

Summary
The RNA-dependent-RNA polymerase (RdRp) from SARS-CoV-2 is an important drug target because it is responsible for viral RNA genome replication. Efficient production of recombinant RdRp is important in screening antivirals to treat COVID-19. Here, we present our protocol for expression of tag-free replication complex proteins in E. coli and subsequent purification. Despite the added complexity of multiple purification steps, our methods provide greater activity, yield at lower cost, and are faster than baculovirus expression systems.
For complete details on the use and execution of this protocol, please refer to Dangerfield et al. (2020).
Subject areas: Artificial intelligence, Human-centered computing, Human-computer interaction
Graphical abstract
Highlights
-
•
Soluble expression of tag-free, highly active SARS-CoV-2 polymerase complex in E. coli
-
•
Purification protocol for NSP-7/8/12 RNA polymerase complex is provided
-
•
Protocol for expression and purification of supplemental NSP8 included
-
•
Purified SARS-CoV-2 RNA polymerase complex catalyzes fast processive RNA replication
The RNA-dependent-RNA polymerase (RdRp) from SARS-CoV-2 is an important drug target because it is responsible for viral RNA genome replication. Efficient production of recombinant RdRp is important in screening antivirals to treat COVID-19. Here, we present our protocol for expression of tag-free replication complex proteins in E. coli and subsequent purification. Despite the added complexity of multiple purification steps, our methods provide greater activity, yield at lower cost, and are faster than baculovirus expression systems.
Before you begin
The SARS-CoV-2 RNA-dependent-RNA polymerase (RdRp) complex is responsible for replication of the viral RNA genome and consists of non-structural proteins (nsp)12, nsp7, and nsp8. The only FDA approved treatment for COVID-19 infections is the nucleoside analog, Remdesivir, highlighting the importance of this enzyme as a target for therapies to combat the viral infection. Many protocols to express tagged RdRp complex proteins in baculovirus-infected insect cells have been published (Gordon et al., 2020; Hillen et al., 2020; Yin et al., 2020) but these methods are slow, expensive, and have a limited yield. Furthermore, tagged proteins can display different activity than their untagged counterparts, and appropriate controls for the effect of the tag are sorely lacking. Here we present our protocol for co-expression of nsp12/7/8 complex and tag-free nsp8 in E. coli and subsequent purification. The procedure yields approximately 7 mg of highly active enzyme per liter of culture that catalyzes RNA polymerization at a rate sufficient to account replication of the 30 kb viral genome in 2 min (Dangerfield et al., 2020).
Prepare electrocompetent E. coli BL21/ pG-Tf2
Timing: 3 days
In this step, electrocompetent E. coli BL21 / pG-Tf2 are prepared which will be used to co-express the RdRp complex in E. coli with the aid of chaperone proteins from the pG-Tf2 plasmid.
-
1.Before starting:
-
a.Autoclave the following items: 2 L dH2O split into 2 flasks, 100 mL of 10% (v/v) glycerol, 4 × 250 mL centrifuge bottles and caps, 1.5 mL microcentrifuge tubes, 250 mL graduated cylinder, 100 mL graduated cylinder, 1 L LB media in 2.8 L baffle-bottomed flasks. Add chloramphenicol to 20 μg/mL after cooling to 25°C.
-
b.Chill the following items to 4°C: 2 L autoclaved dH2O, 100 mL autoclaved 10% (v/v) glycerol, 4 × 250 mL autoclaved centrifuge bottles and caps, 1.5 mL autoclaved microcentrifuge tubes, 250 mL autoclaved graduated cylinder, 100 mL autoclaved graduated cylinder
-
a.
-
2.
Transform BL21 E. coli with the plasmid pG-Tf2 following transformation protocols for the type of competent BL21 E. coli used. Spread the transformation reaction solution onto an LB/Agar plate containing 20 μg/mL chloramphenicol.
-
3.
Incubate the plate for 16 h at 37°C.
-
4.
Select a single colony from the plate with the previous day’s transformation and inoculate a 10 mL starter culture of 2× YT media (defined under Materials and Equipment) with 20 μg/mL chloramphenicol. Grow the culture for 16 h at 37°C with shaking at 200 RPM.
-
5.
Inoculate 1 L of LB media (defined under Materials and Equipment) + 20 μg/mL chloramphenicol with the 10 mL starter culture
-
6.
Incubate cells with shaking at 30°C, 250 RPM. Measure the OD600 periodically (every half hour or so until the OD600 reaches 0.2, then every 15 min).
-
7.
When the OD600 reaches 0.35–0.4, immediately put the cells on ice and chill the culture for 25 min, swirling occasionally to ensure even cooling. Place centrifuge bottles on ice at this time and move the needed rotors to the centrifuge to start cooling to 4°C
CRITICAL: It is important to not let the OD600 get any higher than 0.4. The OD600 should be carefully monitored and checked often, especially when it gets above 0.2 as the cells grow exponentially. It usually takes about 3 h to reach an OD600 of 0.35 when using a 10 mL starter culture added to 1 L of medium. It is also very important to keep the cells at 4°C for the remainder of the procedure following growth. The cells and any bottles or solutions that they come in contact with must be pre-chilled to 4°C.
-
8.
Spin 1: Split the 1 L culture into four parts by pouring about 250 mL into ice cold centrifuge bottles. Harvest the cells by centrifugation at 1,000 × g for 20 min at 4°C.
-
9.
Decant the supernatant and resuspend each pellet in 200 mL of ice cold dH2O by gentle swirling
-
10.
Spin 2: Harvest the cells by centrifugation at 1,000 × g for 20 min at 4°C
-
11.
