

Abstract
Background
Lysophosphatidic acid (LPA) is known to promote cancer cell invasiveness through LPA1, but the downstream signaling cascades are still not fully clarified. The CKLF-like MARVEL transmembrane domain-containing (CMTM) family regulates aggressive phenotype in many cancers.
Methods
We performed LPA1 co-immunoprecipitation combined with mass spectrometry to search for LPA1-associated proteins. The role of CMTM8 in mediating the pro-invasive activity of LPA was investigated in pancreatic cancer.
Results
We identified CMTM8 as an LPA1-interacting protein. LPA1 and CMTM8 were co-localized in pancreatic cancer cells. LPA treatment led to stabilization of CMTM8 protein, which was impaired by knockdown of LPA1. Depletion of CMTM8 significantly suppressed the migration and invasion of pancreatic cancer cells. Conversely, ectopic expression of CMTM8 enhanced the migratory and invasive capacity of pancreatic cancer cells. CMTM8 depletion blocked the formation of metastatic lesions in the lung. Knockdown of CMTM8 attenuated LPA-induced migration and invasion in pancreatic cancer cells. CMTM8 overexpression stimulated β-catenin activation through reduction of GSK3β. In addition, knockdown of β-catenin dramatically antagonized CMTM8-mediated migration and invasion in pancreatic cancer cells.
Conclusions
CMTM8 serves as a key mediator of LPA-induced invasiveness in pancreatic cancer. The interaction between CMTM8 and LPA1 leads to activation of oncogenic β-catenin signaling. CMTM8 represents a potential therapeutic target for pancreatic cancer.
Keywords: β-catenin, CKLF-like MARVEL transmembrane domain-containing 8 (CMTM8), invasion, lysophosphatidic acid 1 (LPA1), pancreatic cancer
Introduction
Lysophosphatidic acid (LPA) is a naturally producing lipid that can modulate cell migration during development and cancer (1-3). In many cancer cell types, such as breast cancer (1), ovarian cancer (4), melanoma (5), prostate cancer (6), and pancreatic cancer (7), LPA exhibits the ability to promote migration. Besides migration, several other aspects of tumor biology including proliferation, and invasion, angiogenesis are also coordinated by LPA in restricted cancer types (8). Six LPA receptors (LPA1–6) have been identified to mediate the pleiotropic effects of LPA. When LPA binds to its specific receptors, multiple downstream signaling cascades are activated, consequently participating in various biological processes (9,10). It has been documented that activation of the oncogenic β-catenin pathway is responsible for LPA-induced proliferation of colon cancer cells (10). The stability of β-catenin is controlled by a multiprotein complex consisting of adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), and axin. GSK3β-mediated phosphorylation of β-catenin leads to ubiquitination and proteasome degradation of β-catenin (11). LPA is known to activate β-catenin via phosphorylation and inactivation of GSK3β (10). However, the signal-transduction pathways responsible for LPA-induced aggressive phenotype in cancer cells have not been fully elucidated.
The CKLF-like MARVEL transmembrane domain-containing (CMTM) family consists of 8 member proteins (CMTM1–8) (12). Compelling evidence has pointed toward the tumor suppressive role of CMTMs (13,14). For instance, CMTM3 acts as a tumor suppressor in gastric cancer (14) and hepatocellular carcinoma (15). CMTM5 displays a pro-apoptotic activity in pancreatic cancer cells (16). CMTM8 inhibits the proliferation and invasion of bladder cancer cells (17). Despite these findings, some studies indicate that CMTMs can also support tumor progression. For instance, CMTM6 is known to enhance the stability of programmed death-ligand 1 (PD-L1) in cancer cells, contributing to escape from T-cell-mediated immunosurveillance (18). An alternatively spliced isoform of CMTM1 shows the ability to promote cell proliferation and survival in breast cancer cells (19).
Pancreatic cancer is one of the most lethal malignancies, with a median overall survival of 6 months and 5-year survival rate of less than 5% (20). Although CMTM5 (16) and CMTM7 (21) have been reported to exert tumor-suppressive effects on pancreatic cancer, the roles of other CMTM members in this malignancy are largely unknown.
In this study, we establish a link between LPA1 and CMTM8 in orchestrating pancreatic cancer progression. We also clarified the role of β-catenin signaling in mediating CMTM8 activity in pancreatic cancer.
We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-1013).
Methods
Cell culture and treatment
Human pancreatic cancer cell lines (BxPC-3, PANC-1, and CFPAC-1) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA). Pancreatic cancer cells were serum-starved for 12 h before treatment with 10 µM LPA (Sigma-Aldrich) for another 24 h and tested for gene expression, migration, and invasion. TGF-β1 recombinant protein (50 ng/mL; PeproTech, Rocky Hill, NJ, USA) was also used to induce pancreatic cancer cell migration and invasion. In some experiments, pancreatic cancer cells were treated with LY2090314 (50 nM; Selleckchem, Houston, TX, USA) for 24 h before transfection with CMTM8 shRNA.
