

Abstract
Heterotaxy syndrome (HS) constitutes a spectrum of anomalies arising from embryological errors that result in abnormalities of lateralization involving thoraco-abdominal viscera and culminate in loss of normal asymmetric arrangement of these organs. Besides the unique challenges involved in planning and execution of surgical procedures aimed at correction or palliation of these anomalies, they have the potential to cause profound physiological and immunological consequences in the individual patient due to their cardiac and extra-cardiac manifestations. This article aims to review the literature on this rare and extraordinary subset of developmental anomalies with the intention of familiarizing the reader on the modes of presentation, manifestations, and the variations thereof while dealing with this anomaly. In our institutional experience with HS, 75 consecutive patients were seen between January 2011 and September 2018. Of these, 48 (64%) were confirmed to have isomerism of right atrial appendages (IRAA) and the rest had isomerism of left atrial appendages (ILAA). The cardiac and extra-cardiac manifestations of these patients were listed out. Fifty-four patients (34 with IRAA and 20 with ILAA) underwent 83 surgical procedures. While 49 patients were palliated on the univentricular pathway, 5 underwent biventricular repair. The in-hospital mortality was 7 (13%) in both groups combined (5 for patients with IRAA and 2 for ILAA). In conclusion, the surgical management of HS is associated with satisfactory outcomes in current era.
Keywords: Heterotaxy syndrome, Right atrial isomerism, Left atrial isomerism, Single ventricle, Biventricular repair
Introduction
The heterotaxy syndrome (HS) occurs in approximately 1 per 10,000 live births. It is characterized by abnormal lateralization of the abdominal and thoracic organs including the cardiac atria. The term heterotaxy is used interchangeably with “visceral heterotaxy” and “heterotaxy syndrome” [1]. In this condition, organs that typically develop asymmetrically are noted to display symmetry and mirror image patterns. The heterotaxy syndrome is usually associated with complex cardiovascular malformations and almost 3% of all congenital cardiac anomalies occur in the setting of HS [2].
Definitions and terminology
Situs solitus: Normal position of thoracic and abdominal organs with the cardiac base to apex axis pointing left. The liver and stomach bubble are on the right and left sides, respectively. In addition, the morphological right (short, wide, and straight) and left (long, narrow, and curved) bronchi are concordant (Fig. 1)
Fig. 1.
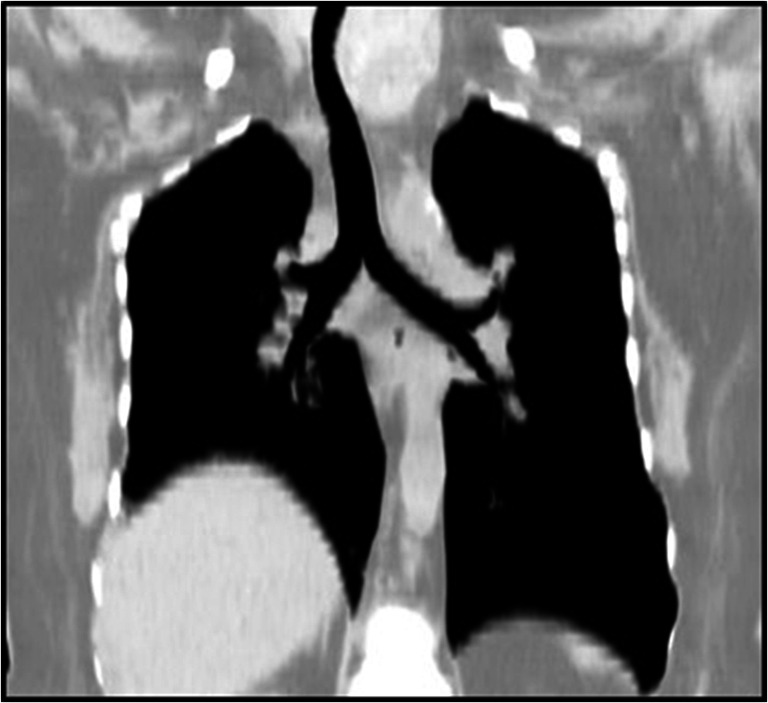
Computerized tomogram showing normal bronchial anatomy, right bronchus is short and broad while left is long and narrow
Situs inversus: Opposite or mirror image of normal
Situs ambiguous: Uncertain, indeterminate, or ambiguous position of thoracic and abdominal organs which are positioned in such a way that they have neither normal nor mirror-imaged arrangement
Heterotaxy: Heteros (Gr.)—other/different, taxis (Gr.)—arrangement. Abnormal development and arrangement of left and right-sided structures
Isomerism: Isos (Gr.)—equal. Meros (Gr.)—part. Morphological symmetry of bilateral structures which are normally asymmetrical
Molecular genetics of heterotaxy syndrome
In a developing fetus, left-right patterning occurs at an early stage during embryogenesis, before organ formation. Heterotaxy syndrome is primarily induced by abnormalities of left-right axis determination during early embryonic development.
During normal embryogenesis, the initial break in symmetry occurs at the primitive node as a leftward “nodal flow” caused by the unidirectional rotation of nodal cell cilia. This flow generates asymmetry signals which are transmitted towards the left lateral plate mesoderm (LPM) and activate the left-side specific growth and transcription factors (including Nodal, Lefty2, and Pitx2c) which in turn instruct the left lateral plate to generate left-sided structures [3]. Right-sided structures appear to develop from the right lateral plate by default.
Pitx2 is a major laterality gene which plays an important role in the asymmetric organogenesis of heart and other visceral organs. Pitx2c is a left-right asymmetric isoform which along with some unidentified factors regulates the asymmetric organ morphogenesis in the left side of body. Its dysfunction is associated with severe cardiac malformations including right isomerism.
In humans, the genes which have been associated with left-right laterality and heterotaxy syndrome are ZIC3, NODAL, CFC1, ACVR2B, LEFTY2, CITED2, and GDF1. Mutations in at least 20 genes have been identified in people with heterotaxy syndrome [3].
Causes of heterotaxy syndrome
Heterotaxy syndrome can occur in isolation or could be a feature of other genetic syndromes like primary ciliary dyskinesia (Kartagener syndrome). Combinations of rare mutations in genes which control early embryonic left-right patterning and environmental insults during early embryogenesis probably cause most of the cases [4].
- Genetic abnormalities:
- Sporadic genetic mutations like in ZIC3 gene
- Non-genetic or environmental etiology: data based on Baltimore-Washington infant study [5]
- Maternal diabetes (odds ratio 5.5)
- Family history of malformations (odds ratio 5.1)
- Cocaine use by mother during first trimester (odds ratio 3.7)
Modes of inheritance
Autosomal dominant inheritance with incomplete penetrance
Autosomal recessive inheritance as evidenced by offspring of unaffected parents and in consanguinity. This pattern is seen in primary ciliary dyskinesia
X-linked inheritance (rare) [4]
Morphological aspects of the heterotaxy syndrome
The HS affects multiple organ systems within the body resulting in a spectrum of anatomical defects that have widespread physiological and immunological consequences. The various morphological patterns are best understood under the headings of cardiac and extra-cardiac anatomical features of the syndrome.
Cardiac manifestations
Understanding the cardiac manifestations of the HS requires in turn, understanding the abnormal anatomical arrangements of the heart with emphasis on the morphology of the atrial appendages, the form and type of ventricular looping, the connections across the atrio-ventricular and ventriculo-arterial junction, and the relationship of the great arteries. Furthermore, position of the cardiac mass and cardiac apex, and abnormal connections of the systemic and pulmonary veins to the respective atria also need to be described.
Cardiac relations and the arrangement of cardiac structures relative to each other are best described by one of two independent and exclusive descriptive systems: the “segmental approach” developed by Van Praagh et al. [6] and the “sequential segmental approach” developed by the European Pediatric Cardiac Code popularized by Anderson et al. [7, 8].
Atrial situs
Atrial situs refers to the arrangement of the atrial chambers on the basis of their sidedness: lateralization. Normal lateralization refers to the situation in situs solitus wherein the normally right-sided and left-sided structures develop on the right and left sides, respectively.
The right atrial appendage is short, broad, and blunt, while the left is long, narrow, and pointed.
Interatrial septum contains the oval fossa which on the right side contains the limbus while on the left it contains the flap valve.
