

Abstract
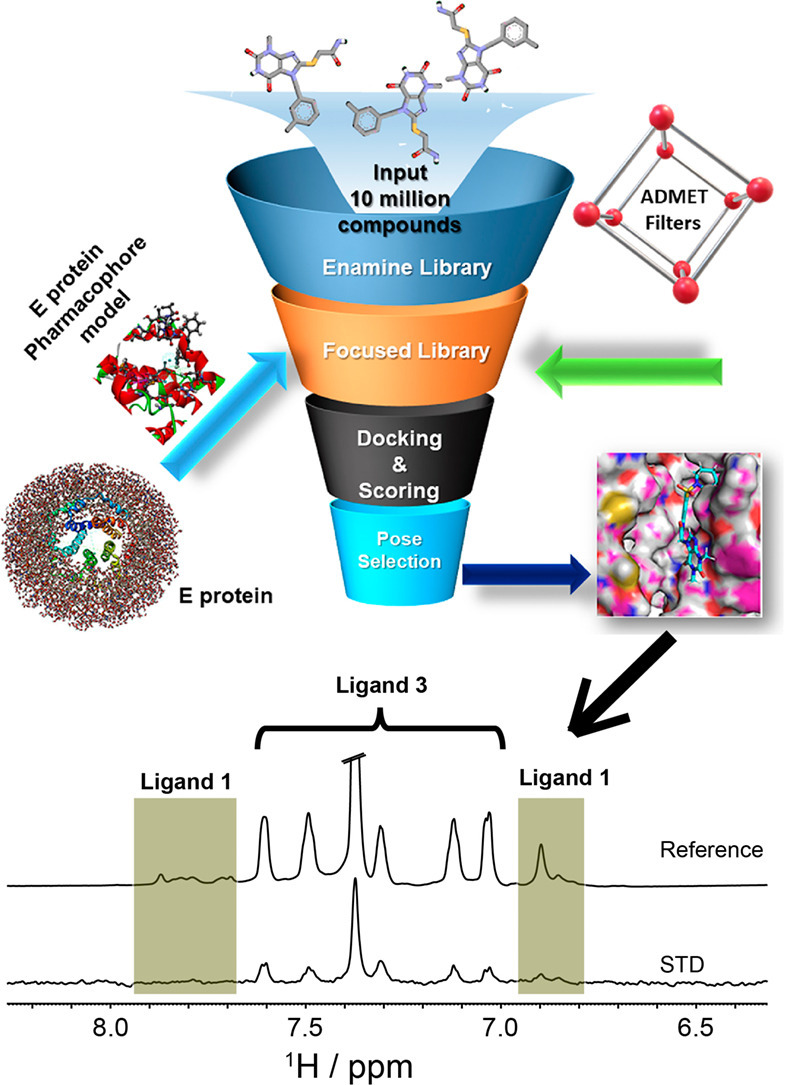
One of the most crucial characteristic traits of Envelope (E) proteins in the severe acute respiratory syndrome SARS-CoV-1 and NCOVID19 viruses is their membrane-associated oligomerization led ion channel activity, virion assembly, and replication. NMR spectroscopic structural studies of envelope proteins from both the SARS CoV-1/2 reveal that this protein assembles into a homopentamer. Proof of concept studies via truncation mutants on either transmembrane (VFLLV), glycosylation motif (CACCN), hydrophobic helical bundle (PVYVY) as well as replacing C-terminal “DLLV” segments or point mutants such as S68, E69 residues with cysteine have significantly reduced viral titers of SARS-CoV-1. In this present study, we have first developed SARS-2 E protein homology model based on the pentamer coordinates of SARS-CoV-1 E protein (86.4% structural identity) with good stereochemical quality. Next, we focused on the glycosylation motif and hydrophobic helical bundle regions of E protein shown to be important for viral replication. A four feature (4F) model comprising of an acceptor targeting S60 hydroxyl group, a donor feature anchoring the C40 residue, and two hydrophobic features anchoring the V47 L28, L31, Y55, and P51 residues formed the protein based pharmacophore model targeting the glycosylation motif and helical bundle of E protein. Database screening with this 4F protein pharmacophore, ADMET property filtering on enamine small molecule discovery collection yielded a focused library of ~7000 hits. Further molecular docking and visual inspection of docked pose interactions at the above mention V47 L28, L31, Y55, P51, S60, C40 residues led to the identification of 10 best hits. Our STD NMR binding assay results demonstrate that the ligand 3, 2-(2-amino-2-oxo-ethoxy)-N-benzyl-benzamide, binds to NCOVID19 E protein with a binding affinity (KD) of 141.7 ± 13.6 μM. Furthermore, the ligand 3 also showed binding to C-terminal peptide (NR25) as evidenced with the STD spectrums of wild type E protein would serve to confirm the involvement of C-terminal helical bundle as envisaged in this study.
Keywords: SARS-CoV-1, NCOVID19, Envelope protein, Protein pharmacophore, Database screen, Virtual screening, STD NMR
Graphical abstract
1. Introduction
Since the last SARS-CoV-1 outbreak in 2002, a tremendous amount of basic and clinical research on coronaviruses has led to understanding its protein machinery and its role in pathogenesis [1,2]. Coronavirus members are enveloped viruses containing positive-sense RNA (29.99 kb) as genetic material and are grouped under the β-coronavirus genus [3]. Both SARS-CoV-1 and NCOVID19 viruses belong to Coronavirinae subfamily/Coronaviridae family [4,5]. The structural proteins such as the spike (S), membrane (M), an envelope protein (E), the nucleocapsid protein (N) are all involved in diverse processes that regulate the viral cell entry into the host, subsequent pathogenesis and replication, respectively [6,7].