Co-immunoprecipitation assay and mass spectrometry
Immunoprecipitation experiments were performed as described previously (22). In brief, cell lysates were pre-cleared with Protein A/G Magnetic Beads (New England Biolabs, Inc., Ipswich, MA, USA) for 1 h, and incubated with 4 µg anti-CMTM8, anti-LPA1, or isotype IgG for 6 h at 4 °C with rotation. Beads were magnetically immobilized and eluted by boiling in loading buffer. Immunoprecipitates were subjected to Western blot analysis. For mass spectrometry assay, the proteins from LPA- and vehicle-treated PANC-1 cells were immunoprecipitated by anti-LPA1 antibody and digested overnight with trypsin (Sigma-Aldrich) at 37 °C. The resultant peptides were desalted and then subjected to liquid chromatography using acetonitrile mobile phase gradients containing 0.1% formic acid. Separated peptides were examined by tandem mass spectrometry with a high-resolution hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA).
Immunofluorescence
BxPC-3 cells were grown on coverslips and fixed with 4% paraformaldehyde. The cells were then blocked with 5% bovine serum albumin and incubated at 4 °C overnight with primary antibodies recognizing CMTM8 and LPA1. Afterwards, Alexa Fluor 594- or Alexa Fluor 488-conjugated secondary antibodies (Thermo Fisher Scientific) were added and incubated for 45 min. The photographs were taken with a confocal laser-scanning microscope.
Western blot analysis
Tissue and cell samples were lysed in radioimmunoprecipitation assay buffer supplemented with a protease inhibitor cocktail (Roche, Mannheim, Germany). Protein samples were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked with 5% non-fat milk and incubated overnight at 4 °C with primary antibodies summarized in Table S1. After washing, the membranes were then incubated with secondary antibodies coupled to horseradish peroxidase for 1 h at room temperature. Protein signals were detected by the Western Lightning Plus ECL kit (PerkinElmer, Waltham, MA, USA) and quantified by densitometry with the Image J software (NIH, Bethesda, MD, USA).
siRNAs, plasmids, and transfections
Small interfering RNAs (siRNAs) targeting LPA1, and β-catenin, as well as nonspecific siRNAs were synthesized by Sangong Biotech (Shanghai, China). The siRNA target sequences are shown in Table S2. CMTM8 cDNA were cloned to pcDNA3.1(+) expression vector. Two independent short hairpin RNAs (shRNAs) targeting CMTM8 was synthesized and cloned into the pLKO.1 vector. The CMTM8 shRNAs are shown as follows: shCMTM8#1: 5'-CCGGCATTTGGCTGGGTCATGTTTGCTCGAGCAAACATGACCCAGCCAAATGTTTTTC-3'; shCMTM8#2: 5'-CCGGCACCGTCTTCTTCCTCATTATCTCGAGATAATGAGGAAGAAGACGGTGTTTTTC-3'. The plasmid constructs were confirmed by DNA sequencing. Transfections were performed using FuGENE (Roche Diagnostics) following the manufacturer’s instructions. If not stated otherwise, transfected cells were harvested for further experiments at 24 h after transfection.
CMTM8 protein stability analysis
The cells were transfected with control or LPA1-targeting siRNAs and then exposed to 10 µM LPA or 5 µM MG132 (Calbiochem, La Jolla, CA, USA). To inhibit protein synthesis, 50 µM cycloheximide (CHX; Sigma-Aldrich) was used. The cells were lysed at 1, 2, and 3 h, and subjected to Western blot analysis.
Quantitative real-time PCR analysis
Total RNA was extracted using TRIZOL (Invitrogen, Carlsbad, CA, USA), and reversely transcribed to cDNA with M-MLV reverse transcriptase (Promega, Madison, WI, USA). Expression of LPA1 mRNA was measured using the SYBR green RT-PCR assay (Takara, Dalian, China). The PCR primers are listed in Table S2. Relative LPA1 mRNA expression was determined using the comparative threshold cycle method (2−ΔΔCt).
Scratch wound healing assay
Cells were seeded onto 6-well plates and allowed to grow to confluence. The monolayer was scratched with a pipette tip. Mitomycin C (2 µg/mL; Sigma-Aldrich) was added to inhibit cell proliferation. Cells were photographed 24 or 48 h after scratching. The percentage of wound closure was determined from three independent experiments.
Transwell invasion assay
Cells (4 × 104) in serum-free medium were seeded into the upper chamber with Matrigel-coated filters (8 µm in pore size). Complete medium containing 10% FBS was added to the lower chamber. After incubation for 24 or 48 h, cells that invaded to the lower compartment were stained with 0.1% crystal violet. Three fields were randomly chosen to count the number of invaded cells.
Cell proliferation assay
Cells were seeded onto 96-well plates (4×103 cells/well) and cultured for 1, 3, and 5 days. Cell viability was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich). Absorbance was measured at 570 nm with a multifunctional microplate reader.
Colony formation assay
Cells were plated onto 6-well plates (500 cells/well). The cells were cultured for 10–14 days. Cell were stained with 0.1% crystal violet. The number of colonies was counted under a microscope.