Decant the supernatant and resuspend each pellet in 100 mL of ice cold dH2O with gentle swirling. Combine the resuspensions into 2 centrifuge bottles
-
12.
Spin 3: Harvest the cells by centrifugation at 1,000 × g for 20 min at 4°C. At this step chill two 50 mL conical tubes on ice
-
13.
Decant the supernatant and resuspend each pellet in 40 mL of ice cold 10% (v/v) glycerol with gentle swirling. Transfer each suspension to a 50 mL conical tube
-
14.
Spin 4: Harvest the cells by centrifugation at 1,000 × g for 20 min at 4°C. Carefully aspirate the supernatant with a sterile Pasteur pipette
-
15.
Resuspend each pellet in 1 mL of ice cold 10% (v/v) glycerol by gently swirling
-
16.
Aliquot 50 μL into sterile and cold 1.5 mL microfuge tubes and snap freeze in liquid nitrogen. Store aliquots at −80°C.
Hazard note: Dispose of E. coli cultures as recommended by your local biosafety protocols
Transform expression plasmids into E. coli BL21
In this step, the expression plasmids are transformed into BL21 E. coli by electroporation. Any transformation protocol can be used for the pcIts,ind+-(NSP8) plasmid into BL21 E. coli but the protocol presented here is for electrocompetent cells prepared as described above.
-
17.
Chill 0.1 mm electroporation cuvettes on ice for at least 15 min
-
18.
Remove one tube of competent E. coli BL21 and one tube of electrocompetent E. coli BL21/pG-Tf2 made in the previous step from the −80°C freezer and place on ice for 10 min
-
19.Add plasmid DNA to appropriate cells
-
a.Pipette 10 ng of pcIts,ind+-(NSP8) in a volume less than 5 μL into the tube of E. coli BL21. Perform transformation following the protocol of the manufacturer of the competent cells.
-
b.Pipette at least 100 ng of pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) in a volume less than 5 μL into the tube of electrocompetent E. coli BL21/pG-Tf2. The rest of the protocol is for transformation into the electrocompetent E. coli BL21/pG-Tf2.
-
a.
-
20.
Transfer the cells/DNA mixture to the chilled electroporation cuvette and gently tap the cuvette on the lab bench to ensure the cells are in the gap in the bottom of the cuvette
-
21.
Wipe off any water or ice that is on the metal parts of the electroporation cuvette with a Kimwipe (paper tissue). Insert the cuvette into the cuvette holder connected to the Gene Pulser and slide into the safety cover.
-
22.
Electroporate at 1.8 kV, 25 μF by holding down the left red button and pressing the right red button until the machine beeps. Immediately release the buttons after you hear the beep
-
23.Check that the time constant given on the Gene Pulser is greater than 3.8.
-
a.If the time constant is less than 3.8, try the transformation again or re-make the competent cells.
-
a.
-
24.
Immediately add 950 μL of sterile SOC media (defined under Materials and Equipment) to the cuvette and pipette up and down to mix
-
25.
Transfer the cells to a 1.5 mL microcentrifuge tube and do outgrowth at 30°C for 1 h.
-
26.Plate transformations on selective plates
-
a.Plate E. coli BL21/pcIts,ind+-(NSP8) on LB plates with 100 μg/mL ampicillin
-
b.Plate E. coli BL21/pG-Tf2/pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) on LB plates with 30 μg/mL kanamycin and 20 μg/mL chloramphenicol
-
a.
-
27.Incubate plates at 30°C
-
a.E. coli BL21/pcIts,ind+-(NSP8) will show colonies after 16–24 h
-
b.E. coli BL21/pG-Tf2/pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) will take longer to see colonies, 24–48 h.
-
a.
Hazard note: Dispose of E. coli cultures as recommended by your local biosafety protocols
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli BL21 | New England Biolabs | Cat. # C253OH |
| Chemicals, peptides, and recombinant proteins | ||
| Chloramphenicol | Fisher Scientific | Cat. # BP904-100 |
| Ampicillin | Fisher Scientific | Cat. # BP1760-25 |
| Kanamycin | Fisher Scientific | Cat. # BP906-5 |
| Tetracycline | Fisher Scientific | Cat. # BP912-100 |
| Isopropyl-beta-D-thiogalactoside (IPTG) | Gold Biotechnology | Cat. # L2481C |
| Nalidixic acid | Sigma-Aldrich | Cat. # N8878 |
| Lysozyme | Sigma-Aldrich | Cat. # L4919 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat. # P7626 |
| Recombinant DNA | ||
| pG-Tf2 | Takara Biosciences | Cat. # 3340 |
| pcIts,ind+-(NSP8) | Dangerfield et al., 2020 | Addgene, 160656 |
| pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) | Dangerfield et al., 2020 | Addgene, 160540 |
| Other | ||
| Gene Pulser II | Bio-Rad | Model # 165-2105 |
| 0.1 mm electroporation cuvettes | Bio-Rad | Cat. # 165-2089 |
| Whatman 0.2 μm PES syringe filters | Millipore Sigma | Cat. # WHA67802502 |
| Branson Sonifier 450 | VWR Scientific | Model # S-450A |
| 5 mL Q FF columns | Cytiva Life Sciences | Cat. # 17515601 |