Among these proteins, Envelope protein (E) is a small (~75 amino acids) integral membrane protein comprising of an N-terminal transmembrane domain, and a long hydrophobic C-terminal domain [8]. Interestingly, accumulating evidence on the less recognized component of the SARS-CoV-1 E protein has pointed to its essential role as a key virulence factor for the SARS-CoV-1 infections [[9], [10], [11], [12]]. The E protein was shown to mediate ion channel/membrane permeabilizing (viroporin) activities [13] and thereby regulate virion assembly morphogenesis [14], maintaining the integrity of the viral pathogenesis, cell stress responses, apoptosis as well as inflammation [15]. The absence of E protein in SARS-CoV-1 displayed lower viral titer, immature, and inefficient progenies [12,16]. Recent studies have shown that the SARS-CoV-1 E protein can alter the host cell membrane permeability by forming oligomeric cation-selective ion channels [17]. More importantly, the transmembrane regions of E protein have been known to interact with M protein and aids in its colocalization [18], thus maintaining the integrity in viral morphogenesis, especially during assembly and budding [14]. Biophysical studies using the “VYVY” motif [19] have shown that this region assumes helical orientation, a characteristic trait of the known amyloidogenic propensity [20], in membrane environment and enable the self-aggregating motif [21] to insert into membrane areas and might contribute to the process of entry into host environment or viral assembly [19,22].
Additionally, the role of C-terminal residues of SARS-CoV-1 E protein in association with nucleocapsid (N) [23], PALS1 PDZ domain [24] was also shown to be implicated in regulation of viral pathogenesis and viral replication by both in vitro and in vivo studies [24]. Several mutations focusing on the transmembrane “VFLV” segment, the glycosylation motif “RLCAYCCN”, and the hydrophobic helical bundle “PSFYVYVYSR” stretches on SARS-CoV-1 E protein have shown a complete reduction in the viral replication. Similarly, point mutation studies replacing the S68-E69 with a single cysteine residue or replacing the C-terminal tail (DLLV) with glycine residues in SARS-CoV-1 had drastically reduced viral counts [25]. More importantly, the genotyping analysis from recent NCOVID19 infections worldwide points to a lower mutational frequency in this E protein, highlighting its suitability for targeted drug discovery [26].
In this study, we have attempted to target C-terminal helical bundle to identify and characterized biophysical binding of small molecule ligands to NCOVID19 Envelope E protein using structure based drug design and STD NMR tools. First, we have developed a 3D structure model for NCOVID19 E protein with reliable stereochemical quality from its closest homolog using homology modeling studies. Next, four feature protein based pharmacophore model was developed by targeting the residues of glycosylation motif and the helical bundle of C-terminal region. Together with database screening with enamine small molecule library [27], ADMET filtering [28], molecular docking, and visual inspection of interactions at the above described epitope residues led to purchase of focused library of 10 molecules. Next, Saturation Transfer Difference (STD) NMR binding studies of these 10 molecules with E protein led to the identification of two ligands 1 and 3 that bind to E protein in high micromolar affinity.
2. Materials & methods
2.1. Protein and ligands
The NCOVID19 E protein and its C-terminal fragment NR25 (N45IVNVSLVKPSFYVYSRVKNLNSSR69-NH2) was obtained from LSBio Inc., WA, USA and Genscript, USA, respectively. The E protein (catalogue No.: LS-G145857) used in this study comprised of 75 amino acids with six histidine residues tagged in a beta-barrel protein (molecular weight ~15 kDa) at the N-terminal. The resultant recombinant protein including tags was predicted to be ~27.5 kDa. SDS 15% Tris-gel was used to check the quality of the protein that was then used without any further purification. The best three selected ligands were purchased from Enamine vendor [27] and the purity of the samples was confirmed by MALDI-TOF studies by comparison to quality data provided by the vendor.
2.2. Peptide synthesis
Selectively 15N-labeled NR25 (N45IVNVSLVKPSFYVYSRVKNLNSSR69-NH2) (the underlined amino acids are 15N labeled) peptide was synthesized on a solid phase peptide synthesizer (Aapptec Endeavor 90, USA) with 15N-labeled Fmoc protected Val and Leu. A solid-phase peptide synthesis protocol was used with a Rink Amide MBHA resin (substitution 0.69 mmol/g; Novabiochem, San Diego, California, USA) [19]. The resultant synthesized peptide was then purified using reverse-phase HPLC (SHIMADZU, Japan) on a Phenomenix C18 column (dimension 250 × 10 mm, with a pore size of 100 Å, 5-μm particle size). Linear gradient elution technique was employed with water and methanol along with 0.1% TFA (serving as the ion-pairing agent) as the solvent. Mass spectrometry and NMR spectroscopy were used to check the purity of the sample. The purified peptide was then shelved at −20 °C. Working stock solutions of 1 mM peptide was prepared either in sterile water or 10 mM phosphate buffer (pH 7.4) and used for the respective experiments and stored at 4 °C was short periods.