Luciferase reporter assay
β-Catenin transcriptional activity was evaluated using TOP/FOP luciferase reporter assay, as described previously (23). In brief, BxPC-3 and PANC-1 cells were transfected with the TOP-FLASH or FOP-FLASH plasmid (carrying a wild-type or mutant TCF specific promoter, respectively) together with CMTM8-expressing plasmid. Luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s instructions. The relative luciferase activity was determined after normalization against Renilla luciferase activity.
Animal studies
Male BALB/c nude mice (4–6 weeks old) were housed under specific pathogen-free conditions and had free access to water and food. PANC-1 cells carrying control or CMTM8-targeting shRNA were stably transfected with a luciferase-expression vector. They were randomly injected to the nude mice (n=6 for each group) through the tail vein at 4×106 cells per mouse, as described previously (24). The mice were imaged and sacrificed 5 weeks after tumor cell inoculation. D-luciferin was injected intraperitoneally 5 min before direct visualization using the IVIS Xenogen imaging system. Lung tissues were removed and fixed in 4% paraformaldehyde. The sections of lung samples were stained with hematoxylin and eosin for histopathologic analysis. All animal experiments were approved by the Animal Care and Use Committee of Fudan University Shanghai Cancer Center, Shanghai, China (approval number: 2019-JS-146).
Tissue specimens
We collected 64 pairs of pancreatic cancer tissues and adjacent normal pancreas tissues from pancreatic cancer patients who were treated between February 2015 and February 2017 at our hospital. All the cases were diagnosed histologically. No patients received any anticancer treatment before collection of the tissue specimens. Written informed consent was obtained from each patient. This study carried out in accordance with the Declaration of Helsinki (as revised in 2013) and approved by the Institutional Review Board of Fudan University, Shanghai, China (approval number: 2018-0120).
Immunohistochemistry
Tissue sections were deparaffinized in xylene and hydrated in serial alcohol solutions. After blocking in 3% hydrogen peroxide for 10 min, tissue sections were stained with anti-CMTM8 antibody (1:50 dilution) followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies. 3,3'-Diaminobenzidine (DAB) was used as the substrate of HRP. The sections were counterstained with hematoxylin. Staining was semiquantitatively scored according to the product of the staining intensity multiplied by the percentage of positive cells. The staining intensity was defined as 0 = negative, 1 = weak, 2 = moderate, and 3 = strong. The percentage of positive cells was defined as 0, <1%; 1, 1–25%; 2, 26–50%; 3, 51–75%; and 4, 75–100%.
Statistical analysis
All values are reported as mean ± standard deviation (SD) and analyzed by the Student’s t-test or one-way analysis of variance followed by the post hoc Tukey's test. The difference in CMTM8 expression between pancreatic cancer and adjacent normal tissues was analyzed by the Mann-Whitney U test. The relationship between CMTM8 expression and clinicopathologic parameters of pancreatic cancer was analyzed by the chi-square test. Differences were considered statistically significant at P<0.05.
Results
Identification of CMTM8 as an LPA1-associated protein
It has been documented that LPA1 is a major receptor mediating the pro-invasive effect of LPA on pancreatic cancer cells (25,26). To identify LPA1 interacting proteins, we performed LPA1 co-immunoprecipitation combined with mass spectrometry in PANC-1 cells after LPA treatment. In this study, we selected 5 top protein candidates (PRDX2, CAP1, CMTM8, ALKBH5, and RACK1) for validation experiments. Western blot analysis revealed the presence of CMTM8 protein in LPA1 immunoprecipitates from BxPC-3 and PANC-1 cells, especially after LPA (10 µM) treatment (Figure 1A). However, the other 4 candidates were undetectable in the protein complex immunoprecipitated by anti-LPA1 antibody (data not shown). We also performed immunoprecipitations with an anti-CMTM8 antibody. As a consequence, LPA1 protein was detected in CMTM8 immunoprecipitates from pancreatic cancer cells (Figure 1B). Confocal microscopy indicated that LPA1 and CMTM8 were co-localized in pancreatic cancer cells (Figure 1C). Both LPA1 and CMTM8 protein were consistently observed in multiple pancreatic cancer cell lines (Figure 1D). These data suggest an interaction between LPA1 and CMTM8 proteins in pancreatic cancer cells.
Figure 1.