| 5 mL SP-FF columns | Cytiva Life Sciences | Cat. # 17515701 |
| 5 mL HiTrap Blue-HP columns | Cytiva Life Sciences | Cat. # 17041301 |
| Superdex 200 10/300 GL | Cytiva Life Sciences | Model # 17-5175-0 |
| Q Sepharose-FF Bulk Resin | Cytiva Life Sciences | Cat. # 17051001 |
| Heparin Sepharose 6 FF bulk resin | Cytiva Life Sciences | Cat. # 17099801 |
| Amicon 10 kDa MWCO centrifugal concentrators | Millipore Sigma | Cat. # UFC901024 |
| Amicon 30 kDa MWCO centrifugal concentrators | Millipore Sigma | Cat. # UFC903024 |
| SpectraPor 1 kDa MWCO dialysis tubing | Spectrum Chemical | Cat. # 888-10966 |
| NanoDrop One | Thermo Fisher | Cat. # ND-ONE-W |
| Roche cOmplete, EDTA-free protease inhibitor tablets | Millipore Sigma | Cat. # 11873580001 |
| AKTA Pure FPLC | Cytiva Life Sciences | Cat. # 29018226 |
Materials and equipment
| Solution | Contents | Final volume | Storage |
|---|---|---|---|
| LB media | 0.5% (w/v) yeast extract, 1% (w/v) Bacto Tryptone, 1% (w/v) NaCl, appropriate antibiotic after cooling | 1 L | 25°C, 1 week |
| LB + Amp. + agar plates | 0.5% (w/v) yeast extract, 1% (w/v) Tryptone, 1% (w/v) NaCl, 1.5% (w/v) Bacto agar, 100 μg/mL ampicillin after cooling | 500 mL (~12 plates) | 4°C, 1 month |
| LB + Chl. + agar plates | 0.5% (w/v) yeast extract, 1% (w/v) Tryptone, 1% (w/v) NaCl, 1.5% (w/v) Bacto agar, 20 μg/mL chloramphenicol after cooling | 500 mL (~12 plates) | 4°C, 1 month |
| LB + Chl. + Kan. + agar plates | 0.5% (w/v) yeast extract, 1% (w/v) Tryptone, 1% (w/v) NaCl, 1.5% (w/v) Bacto agar; 20 μg/mL chloramphenicol and 30 μg/mL kanamycin after cooling | 500 mL (~12 plates) | 4°C, 1 month |
| 2× YT media | 1% (w/v) yeast extract, 1.6% (w/v) Bacto Tryptone, 0.5% (w/v) NaCl | 10 mL | 25°C, 1 week |
| SOC media | 0.5% (w/v) yeast extract, 2% (w/v) Bacto Tryptone, 0.05% (w/v) NaCl, 20 mM glucose, 10 mM MgCl2, 2.5 mM KCl | 10 mL | 25°C, 1 week |
| Terrific broth | 2.4% (w/v) yeast extract, 2% (w/v) Bacto Tryptone, 0.4% (v/v) glycerol, 17.2 mM KH2PO4, 72 mM K2HPO4 | 4 L | 25°C, 1 week |
| 10% glycerol | 10% (v/v) glycerol | 150 mL | 4°C, 1 month |
| 6× SDS sample buffer | 12% (w/v) SDS, 0.06% (w/v) bromophenol blue, 47% (v/v) glycerol, 42 mM Tris-HCl pH 6.8, 9.3% (w/v) DTT | 5 mL | −20°C, 1 month |
| 100 mg/mL ampicillin | 10% (w/v) ampicillin, solvent: dH2O | 3 mL | −20°C, 2 weeks |
| 30 mg/mL kanamycin | 3% (w/v) kanamycin sulfate, solvent: dH2O | 2 mL | −20°C, 2 weeks |
| 20 mg/mL chloramphenicol | 2% (w/v) chloramphenicol, Solvent: Ethanol | 3 mL | −20°C, 2 weeks |
| 1 M IPTG | 23.8% (w/v) IPTG, solvent: dH2O | 1 mL | 4°C, make fresh |
| 25 mg/mL nalidixic acid | 2.5% (w/v) nalidixic acid, solvent: 0.3 M NaOH | 3 mL | make fresh |
| 10 μg/mL tetracycline | 0.001% (w/v) tetracycline, solvent: dH2O | 1 mL | make fresh |
| 1 M DTT | 15.4% (w/v) DTT, solvent: dH2O | 50 mL | −20°C, 1 month |
| 30 mg/mL lysozyme | 3% (w/v) lysozyme, solvent: lysis buffer | 3 mL | 4°C, make fresh |
| 800 mM PMSF | 13.9% (w/v) PMSF, solvent: acetone | 3 mL | make fresh daily |
| 4% Sodium deoxycholate | 4% (w/v) sodium deoxycholate, solvent: dH2O | 5 mL | 4°C, 1 month |
| Expression test sample Buffer | 66% (v/v) 2× SDS sample buffer, 33% (v/v) 10% glycerol | 5 mL | −20°C, 1 month |
| Expression test buffer A | 50 mM Tris-HCl pH 8, 2 mM EDTA | 10 mL | 4°C, 1 month |
| Expression test buffer B | 50 mM Tris-HCl pH 8, 2 mM EDTA, 500 mM NaCl | 10 mL | 4°C, 1 month |
| Lysis buffer-1 | 50 mM Tris-HCl pH 8.0, 2.5 mM EDTA, 150 mM NaCl, 1 mM DTT | 100 mL | 4°C, make fresh |
| Lysis buffer-2 | 50 mM Tris-HCl pH 8.0, 2 mM EDTA, 250 mM NaCl, 10% glycerol, 5 mM DTT | 100 mL | 4°C, make fresh |
| Dilution buffer-1 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 10% glycerol, 1 mM DTT | 3 L | 4°C, make fresh |
| Dilution buffer-2 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 10% glycerol, 5 mM DTT | 3 L | 4°C, make fresh |
| Buffer A-1 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 50 mM NaCl, 10% glycerol, 1 mM DTT | 4 L | 4°C, make fresh |
| Buffer A-2 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 50 mM NaCl, 10% glycerol, 2 mM DTT | 4 L | 4°C, make fresh |
| Buffer B-1 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 1 M NaCl, 10% glycerol, 1 mM DTT | 3 L | 4°C, make fresh |
| Buffer B-2 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 1 M NaCl, 10% glycerol, 2 mM DTT | 3 L | 4°C, make fresh |
| Storage buffer-1 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 20 mM KCl, 50% glycerol, 1 mM DTT) | 2 L | 4°C, make fresh |
| Storage buffer-2 | 50 mM Tris-HCl pH 7.5, 0.1 mM EDTA, 50 mM NaCl, 50% glycerol, 1 mM DTT | 2 L | 4°C, make fresh |
∗ The pH of all buffers was measured at 25°C
-
•
EDTA – Harmful if inhaled. Eye irritant. Wear goggles and weigh powder in fume hood.