The terminal (supra diaphragmatic) portion of the inferior caval vein (IVC) is a reliable (though not infallible) guide to the situs of the atria. In normal hearts with situs solitus, the IVC lies to the right side of the spine and opposite the location of the descending thoracic aorta that lies on the left of the spine. This relationship is obviously reversed in the heart with mirror image arrangement (situs inversus). However in hearts with heterotaxy, this methodical relationship is altered. In the setting of isomerism of right atrial appendage (IRAA), the IVC lies on the same side as the descending thoracic aorta, while in isomerism of left atrial appendage (ILAA), the IVC is frequently interrupted with no connection to the atrium; the azygous or hemiazygos veins draining the lower body to the heart.
Another method to assess the situs of the atrial chambers is the morphological characteristics of the pectinate muscles and their extent within the vestibules of the respective atrioventricular junctions [9]. In the morphological right atrium, the pectinate muscles surround the vestibule and reach the crux of the heart while they are confined within the appendage on the left side.
As described above, in hearts with heterotaxy, two possibilities exist: namely IRAA or that of ILAA, respectively; it is the appendages that are isomeric, and not the atria themselves. However, Van Praagh et al. consider that the concept of atrial isomerism is erroneous both anatomically and conceptually, because in their view partial mirror imagery does not qualify for isomerism [10].
Though IRAA is closely associated with absence of the spleen (asplenia syndrome) and ILAA with multiple spleens (polysplenia syndrome), these do not always constitute a reliable correlation.
Ventricular looping
The interrelationship of the ventricles, their constituent parts like the atrioventricular junction, the ventriculo-arterial connection, the ventricular mass, and the apex together make up the topology of the ventricles.
In HS, particularly in the setting of IRAA, a univentricular atrioventricular connection is common. However, when the atrioventricular connection (alignment) in the setting of heterotaxy is biventricular in nature, the heart is necessarily situs ambiguous or “mixed” in alignment [1]. This is because, when the atrial appendages are isomeric and one of the atrioventricular connections is concordant in nature, the other has to be necessarily discordant. The corresponding atrioventricular valve determines the respective ventricle in this instance. A common atrioventricular valve is frequent in these situations particularly in IRAA.
Similarly, in HS the nature of the ventriculo-arterial connection is disturbed and variable. While all combinations and patterns are possible, pulmonary stenosis/atresia and discordant ventriculo-arterial connections are more frequent with IRAA while concordant connections with varying levels of left ventricular outflow obstruction are common with ILAA.
In heterotaxy, it is also mandatory to describe the position of the cardiac mass in the chest as well as the orientation of the cardiac apex. “Dextrocardia” refers to the situation when the cardiac mass is on the right side while “dextroversion” is defined as when the cardiac apex points right. Similarly, “levocardia” and “levoversion” refers to the left-sided orientation of the cardiac mass and apex, respectively. “Mesocardia” is present when the cardiac mass occupies the midline of the chest cavity.
Veno-atrial connections
The anomalies of veno-atrial connections constitute the hallmark of HS. All combinations are possible with either type of isomerism; in many instances these anomalies dominate the clinical picture and influence surgical decisions.
Total anomalous pulmonary venous connection (TAPVC) is frequent in hearts with IRAA and so is the absence of coronary sinus. Interruption of the inferior caval vein with continuity through the azygous or hemiazygous system is likewise frequent in the setting of ILAA. Superior caval veins (SVC) occurring bilaterally are frequent in both subsets of HS, though presence of a coronary sinus in this setting occurs only in the subset of ILAA.
Extra-cardiac manifestations
While heterotaxy may exist independent of any congenital heart defect, it has traditionally been the cardiac manifestations that have attracted most attention. However, systemic involvement in HS is the rule; autopsy studies have shown a particular propensity for midline defects in patients with asplenia and polysplenia syndromes [11].
Immunological system
Immunological derangements expose patients with HS to infections particularly with encapsulated organisms and require prophylactic antibiotics and vaccinations [12]. Risk estimation models have identified that asplenia was associated with increased risk following cardiac surgery [13]. Howell-Jolly bodies and percentage of pitted erythrocytes are sensitive tests to assess splenic function.
Gastrointestinal system
Intestinal malrotation, biliary atresia, and midline liver are the commonest anomalies though they may also have anomalies like annular pancreas, duodenal atresia, trachea-esophageal fistula, and anal atresia.
Respiratory system
Primary ciliary dyskinesia may cause pulmonary complications in patients with HS [14]. This may be related to inadequate clearance of secretions, atelectasis, and recurrent chest infections.
Genitourinary system
Almost 26% of patients with HS have anomalies of the genitourinary system [11]. Anomalies like horseshoe kidney, hypoplastic or dysplastic kidney are common.
Central nervous system
Abnormalities of the central nervous system like hydrocephalus, absence of the corpus callosum, meningomyelocele, etc. can be encountered in HS.
Other anomalies
Higher platelet counts and increased incidence of thromboembolism have been mentioned in association with asplenia syndrome [15].
Congenital porto-systemic venous connections (Abernethy malformation) are frequently associated with polysplenia syndrome and univentricular heart [16]. These patients are also prone to develop pulmonary arteriovenous malformations after total cavo-pulmonary shunt (Kawashima operation).
Classification of HS
Patients with heterotaxy are classified into two subsets based on the morphological symmetry (isomerism) of their atrial appendages.
Isomerism of right atrial appendage (asplenia syndrome)
“Right isomerism is defined as a subset of heterotaxy where some paired structures on opposite sides of the left-right axis of the body are symmetrical mirror-images of each other, and have the morphology of the normal right sides structures” [1]. Right isomerism is also called Ivemark syndrome. Several studies have demonstrated a strong predilection (80%) for IRAA in the Asian population [17, 18].
The features of right isomerism can be summarized as follows:
1. Cardiac position: Usually abnormal, displaying either dextrocardia or mesocardia. However, levocardia may also be seen.
2. Morphology of the atrial appendage: Both atrial appendages display the morphological features of the right atrial appendage (broad-based, triangular shape) (Fig. 2).
Fig. 2.
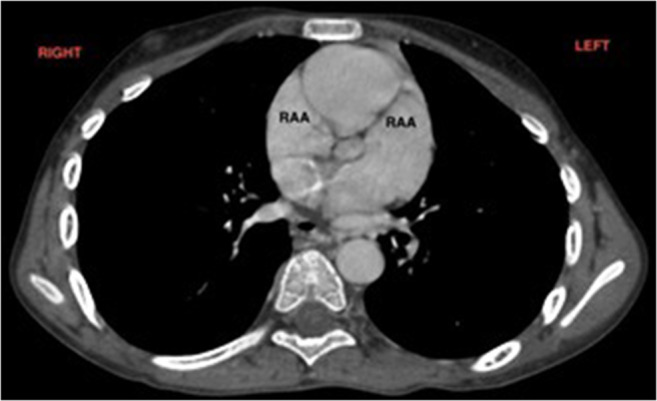
Computerized tomogram showing bilateral appendages of right atrial morphology
3. Systemic venous return: Bilateral SVC occurs in almost 71% of cases. When two SVCs are present, they typically connect to the top corner of corresponding atrium. IVC drains into one of the atria; hepatic venous drainage may well be to the contralateral atrium. Coronary sinus is absent in almost 100% cases.
4. Pulmonary venous drainage: Frequent occurrence of extra-cardiac TAPVC with more than 50% incidence. May be obstructed frequently.
5. Inter artrial septum: Common atrium frequent.
6. Atrioventricular junction: Complete atrioventricular septal defect (AVSD) with common atrioventricular valve (93%).
7. Atrioventricular connection: Frequently univentricular in nature (70%) with a solitary ventricular chamber.
8. Ventricular outflow: Pulmonary stenosis or atresia is frequent (96%); systemic arterial outflow obstruction is rare.
9. Ventriculo-arterial connection: Frequently abnormal (95%). Discordant connections and double outlet right ventricle are common (82%).
10. Conduction system: Bilateral sino-atrial node, one in each atrium. Two atrioventricular (AV) nodes may be present with sling of conduction tissue. Atrial tachyarrhythmias are common.
11. Extra-cardiac anomalies
Spleen: Characteristically absent. If present (10–15%), abnormal and hypo functional.
Lungs and bronchi: Two trilobed lungs with bilateral eparterial bronchi (Fig. 3)
Liver: Central and transverse (Fig. 4)
Bowel: Malrotation less frequent.
Fig. 3.