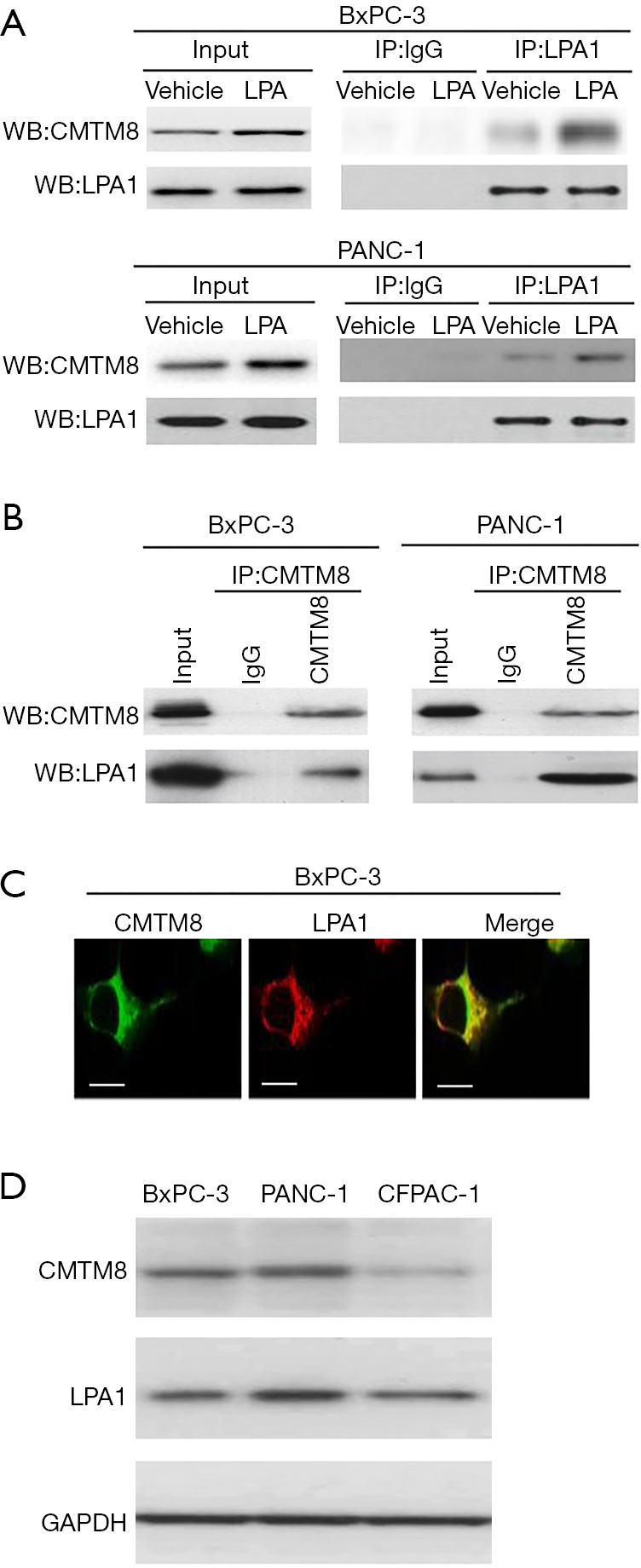
Identification of CMTM8 as an LPA1-interacting protein. (A) PANC-1 and BxPC-3 cells were exposed to 10 μM lysophosphatidic acid (LPA) and immunoprecipitated with anti-LPA1 antibody or isotope control IgG and probed for indicated proteins by Western blotting. (B) Detection of LPA1 protein by Western blotting in CMTM8 immunoprecipitates from BxPC-3 and PANC-1 cells. (C) Confocal microscopy indicated that LPA1 and CMTM8 were co-localized in pancreatic cancer cells. Scale bar =10 μm. (D) Western blot analysis of LPA1 and CMTM8 in pancreatic cancer cell lines. All experiments were performed three times.
LPA treatment stabilized CMTM8 protein through LPA1
Next, we tested whether LPA treatment affects the expression of CMTM8 in pancreatic cancer cells. Interestingly, we found that CMTM8 protein expression was markedly increased in pancreatic cancer cells after treatment with 1 and 10 µM LPA (Figure 2A). In contrast, CMTM8 mRNA abundance remained unchanged after LPA treatment (Figure S1). These results imply that LPA treatment may induce CMTM8 expression at the post-translational level. When CHX was utilized to block de novo protein synthesis, CMTM8 protein turnover was mitigated by LPA treatment (Figure 2B), indicating increased protein stability. Treatment with the proteasome inhibitor MG132 also stabilized CMTM8 protein in pancreatic cancer cells (Figure 2B), suggesting that CMTM8 is regulated by the proteasome-dependent pathway. To determine whether LPA1 activation is required for the stabilization of CMTM8 by LPA, we selectively knocked down LPA1 using siRNA technology (Figure 2C). Notably, silencing of LPA1 impaired LPA-mediated stabilization of CMTM8 protein in BxPC-3 pancreatic cancer cells (Figure 2D). Taken together, LPA exposure enhances the stability of CMTM8 protein through LPA1, consequently leading to increased expression of CMTM8.
Figure 2.

Lysophosphatidic acid (LPA) treatment stabilizes CMTM8 protein through LPA1. (A) Western blot analysis of CMTM8 protein levels in BxPC-3 and PANC-1 cells after treatment with 1 or 10 µM LPA. (B) Cells were exposed to 10 µM LPA or 5 µM MG132 and treated with 50 µM cycloheximide (CHX). CMTM8 protein levels were detected by Western blot analysis at 1, 2, and 3 h. (C) BxPC-3 cells were either transfected with nonspecific control siRNA (siCtrl) or the indicated LPA1 siRNAs. LPA1 mRNA levels were measured using quantitative real-time PCR analysis. *P<0.05 vs. the siCtrl group. (D) Analysis of CMTM8 protein turnover in LPA1-depleted and control BxPC-3 cells after treatment with 10 μM LPA. *P<0.05 vs. the siCtrl group. Data are expressed as mean ± SD (n=3).