-
•
SDS – Harmful if swallowed or inhaled. Skin and eye irritant. Wear protective gloves and weigh chemical in a fume hood.
-
•
Ampicillin – Harmful if inhaled. Potential skin irritant. Wear gloves.
-
•
Kanamycin – Harmful if inhaled or ingested. Potential skin irritant. Wear gloves.
-
•
Chloramphenicol – Harmful if inhaled. Potential skin irritant. Wear gloves.
-
•
IPTG – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves.
-
•
Tetracycline – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves.
-
•
Nalidixic acid – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves.
-
•
DTT – Harmful if swallowed or inhaled. Potential skin irritant. Wear gloves.
-
•
PMSF – Very toxic. Harmful if swallowed, inhaled, or applied to skin. Wear gloves, weigh powder in fume hood.
-
•
Sodium deoxycholate – Harmful if swallowed or inhaled.
Step-by-step method details
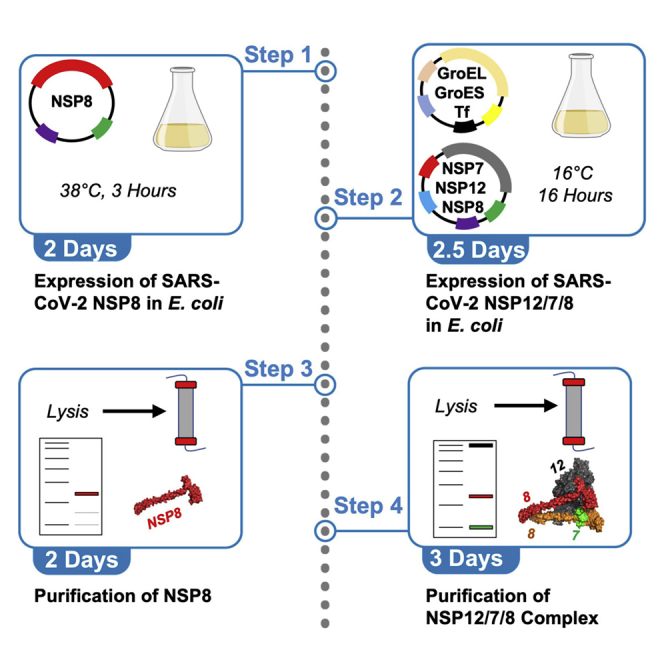
Expression of SARS-CoV-2 nsp8 in E. coli
In this step, a 2 L culture of E. coli harboring the plasmid pcIts,ind+-(NSP8) is grown to a high density. Protein expression is then induced by shifting the temperature from 30°C to 42°C for 20 min then to 38°C for 3 h. Finally, the cells are harvested and frozen for later use in protein purification.
-
1.
Inoculate a single colony from the plate containing E. coli BL21/ pcIts,ind+(NSP8) into 8 mL of Terrific broth with 100 μg/mL ampicillin. Incubate for 16 h with shaking at 30°C, 200 rpm.
-
2.
The following morning pellet the cells by centrifugation at 5,000 × g for 10 min.
-
3.
Decant the supernatant and resuspend the cells in 6 mL of fresh Terrific broth with 100 μg/mL ampicillin.
-
4.
Inoculate each of 2 × 1 L of Terrific broth with 100 μg/mL ampicillin, split into 2 × 2.8 L baffle-bottomed flasks, with 3 mL of the resuspended culture from step 3. Incubate the culture at 30°C with shaking at 250 rpm.
-
5.
Measure the OD600 of the culture every 30 min until the OD600 reaches 2.
-
6.
When the OD600 reaches 2, remove a “pre-induction” sample by pelleting (10,000 × g, 5 min, 20°C–25°C) a volume of cells equal to 1 mL at an OD600 of 6. Aspirate the supernatant then store the pellet at −80°C.
-
7.
Induce expression by moving the cultures to an incubator preheated to 42°C for 20 min and incubate with shaking at 250 rpm.
-
8.
Move the cultures to an incubator preheated to 38°C and incubate with shaking at 250 rpm for 3 h.
-
9.
Measure the OD600 of the cultures. Take a post-induction sample by pelleting a volume of cells equal to 1 mL at an OD600 of 6. Decant then aspirate the remaining supernatant. Store the pellet at −80°C.
-
10.
Harvest the cells by centrifugation at 6,000 × g for 20 min at 4°C Decant the supernatant then transfer the pellet to a pre-weighed 50 mL conical tube with a spatula.
-
11.
Determine the mass of the tube plus pellet then subtract the mass of the empty tube to get the yield of cell pellet. Record the mass on the tube as well as in your notes. Typical yields are 15–20 g of cell pellet per 2 L of growth media.
-
12.
Store the pellet at −80°C. The pellet is stable for at least one month at −80°C but likely is stable for longer than we have tested.