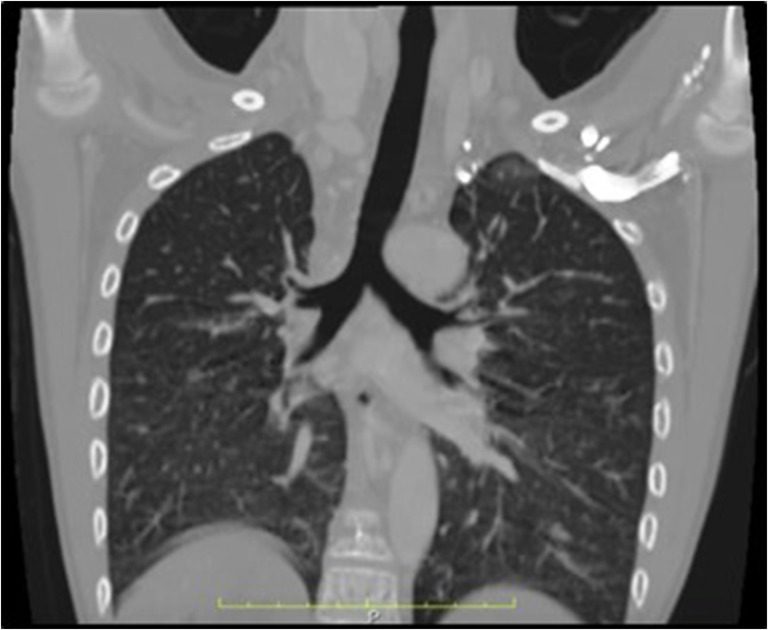
Computerized tomogram showing bilateral right bronchial anatomy in a patient with right atrial isomerism
Fig. 4.
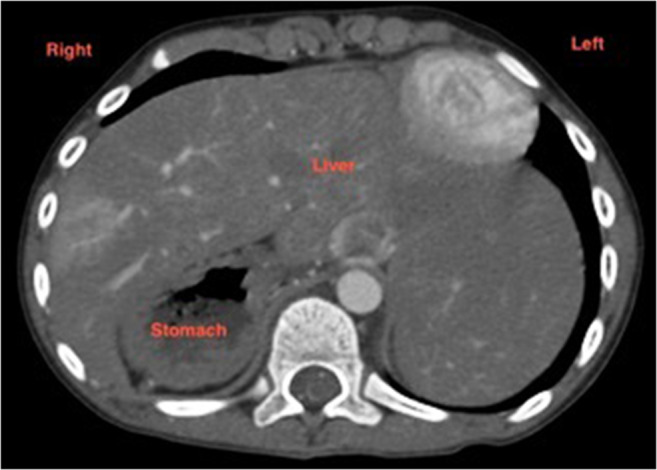
Computerized tomogram of abdomen showing transverse liver and right-sided stomach in a patient with right atrial isomerism. Spleen is absent
Isomerism of left atrial appendage (polysplenia syndrome)
It is defined as “a subset of Heterotaxy where some paired structures on opposite sides of the left-right axis of the body are symmetrical mirror images of each other and have the morphology of normal left sided structures” [1].
These patients have bilateral bi-lobed lungs with hyparterial bronchi and multiple spleens (polysplenia). The cardiac anomalies associated with left isomerism are as follows:
1. Cardiac position: Abnormal in 40–50% cases.
2. Atrial appendage: Both display morphology of left atrial appendage.
3. Systemic veins
Bilateral SVC occurs in two thirds of patients; one may connect to the coronary sinus.
IVC interruption and azygous extension to right or left SVC occurs exclusively in patients with left atrial isomerism; the incidence is about 80%. When the IVC connects directly to atria from below, it may connect to the left- or right-sided atrium. Similarly, hepatic veins may connect to both atria or to both sides of a common atrium.
4. Pulmonary venous return: Partial anomalous pulmonary venous connection (PAPVC) can be seen in > 50% cases due to bilateral atrial connection of ipsilateral pulmonary veins or due to septal malalignment. Rarely TAPVC may be present.
5. Interatrial septum: AV septal defect is common (but prevalence is higher in right atrial isomerism), most patients have a common atrioventricular orifice. Very rarely the atrial septum is well formed and intact.
6. Atrioventricular connection: The atrioventricular connection is biventricular but ambiguous in 75%.
7. Ventriculo-arterial connection: Ventriculo-arterial connection is either discordant or double outlet right ventricle in 20–30%.
8. Ventricular outflow: Unobstructed pulmonary outflow is common, 50% have pulmonary stenosis and < 10% may have pulmonary atresia. Sub-aortic obstruction may occur in 22%.
9. Conduction system
Absence of sinus node in most cases; if present, it is abnormally positioned and hypoplastic. Clinically manifests as slow atrial or junctional rhythm and abnormal axis of P waves on electrocardiogram (ECG)
Complete heart block may occur in some neonates with left atrial isomerism.
Disharmonious patterns of heterotaxy and isomerism
The classification of heterotaxy in left and right atrial isomerism relies on the morphology of atrial appendages which sometimes is challenging to assess in vivo by echocardiography and other cross-sectional imaging. Therefore in clinical setting, sometimes the atrial situs determination is based on the general pattern of associated cardiovascular and other non-cardiac findings rather than actual and accurate assessment of atrial morphology.
However, there is growing evidence that the two most common methods of classifying HS (based on morphology of atrial appendage and status of spleen) are frequently inconsistent with the expected pattern of cardiac and non-cardiac anomalies. Yim et al. [19] in a retrospective analysis of noninvasive imaging data (based on cardiac magnetic resonance imaging, computerized tomographic scan, or both) of 114 patients with heterotaxy have demonstrated that the general features of classic isomerism were breached in > 20% patients and the inconsistency was more prevalent with polysplenia compared to asplenia syndrome. The study demonstrated that polysplenia was associated with inconsistent anatomy of atrial appendages in 21% and inappropriate bronchopulmonary branching pattern in 25% cases.
They further observed that the determination of bronchopulmonary relationship (eparterial or hyparterial) based on computerized tomography (CT) or magnetic resonance imaging (MRI) was more consistent with the right or left atrial isomeric pattern.
This study emphasized that the cardiac and extra-cardiac manifestations which are classically described with left and right atrial isomerism are mere statistical associations and not rules [20]. Accurate diagnosis should take into account the variability and unpredictability of this complex heart condition and should include independent description of each organ system to dispel any ambiguity; particularly when there is disharmony between different systems of organs.
Clinical presentation and diagnosis
There are no clinical features which are specific to atrial isomerism; the manifestations depend on the specific cardiac anomaly present in a particular case.
Radiography
Although the precise intra-cardiac diagnosis cannot be made by the radiologist, characteristic radiologic findings of specific cardiovascular malformations may be apparent on the chest radiograph. Position of the heart such as levocardia, mesocardia, or dextrocardia is evident on chest X-ray.
A transverse liver seen as a midline shadow in an X-ray would point to visceral heterotaxy.
The evaluation of pulmonary vasculature and lung fields may reveal oligemia or hyperemia based on the physiology resulting from a particular cardiovascular malformation.
The chest radiograph can also be useful to raise the suspicion of TAPVC, particularly if the drainage is obstructed.
Bronchial isomerism can be assessed by comparing bronchial lengths on chest X-ray. A ratio of bronchial lengths less than 1.5 is indicative of bronchial isomerism. Once bronchial isomerism is confirmed, right isomerism can be distinguished from left isomerism by measuring the bronchial angles. Angles less than 135° are consistent with left isomerism while angles greater indicate right isomerism [21].
Electrocardiography
Though the majority of patients with heterotaxy may have sinus rhythm, many would not always show sinus rhythm on 24 h Holter monitoring. In some patients with right isomerism, P waves of both right and left atrial origin may be seen at different times due to presence of bilateral sinus nodes [22].
In left isomerism, the sinus node may be hypoplastic and slowing of heart rate may be noticed with age; pacemaker may be required later in life [23, 24]. Patients with left isomerism may also exhibit varying degrees of atrioventricular block especially when associated with atrioventricular septal defect, which is rarely seen in patients with right isomerism.
Although rare, a variety of supraventricular arrhythmia substrates are found in adults with atrial isomerism. The types of supraventricular tachycardia reported are macro reentrant atrial tachycardia, focal atrial tachycardia, and atrial fibrillation. Unusual tachycardia mechanisms have also been occasionally reported, such as twin AV nodal reentrant tachycardia [25].