CMTM8 augmented the migratory and invasive capacity of pancreatic cancer cells
Next, we sought to explore the function of CMTM8 in pancreatic cancer. shRNA-mediated CMTM8 gene silencing (Figure 3A) significantly inhibited the migration activity of BxPC-3 and PANC-1 cells, as assessed by the scratch wound healing assay (Figure 3B). Transwell invasion assay confirmed that the invasive ability of BxPC-3 and PANC-1 cells was attenuated when CMTM8 was silenced (Figure 3C). Conversely, overexpression of CMTM8 (Figure 3D) led to a significant increase in the migratory and invasive capacity of BxPC-3 and PANC-1 cells (Figure 3E,F). However, the proliferative and clonogenic potential of BxPC-3 and PANC-1 cells was not affected by overexpression of CMTM8 (Figure S2). These results indicate the ability of CMTM8 to accelerate the migration and invasion of pancreatic cancer cells in vitro.
Figure 3.

CMTM8 augments the migratory and invasive capacity of pancreatic cancer cells. (A) Quantitative PCR analysis of CMTM8 mRNA levels in BxPC-3 and PANC-1 cells transfected with control or CMTM8 short hairpin RNAs (shRNAs). (B) In vitro wound-healing assay. The percentage of wound closure was evaluated 48 h after scratching. (C) Transwell invasion assay. After incubation for 48 h, invaded cells were stained with 0.1% crystal violet and counted using a microscope (×20). (D) Western blot analysis of CMTM8 protein levels in pancreatic cancer cells transfected with indicated constructs. Numbers indicate fold change. (E) In vitro wound-healing and (F) Transwell invasion assays were performed to assess the migration and invasion capacity, respectively, of CMTM8-overexpressing and control cells. *P<0.05. Data are expressed as mean ± SD (n=3).
CMTM8 knockdown inhibited pancreatic cancer metastasis in vivo
To further explore the effects of CMTM8 on metastasis of pancreatic cancer, CMTM8-depleted and control PANC-1 cells were inoculated to nude mice via the tail vein. Lung metastases were then examined. We found that CMTM8 deficiency significantly inhibited lung metastasis of PANC-1 cells (Figure 4A,B). Histological examination confirmed the reduction of metastatic lesions in the lung (Figure 4C,D). The animal findings suggest that CMTM8 is required for pancreatic cancer progression. Clinically, we showed that CMTM8 was significantly upregulated in PDAC tissues relative to adjacent noncancerous pancreatic tissues (P<0.0001; Figure 4E). The expression of CMTM8 was not associated with tumor size, TNM stage, or lymph node metastasis (Table S3).
Figure 4.

CMTM8 knockdown inhibits pancreatic cancer metastasis in vivo. (A) CMTM8-depleted and control PANC-1 cells were inoculated to nude mice via the tail vein, and lung metastasis was analyzed by in vivo bioluminescence imaging. (B) Quantification of metastatic lesions (n=6 mice per group). (C) Representative hematoxylin and eosin stained sections of the lung with metastatic nodules. Scale bar =50 µm. (D) Determination of the numbers of metastatic nodules in the lung. *P<0.05. (E) Immunohistochemical analysis of CMTM8 in 64 pairs of pancreatic cancer and adjacent noncancerous pancreatic tissues. Right panels show semi-quantitative analysis of CMTM8 staining. Scale bar =100 µm.
CMTM8 was involved in LPA-mediated invasiveness in pancreatic cancer cells
Next, we examined whether CMTM8 is involved in LPA-induced invasiveness. We found that depletion of CMTM8 prevented LPA-induced migration (Figure 5A) and invasion (Figure 5B) in both BxPC-3 and PANC-1 cells. In contrast, silencing of CMTM8 had no significant impact on the migration and invasion of pancreatic cancer cells induced by TGF-β1 (Figure S3), a positive regulator of pancreatic cancer invasiveness (27). These results indicate that CMTM8 is specific to LPA-induced aggressive phenotype in pancreatic cancer. LPA has been known to activate β-catenin signaling in different cell types (28,29). Therefore, we examined the activity of CMTM8 in modulating β-catenin activation in pancreatic cancer cells. Notably, depletion of CMTM8 blocked LPA-induced β-catenin activation (Figure 5C,D).
Figure 5.

CMTM8 is involved in lysophosphatidic acid (LPA)-mediated invasiveness in pancreatic cancer cells. (A,B) BxPC-3 and PANC-1 cells were transfected with control or CMTM8-targeting shRNA (shCMTM8#1) before exposure to LPA, and then subjected to (A) wound-healing and (B) Transwell invasion assays. *, P<0.05. (C) Western blot analysis of indicated protein levels. (D) BxPC-3 and PANC-1 cells were transfected with the TOP-FLASH or FOP-FLASH plasmid together with shCMTM8#1 before treatment with LPA. Luciferase activities were then measured. *, P<0.05. Data are expressed as mean ± SD (n=3).