2.3. In-silico studies
2.3.1. Homology modeling
Sequence analysis has revealed that NCOVID19 E protein with SARS-CoV-1 had almost 96 percent sequence identity with only four varied residues at the C-terminal region. Protein blast search for homologous structures (from protein data bank database) pointed out the recently characterized pentameric ion channel NMR structure (PDB: 5X29) [22] of SARS-CoV-1 was the closest homolog with 84.6% sequence identity. It is to be noted that this structure has cysteine residues mutated to alanine residues and also lacks the C-terminal ten residues. It was utilized as a template to build the protein for NCOVID19 Envelope small membrane protein. Phyre2 [29] was employed to construct the 3D model of NCOVID19 E protein comprising of 1–65 amino acid residues. Later, the pentameric assembly of the NCOVID19 ion channel was built by superimposing the 3D protein models on the structure template (5 × 29).
2.3.2. Protein preparation
Next, the pentameric assembly of NCOVID19 E Protein was checked for stereochemical clashes within the arrangement. The sterically clashing residues (F23) with V25 of adjacent subunit at the central cavity in the pentamer arrangement were adjusted using the rotamer library. The heavy atoms of the pentameric assembly were constrained, and the added hydrogen atoms were energy minimized using steepest descent and conjugate algorithms for 2000 steps using prepare protein module in Discovery studio [30]. Upon satisfying the energy/rmsd convergence criterion, the energy minimized structures were saved for further modeling studies.
2.3.3. Structure-based pharmacophore model
The first step in developing the structure-based pharmacophore model on this domain was to enumerate the possible hotspot features such as donor (green), acceptor (magenta), and hydrophobic (cyan) feature vector site points at CTD using the interaction site generation module in Discovery Studio 2020 [[30], [31], [32], [33], [34]]. These numerous three feature vector site points are hierarchically clustered based on their rmsd to their respective feature type and only cluster centers for each of the three feature(s) were included in the study. Finally, a donor feature mapping the carbonyl atoms of C40 residue, an acceptor partially mapped to the hydroxyl group of S60, and two hydrophobic features in the near vicinity of hydrophobic residues V47, L28, Y57, P54 were included in the structure-based pharmacophore model. Site points that were further away from the above-mentioned residues of CTD were discarded.
2.3.4. 3D-database preparation
3D databases of enamine datasets from Zinc library [35] were employed in this study. 3D coordinates were generated using prepare ligands tools. Next, ADMET [28] property filters were applied for enamine libraries. Ligands that are likely to induce the liver enzyme CYPD6 inhibition, hepatotoxicity, high plasma protein binding, and ligands with low solubility and absorption were excluded from further consideration. Thus, the resulting clean ligands were utilized to build 3D databases using the “CEASER” search algorithm with an option to generate 50 conformations per ligand in Discovery studio 2020 [30]. Next, 4F pharmacophores were used to search the 3D database resulting in a focused library of <7000 hits.
2.3.5. Docking
Gold molecular docking program 2020 was utilized to dock the obtained focused library at the previously discussed C-terminal domain using default parameters available with the GOLD docking program [36]. Together with help the GOLD PLP fitness scores and visual inspection of interaction with the intended C-terminal residue (S60) led us to identify the 10 best poses that could dock at the C-terminal helical bundle of NCOVID19 E protein that could potentially inhibit the viral pathogenesis and viral replication as suggested in SARS-CoV-1 E protein knock out studies.
2.4. STD NMR spectroscopy
The NMR sample for STD NMR experiments was prepared with 5 μM NCOVID19 E protein/NR25 peptide in 10 mM PBS (pH 7.4). The ligands to be screened were subsequently added from a concentrated d6-DMSO stock solution to the protein sample yielding a final concentration of 500 μM in 0.3% d6-DMSO (molar ratio of protein:ligand = 1:100).
The experiments were performed at 25 °C in a Bruker Avance III 700 MHz, equipped with an RT probe and the Topspin v3.2 software [37] for data acquisition, processing, and analysis. The STD NMR spectra were acquired using a double pulsed-field gradient spin-echo (DPFGSE) pulse sequence, providing a better baseline and improved water suppression. The STD NMR spectra (on-resonance = 0 ppm and off-resonance = 40 ppm) for each ligand was performed with the total saturation time of 2 s (a train of 40 selective Gaussian pulses, 49 ms each-with a 1 ms intervals) at 50 dB for 4 K scans while the reference spectra were acquired with 2 K scans. The transfer of saturation from the E protein to the respective ligands was generated upon subtraction of the on-resonance spectra from the off-resonance by phase cycling. The competitive inhibition was performed with a molar ratio of protein:ligand 1:ligand 3 = 1:100:100.
2.5. Determination of equilibrium dissociation constant (KD) by STD
A series of one-dimensional STD spectra were acquired by stepwise additions of ligand 3 from a stock solution of 88 mM to the sample containing 5 μM E protein. To determine the dissociation constant (KD) values of Protein/ligand interactions, the STD effects for aromatic ring protons of ligand were converted to STDtotal. The equations used to calculate the dissociation constants can be derived as follows.
| (1) |
where, Io is the intensity of the off-resonance spectrum and Isat is the intensity of the on-resonance spectrum.