Abdominal ultrasound
In the past, heterotaxy was typically segregated into the subsets of so-called asplenia and polysplenia syndromes. Though HS cannot be strictly classified thus, it is important nonetheless to specifically delineate splenic anatomy by abdominal ultrasound. In right isomerism, typically the spleen is absent and in left isomerism multiple splenules may be present.
Abdominal ultrasonography identifies position of aorta, IVC, and position of the liver. Major liver mass is on the right side in situs solitus, on the left in situs inversus, and transverse in situs ambiguous. Ultrasound of the abdomen would also identify the presence or absence of abnormalities of the biliary tree and the orientation of mesenteric vessels.
Echocardiography
The important anatomical features of cardiac anomaly associated with HS can be delineated by Doppler echocardiography even in newborns and infants.
Approximately one third of the patients with HS have dextrocardia. In left isomerism, the suprarenal segment of IVC is interrupted with azygous continuation to either a left or right SVC. A cross-sectional view of the abdomen at the level of the spine will determine the arrangement of the aorta, IVC, and azygous veins. In right isomerism, the IVC is to the same side of the spine as aorta, either slightly anterior or to the left of the aorta and in left isomerism, the azygous vein is posterior to the aorta in the paravertebral space [26].
It is important to clearly delineate the connections and drainage of all systemic and pulmonary veins in patients with HS. TAPVC to a systemic vein has been found in about two thirds of patients with right isomerism, whereas it is rare with left isomerism. Presence of obstruction to pulmonary venous drainage is an important finding to necessitate early surgical intervention.
An abnormality of the atrioventricular canal portion of the heart is common to all the patients with HS. The echocardiography must clearly delineate the status of interatrial septum, atrioventricular valves, inter-ventricular septum, and atrioventricular connections. Presence or absence and the degree of atrioventricular valve incompetence have implications for surgical strategy as well as long-term prognosis of the patient.
The ventriculo-arterial alignment as seen by echocardiography defining the relative alignment of the great vessels to a ventricular septal defect is essential to plan surgical strategy. Many of the heterotaxy patients have double outlet right ventricle and transposition of great vessels. Pulmonary stenosis or atresia is fairly common in right isomerism and is also seen in almost half the patients with left isomerism. Systemic outflow obstruction should also be looked for in left isomerism patients.
CT and MRI
Although echocardiography is the first-line investigation, cardiovascular magnetic resonance (CMR) is now the gold standard imaging modality and is useful for assessment of cardiac and vascular structures and function. CMR and CT are also useful for the study of other organs within the chest and abdominal cavities. Echocardiography and CMR are preferred as they involve no radiation hazard. However, the newer multi-detector computerized tomography (MDCT) scanners have the advantage of evaluating cardiac morphology, great vessel angiography, and abdominal viscera at acceptable radiation doses.
Cardiac catheterization
If cardiac CT or CMR are not available or inconclusive, cardiac catheterization can be performed to delineate the cardiac anatomy and study the hemodynamics for surgical planning. It helps in delineation of pulmonary artery anatomy, measurement of pulmonary artery pressure, and calculation of pulmonary vascular resistance for single ventricle palliation.
It is also useful in identification and treatment of rare extra-cardiac anomalies such as pulmonary arteriovenous malformation (Fig. 5) and congenital extra-hepatic porto-systemic shunt (Abernethy malformation) found in association with polysplenia.
Fig. 5.
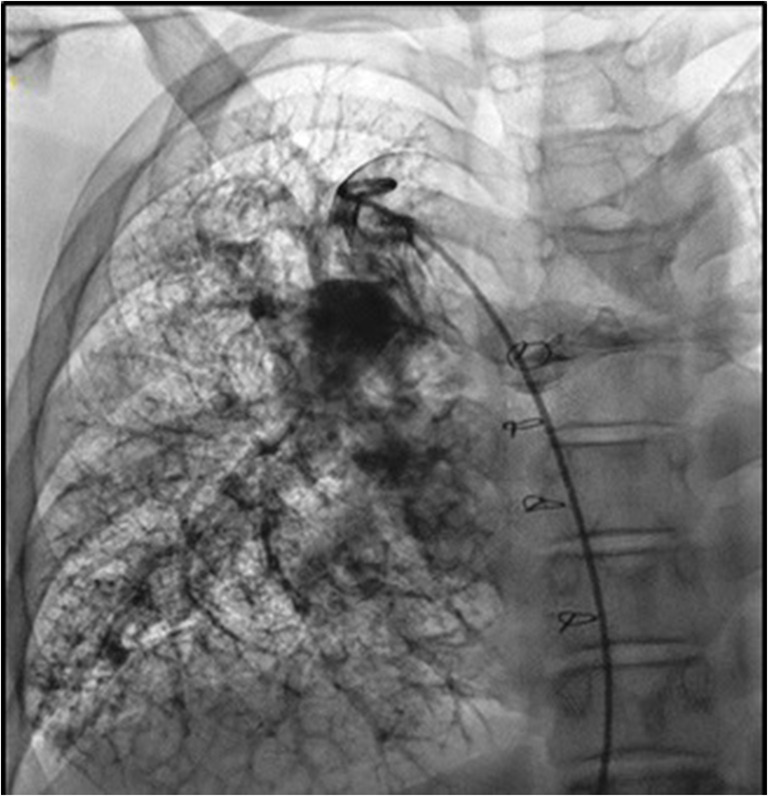
Pulmonary angiogram depicting extensive pulmonary arterio-venous malformation in the right lung of a patient with left atrial isomerism post Kawashima operation
Prenatal diagnosis
1. Fetal echocardiography should be carried out in a segmental approach using standardized anatomical planes along with pulsed wave and color Doppler imaging.
Left isomerism is diagnosed when two of the following findings are present: [27].
-
i.
Situs ambiguous (discordant laterality of stomach, portal sinus, or gallbladder)
-
ii.
Azygous continuation of interrupted inferior vena cava
-
iii.
Early fetal heart block
-
iv.
Complete AV septal defect or other structural heart disease
Right isomerism is diagnosed in presence of at least two of the following findings: [27].
-
i.
Situs ambiguous
-
ii.
Structural heart defect i.e. complete AV septal defect
-
iii.
Juxtaposition of inferior vena cava and descending aorta
Although most features of heterotaxy syndrome can be diagnosed accurately by fetal echocardiography, pulmonary venous obstruction and coarctation of aorta are difficult to diagnose prenatally [28].
2. Fetal MIR: While fetal echocardiography is still the most commonly used modality to image the fetus, MRI has advantages for evaluation of the central nervous system, bronchopulmonary, and abdominal malformations. More recently, cardiac malformations have increasingly been identified using fetal MRI. The technique offers improved delineation of anatomy when compared to fetal echocardiography; it provides multiple viewing planes and is not limited by maternal obesity, oligohydramnios, or the fetal lie [29].
Surgical management
The syndrome of heterotaxy constitutes a spectrum of anomalies that involve multiple organ systems. Treatment of these patients is therefore determined by the nature of various cardiac and extra-cardiac abnormalities. The associated extra-cardiac anomalies, such as ciliary dysfunction, intestinal malrotation, and asplenia, are known to influence the early postoperative morbidity as well as the long-term outcomes.
In many instances, in fact the majority, surgical treatment is palliative in nature due to failure to achieve complete anatomical reconstruction. As with all patients having congenital cardiac anomalies, patients with HS too have two modes of surgical options available to them, i.e., biventricular repair or univentricular type operations.
A significant number of these patients require initial palliation during the neonatal period dictated by the anatomy as well as the degree of pulmonary or systemic outflow obstruction. Accordingly, the baby may require a systemic to pulmonary artery shunt, pulmonary artery banding, TAPVC repair, or repair of coarctation/Norwood operation. Subsequently, they may undergo a definitive surgical procedure or may require multiple palliative operations along their eventual single ventricle pathway.
Biventricular repair
A significant minority of patients born with HS are considered to be suitable candidates for biventricular repair.
Patients with left isomerism in general are more suited for possible biventricular repair strategies. These patients more frequently have balanced atrioventricular septal defects and concordant atrioventricular connections albeit with anomalies of systemic and pulmonary venous return. Biventricular anatomical repair though difficult can be achieved by complex intra-atrial baffling along with the repair of AV septal defect which is usually associated in these cases [30]. It must however be borne in mind that many of these patients with left isomerism have propensity to develop left ventricular outflow tract narrowing leading to sub-aortic stenosis that may require reoperations in future.