Hazard note: Dispose of E. coli cultures as recommended by your local biosafety protocols
Expression of SARS-CoV-2 nsp12/7/8 in E. coli
In this step, E coli harboring the chaperone plasmid pG-Tf2 and the RdRp complex co-expression plasmid pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) are grown to a high density before inducing expression for 16 h and harvesting the cells by centrifugation.
-
13.
Inoculate a single colony from the plate with E. coli BL21/pG-Tf2/pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) into an 8 mL starter culture of Terrific broth with 30 μg/mL kanamycin and 20 μg/mL chloramphenicol. Incubate at 30°C with shaking at 200 rpm for 16 h.
-
14.
The following morning inoculate the starter culture into 1 L of Terrific broth with 30 μg/mL kanamycin and 20 μg/mL chloramphenicol in a 2.8 L baffle-bottomed flask and incubate with shaking at 30°C, 250 rpm.
-
15.
Periodically check the OD600 of the culture. When the OD600 reaches 2, take a pre-induction sample by harvesting a volume of cells equal to 1 mL at an OD600 of 6 and storing the pellet at −80°C.
-
16.
Add tetracycline from a 10 μg/mL stock to a final concentration of 10 ng/mL and move the culture to an incubator pre-cooled to 16°C.
-
17.
Incubate with shaking at 16°C, 250 rpm for 20 min.
-
18.
Add IPTG to 0.5 mM from a 1 M stock solution. Add nalidixic acid to 50 μg/mL from a 25 mg/mL stock.
-
19.
Express for 16 h at 16°C with shaking at 250 rpm.
-
20.
Measure the OD600. Pellet a volume of cells equal to 1 mL at an OD600 of 6 and store the pellet at −80°C.
-
21.
Harvest the cells by centrifugation at 6,000 × g for 20 min at 4°C.
-
22.
Decant the supernatant and transfer the pellet to a pre-weighed 50 mL conical tube with a spatula. Determine the mass of the pellet then store at −80°C.
Hazard note: Dispose of E. coli cultures as recommended by your local biosafety protocols
Testing expression levels from large-scale E. coli expressions
In this step, pre- and post-induction samples collected during protein expression are lysed and separated by SDS-PAGE to determine the level of overexpression and solubility of the proteins of interest before pursuing further purification from the large pellets. The method described here is adapted from (Brandis and Johnson, 2009).
-
23.For each sample to be analyzed, set up a series of 3 tubes for each time point, each containing 45 μL of Expression Test Sample Buffer. Label the tubes in each set of three as follows:
-
a.TCP = Total Cell Protein
-
b.SOL = Soluble Protein
-
c.INS = Insoluble Protein
-
a.
-
24.
Thaw pellets collected pre- and post-induction from the large-scale expressions above on ice.
-
25.
Add 200 μL of Expression Test Buffer A containing 0.3 mg/mL Lysozyme to each pellet. Vortex until the pellets have been uniformly resuspended.
-
26.
Incubate samples at 37°C for 10 min then place on ice. The solutions should be very viscous due to the chromosomal DNA that is released upon lysis.
-
27.
Sonicate with the micro-tip (20% duty cycle, 0% output) for 30 – 40 bursts, ensuring the tip is always under the level of the solution in the tube. After sonication, the viscosity of the sample should be much lower.
-
28.
Add 22.4 μL of 5 M NaCl to each sample. Seal the tubes and vortex.
-
29.
Remove 15 μL of the sonicate and transfer it to the appropriate “TCP” sample tube, containing 45 μL of Expression Test Sample Buffer.
-
30.
Spin the remainder of the sonicate in a microcentrifuge tube at 16,000 × g in a tabletop microcentrifuge for 10 min at 20°C–25°C.
-
31.
Remove 15 μL of the supernatant to the appropriate “SOL” sample tube already containing Expression Test Sample Buffer.
-
32.
Aspirate off the remaining supernatant.
-
33.
Add 400 μL of Expression Test Buffer B and resuspend the pellet using the same sonication procedure as in step 27. This is a wash step to remove any residual soluble protein from the pellet and from the walls of the tube.
-
34.
Spin the suspension at 16,000 × g for 10 min at 20°C–25°C in a tabletop microcentrifuge.
-
35.
Aspirate and discard the supernatant.
-
36.
Add 192 μL of Expression Test Buffer B to each sample and resuspend the pellets by sonication as in step 27. Usually only 10 – 12 bursts are needed to resuspend the pellet.
-
37.
Remove 15 μL of the resuspended pellet solution to the appropriate “INS” tube with Expression Test Sample Buffer.
-
38.
Heat all samples at 80°C–85°C for 5 min prior to loading 10 – 15 μL per lane for SDS-PAGE.
-
39.
Run SDS-PAGE gels according to the manufacturers protocol, then stain with Coomassie brilliant blue. We recommend 15% acrylamide gels for resolving overexpressed nsp7 (9.4 kDa) and nsp8 (22 kDa), and 6% acrylamide gels for resolving overexpressed nsp12 (107 kDa).
Purification of SARS-CoV-2 nsp8
In this step, the frozen pellet obtained from the bacterial expression of nsp8 is lysed, and nsp8 is purified by a number of chromatography steps.
-
40.
Thaw the bacterial pellet from the expression above, on ice.
-
41.
Resuspend the cell pellet in Lysis Buffer-1 at 5 mL buffer per gram of E. coli.
-
42.
Add 800 mM phenylmethylsulfonyl fluoride (PMSF) to a final concentration of 10 mM. Add 30 mg/mL Lysozyme to a final concentration of 0.3 mg/mL and stir the lysate at 25°C for 15 min on a stir plate.