| (2) |
| (3) |
To determine the STD build-up curves:
| (4) |
where, STDmax is the maximal STD intensity, t is the saturation time and kst is the saturation rate constant. From Eq. (4), the initial slope, which is known as the total STD value, can be obtained using Eq. (5):
| (5) |
The initial slope of the STDtotal isotherm can be plotted as a function of ligand concentration to obtain the dissociation constant, KD:
| (6) |
3. Results and discussion
3.1. Sequence analysis
Comparison of E protein of NCOVID19 to SARS-CoV-1 revealed a very high sequence identity (96% at sequence level) with prominent variations occurring at the C-terminal domain sequence (* highlighted residues, Fig. 1 ). The striking variations include (i) deletion of a C-terminal glycine (G71) residue; (ii) incorporation of serine (S55), aromatic phenylalanine (F56) residue and positively charged R69 residue in place of threonine (T55) valine (V56), and glutamic acid (E69) residues, respectively in comparison to SARS-CoV-1. Despite the slight alteration in protein parameters due to these subtle variations in the NCOVID19 E protein sequence, it essentially seems to maintain the viral titers needed for its dominant infectivity rates as seen through this NCOVID19 pandemic in comparison to SARS-CoV-1. Knowledge gained from homology to SARS-CoV-1 via proof of concept studies on transmembrane (VFLV segment), glycosylation motif (CAYCCN), helical bundle stretch (VYVYSR), form the putative sites that could anchor the ligand binding sites and potentially inhibit the viral titer [25].
Fig. 1.

Sequence alignment of NCOVID19 E protein with its closest homolog SARS-CoV-1. TM stands for trans-membrane domain. Image prepared using ESPript 3.0 [38].
Blast protein search on protein databank revealed that the NCOVID19 E protein sequence harbors 86.4% identity with SARS-CoV-1 E protein (pdb 5X29) as their closest homolog structure. So far, only NMR structure studies were elucidated, describing the monomer (wild type protein [13]), and pentameric structural arrangements using Cys to Ala mutated constructs of SARS-CoV-1, respectively. Despite the lack of structural information on last 10 C-terminal residues, the pentameric arrangement of E protein provided the structural basis for its ion channel activity. Very recently, the NMR structure of NCOVID19 E protein trans-membrane (TM) domain (pdb 7K3G) in homo-pentameric assembly was also determined highlighting the importance of TM residues in mediating ion channel [39]. However, lack of the C-terminal self-aggregating helical domain and that of PDZ domain structural details (Fig. 1) eclipses our understanding on these domains at molecular or atomistic level. Therefore, in this study, we have utilized the NMR structure (PDB 5X29) [22] with 84.6% sequence identity as a template to build the 3D structure of NCOVID19 E protein (1–65 amino acids) using the Phyre2.0 program [29]. Next, our Ramachandran plot results suggested the model was of excellent stereochemical quality, with several residues in the most favored regions (Supplementary Fig. 1). Superimposition of five copies of this E protein onto the individual subunits of SARS-CoV-1 pentamer using the pymol program provided a similar pentameric assembly for NCOVID19 E protein.
Binding site analysis with the pentamer structural arrangement revealed prominent binding cavities at the interface of C-terminal stretch and NTD regions as possible sites for ligand binding, besides the main central cavity of the ion channel (Fig. 2A and B). The C-terminal “YVYSRVKNLNSSRV” stretch from one subunit juxtaposes in the vicinity of the CAYCCN segment harboring the glycosylation site (N45) and the central channel site of the second subunit at the dimeric interface. This arrangement provided a prominent binding cavity that serves as potential anchoring motifs for targeted ligand-based therapeutics. This structural pattern was visualized across all the five subunits of the pentamer assembly (Fig. 3 ).
Fig. 2.
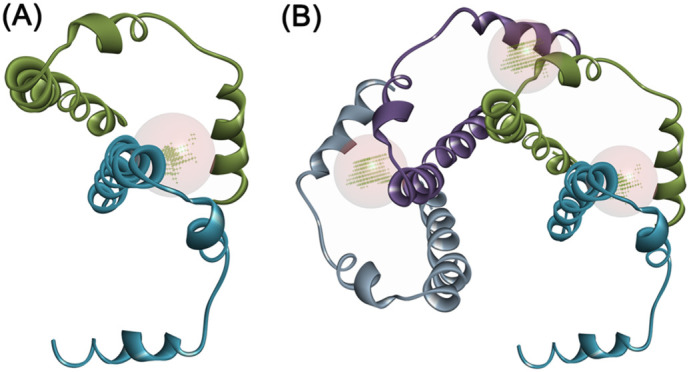
Assembly of the E protein. The (A) dimeric arrangement of E protein reveals the binding pocket at the C-terminal region in anchorage with the second subunit. (B) This arrangement was also evidenced in the tetramer.
Fig. 3.

Pentameric assembly of the E protein. (A) The site point at the interface of CTD and the NTD are highlighted. (B) The CTD stretch S50-R61 was in close anchorage with the NTD L28-V47 region that forms a binding cavity.
3.2. Structure-based pharmacophore model
As highlighted in the earlier section, the C-terminal helical bundle, glycosylation motif, and the central channel residues (NTD) are nicely juxtaposed in a pentameric assembly to form a ligand-binding site. Residues such as C40, C44, V47, I48, L28, L31 from NTD of one subunit and the C-terminal residues P54, F56, S60, R61 from another subunit form the ligand-binding site (Fig. 3B). Mainly, three feature probes comprising of a donor, an acceptor, and a hydrophobe site points are used to enumerate the interaction hot spots (vector site points). Hierarchical clustering based on rms distance on each of the three feature types provided the pharmacophore features targeting the above-mentioned residues (Fig. 4A). Feature or site points that are further away from these reside are discarded from further model development. Finally, a four-feature model comprising of donor feature anchored to carbonyl atoms of C49 residue, an acceptor feature near S60 hydroxyl atoms and two hydrophobic features in the vicinity of L28, V47, P54, Y57 (Fig. 4B) were evolved to screen the enamine databases.
