivery.
In the final 2/9 (22%) of cases, suspicion for a skeletal dysplasia did not occur until the third trimester ultrasound. Each of these cases (8 and 9) were prenatally suspected to have achondroplasia vs hypochondroplasia based on the fetal findings of widened metaphysis and mildly foreshortened ribs. This diagnosis was confirmed in both neonates based on clinical and radiologic evaluation, and in case 9 through molecular analysis; testing for FGFR3 was offered and declined in case 8. For all nine cases except for the case of perinatal hypophosphatasia (case 2), the severity of the ultrasonographic fetal findings worsened with advancing gestational age.
In summary, we successfully obtained molecular diagnoses in 88.9% of our prenatally referred cases with suspected skeletal dysplasia. One case declined to pursue molecular testing, however, the clinical prenatal and postnatal phenotype was consistent with the disorder for which the single gene testing was recommended. In 5 cases, the diagnosis was made from targeted single gene sequencing based on fetal phenotype, and multi-gene panel testing was performed in an additional 3 cases. Per patient request, the molecular diagnosis was obtained by neonatal blood in 4 cases; in all of these, the third trimester prenatal diagnosis was consistent with the postnatal diagnosis. Although there is low yield for CMA in the setting of skeletal dysplasia, CMA was helpful in identifying areas of homozygosity on one of our cases, demonstrating utility for CMA particularly if the parents have known consanguinity. Findings suggestive of a skeletal dysplasia were visualized on prenatal ultrasound prior to 21 weeks gestation in all lethal cases and in 77.8% of all cases.
Discussion
Principal findings
Skeletal abnormalities are encountered relatively commonly on prenatal sonography, however a diagnosis of skeletal dysplasia is rare. As a tertiary care center, our incidence of 0.2% is higher than the reported skeletal dysplasia incidence of 1 per 4000 at birth. The combination of short long bones with additional features narrows the differential diagnosis to that of a skeletal dysplasia. In our series these additional features included bowing, fractures, metaphyseal irregularities, club feet, polydactyly, bell shaped thorax, short ribs, abnormal spine, abnormal scapula and frontal bossing. The specific finding of an abnormal scapula assisted in the diagnosis of a ciliopathy for which panel testing revealed homozygous NEK1 mutations in one case and compound heterozygous DYNC2H1 in a second. A molecular diagnosis for a prenatally suspected skeletal dysplasia is feasible and facilitated by recognition of characteristic osseous abnormalities that may be visualized on prenatal ultrasound. The present study adds to our understanding of the features and their timing during gestation for seven such disorders. It also includes one case affirmed as Russell-Silver syndrome which is classified as a disorder of fetal growth.
In our series of 9 prenatally identified cases with complex skeletal findings concerning for skeletal dysplasia, a confirmative molecular diagnosis was established in all but one case in which testing was declined. This high diagnostic yield is partially due to the collaborative efforts of experienced sonographers, certified genetic counselors, prenatal geneticists and maternal-fetal medicine specialists working with an experienced pediatric geneticist with expertise in skeletal dysplasias. We were able to provide a molecular diagnosis in all cases in which the parents elected to pursue molecular testing. When a fetal skeletal dysplasia is suspected, based on our criteria, targeted molecular testing via gene panels or single-gene testing is more effective in achieving a diagnosis than karyotype or CMA, however, the latter may prove helpful in cases of known consanguinity as in our Case 1.
Abnormalities were visualized between 11–14 weeks in 71.4% of those with an ultrasound performed at this gestational age in our series of skeletal dysplasias. Although in the majority of these cases these findings were not specific enough for a diagnosis, the abnormal first trimester findings did generally involve the fetal skeletal system rather than an increased NT. When early abnormalities are detected, a detailed second trimester ultrasound is recommended [7].
Our series included 2 cases of hypochondroplasia, which is not typically diagnosed prenatally. In these cases the diagnoses were suspected prenatally based on third trimester ultrasound findings. None of the parents in our series had a known or suspected diagnosis of skeletal dysplasia. In our series, fetal skeletal dysplasias were not associated with preterm birth in an unaffected mother; although this study is small, all patients who elected to continue their pregnancy delivered at term. While only 2 of these 7 term infants were delivered vaginally, there is no evidence to support a primary cesarean in the absence of standard obstetric indications [8].