Unlike left isomerism, patients with right isomerism have less likelihood of having anatomical features conducive to biventricular repair. Successful biventricular repair has however been reported in this subset as well [31–33] and can be considered in carefully selected patients. Biventricular repair in this group of patients may require complex intraventricular baffle, repair of AV septal defect, placement of right ventricle (RV) to pulmonary artery (PA) conduit, and TAPVC repair.
1.1 Patient selection for biventricular repair: the morphological characteristics of patients with HS are so variable that there are no clear guidelines to determine suitability for biventricular repair and each case has to be considered on its own merits. However, presence of few favorable anatomic features is essential to attempt biventricular repair [32].
Two ventricles of adequate volume and function. In a publication from Children’s Hospital Boston [32], presence of unbalanced ventricle was identified as an independent risk factor for death (hazard ratio 29.6; p = 0.006)
Septatable atrioventricular valves
Veno-atrial connections which can be safely baffled to respective ventricles
1.2 Outcome of biventricular repair:
Mortality: rate ranges from 4 to 20% in different series [32, 33]
Presence of atrioventricular valve incompetence and anomalous pulmonary venous drainage should not be considered absolute contraindications for biventricular repair and they have not been found to contribute to mortality.
- Arrhythmias:
-
i.Postoperative atrioventricular block may occur in 30–40% patients after biventricular repair due to morphological variations and complexity of ventricular septal defect (VSD) closure.
-
ii.Tachyarrhythmias including junctional tachycardia, ectopic atrial tachycardia, and ventricular tachycardia may occur in 16.5% patients [32]
-
i.
Reinterventions and reoperation: Catheter-based reintervention and reoperations are required for 23 and 30% patients, respectively. Pulmonary stenosis, pulmonary atresia, and common AV valve were identified as independent predictors for the need of reintervention and reoperation [32].
Gastrointestinal complications: Perioperative bowel perforation due to gut malrotation has been reported [33] and is one of the causes of perioperative morbidity and mortality. High index of suspicion should be maintained in babies having prolonged ileus in postoperative period.
To summarize, the survival after biventricular repair for babies with HS is excellent in patients who have favorable anatomy but they are prone for arrhythmias and reinterventions/reoperations. It has been observed that the survival reported after single ventricle palliation for HS is comparable to that reported for biventricular repair, but the functional status was better preserved for patients who had biventricular repair [32].
Univentricular repair
The majority of patients with heterotaxy (almost all with right atrial isomerism and significant numbers with left atrial isomerism) do not have anatomy amenable to biventricular repair. Complex malformations characterized by combination of situs ambiguous, double inlet ventricle, straddling atrioventricular valves, unbalanced ventricles with common atrioventricular valve, and malformations like criss-cross atrioventricular relationship preclude biventricular repair in these patients.
Univentricular palliation for babies with HS and single ventricle physiology becomes more challenging due to the presence of additional morphological features like TAPVC, atrioventricular valve anomalies, pulmonary atresia, and conduction abnormalities including tachyarrhythmias and heart block; all of which are considered to be risk factors for increased morbidity and mortality.
Many of these children require palliative procedures in early infancy to achieve balanced pulmonary blood flow, unobstructed pulmonary venous return, and systemic outflow. The aim of these early interventions is to prepare them for bidirectional cavo-pulmonary shunt and eventual Fontan type final palliation.
2.1 Indications for single ventricle palliation in HS:
Extreme hypoplasia of either ventricle
Inability to achieve two separate AV valves of adequate size and competence
Veno-atrial connections which preclude complete separation of systemic and pulmonary venous return
Non-committed or remote VSD in the setting of double outlet right ventricle (relative contraindication for biventricular repair)
2.2 Complexities, risk factors, and outcome
a. Anomalous pulmonary venous connection: Neonates with HS, single ventricle physiology, and anomalous pulmonary venous connection are a particularly challenging subset to manage; especially when the pulmonary venous drainage is significantly obstructed. Various series have reported a mortality of 33 to 64% during initial palliation [34, 35]. In this situation it is very difficult to assess the amount of antegrade pulmonary blood flow (except in patients with pulmonary atresia) and a surgical procedure involving the repair of TAPVC combined with a systemic to pulmonary artery shunt is most likely to yield a predictable outcome [36].
The outcome of TAPVC repair in patients with HS was studied in a multi-institutional cohort selected from the STS database which included patients from the year 2000 to January 2014 and the results were published in 2015 [37]. The authors concluded that there is substantial early mortality in patients with HS undergoing repair of TAPVC and that the outcomes are worse when the patients have functionally univentricular physiology (mortality rate 43% for single ventricle group vs. 30% for non-single ventricle group, p = 0.03).
b. Atrioventricular valve incompetence: Many patients with HS have a common atrioventricular valve which is prone to develop progressive regurgitation over time. In this situation, early reduction of volume overload on the single ventricle is very important and superior cavo-pulmonary anastomosis should be performed at an early age.
Significant incompetence of AV valves is a factor associated with poor long-term outcome and AV valve repair should be performed whenever indicated, even during neonatal palliation. Leaflet apposition, cleft closure, Alfieri type of repair, and segmental annuloplasty techniques have been employed to achieve competence of regurgitant common AV valves. If one component of the valve is grossly incompetent and the other is of adequate size and competence, the leaking valve can be closed completely using a pericardial patch.
c. IVC interruption and azygous or hemiazygous continuation: Particularly important in the setting of left atrial isomerism is the issue of interruption of the IVC, in which instance, the bidirectional cavo-pulmonary connection (Kawashima operation) in effect routes all systemic venous blood to the pulmonary circulation with the only exception of hepatic veins. This may result in significant long-term morbidity due to development of pulmonary arteriovenous malformations (PAVMs) caused by non-inclusion of hepatic factor in the pulmonary circulation. In a follow-up study of children who underwent Kawashima operation for HS and IVC interruption, 58% developed pulmonary AVMs within a median follow-up duration of 5 years [38]. These patients necessarily require subsequent completion of a Fontan procedure to redirect the hepatic venous blood to the lungs [39].
Although these PAVMs regress in most cases after redirection of hepatic venous blood to the pulmonary circulation, the surgery carries high operative risk and sometimes the PAVMs may not regress completely. The emphasis now is not to treat but to prevent the development of PAVMs in such cases and the following recommendations have been made by Vouhe in his editorial commentary [40]:
-
i.
At the time of Kawashima procedure, antegrade pulmonary blood flow should be maintained if pulmonary stenosis is severe enough to allow low pulmonary pressure. This may be particularly relevant if the patient is not an ideal candidate for eventual Fontan completion due to atrioventricular valve incompetence or ventricular dysfunction.
-
ii.
Fontan completion should be performed electively 1–2 years after the Kawashima procedure, when the criteria for a successful Fontan procedure are met. Care should be taken to avoid streaming of hepatic venous blood to a single lung.
-
iii.
In an ideal Fontan candidate, single stage total cavo-pulmonary connection without intervening Kawashima procedure could be an option.
d. Hepatic venous drainage and its inclusion in Fontan pathway: Many times in patients with HS, the hepatic veins may drain separate from the IVC to the contralateral atrium; or in patients with interrupted IVC the hepatic veins may drain individually to both sides of the common atrium. This anatomy should be carefully looked for and if required the atrium should be opened after administration of cardioplegia to identify and include all the hepatic veins in the Fontan pathway. Intra/extra-cardiac technique of Fontan completion can be employed by suturing the intra-cardiac portion of the conduit around the IVC and hepatic veins [41]. Alternatively, if extra-cardiac Fontan is being performed, the conduit can be beveled to include the IVC and hepatic veins which have been separated from the atrium with a cuff of atrial tissue.
e. Outcome of single ventricle palliation for HS: The overall outcome of children with HS is generally poor and there is significant mortality before or at the time of initial palliation. The results of first stage palliation in neonates with HS and single ventricle physiology when compared with that of non-heterotaxy babies revealed that the heterotaxy babies exhibit more resource utilization (duration of ventilation and stay in intensive care unit), more complicated post-surgical course, higher operative mortality, and lower overall survival [42, 43].
It was observed in one study that the risk of mortality in patients with HS is highest at the time of first stage palliation and during the inter-stage period before second stage palliation. No mortality was reported after cavo-pulmonary anastomosis, Kawashima procedure, and Fontan completion [44]. The authors state that they rely on early identification and treatment of residual or recurrent cardiac lesions, complete gastrointestinal system workup to identify and surgically correct patients with malrotation of gut, and aggressive home monitoring program to achieve this successful outcome.