CMTM8 promoted pancreatic cancer invasiveness by stimulating β-catenin activation
We also explored the ability of CMTM8 overexpression on β-catenin activation. As shown in Figure 6A, overexpression of CMTM8 substantially raised the level of non-phosphorylated (active) β-catenin and phosphorylated GSK3β in BxPC-3 and PANC-1 cells. The total level of GSK3β was reduced when CMTM8 was overexpressed. The TOPflash reporter assay revealed that the β-catenin transcriptional activity was significantly increased in CMTM8-overexpressing cells (Figure 6B). Inhibition of GSK3β using an inhibitor LY2090314 blocked CMTM8 overexpression-induced β-catenin activation (Figure 6A,B). Most importantly, knockdown of β-catenin abrogated CMTM8-mediated migration and invasion in BxPC-3 and PANC-1 cells (Figure 6C,D,E). Overall, activation of β-catenin signaling accounts for the pro-invasive activity of CMTM8 in pancreatic cancer cells.
Figure 6.

CMTM8 promotes pancreatic cancer invasiveness by stimulating β-catenin activation. (A) Western blot analysis of indicated protein levels in pancreatic cancer cells transfected with CMTM8-expressing plasmid or empty vector. Numbers indicate fold change. (B) BxPC-3 and PANC-1 cells were transfected with the TOP-FLASH or FOP-FLASH plasmid together with CMTM8-expressing plasmid, and luciferase activities were measured. (C) Quantitative PCR analysis of β-catenin mRNA levels in BxPC-3 and PANC-1 cells transfected with control or β-catenin shRNAs. (D) In vitro wound-healing and (E) Transwell invasion assays were performed to assess the migration and invasion capacity, respectively, of pancreatic cancer cells transfected with indicated constructs. *P<0.05. Data are expressed as mean ± SD (n=3). (F) A schematic model showing the mechanism by which CMTM8 mediates lysophosphatidic acid (LPA)-induced invasiveness in pancreatic cancer cells.
Discussion
LPA is a ubiquitously present bioactive lipid participating in various biological processes including cell proliferation, migration, and invasion (1-4). LPA receptors including LPA1 play an important role in mediating LPA activity in pancreatic cancer. Yamada et al. reported that high expression of LPA1 is associated with an increased migration activity of pancreatic cancer cells (30). Silencing of LPA1 reduces the migration response to ascites in pancreatic cancer cells. Komachi et al. demonstrated that LPA1 is required for LPA-induced migration in pancreatic cancer cells (25). These studies point toward an essential role for LPA1 in LPA-dependent invasiveness. Previous studies have reported that the cross-talk between LPA1 and tropomyosin receptor kinase A facilitates lung epithelial cell migration (31). In the present work, we perform co-immunoprecipitation experiments and identify CMTM8 as a novel LPA1-associated protein. The 2 proteins are co-localized in pancreatic cancer cells. In response to LPA treatment, the interaction between LPA1 and CMTM8 was enhanced in pancreatic cancer cells. These data suggest a cross-talk between CMTM8 and LPA-mediated signaling. However, a direct interaction has not yet been validated for LPA1 and CMTM8 proteins. A deletion mapping of the LPA1-interacting domains on CMTM8 protein will help to clarify this issue. In addition to CMTM8, mass spectrometry analysis of LPA1 immunoprecipitates also identified PRDX2, CAP1, ALKBH5, and RACK1 as candidate LPA1-interacting proteins. Previous studies have indicated the involvement of these proteins in pancreatic cancer progression (32-35). Therefore, we suggest that several other proteins such as PRDX2, CAP1, ALKBH5, and RACK1 may be indirectly associated with LPA1 and modulate LPA signaling in pancreatic cancer.
It has been documented that prolonged LPA stimulation induces the association of Siva-1 with LPA2, which leads to ubiquitination and degradation of Siva-1 (36). In contrast, our data demonstrate the increase of CMTM8 protein in pancreatic cancer cells upon LPA exposure. To understand the mechanism by which LPA increases the protein level of CMTM8, we examined the effect of LPA on the stability of CMTM8 protein. Of note, we show that LPA treatment reduces the turnover of CMTM8 protein, which provides an explanation for the enhancement of CMTM8 expression. We further reveal that LPA1 depletion blocks LPA-mediated stabilization of CMTM8 protein. In the absence of LPA treatment, inhibition of LPA1 activity using the LPA1/LPA3 antagonist Ki16425 also reduces the expression of CMTM8 protein in PANC-1 cells (Figure S4). In addition, overexpression of LAP1 increased the level of CMTM8 protein in pancreatic cancer cells (Figure S4). These results suggest the association with LPA1 enhances the stabilization of CMTM8.
There is overwhelming evidence that CMTMs are frequently downregulated in cancer tissues and exert tumor-suppressive effects (14-17). Downregulation of CMTM8 occurs in many cancers, including bladder, liver, lung, colon, and gastric cancers (17). Overexpression of CMTM8 was reported to inhibit carcinogenesis and progression in bladder cancer (17). Unlike these studies, we provide evidence that CMTM8 plays an oncogenic role in pancreatic cancer. Reduction of CMTM8 upon treatment with Ki16425 is associated with inhibition of PANC-1 cell migration and invasion (Figure S4). Overexpression of CMTM8 augments the migratory and invasive capacity of pancreatic cancer cells, whereas depletion of CMTM8 yields an opposite effect. In vivo studies confirm that depletion of CMTM8 suppresses pancreatic cancer metastasis. When CMTM8 is depleted, LPA-induced pancreatic cancer invasiveness is impaired. These data suggest that maintenance of CMTM8 expression is of importance for pancreatic cancer invasion and metastasis.