Fig. 4.

Structure-based pharmacophore model. (A) Interaction feature (acceptor (green), the donor (magenta), hydrophobic (cyan)) clusters at the defined active site on E protein. (B) Four feature models comprising of (i) an acceptor targeting S60 hydroxyl atoms, (ii) donor anchoring the main chain atoms of C40, and (iii and iv) two hydrophobic features in the vicinity of V47 L28, L31, Y55, and P51 residues were included in the pharmacophore model.
Database screening with the 4F pharmacophore model was performed with 133 FDA drug molecules that have been recently reported to be able to inhibit NCOVID19 infection in Vero cell line screening [40] to assess whether any of these ligands mapped to the E protein pharmacophore. Interestingly, 13 ligands (Supporting information, Table 1) were mapped to the NCOVID19 E protein-based pharmacophore. With this kind of blind control that could map the known active NCOVID19 ligands, we proceeded to screen the enamine small molecule commercial library. ADMET property calculations enabled to filter ligands that are likely to inhibit to induce liver enzyme CYPD6 inhibition, hepatoxicity, high plasma protein binding as well as ligands with unfavorable solubility and absorption predictions were excluded from docking studies.
3.3. STD NMR screening identifies ligands that bind to NCOVID19 E protein
Saturation Transfer Difference (STD) NMR spectroscopy is a versatile and widely used tool [41] that not only identifies the potential binders (high micromolar to low millimolar affinity) but also enables information on the ligand's binding epitope for the protein (NCOVID19 E Protein). STD NMR experiments with the three ligands helped us to clearly delineate their divergent binding affinities for the NCOVID19 E protein. Of the 3 ligands purchased, only ligands 1 and 3 (Table 1 ) showed weak to moderate affinity, respectively. With respect to the reference spectrum, the STD spectrum of ligand 1 with 4096 scans shows low-intensity peaks from 6.8 to 7.0 ppm and 7.8 to 8.0 ppm (data not shown). Increasing the STD scans to 8192 did reveal conspicuous ligand peaks from aromatic phenyl rings from 6.8 to 7.0 ppm and 7.8 to 8.0 ppm region. This data suggests that the aromatic ring protons of ligand 1 were involved in its binding to the E protein (Fig. 5A). The ligand 2, however, did not show any STD signal, confirming that it does not bind to the E protein (Supporting information Fig. 2). In contrast, ligand 3, with a much smaller molecular mass than ligand 1 showed moderate binding for the protons from 7.0 to 7.7 ppm, signifying the involvement of aromatic benzene rings in its binding to the E protein (Fig. 5B). These observations indicated the importance of the aromatic interactions in mediating the E protein association for both ligand 1 and ligand 3.
Table 1.
The best three molecules with good binding Fitness scores.
| Ligand no. | Zinc Id number (enamine ID) | Structure | GOLD PLP fitness | ClogP |
|---|---|---|---|---|
| 1 | ZINC23221929 (Z51148611) |  |
61 | 3.27 |
| 2 | ZINC0969702 (Z274601552) | 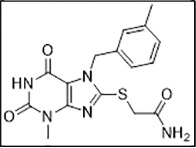 |
57 | 0.77 |
| 3 | ZINC06220062 (Z24937170) | 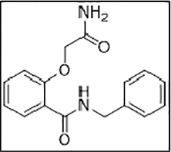 |
53 | 1.45 |
Fig. 5.

STD results showing the evidence that ligands bind to the E protein. (A) Ligand 1 shows weak binding with only aromatic protons from 6.8 and 7.8 ppm region; (B) ligand 3 also shows binding with peaks from aromatic protons from phenyl rings indicated at 7.0–7.6 ppm range. The experiment was performed at 298 K using Bruker Avance III 700 MHz and at a molar ratio of E protein:ligands = 1:100.
3.4. Insight into molecular interactions of ligand 1, 3
Our pose selection criterion was based on PLP fitness scores as well as interactions with the T32, S60, R62, and the hydrophobic interactions involving the L28, L31 V42, I46, L51, P54 residues from the E protein that led to identifying three hits (Table 1 and Supporting information Fig. 3). Among the three hits verified from STD experiments, ligand 1 (ZINC23221929/Z51148611) with a chemical name, [2-[[(3S)-2,3-dihydro-1,4-benzodioxin-3-yl]methylamino]-2-oxo-ethyl] (2S)-2-(1,3-dioxoisoindolin-2-yl)-4-methylsulfanyl-butanoate, manifested in a PLP fitness of 61. This ligand comprises two ring systems: benzodioxin-3-yl and 1,3-dioxoisoindoline groups linked by a 7-atom linker fragment (Supporting information Fig. 3B). Among the two aryl groups, the benzodioxine moiety (at one end of the molecule) was found to be engaged in hydrophobic interactions with I46, L51, P54, residues of the E protein (light yellow arrows). The other aryl group, i.e., 1,3-dioxoisoindoline moiety (at the other end of the molecule), was engaged in both H-bonding interactions with S60 as well as partakes in hydrophobic interactions with C44 and V47 (residues not shown). The 4-methylthiobutanoate fragment, on the other hand, maintained alkyl hydrophobic interactions with L31, L34, C40 (Fig. 6 ) in E protein.