-
43.
Sonicate the lysate (20% duty, power 5) on ice for 20 min with constant stirring from a stir bar. During sonication, check the sample every 3–5 min to ensure efficient mixing and ice cooling as the sonicator produces heat.
-
44.
Add 4% sodium deoxycholate to a final concentration of 0.1%. Add 5 M NaCl to a final concentration of 0.5 M. Stir on ice for 30 min.
-
45.
Centrifuge the lysate at 75,000 × g for 45 min at 4°C.
-
46.
Filter the supernatants through a 0.2 μm syringe filter.
-
47.
Measure the conductivity of Buffer A-1 containing 10% Buffer B-1. Dilute the filtered sample with Dilution Buffer-1 until the conductivity of the sample is equal to the conductivity of Buffer A-1 with 10% Buffer B-1 (approximately a 5-fold dilution). Remove an aliquot of the diluted sample into 6× SDS Sample Buffer tor later analysis by SDS-PAGE.
-
48.
HiTrap Q-FF / HiTrap SP-FF Chromatography: Flow the lysate on to two sequential 5 mL HiTrap Q FF column with the outlet connected to two sequential 5 mL HiTrap SP-FF column, equilibrated in Buffer A-1, at 10 mL/min. Monitor absorbance at 280 nm. Collect the flow through in a separate, labeled container and store at 4°C as a backup.
-
49.
Wash with 10% Buffer B-1 until the UV280 reaches baseline (approximately 100 mL). Collect the eluate in a separate container, save a sample for SDS-PAGE analysis, and store at 4°C.
-
50.
Disconnect the Q columns, leaving the two SP columns connected to the FPLC.
-
51.
Elute proteins bound to the SP column with a 200 mL gradient from 10%–80% Buffer B at 5 mL/min. Collect 5 mL fractions and store fractions on ice.
-
52.
Remove aliquots of various fractions with a large A280 peak in the chromatogram into 6× SDS Sample Buffer. Heat samples at 85°C for 5 min. Load samples onto 15% SDS-PAGE gels, then run the gel until the bromophenol blue reaches the bottom of the gel following the manufacturers protocol. Stain and destain the gel with Coomassie brilliant blue.
-
53.
Pool fractions containing nsp8.
Pause point: At this point, fractions can be left at 4°C for up to 16 h.
-
54.
HiTrap Blue-HP Chromatography: Dilute the pooled fractions with Dilution Buffer-1 until the conductivity of the sample is equal to the conductivity of Buffer A-1.
-
55.
Load the sample onto a 5 mL HiTrap Blue-HP column, equilibrated in Buffer A-1 at 10 mL/min. Collect the flow through in a separate, labeled container, save a sample for SDS-PAGE, and store at 4°C.
-
56.
Wash the column with 10% Buffer B at 5 mL/min until the UV280 reaches baseline, collecting the eluate in a separate, labeled container. Save a sample for SDS-PAGE and store at 4°C.
-
57.
Elute bound proteins with a 50 mL linear gradient from 10%–80% Buffer B at 5 mL/min, collecting 2 mL fractions.
-
58.
Analyze samples from the Blue column by SDS-PAGE as described above.
-
59.
Pool fractions containing nsp8.
-
60.
Concentrate the pooled fractions by ultrafiltration with Amicon Ultra centrifugal concentrators with a 10 kDa molecular weight cutoff (MWCO) at 4°C until the sample volume is less than 1 mL, following the manufacturer’s instructions.
-
61.
Size Exclusion Chromatography: Inject the sample onto the Superdex 200 10/300 GL column, equilibrated in 10% Buffer B with the sample loop on the HPLC.
-
62.
Elute proteins with 10% Buffer B-1, collecting 1 mL fractions.
-
63.
Analyze fractions by SDS-PAGE as described above.
-
64.
Pool fractions containing nsp8.
-
65.
Concentration / Storage: Determine the concentration of nsp8 by absorbance at 280 nm using the extinction coefficient ε280: 19,940 M−1 cm−1 (Dangerfield et al., 2020).
-
66.
If necessary, concentrate the sample with Amicon Ultra centrifugal concentrators (10 kDa MWCO) following the manufacturer’s protocol . We concentrated the protein to 300 μM at this step without precipitation problems.
-
67.
Dialyze the sample into Storage Buffer-1 with 1 kDa MWCO dialysis tubing at 4°C with 2 × 500 mL × 1 h buffer changes.
-
68.
Remove the sample from the dialysis tubing and determine the concentration of nsp8 by absorbance at 280 nm using the extinction coefficient above.
-
69.
Divide the purified protein into aliquots, flash freeze into liquid nitrogen and store at −80°C until use.
Purification of nsp12/7/8
In this step, pellets of E. coli harboring overexpressed nsp12/7/8 complex are lysed and subsequently purified over several chromatography steps.
-
70.
Thaw cells from previous expression and resuspend in Lysis Buffer-2 at 5 mL buffer per gram of E. coli cells.
-
71.
For each 50 mL of lysis buffer used above in step 70, dissolve one EDTA-free protease inhibitor tablet into 2 mL of Lysis Buffer-2 and add to the sample. Add 800 mM PMSF to a final concentration of 10 mM. Add 4% sodium deoxycholate to a final concentration of 0.1%. Add 30 mg/mL lysozyme to a final concentration of 0.3 mg/mL. Stir at 25°C for 15 min on a stir plate.
-
72.
Sonicate the lysate on ice for 20 min with stirring (20% duty cycle, power 5). Check ice cooling every 5 min.
-
73.
Clarify the lysate by centrifugation at 75,000 × g at 4°C for 1 h.
-
74.