Strengths and limitations
By its retrospective nature, our study has limitations. Although we included all cases determined to have a certain or high likelihood for a fetal skeletal dysplasia during the 2-year time period, it is possible, though unlikely, that cases may have been missed. Clearly our incidence of 0.2% of all referrals for prenatal ultrasonography is not applicable to the overall 1 per 4000 incidence of fetal skeletal dysplasias at birth. Incidentally absent from our two-year series were any cases of the two most common skeletal dysplasias, thanatophoric dysplasia and achondroplasia. Thus, we are unable to comment on the diagnostic yield of ultrasound and different avenues pursuant to molecular diagnostic testing for in the detection of these two specific skeletal dysplasias. The mild and most severe forms of skeletal dysplasia were presumably less likely to be referred, thus decreasing the number of cases evaluated a tertiary care center.
This study has several strengths. This is a relatively large series of fetal skeletal dysplasias studied at a time when prenatal gene sequencing options were accessible yet in an era which precedes the availability of whole exome sequencing as a diagnostic tool for use in this setting. It adds to the limited data in the literature about the nature of the associated fetal sonographic findings and the gestational age of presentation. It may help inform future clinical protocols for cost-effective diagnostic evaluations. As a referral center for congenital skeletal anomalies, we had access to a relatively high number of fetuses referred for suspected skeletal dysplasias, with clinical expertise readily available. This study describes the fetal sonographic phenotype of these nine cases, concomitant with the confirmed genetic diagnosis, and includes one case we felt seemed less consistent with an osteochondrodysplasia versus a fetal growth disorder, correctly identified as Russell-Silver syndrome. The sonographic findings visualized in our series are generalizable to most centers that perform obstetrical ultrasound.
Clinical implications
In conclusion, the specific sonographic findings in suspected skeletal dysplasias can inform the laboratory testing strategy employed in pursuit of a definitive molecular diagnosis.
This study adds to further our expertise in these regards, and will assist with prenatal counseling for abnormal skeletal findings, osteochondrodysplasias in particular. Targeted molecular testing is of high diagnostic yield, but only in certain settings. Skeletal gene panels are indicated and beneficial when nonspecific findings are present or there is considerable overlap of features that may be common to several molecularly distinct disorders. While chromosomal analysis appears to have limited diagnostic yield when a fetal skeletal dysplasia is suspect, CMA may be beneficial in targeting specific candidate genes located within areas of identified homozygosity, particularly when there is consanguinity between the parents. Increasing clinical acumen and experiences in recognizing key characteristics of specific skeletal dysplasias will improve the prenatal diagnostician – sonologist’s capability to discriminate between the different types and categories of this large spectrum of disorders to thereby guide the optimal testing strategy.
Funding
This work was supported by Johns Hopkins Women’s Health Scholarship.
Footnotes
Disclosure statement
The authors have no conflicts of interest.
References
- [1].Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393–2419. [DOI] [PubMed] [Google Scholar]
- [2].Aitken GW, Cohen A, Verco PW. Osteogenesis imperfecta a case discovered in utero. J Fac Radiol. 1954; 6(1):62–66. [DOI] [PubMed] [Google Scholar]
- [3].Scheer K Sonography as a routine obstetrical diagnostic procedure. J Clin Ultrasound. 1977;5(2): 101–102. [DOI] [PubMed] [Google Scholar]
- [4].Schramm T, Gloning KP, Minderer S, et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound Obstet Gynecol. 2009;34(2):160–170. [DOI] [PubMed] [Google Scholar]
- [5].Parilla BV, Leeth EA, Kambich MP, et al. Antenatal detection of skeletal dysplasias. J Ultrasound Med. 2003;22(3):255–258. [DOI] [PubMed] [Google Scholar]
- [6].Forster KR, Hooper JE, Blakemore KJ, et al. Prenatal diagnosis of Desbuquois dysplasia Type 1: Utilization of high-density SNP array to map homozygosity and identify the gene. Am J Med Genet A. 2019;179(12):2490–2493. [DOI] [PubMed] [Google Scholar]
- [7].Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med. 2009;11(2):127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bellur S, Jain M, Cuthbertson D, et al. Cesarean delivery is not associated with decreased at-birth fracture rates in osteogenesis imperfecta. Genet Med. 2016; 18(6):570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]