Clinically, the expression of CMTM8 is significantly increased in pancreatic cancer tissues compared to adjacent noncancerous tissues. However, we did not observe the significant association between CMTM8 expression and pancreatic cancer features, which seems to be inconsistent with our animal findings. It should be noted that CMTM8 has 2 alternative splice variants (37). In this study, we did not validate which or both variant(s) contributes to pancreatic cancer progression. Therefore, we cannot exclude the possibility that one of the CMTM8 variants may be significantly associated with the invasive parameters of pancreatic cancer. Ongoing studies are conducted to test this hypothesis.
LPA1 is required for activation of multiple signaling pathways during cancer progression (30,38). For instance, LPA1 is involved in hepatocellular carcinoma cell invasion through activation of the PI3K and p38 MPAK pathways, consequently inducing MMP9 expression (38). Inhibition of LPA1 suppresses breast cancer metastasis in mouse models by modulating ERK and p38 MAPK signaling (39). Zhang et al. reported that CMTM8 modulates the epithelial-to-mesenchymal transition in hepatocellular carcinoma cells through c-MET/ERK signaling (40). However, we did not observe the alteration of ERK signaling in pancreatic cancer cells by CMTM8 (data not shown). Instead, we reveal that CMTM8 overexpression promotes the activation of β-catenin signaling via inactivation of GSK3β in pancreatic cancer cells. LPA1-mediated activation of β-catenin signaling has also been noted in non-malignant cells such as human lung-resident mesenchymal stem cells (29). The β-catenin pathway is known to participate in pancreatic cancer development and progression (41). Rescue experiments show that knockdown of β-catenin hampers CMTM8-induced pancreatic cancer cell migration and invasion. Taken together, these findings indicate that CMTM8 as an LPA responsive gene regulates LPA1-dependent β-catenin signaling in a positive feedback manner, consequently enhancing pancreatic cancer invasiveness (Figure 6E). However, the exact mechanism by which CMTM8 activates β-catenin signaling remains to be further clarified.
A previous study has reported that LPA can promote the migration of pancreatic cancer cells through induction of EGFR transactivation (42). There is evidence that LPA1 is involved in hypoxia-induced EGFR transactivation due to the cooperation between LPA1 and EGFR (43). Therefore, it is possible that the interplay between LPA1 and EGFR may also mediate the invasion-stimulating effect of LPA in pancreatic cancer.
In conclusion, our results support CMTM8 as a novel LPA1-associated protein in pancreatic cancer. LPA treatment leads to stabilization of CMTM8 protein through LPA1, which in turn promotes the invasion and metastasis of pancreatic cancer cells by stimulating β-catenin signaling. Therefore, CMTM8 may represent a potential therapeutic target for pancreatic cancer.
Supplementary
The article’s supplementary files as
Acknowledgments
We would like to thank members of Hanyu Biomed Center (Beijing, China) for technical help in preparing plasmids and performing Western blotting.
Funding: This study was supported by the National Science Foundation of China (81673746, 81774063, and 81673749).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Written informed consent was obtained from each patient. This study carried out in accordance with the Declaration of Helsinki (as revised in 2013) and approved by the Institutional Review Board of Fudan University, Shanghai, China (approval number: 2018-0120). All animal experiments were approved by the Animal Care and Use Committee of Fudan University Shanghai Cancer Center, Shanghai, China (approval number: 2019-JS-146).
Footnotes
Reporting Checklist: The authors have completed the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-1013).
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-1013
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-1013). The authors have no conflicts of interest to declare.