Filter the supernatant through at 0.2 μm syringe filter.
-
75.
Measure the conductivity of Buffer A-2. Measure the conductivity of the sample and dilute with Dilution Buffer-2 until the conductivity is equal to the conductivity of Buffer A-2 (~ 6 fold dilution). Save a sample for SDS-PAGE analysis.
-
76.
Q Sepharose-FF Chromatography: Load the sample at 5 mL/min onto a 30 mL Q Sepharose-FF column equilibrated in Buffer A-2, collecting the flow through in a separate, labeled container. Store the flow through at 4°C.
-
77.
Wash with Buffer A until the UV reaches baseline, collecting the eluate in a separate container. Store at 4°C.
-
78.
Elute bound proteins with a 300 mL linear gradient from 0%–100% Buffer B-2, collecting 10 mL fractions.
-
79.
Analyze fractions by SDS-PAGE.
-
80.
Pool fractions containing nsp12/7/8.
-
81.
HiTrap Blue-HP Chromatography: Filter the pooled fractions with a 0.2 μm syringe filter.
-
82.
Dilute the pooled fractions with Dilution Buffer-2 until the conductivity is equal to the conductivity of Buffer A-2.
-
83.
Load the sample onto the column at 5 mL/min and collect the flow through, containing nsp12/7/8.
-
84.
Heparin Sepharose 6-FF Chromatography: Load the flow through from the blue column directly on to a 5 mL Heparin Sepharose 6-FF column equilibrated in Buffer A-2 at 5 mL/min and collect the flow through in a separate labeled container. Store at 4°C.
-
85.
Wash with Buffer A-2 until the UV reaches baseline, collecting the eluate in a separate labeled container. Store at 4°C.
-
86.
Elute bound proteins with a 50 mL linear gradient of 0%–100% Buffer B-2 at 2.5 mL/min, collecting 5 mL fractions.
-
87.
Analyze results of column by SDS-PAGE as described above. Pool fractions containing nsp12/7/8 complex.
-
88.
HiTrap SP-FF Chromatography: Filter the pooled fractions with a 0.2 μm syringe filter. Dilute the pooled fractions with Dilution Buffer-2 until the conductivity is equal to the conductivity of Buffer A-1.
-
89.
Load the diluted sample onto a 5 mL HiTrap SP-FF column at 5 mL/min, equilibrated in Buffer A-2, collecting the flow through in a container on ice.
-
90.
Concentrate the flow through from the SP column with Amicon Ultra centrifugal concentrators (30 kDa MWCO) following the manufacturers protocol until the volume is less than 0.5 mL.
-
91.
Size Exclusion Chromatography: Inject the concentrated sample onto a Superdex 200 10/300 GL column, equilibrated in Buffer A-2 with the sample loop on the FPLC at 0.75 mL/min.
-
92.
Separate the complexes in the same buffer, collecting 1.5 mL fractions.
-
93.
Analyze fractions by SDS-PAGE. Pool fractions containing nsp12/7/8 complex.
-
94.
Concentration / Storage: Concentrate the pooled fractions using Amicon Ultra (30 kDa MWCO) centrifugal concentrators until the volume is approximately 1 mL, following the manufacturers protocols.
Note: We have concentrated the nsp12/7/8 complex up to 165 μM without precipitation, although the maximum concentration obtainable without precipitation is not known.
-
95.
Dialyze the concentrated sample into Storage Buffer-2 with 1 kDa MWCO dialysis tubing at 4°C with two 500 mL buffer changes, 1 h of equilibration for each.
-
96.
Remove sample from dialysis tubing, determine the final volume, and measure the concentration by absorbance at 280 nm using the extinction coefficient ε280: 181,300 M−1 cm−1 calculated from the amino acid sequence and assuming one nsp12, one nsp7, and two nsp8 polypeptides per complex.
-
97.
Flash freeze aliquots in liquid nitrogen and store at −80°C.
Expected outcomes
The expected outcomes are shown in Figures 1, 2, and 3.
Figure 1.

Expression and purification of nsp8
The soluble fraction of the E. coli lyase is shown in the left most lane, followed by a sample taken before loading onto the Q/SP columns, and blue column. The lane labeled MW ladder contains PAGE Ruler Plus molecular weight ladder, with corresponding sizes as follows from small to large: 10 kDa (green), 15 kDa (blue), 25 kDa (red), 35 kDa (blue), 55 kDa (blue), 70 kDa (red), 100 kDa (blue). Lanes to the right of the molecular weight ladder are different dilutions of the final purified protein. Figure reprinted with permission from Dangerfield et al., 2020.
Figure 2.

Expression test for nsp12/7/8
Samples are from an expression test from a large-scale expression of the nsp12/7/8 complex. Molecular weight markers are in the leftmost lane of each gel, with their corresponding molecular weights (in kDa) given to the left of each gel.
(A) 15% SDS-PAGE gel of pre- and post-induction samples. Samples corresponding to the total cell protein (TCP), soluble protein (Sol.) and insoluble protein (Ins.) are shown both before induction (Pre-induction), and after induction (post-induction).
(B) 6% SDS-PAGE gel of pre-and post-induction samples. Samples are labeled as in (A), but higher molecular weight bands can be seen. Figure reprinted with permission from Dangerfield et al. (2020).
Figure 3.

Gel from nsp12/7/8 purification
The leftmost lane contains a molecular weight marker, with corresponding sizes in kDa given to the left of the gel. Two dilutions of the final purified nsp12/7/8 complex are shown in the right two lanes. Bands of interest are labeled to the right of the gel. Figure reprinted with permission from Dangerfield et al. (2020).