Factors affecting early outcomes and quality of life following single ventricle palliation along the Fontan pathway include presence of anomalous pulmonary venous return (particularly obstructed) and/or presence of more than moderate atrioventricular valve regurgitation. Significant atrioventricular valve insufficiency is usually multifactorial and it is difficult to achieve satisfactory long-term surgical outcome. Aggressive approach towards repair of these has been shown to improve the outcome following Fontan completion [44].
There are conflicting reports in literature about the mid- and long-term results of Fontan operation in patients with single ventricle physiology associated with HS compared to those who do not have HS. Use of extra-cardiac conduit, early detection, and repair of associated anomalies before the Fontan procedure have resulted in improved early and mid-term outcomes [45]. Mayo clinic group has also reported improved survival of this group of patients in the latter part of their experience (1995 to 2004), but during a median follow-up of 15 years there was a 46% incidence of arrhythmias and 22% required permanent pacemaker [46]. Kim et al. retrospectively reviewed 185 patients who had undergone extra-cardiac Fontan procedure from 1996 to 2005 and compared the outcome of patients who had HS with non-heterotaxy patients. The outcome was not found to be different between two groups except higher incidence of early and late arrhythmias in patients with HS [47]. The Pediatric Heart Network conducted a multicentric cross-sectional study of 546 survivors of Fontan procedure including HS and non-HS patients. The patients with HS had greater degree of AV valve incompetence on echocardiography and higher incidence of arrhythmias; but there were no important differences in exercise performance and functional health status between the two groups [48].
2.3 Heart transplantation for single ventricle physiology with HS: A systematic review and meta-analysis was conducted using all the citations from January 1998 to December 2017 dealing with the risk factors for death or heart transplantation among patients with single ventricle physiology including HS. The authors found that the rate of heart transplantation was low in the high risk non-hypoplastic left heart syndrome (HLHS) single ventricle population. Ventricular dysfunction and systemic AV valve regurgitation were identified as the risk factors for death and heart transplantation in patients with single ventricle physiology and HS. They recommend that these simple predictors should be used to guide early referral of these patients for advanced therapy including heart transplantation [49].
Our institutional experience
The medical records of all patients who presented from 2011 to 2018 with a diagnosis of HS, situs ambiguous, and left or right atrial isomerism were reviewed.
There were 75 patients with a diagnosis of HS; 48 were identified as right atrial isomerism and 27 had features of left atrial isomerism.
In patients with right atrial isomerism, there were 36 males and 12 females with age ranging from 15 days to 31 years (median age 3.5 years). The patients with left atrial isomerism had 17 males and 10 females with age range from 1 month to 22 years (median age 3 years). The morphological features of these patients are presented in Table 1.
Table 1.
Morphological characteristics of right and left atrial isomerism
| Serial no. | Morphological feature | Right isomerism (n = 48) | Left isomerism (n = 27) |
|---|---|---|---|
| 1 | Gender | ||
| Male | 36 (75%) | 17 (63%) | |
| Female | 12 (25%) | 10 (37%) | |
| 2 | Cardiac position | ||
| Levocardia | 29 (60.4%) | 17 (63%) | |
| Dextrocardia | 18 (37.5%) | 07 (26%) | |
| Mesocardia | 01 (2.08%) | 03 (11%) | |
| 3 | SVC position | ||
| Bilateral | 32 (66.67%) | 12 (44.44%) | |
| Right SVC | 13 (27.08%) | 12 (44.44%) | |
| Left SVC | 03 (6.25%) | 03 (11.11%) | |
| 4 | IVC drainage | ||
| To right atrium | 26 (54.17%) | Nil | |
| To left atrium | 14 (29.17%) | Nil | |
| Hepatic veins to contralateral atrium | 08 (16.66%) | N/A | |
| Interrupted IVC | 27 (100%) | ||
| 5 | Aorto-caval juxtaposition | 48 (100%) | Nil |
| 6 | Coronary sinus | ||
| Absent | 48 (100%) | 16 (59.3%) | |
| Present | – | 11 (40.7%) | |
| 7 | Pulmonary veins | ||
| TAPVC | 14/48 (29.1%) | 02/27 (7.4%) | |
| Obstructed | 03/14 (21.4%) | 0 | |
| Unobstructed | 11/14 (78.6%) | 02/02 (100%) | |
| HAPVC | Nil | 02/27 (7.4%) | |
| PAPVC | 01/48 | Nil | |
| 8 | Interatrial septum | ||
| Common atrium | 45 (93.75%) | 22 (81.5%) | |
| Small ASD/PFO | 02 (4.16%) | 04 (14.8%) | |
| Intact atrial septum | 01 (2.08%) | 01 (3.7%) | |
| 9 | Atrioventricular Valve | ||
| Common AV valve | 37 (77.1%) | 13 (48.1%) | |
| Two AV valves | 11 (22.9%) | 14 (51.9%) | |
| Hypoplastic left AV valve | Nil | 02/14 (14.3%) | |
| 10 | AV valve incompetence | ||
| No | 08 (16.6%) | 04 (14.8%) | |
| Trivial/mild | 25 (52.1%) | 15 (55.5%) | |
| Moderate | 11 (22.9%) | 06 (22.2%) | |
| Severe | 04 (8.3%) | 02 (7.4%) | |
| 11 | Inter-ventricular septum | ||
| Intact | 01 (2.1%) | 06 (22.2%) | |
| Large VSD | 47 (97.9%) | 21 (77.8%) | |
| 12 | Atrioventricular connection | ||
| Univentricular | 41 (85.4%) | 17 (63%) | |
| Biventricular | 07 (14.5%) | 10 (37%) | |
| 13 | Ventriculo-arterial connection | ||
| NRGA | 09 (18.75%) | 10 (37.03%) | |
| DORV | 37 (77.08%) | 16 (59.26%) | |
| TGA | 02 (4.17%) | 01 (3.7%) | |
| 14 | Ventricular outflow | ||
| Pulmonary atresia | 06/48 (12.5%) | 04/27 (14.8%) | |
| Pulmonary stenosis | 38/48 (79.2%) | 13/27 (48.1%) | |
| Sub-aortic obstruction | Nil | 01/27 (3.7%) | |
| 15 | Rhythm | ||
| Normal sinus rhythm | 41/48 (85.4%) | 11/27 (40.7%) | |
| Ectopic atrial rhythm | 05/48 (10.4%) | 12/27 (44.4%) | |
| Atrial flutter | 01/48 (2.1%) | Nil | |
| Supraventricular arrhythmia | 01/48 (2.1%) | 02/27 (7.4%) | |
| CHB | Nil | 02/27 (7.4%) | |
| 16 | Pulmonary AV malformations | Nil | |
| Mild | 01/27 (3.7%) | ||
| Extensive | 04/27 (14.8%) | ||
| 17 | Liver | ||
| Midline | 48 (100%) | 16 (59.25%) | |
| Right side | Nil | 09 (33.3%) | |
| Left side | Nil | 02 (7.4%) | |
| 18 | Spleen | ||
| Asplenia | 40 (83.3%) not reported in 8 cases | N/A | |
| Polysplenia | Nil | Data not available | |
| 19 | Abernathy malformation | Nil | 2/27 (7.4%) |
Management strategies and outcome
Among 48 patients with right atrial isomerism, 34 patients underwent 57 surgical procedures. One patient (2.08%) underwent biventricular repair (repair of partial AV septal defect) and 33 (68.75%) underwent single ventricle palliation; the procedures performed are presented in Table 2.
Table 2.
Management of patients with right atrial isomerism (n = 48)
| Serial no. | Procedure performed | No. of patients | Remarks |
|---|---|---|---|
| 1 | Biventricular repair (partial AV canal repair) | 1 | Alive, on follow-up moderate MR |
| 2 | Systemic artery to pulmonary artery shunt | 3 | Survived to next stage |
| 3 | Bidirectional cavo-pulmonary shunt | 30 | Survived to next stage |
| With TAPVC repair | 4/30 | ||
| 4 | Extra-cardiac Fontan completion | 20 | 2/20 expired |
| With AV valve repair | 2/20 | ||
| With AV valve replacement | 1/20 | ||
| With pacemaker implantation | 1/20 | ||
| 5 | AV valve re-replacement in a patient with Fontan + AV valve replacement | 1 | Expired |
| 6 | TAPVC repair | ||
| With PA banding | 1 | Expired | |
| BT shunt | 1 | Expired | |
| 7 | Catheter-based interventions | 2 | |
| Balloon pulmonary valvotomy | 1/2 | Unsuitable for single ventricle repair strategy | |
| RVOT stenting | 1/2 | ||
| 8 | No interventions performed | 12 | |
| High PA pressure | 6 | ||
| Parents refused surgery | 5 | ||
| Absent central PA’s | 1 | ||
Among the patients routed through single ventricle pathway, six patients underwent concomitant TAPVC repair. Two of them were performed in early infancy; concomitant PA banding or aorto-pulmonary shunt was performed in one case each. Both these patients expired. Four patients had TAPVC repair later in life (age 4.5–7 years) and were performed at the time of bidirectional cavo-pulmonary shunt. All four survived to the next stage.