Fig. 6.

Cartoon and surface representation of ligand 1 (ZINC23221929) binding mode on E proteins. (A) 1,3-dioxoisoindoline moiety of ZINC096322622 was involved in key H-bonding contact with S60 (black dotted line), while the other two substitutions - benzodioxine ring and 4-methylthiobutanoate moieties mediate the hydrophobic interactions with I46, L51, P54, and L31, L34, C40 residues respectively (light yellow arrows). (B) Surface View of the binding mode of ZINC096322622 in E protein.
In sharp contrast, ligand 3 (ZINC06220062) with a chemical name - 2-(2-amino-2-oxo-ethoxy)-N-benzyl-benzamide had a PLP fitness of 53 (Supporting information Fig. 3D). The 2-oxo-functional group in the 2-amino-2-oxo-ethoxy fragment was plausibly engaged in the crucial H-bonding interaction with the S60 residue of CTD of the E protein. On the other hand, the phenyl moiety anchoring the ethoxy amine fragment was involved in hydrophobic interactions with V47, C40 residues. Additionally, the N-phenyl moiety was engaged in hydrophobic interactions with the E protein L28, L31, and Y57 residues (light yellow arrows, Fig. 7 ). The carbonyl atoms of amide linker, linking the aromatic moieties, in turn, were engaged in a hydrogen bonding interaction with T35 (black dotted lines) on the N-terminal side of another subunit. Taken together, the hydrophobic interactions with L28, L31, P51, Y57 on one subunit and V42, C40, C44, T35 on another (light yellow arrows) along with the key hydrogen bonding interactions involving the S60 residue stabilized the ligand binding. This stable binding to the E protein can potentially inhibit the protein's functioning, preventing its viral assembly and replication.
Fig. 7.

Binding mode of ZINC06220062 on E protein. (A) Cartoon representation highlighting the crucial H-bonded interaction of 2-amino-2-oxo-ethoxy fragment with S60 residue of E protein. The linker carbonyl atoms linking the two-aryl moieties were engaged in H-bonded contact T35 (black dotted lines). The two aryl groups - benzyl and benzamide rings were involved in hydrophobic interaction L28, L31, P51, Y57 on one subunit, and V42, C40, C44, T35 on another subunit (light yellow arrows). (B) Surface view of the binding mode of the ligand.
These molecular insights prompted us to evaluate whether ligands 1 and 3 compete for similar site and led us to carry out a STD experiment using an equimolar mixture of ligands 1 and 3 were incubated with the E protein at a molar ratio of 1:100:100 (protein:ligand 1:ligand 3) to assess a probable competitive binding for a common binding site (Fig. 8A). Interestingly, the STD profile obtained for the ligand mixture manifested in better signal intensities between 7.0 and 7.7 ppm, suggesting a stronger binding affinity from ligand 3 (Fig. 8A). Parallelly, only very weak signals were obtained around 7.8 ppm and further upstream, between 6.8 and 7.0 ppm corresponding to the resonances of ligand 1 (highlighted in Fig. 8A). This data clearly suggested a common or comparable binding site in E protein for either of the two ligands (Fig. 8B).
Fig. 8.

(A) The competitive binding assay shows most of the peaks involved with ligand 3 than ligand 1, signifying that ligand 3 had a comparatively more substantial binding. (B) Molecular overlay of ligands 1 and 3 binding poses onto E protein. Shown in subset box is the superimposition of aryl groups in ligand 1 and 3 in close proximity (protein E cartoon structure was hidden for clarity).
Nevertheless, the sharper signals from ligand 3 indicated a relatively stronger binding affinity that further attenuates the 1st ligands weak binding in this competitive interaction for the protein site. The fact that the aromatic rings partake in the binding interactions with the E protein suggests that the difference in the aromatic moieties' structural orientation could be directly correlating to the divergent functional association of the two ligands (Fig. 8B). This alternatively indicates the vantage of the composite benzene rings in the two ligands in moderating a plausible hydrophobicity mediated stable interaction. This understanding further prompted us to focus our studies on the hydrophobic-rich segment from the E protein that might be serving as the key residues that define the binding site. We next determined the binding affinity (KD) of ligand 3 using five different molar ratios of E protein/ligand 3 complex. Our binding affinity titrations (Fig. 9A) depicted the graded increase of STD amplification factor in the proton resonances of phenyl moiety with increasing concentration of ligand 3 in the presence of 5 μM of protein. The STDtotal isotherms (Fig. 9B) fit nicely to the specific binding mode with hill slope Eq. (6) and provided the estimate of KD to be 141.7 ± 13.6 μM (Fig. 9B).
Fig. 9.

Binding affinity of ligand 3 to E protein. (A) STD build up curves at different saturation time to determine STDtotal using Eqs. (4), (5). (B) Dissociation constant (KD) were calculated for E protein/ligand 3 complex using hill equation (Eq. (6)) using prism 9. (C) Binding of ligand 3 to C terminal E protein NR25 (N45-R69) at a molar ratio of NR25:ligand 3 = 1:100. The experiment was performed at 298 K using Bruker Avance III 700 MHz.