Quantification and statistical analysis
Calculating the final yield in mg of each protein preparation based on the volume and molar concentration measured by absorbance:
To determine the yield of each protein preparation in mg, use the following equation:
For the nsp8 preparation, the molecular weight is 22,000 g/mol and the typical yield is 25 mg per liter of culture. For the nsp12/7/8 complex preparation, the combined molecular weight of the complex (assuming 2 molecules of nsp8 per molecule of nsp7 and nsp12) is 160,000 g/mol and the typical yield is 7 mg per liter of culture.
Limitations
While our bacterial expression protocol is significantly less time consuming that baculovirus protein expression, our purification protocols require many more steps than traditional purification of tagged proteins. Also even after multiple chromatography steps, there are still some proteolysis products, visible by SDS-PAGE, in the preparations of both enzymes. Both protein preparations are free from contaminating nuclease activity. Single turnover kinetic analysis shows that the enzyme is greater than 60% active. The purification procedures can likely be further optimized to eliminate these contaminating bands.
Troubleshooting
Problem 1
No colonies after transforming expression plasmids into E. coli (before you begin, step 27).
Potential solution
Ensure E. coli cells are kept as cold as possible after the growth phase (before you begin, steps 7–16) when preparing competent cells. Add more plasmid DNA before electroporation of competent cells (before you begin, step 19).
Problem 2
Inconsistent band intensity / same bands in soluble fraction and insoluble fraction (step-by-step method details, step 39)
Potential solution
Ensure all media is aspirated from cell pellets harvested for expression test before freezing samples (step-by-step method details, steps 6, 9, 15, 20). Ensure pellets are completely resuspended during lysis steps (step-by-step method details, steps 25–27). Ensure all buffer is aspirated after removing the soluble fraction sample (Step-by-step method details, step 32). Perform an additional “wash step” before removing insoluble fraction sample (step-by-step method details, repeat steps 33–35). Load the same volume of each sample when analyzing results by SDS-PAGE (step-by-step method details, step 38).
Problem 3
No/low expression of nsp8 (step-by-step method details, step 39) .
Potential solution
Always use a fresh transformation of pcIts,ind+-(NSP8) (before you begin, steps 17–27). Ensure ampicillin plates are made fresh as ampicillin is prone to degradation. Preheat incubators to 30°C, 42°C, and 38°C to ensure efficient heat induction (step-by-step method details, steps 4, 7, 8). The 42°C step can be performed in a preheated water bath for better heat transfer to the cells (step-by-step method details, step 7).
Problem 4
No/low expression of nsp12/7/8 complex (step-by-step method details, step 39).
Potential solution
Always use a fresh transformation of pQE-(NSP12)-pcIts,ind+-(NSP7-NSP8) into BL21/pG-Tf2 cells (before you begin, steps 17–27). Make LB/Chl./Kan. plates fresh to prevent satellite colonies. Ensure the nalidixic acid stock is made fresh daily. Ensure antibiotics and IPTG solutions are prepared as fresh as possible.
Problem 5
RdRp complex proteins do not bind to a column (step-by-step method details, steps 48, 55, 76, 83, 84, 89).
Potential solution
Add more of the appropriate Dilution Buffer (step-by-step method details, steps 47, 54, 75, 82, 88) and check to the conductivity of the sample to ensure it is below the conductivity of the appropriate Buffer A. Re-make FPLC buffers, taking care to add the correct concentration of NaCl.
Resource availability
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Kenneth A. Johnson (kajohnson@utexas.edu).
Materials availability
All materials in this study can be obtained from sources given in the key resources table.
Data and code availability
The published article includes all datasets generated or analyzed during this study
Acknowledgments
This work was supported by grants from NIAID (1R01AI110577 to K.A.J.),NIH (R01 GM114223 114223 to K.A.J.), and the Welch Foundation (F-1604 to K.A.J.).
Author contributions
Conceptualization, K.A.J., T.L.D., and N.Z.H.; Methodology, K.A.J., T.L.D., and N.Z.H.; Formal Analysis, K.A.J. and T.L.D.; Investigation, T.L.D. and N.Z.H.; Resources, K.A.J.; Writing—Original Draft, T.L.D.; Writing—Review and Editing, K.A.J., T.L.D., and N.Z.H.; Visualization, T.L.D.; Supervision, K.A.J.; Project Administration, K.A.J.; Funding Acquisition, K.A.J.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Tyler L. Dangerfield, Email: tylerdangerfield@utexas.edu.
Kenneth A. Johnson, Email: kajohnson@mail.utexas.edu.
References
- Brandis J.W., Johnson K.A. High-cell density shake-flask expression and rapid purification of the large fragment of Thermus aquaticus DNA polymerase I using a new chemically and temperature inducible expression plasmid in Escherichia coli. Protein Expr. Purifi. 2009;63:120–127. doi: 10.1016/j.pep.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangerfield T.L., Huang N.Z., Johnson K.A. Remdesivir is effective in combating COVID-19 because it is a better substrate than ATP for the viral RNA-dependent RNA polymerase. iScience. 2020;23:101849. doi: 10.1016/j.isci.2020.101849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C.J., Tchesnokov E.P., Woolner E., Perry J.K., Feng J.Y., Porter D.P., Götte M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020;295:6785–6797. doi: 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillen H.S., Kokic G., Farnung L., Dienemann C., Tegunov D., Cramer P. Structure of replicating SARS-CoV-2 polymerase. Nature. 2020;584:154–156. doi: 10.1038/s41586-020-2368-8. [DOI] [PubMed] [Google Scholar]
- Yin W., Mao C., Luan X., Shen D.-D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020;368:1499. doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published article includes all datasets generated or analyzed during this study