Six patients did not qualify for surgical palliation due to high pulmonary artery pressure and one due to absence of central pulmonary arteries; five patients refused surgery (three of them were high risk due to TAPVC with or without AV valve incompetence). Two patients had catheter-based intervention for palliation.
Out of 34 patients who underwent surgery, there were five in-hospital deaths (14.7%). Two patients expired after TAPVC repair in early infancy and two after staged Fontan completion. One patient who had undergone Fontan completion with AV valve replacement and permanent pacemaker implantation earlier died after surgery for re-replacement of degenerated bio prosthetic valve.
Twenty-seven patients were diagnosed as left atrial isomerism. Twenty of them underwent 26 surgical procedures out of which 4 (20%) were biventricular repairs. The details of surgical procedures are presented in Table 3.
Table 3.
Management of patients with left atrial isomerism (n = 27)
| Serial no. | Procedure performed | No. of patients | Remarks |
|---|---|---|---|
| 1 | Biventricular repair | 4 | All alive and under follow-up |
| Atrial septation | 1 | ||
| Atrial septation + AV valve repair | 2 | ||
| Repair of eventration of diaphragm + ASD closure | 1 | ||
| 2 | Systemic artery to pulmonary artery shunt | 1 | Progressed to next stage |
| 3 | Pulmonary artery banding | 3 | 1 progressed to next stage; 2 unsuitable for cavo-pulmonary shunt due to high PA pressure |
| 4 | Total cavo-pulmonary shunt (Kawashima) | 14 | 1 late death |
| 5 | Extra-cardiac Fontan completion | 4 |
All 4 had extensive PAVMs 1 postoperative death (cardiac tamponade) |
| 6 | No interventions performed | ||
| Awaiting Kawashima procedure | 4 | ||
| High pulmonary artery pressure | 2 | ||
| No intra-cardiac defect (only atrial tachycardia) | 1 | ||
Two patients did not qualify for single ventricle palliation due to high pulmonary artery pressure and one had structurally normal heart except for abnormal systemic venous connections and atrial tachycardia in the setting of left atrial isomerism. Four patients are awaiting cavo-pulmonary anastomosis.
There were two in-hospital deaths (10%); one patient died of cardiac tamponade following Fontan completion for extensive bilateral pulmonary arteriovenous malformations. Second patient had severe AV valve incompetence and ventricular dysfunction with complete heart block late after Kawashima procedure. This patient expired after epicardial permanent pacemaker implantation.
Summary
To summarize, HS is a generalized somatic laterality disorder characterized by abnormal arrangement of thoracic and abdominal viscera. It is associated with complex congenital cardiac malformations which are usually grouped as isomerism of right or left atrial appendages. Most of the cardiac anomalies associated with HS are managed by univentricular pathway, albeit some are amenable for biventricular repair; more so in patients with left atrial isomerism. Biventricular repair strategy provides excellent long-term functional outcome but with a higher incidence of reinterventions and reoperations. The results of univentricular palliation in this setting have improved due to thoughtful early preparation and careful conduct of modified Fontan procedure. Presence of obstructed TAPVC and significant atrioventricular valve incompetence has been associated with poor outcome in patients with single ventricle physiology and aggressive approach towards their repair is recommended.
Funding
The authors declare they have not received any funding.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statements, human and animal rights consent
Not applicable being a review article.
Informed consent
Not applicable being a review article.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jacobs JP, Anderson RH, Weinberg PM, et al. The nomenclature, definition, and classification of cardiac structures in the setting of heterotaxy. Cardiol Young. 2007;17:1–28. doi: 10.1017/S1047951107001138. [DOI] [PubMed] [Google Scholar]
- 2.Sutherland MJ, Ware SM. Disorders of left–right asymmetry: heterotaxy and situs inversus. Am J Med Genet C: Semin Med Genet. 2009;151C:307–317. doi: 10.1002/ajmg.c.30228. [DOI] [PubMed] [Google Scholar]
- 3.Shiraishi I, Ichikawa H. Human heterotaxy syndrome—from molecular genetics to clinical features, management and prognosis. Circ J. 2012;76:2066–75. [DOI] [PubMed]
- 4.Belmont JW, Mohapatra B, Towbin JA, Ware SM. Molecular genetics of heterotaxy syndromes. Curr Opin Cardiol. 2004;19:216–220. doi: 10.1097/00001573-200405000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Kuehl KS, Loffredo C. Risk factors for heart disease associated with abnormal sidedness. Tetralogy. 2002;66:242–248. doi: 10.1002/tera.10099. [DOI] [PubMed] [Google Scholar]
- 6.Van Praagh R. Terminology of congenital heart disease: glossary and commentary. Circulation. 1977;56:139–143. doi: 10.1161/01.cir.56.2.139. [DOI] [PubMed] [Google Scholar]
- 7.Anderson RH, Becker AE, Freedom RM, et al. Sequential segmental analysis of congenital heart disease. Pediatr Cardiol. 1984;5:281–287. doi: 10.1007/BF02424973. [DOI] [PubMed] [Google Scholar]
- 8.Anderson RH, Ho SY. Sequential segmental analysis—description and categorization for the millennium. Cardiol Young. 1997;7:98–116. [Google Scholar]
- 9.Uemura H, Ho SY, Devine WA, Kilpatrick LL, Anderson RH. Atrial appendages and venoatrial connections in hearts with patients with visceral heterotaxy. Ann Thorac Surg. 1995;60:561–569. doi: 10.1016/0003-4975(95)00538-V. [DOI] [PubMed] [Google Scholar]
- 10.Van Praagh R, Van Praagh S. Atrial isomerism in the heterotaxy syndrome with asplenia, or polysplenia, or normally formed spleen: an erroneous concept. Am J Cardiol. 1990;66:1504–1506. doi: 10.1016/0002-9149(90)90543-a. [DOI] [PubMed] [Google Scholar]
- 11.Ticho BS, Goldstein AM, Van Praagh R. Extracardiac anomalies in the heterotaxy syndrome with focus on anomalies of midline-associated structures. Am J Cardiol. 2000;85:729–734. doi: 10.1016/s0002-9149(99)00849-8. [DOI] [PubMed] [Google Scholar]
- 12.Price VE, Blanchette VS, Ford-Jones EL. Prevention and management of infections in children with asplenia or hyposplenia. Infect Dis Clin N Am. 2007;21:697–710. doi: 10.1016/j.idc.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Barker GM, O’Brien SM, Welke KF, et al. Major infection after pediatric cardiac surgery: a risk estimation model. Ann Thorac Surg. 2010;89:843–850. doi: 10.1016/j.athoracsur.2009.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakhleh N, Francis R, Giese RA, et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125:2232–2242. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamura K, Joo K, Ohga S, et al. Thrombocytosis in asplenia syndrome with congenital heart disease: a previously unrecognized risk factor for thromboembolism. Int J Cardiol. 2013;167:2259–2263. doi: 10.1016/j.ijcard.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 16.McElhinney DB, Marx GR, Newbeuger JW. Congenital portosystemic venous connections and other abdominal venous abnormalities in patients with polysplenia and functionally univentricular heart disease: a case series and literature review. Congenit Heart Dis. 2011;6:28–40. doi: 10.1111/j.1747-0803.2010.00478.x. [DOI] [PubMed] [Google Scholar]