3.5. Focus on the C-terminal helical bundle segment exclusively in the binding site for the ligands
To define any involvement of the hydrophobic stretch from the C-terminal region of the E protein, peptide fragment-based studies were performed with a 25 residue peptide stretch, involving the C-terminal segment ranging from N45 to R69 (NR25). The NR25 peptide was synthesized with selectively 15N labeled Val and Leu residues to gain specific insight into any possible role for the hydrophobicity-mediated interaction. Two-dimensional 1H-15N HSQC spectra as well as STD titration were recorded for the NR25 peptide in the presence of either ligands 1, 3. Though, our HSQC results of the ligands 1, 3 with NR25 peptide did not reveal any chemical shift perturbation on amide resonances for the Val and Leu residues (Supporting information Fig. 4). On the other hand, surprisingly, when we employed STD NMR titration of ligand 3 with NR25 (N45-R69) showed the peaks of ligand aromatic moieties at 7.0–7.7 ppm in STD spectrum (Fig. 9C) in a manner similar to E protein. This crucial data further maps the involvement of C-terminal helical bundle as binding epitope for ligand binding and corroborates our hypothesis that C-terminal helical bundle could mediate important role in viral replication.
4. Conclusions
Our present study identified a micromolar affinity hit molecule by targeting NCOVID19 E protein by employing protein-based pharmacophore generation, database screening, docking fitness, and ADMET filters. Further, our STD NMR based biophysical experiments led us to identify that the aromatic benzene rings in ligands 3 form the epitope requisite for binding to the C-terminal helical bundle of NCOVID19 E protein. This study could serve as a stepping-stone to open new avenues for small molecule inhibitor development of NCOVID19 E protein. Future study, employing structure guided hit optimization via medicinal chemistry efforts on ligand 3 scaffold would be the ideal way forward for us to characterize and improve the potency of newer derivatives in cell based viral infectivity assays or suitable animal based models.
Declaration of competing interest
The authors do not have any conflict of interest.
Acknowledgments
Acknowledgement
AB would like to thank Professor Uday Bandyopadhyay, The Director of Bose Institute, India, for his constant support to boost this research work. This study was supported by Dassault Systems and BIOVIA evaluation license during pandemic period. This research was also partly supported by Science and Engineering Research Board (File No. EMR/2017/003457 to AB), Government of India, partly by the Department of Biotechnology (BT/PR29978/MED/30/2037/2018 to AB) Govt. of India and partly by Bose Institute intramural research fund for this research. Authors thank Dipita Bhattacharyya for critical reading of this manuscript. SM thanks UGC, Govt. of India for providing Senior Research Fellowship.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijbiomac.2021.02.011.
Appendix A. Supplementary data
Supplementary data
References
- 1.Cheng V.C., Lau S.K., Woo P.C., Yuen K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007;20(4):660–694. doi: 10.1128/CMR.00023-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pal M., Berhanu G., Desalegn C., Kandi V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12(3) doi: 10.7759/cureus.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.S. Payne, Family Coronaviridae, Viruses2017, pp. 149–158.
- 6.Belouzard S., Millet J.K., Licitra B.N., Whittaker G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4(6):1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Astuti I. Ysrafil, Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): an overview of viral structure and host response. Diabetes Metab. Syndr. 2020;14(4):407–412. doi: 10.1016/j.dsx.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prajapat M., Sarma P., Shekhar N., Avti P., Sinha S., Kaur H., Kumar S., Bhattacharyya A., Kumar H., Bansal S., Medhi B. Drug targets for corona virus: a systematic review. Indian J. Pharmacol. 2020;52(1):56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nieto-Torres J.L., DeDiego M.L., Verdiá-Báguena C., Jimenez-Guardeño J.M., Regla-Nava J.A., Fernandez-Delgado R., Castaño-Rodriguez C., Alcaraz A., Torres J., Aguilella V.M., Enjuanes L. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014;10(5) doi: 10.1371/journal.ppat.1004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeDiego M.L., Alvarez E., Almazan F., Rejas M.T., Lamirande E., Roberts A., Shieh W.J., Zaki S.R., Subbarao K., Enjuanes L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007;81(4):1701–1713. doi: 10.1128/JVI.01467-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo L., Masters P.S. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 2003;77(8):4597–4608. doi: 10.1128/JVI.77.8.4597-4608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortego J., Ceriani J.E., Patiño C., Plana J., Enjuanes L. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology. 2007;368(2):296–308. doi: 10.1016/j.virol.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parthasarathy K., Lu H., Surya W., Vararattanavech A., Pervushin K., Torres J. Expression and purification of coronavirus envelope proteins using a modified β-barrel construct. Protein Expr. Purif. 2012;85(1):133–141. doi: 10.1016/j.pep.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruch T.R., Machamer C.E. The coronavirus E protein: assembly and beyond. Viruses. 2012;4(3):363–382. doi: 10.3390/v4030363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeDiego M.L., Nieto-Torres J.L., Jiménez-Guardeño J.M., Regla-Nava J.A., Alvarez E., Oliveros J.C., Zhao J., Fett C., Perlman S., Enjuanes L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011;7(10) doi: 10.1371/journal.ppat.1002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ortego J., Escors D., Laude H., Enjuanes L. Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 2002;76(22):11518–11529. doi: 10.1128/JVI.76.22.11518-11529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilson L., McKinlay C., Gage P., Ewart G. SARS coronavirus E protein forms cation-selective ion channels. Virology. 2004;330(1):322–331. doi: 10.1016/j.virol.2004.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen S.C., Lo S.Y., Ma H.C., Li H.C. Expression and membrane integration of SARS-CoV E protein and its interaction with M protein. Virus Genes. 2009;38(3):365–371. doi: 10.1007/s11262-009-0341-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh A., Bhattacharyya D., Bhunia A. Structural insights of a self-assembling 9-residue peptide from the C-terminal tail of the SARS corona virus E-protein in DPC and SDS micelles: a combined high and low resolution spectroscopic study. Biochim. Biophys. Acta Biomembr. 2018;1860(2):335–346. doi: 10.1016/j.bbamem.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A., Pithadia A.S., Bhat J., Bera S., Midya A., Fierke C.A., Ramamoorthy A., Bhunia A. Self-assembly of a nine-residue amyloid-forming peptide fragment of SARS corona virus E-protein: mechanism of self aggregation and amyloid-inhibition of hIAPP. Biochemistry. 2015;54(13):2249–2261. doi: 10.1021/acs.biochem.5b00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mukherjee S., Bhattacharyya D., Bhunia A. Host-membrane interacting interface of the SARS coronavirus envelope protein: immense functional potential of C-terminal domain. Biophys. Chem. 2020;266:106452. doi: 10.1016/j.bpc.2020.106452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Surya W., Li Y., Torres J. Structural model of the SARS coronavirus E channel in LMPG micelles. Biochim. Biophys. Acta Biomembr. 2018;1860(6):1309–1317. doi: 10.1016/j.bbamem.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng Y.T., Wang S.M., Huang K.J., Wang C.T. SARS-CoV envelope protein palmitoylation or nucleocapid association is not required for promoting virus-like particle production. J. Biomed. Sci. 2014;21(1):34. doi: 10.1186/1423-0127-21-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teoh K.T., Siu Y.L., Chan W.L., Schlüter M.A., Liu C.J., Peiris J.S., Bruzzone R., Margolis B., Nal B. The SARS coronavirus E protein interacts with PALS1 and alters tight junction formation and epithelial morphogenesis. Mol. Biol. Cell. 2010;21(22):3838–3852. doi: 10.1091/mbc.E10-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castaño-Rodriguez C., Honrubia J.M., Gutiérrez-Álvarez J., DeDiego M.L., Nieto-Torres J.L., Jimenez-Guardeño J.M., Regla-Nava J.A., Fernandez-Delgado R., Verdia-Báguena C., Queralt-Martín M., Kochan G., Perlman S., Aguilella V.M., Sola I., Enjuanes L. 2018. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis, mBio 9(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang R., Hozumi Y., Yin C., Wei G.-W. 2020. Decoding SARS-CoV-2 transmission and evolution and ramifications for COVID-19 diagnosis, vaccine, and medicine, Journal of Chemical Information and Modeling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enamine, 1 Distribution Way, Monmouth Jct., NJ 08852.
- 28.B. Chandrasekaran, S.N. Abed, O. Al-Attraqchi, K. Kuche, R.K. Tekade, Computer-aided prediction of pharmacokinetic (ADMET) properties, Dosage Form Design Parameters2018, pp. 731–755.
- 29.Kelley L.A., Mezulis S., Yates C.M., Wass M.N., Sternberg M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biovia D.S. Dassault Systèmes; San Diego: 2020. Discovery Studio 2020. [Google Scholar]
- 31.A. Hotra, P. Ragunathan, P.S. Ng, P. Seankongsuk, A. Harikishore, J.P. Sarathy, W.G. Saw, U. Lakshmanan, P. Sae-Lao, N.P. Kalia, J. Shin, R. Kalyanasundaram, S. Anbarasu, K. Parthasarathy, C.N. Pradeep, H. Makhija, P. Dröge, A. Poulsen, J.H.L. Tan, K. Pethe, T. Dick, R.W. Bates, G. Grüber, Discovery of a novel mycobacterial F-ATP synthase inhibitor and its potency in combination with diarylquinolines, Angewandte Chemie (International ed. in English) (2020). [DOI] [PubMed]
- 32.Harikishore A., Niang M., Rajan S., Preiser P.R., Yoon H.S. Small molecule Plasmodium FKBP35 inhibitor as a potential antimalaria agent. Sci. Rep. 2013;3:2501. doi: 10.1038/srep02501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amaravadhi H., Baek K., Yoon H.S. Revisiting de novo drug design: receptor based pharmacophore screening. Curr. Top. Med. Chem. 2014;14(16):1890–1898. doi: 10.2174/1568026614666140929115506. [DOI] [PubMed] [Google Scholar]
- 34.Kirchhoff P., Brown R., Kahn S., Waldman M., Venkatachalam C. Application of structure-based focusing to the estrogen receptor. J. Comput. Chem. 2001;22:993–1003. [Google Scholar]
- 35.Irwin J.J., Shoichet B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45(1):177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267(3):727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Topspin v3.2, Bruker Biospin GmbH, Switzerland.
- 38.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandala V.S., McKay M.J., Shcherbakov A.A., Dregni A.J., Kolocouris A., Hong M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020;27(12):1202–1208. doi: 10.1038/s41594-020-00536-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaulton B.A. ChEMBL_27 SARS-CoV-2 release. May. 2020;22 [Google Scholar]
- 41.Bhunia A., Bhattacharjya S., Chatterjee S. Applications of saturation transfer difference NMR in biological systems. Drug Discov. Today. 2012;17(9–10):505–513. doi: 10.1016/j.drudis.2011.12.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data