- 17.Lin JH, Chang CI, Wang JK, et al. Intrauterine diagnosis of heterotaxy syndrome. Am Heart J. 2002;143:1002–1008. doi: 10.1067/mhj.2002.122873. [DOI] [PubMed] [Google Scholar]
- 18.Yan YL, Tan KB, Yeo GS. Right atrial isomerism: preponderance in Asian fetuses. Using the stomach-distance ratio as a possible diagnostic tool for prediction of right atrial isomerism. Ann Acad Med Singap. 2008;37:906–912. [PubMed] [Google Scholar]
- 19.Yim D, Nagata H, Lam CZ, et al. Disharmonious patterns of heterotaxy and isomerism: how often are the classic patterns breached? Circ Cardiovasc Imaging. 2018. 10.1161/CIRCIMAGING.117.006917. [DOI] [PubMed]
- 20.Sanders SP, Geva T. Classifying heterotaxy syndrome: time for a new approach. Circ Cardiovasc Imaging. 2018. 10.1161/CIRCIMAGING.118.007490. [DOI] [PubMed]
- 21.Loomba R, Shah PH, Anderson R, Arora Y. Radiologic considerations in Heterotaxy: need for detailed anatomic evaluation. Cureus. 2016. 10.7759/cureus.470. [DOI] [PMC free article] [PubMed]
- 22.Blieden LC, Moller JH. Analysis of the P wave in congenital cardiac malformations associated with splenic anomalies. Am Heart J. 1973;85:439–444. doi: 10.1016/0002-8703(73)90487-0. [DOI] [PubMed] [Google Scholar]
- 23.Ho SY, Seo JW, Brown NA, Cook AC, Fagg NL, Anderson RH. Morphology of the sinus node in human and mouse hearts with isomerism of the atrial appendages. Br Heart J. 1995;74:437–442. doi: 10.1136/hrt.74.4.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frogoudaki A, Sutton R, Gatzoulis MA. Pacing for adult patients with left atrial isomerism: efficacy and technical considerations. Europace. 2003;5:189–193. doi: 10.1053/eupc.2002.0291. [DOI] [PubMed] [Google Scholar]
- 25.Suman-Horduna I, Babu-Narayan SV, Ueda A, et al. Magnetic navigation in patients with atrial isomerism (heterotaxy syndrome) and supraventricular arrhythmias. EP Europace. 2013;15:877–885. doi: 10.1093/europace/eus384. [DOI] [PubMed] [Google Scholar]
- 26.Marx GR. Echocardiography in heterotaxy syndrome. World J Pediatr Congenit Heart Surg. 2011;2:253–257. doi: 10.1177/2150135110397671. [DOI] [PubMed] [Google Scholar]
- 27.Berg C, Geipel A, Smrcek J, et al. Prenatal diagnosis of cardiosplenic syndromes: a 10 year experience. Ultrasound Obstet Gynecol. 2003;22:451–459. doi: 10.1002/uog.904. [DOI] [PubMed] [Google Scholar]
- 28.Hamasaka N, Matsushita M, Takeda S, et al. Prenatal diagnosis of heterotaxy syndrome: an 11 year experience. Ultrasound Obstet Gynecol. 2010;36:179. [Google Scholar]
- 29.Loomba R, Shah PH, Anderson RH, et al. Fetal magnetic resonance imaging of malformations associated with heterotaxy. Cureus. 2015;7:e269. [DOI] [PMC free article] [PubMed]
- 30.Uemura H, Yagihara T, Kawahira Y, Yoshikawa Y. Anatomic biventricular repair by intraatrial and intraventricular re-routing in patients with left isomerism. Cardiol Young. 2001;11:12–16. doi: 10.1017/s1047951100012361. [DOI] [PubMed] [Google Scholar]
- 31.Koh M, Yagihara T, Uemura H, et al. Biventricular repair for right atrial isomerism. Ann Thorac Surg. 2006;81:1808–1816. doi: 10.1016/j.athoracsur.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Lim HG, Bacha EA, Marx GR, et al. Biventricular repair in patients with heterotaxy syndrome. J Thorac Cardiovasc Surg. 2009;137:371–379. doi: 10.1016/j.jtcvs.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 33.Makhija Z, Marwah A, Mishra S, Kumar J, Goel A, Sharma R. Biventricular repair in heterotaxy patients. World J Pediatr Congenit Heart Surg. 2015;6:195–202. doi: 10.1177/2150135114563772. [DOI] [PubMed] [Google Scholar]
- 34.Heinemann MK, Hanley FL, Van Praagh S, et al. Total anomalous pulmonary venous drainage in newborns with visceral heterotaxy. Ann Thorac Surg. 1994;57:88–91. doi: 10.1016/0003-4975(94)90370-0. [DOI] [PubMed] [Google Scholar]
- 35.Sadiq M, Stumper O, De Giovanni JV, et al. Management and outcome of infants and children with right atrial isomerism. Heart. 1996;75:314–319. doi: 10.1136/hrt.75.3.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobs ML. Complications associated with heterotaxy syndrome in Fontan patients. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2002;5:25–35. doi: 10.1053/pcsu.2002.31500. [DOI] [PubMed] [Google Scholar]
- 37.Khan MS, Bryant R 3rd, Kim SH, et al. Contemporary outcomes of surgical repair of total anomalous pulmonary venous connection in patients with heterotaxy syndrome. Ann Thorac Surg 2015;99:2134–9. [DOI] [PubMed]
- 38.Brown JW, Ruzmetov M, Vijay P, Rodefeld MD, Turrentine MW. Pulmonary arteriovenous malformations in children after the Kawashima operation. Ann Thorac Surg. 2005;80:1592–1596. doi: 10.1016/j.athoracsur.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 39.Kim SJ, Bae EJ, Lee JY, Lim HG, Lee C, Lee CH. Inclusion of hepatic venous drainage in patients with pulmonary arteriovenous fistulas. Ann Thorac Surg. 2009;87:548–553. doi: 10.1016/j.athoracsur.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 40.Vouhe PR. Kawashima procedure: can pulmonary arteriovenous malformations be avoided? Eur J Cardiothorac Surg. 2012;41:579–580. doi: 10.1093/ejcts/ezr091. [DOI] [PubMed] [Google Scholar]
- 41.Jonas RA. Three-stage management of single ventricle. In: Jonas RA. Comprehensive surgical management of congenital heart disease. 2nd edition. CRC Press 2014. pp 501.
- 42.Alsoufi B, McCracken C, Schlosser B, et al. Outcomes of multistage palliation of infants with functional single ventricle and heterotaxy syndrome. J Thorac Cardiovasc Surg. 2016;151:1369–1377. doi: 10.1016/j.jtcvs.2016.01.054. [DOI] [PubMed] [Google Scholar]
- 43.Swisher M, Jonas R, Tian X, Lee ES, Lo CW, Leatherbury L. Increased postoperative and respiratory complications in patients with congenital heart disease associated with heterotaxy. J Thorac Cardiovasc Surg. 2011;141:637–644. doi: 10.1016/j.jtcvs.2010.07.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anagnostopoulos PV, Pearl JM, Octave C, et al. Improved current era outcomes in patients with heterotaxy syndromes. Eur J Cardiothorac Surg. 2009;35:871–7. [DOI] [PubMed]
- 45.Azakie A, Merklinger SL, Williams WG, Van Arsdell GS, Coles JG, Adatia I. Improving outcomes of the Fontan operation in children with atrial isomerism and heterotaxy syndromes. Ann Thorac Surg. 2001;72:1636–1640. doi: 10.1016/s0003-4975(01)03039-9. [DOI] [PubMed] [Google Scholar]
- 46.Bartz PJ, Driscoll DJ, Dearani JA, et al. Early and late results of the modified Fontan operation for the heterotaxy syndrome 30 years of experience in 142 patients. J Am Coll. 2006;48:2301–2305. doi: 10.1016/j.jacc.2006.07.053. [DOI] [PubMed] [Google Scholar]
- 47.Kim SJ, Kim WH, Lim HG, Lee CH, Lee JY. Improving results of the Fontan procedure in patients with heterotaxy syndrome. Ann Thorac Surg. 2006;82:1245–1251. doi: 10.1016/j.athoracsur.2006.04.082. [DOI] [PubMed] [Google Scholar]
- 48.Am A, Cohen MS, Sleeper LA, et al. Functional state of patients with heterotaxy syndrome following the Fontan operation. Cardiol Young. 2007;17:44–53. doi: 10.1017/S1047951107001151. [DOI] [PubMed] [Google Scholar]
- 49.Kulkarni A, Patel N, Singh TP, Mossialos E, Mehra MR. Risk factors for death or heart transplantation in single-ventricle physiology (tricuspid atresia, pulmonary atresia and heterotaxy): a systematic review and meta-analysis. J Heart Lung Transplant. 2019;38:739–747. doi: 10.1016/j.healun.2019.04.001. [DOI] [PubMed] [Google Scholar]