References
- 1.Stuelten CH, Lee RM, Losert W, et al. Lysophosphatidic acid regulates the motility of MCF10CA1a breast cancer cell sheets via two opposing signaling pathways. Cell Signal 2018;45:1-11. 10.1016/j.cellsig.2018.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hao F, Zhang F, Wu DD, et al. Lysophosphatidic acid-induced vascular neointimal formation in mouse carotid arteries is mediated by the matricellular protein CCN1/Cyr61. Am J Physiol Cell Physiol 2016;311:C975-84. 10.1152/ajpcell.00227.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jethwa SA, Leah EJ, Zhang Q, et al. Exosomes bind to autotaxin and act as a physiological delivery mechanism to stimulate LPA receptor signalling in cells. J Cell Sci 2016;129:3948-57. 10.1242/jcs.184424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J, Jang JH, Oh S, et al. LPA-induced migration of ovarian cancer cells requires activation of ERM proteins via LPA1 and LPA2. Cell Signal 2018;44:138-47. 10.1016/j.cellsig.2018.01.007 [DOI] [PubMed] [Google Scholar]
- 5.Susanto O, Koh YWH, Morrice N, et al. LPP3 mediates self-generation of chemotactic LPA gradients by melanoma cells. J Cell Sci 2017;130:3455-66. 10.1242/jcs.207514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin SH, Kwon YW, Heo SC, et al. Kruppel-like factor 4 mediates lysophosphatidic acid-stimulated migration and proliferation of PC3M prostate cancer cells. Exp Mol Med 2014;46:e104. 10.1038/emm.2014.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner JA, Ha JH, Jayaraman M, et al. The gep proto-oncogene Galpha13 mediates lysophosphatidic acid-mediated migration of pancreatic cancer cells. Pancreas 2013;42:819-28. 10.1097/MPA.0b013e318279c577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leblanc R, Peyruchaud O. New insights into the autotaxin/LPA axis in cancer development and metastasis. Exp Cell Res 2015;333:183-9. 10.1016/j.yexcr.2014.11.010 [DOI] [PubMed] [Google Scholar]
- 9.Saunders JA, Rogers LC, Klomsiri C, et al. Reactive oxygen species mediate lysophosphatidic acid induced signaling in ovarian cancer cells. Free Radic Biol Med 2010;49:2058-67. 10.1016/j.freeradbiomed.2010.10.663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang M, Zhong WW, Srivastava N, et al. G protein-coupled lysophosphatidic acid receptors stimulate proliferation of colon cancer cells through the {beta}-catenin pathway. Proc Natl Acad Sci U S A 2005;102:6027-32. 10.1073/pnas.0501535102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pez F, Lopez A, Kim M, et al. Wnt signaling and hepatocarcinogenesis: molecular targets for the development of innovative anticancer drugs. J Hepatol 2013;59:1107-17. 10.1016/j.jhep.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 12.Han W, Ding P, Xu M, et al. Identification of eight genes encoding chemokine-like factor superfamily members 1-8 (CKLFSF1-8) by in silico cloning and experimental validation. Genomics 2003;81:609-17. 10.1016/S0888-7543(03)00095-8 [DOI] [PubMed] [Google Scholar]
- 13.Delic S, Thuy A, Schulze M, et al. Systematic investigation of CMTM family genes suggests relevance to glioblastoma pathogenesis and CMTM1 and CMTM3 as priority targets. Genes Chromosomes Cancer 2015;54:433-43. 10.1002/gcc.22255 [DOI] [PubMed] [Google Scholar]
- 14.Yuan W, Liu B, Wang X, et al. CMTM3 decreases EGFR expression and EGF-mediated tumorigenicity by promoting Rab5 activity in gastric cancer. Cancer Lett 2017;386:77-86. 10.1016/j.canlet.2016.11.015 [DOI] [PubMed] [Google Scholar]
- 15.Li W, Zhang S. CKLF-Like MARVEL Transmembrane Domain-Containing Member 3 (CMTM3) Inhibits the Proliferation and Tumorigenisis in Hepatocellular Carcinoma Cells. Oncol Res 2017;25:285-93. 10.3727/096504016X14732523471442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo X, Li T, Wang Y, et al. CMTM5 induces apoptosis of pancreatic cancer cells and has synergistic effects with TNF-alpha. Biochem Biophys Res Commun 2009;387:139-42. 10.1016/j.bbrc.2009.06.148 [DOI] [PubMed] [Google Scholar]
- 17.Gao D, Hu H, Wang Y, et al. CMTM8 inhibits the carcinogenesis and progression of bladder cancer. Oncol Rep 2015;34:2853-63. 10.3892/or.2015.4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mezzadra R, Sun C, Jae LT, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017;549:106-10. 10.1038/nature23669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Zhang G, Zhang Y, et al. CMTM1_v17 is a novel potential therapeutic target in breast cancer. Oncol Rep 2014;32:1829-36. 10.3892/or.2014.3429 [DOI] [PubMed] [Google Scholar]
- 20.Hidalgo M. Pancreatic cancer. N Engl J Med 2010;362:1605-17. 10.1056/NEJMra0901557 [DOI] [PubMed] [Google Scholar]
- 21.Li H, Li J, Su Y, et al. A novel 3p22.3 gene CMTM7 represses oncogenic EGFR signaling and inhibits cancer cell growth. Oncogene 2014;33:3109-18. 10.1038/onc.2013.282 [DOI] [PubMed] [Google Scholar]
- 22.Dan HC, Adli M, Baldwin AS. Regulation of mammalian target of rapamycin activity in PTEN-inactive prostate cancer cells by I kappa B kinase alpha. Cancer Res 2007;67:6263-9. 10.1158/0008-5472.CAN-07-1232 [DOI] [PubMed] [Google Scholar]
- 23.Kaler P, Augenlicht L, Klampfer L. Activating mutations in beta-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS One 2012;7:e45462. 10.1371/journal.pone.0045462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagarajan A, Dogra SK, Sun L, et al. Paraoxonase 2 Facilitates Pancreatic Cancer Growth and Metastasis by Stimulating GLUT1-Mediated Glucose Transport. Mol Cell 2017;67:685-701.e6. 10.1016/j.molcel.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komachi M, Tomura H, Malchinkhuu E, et al. LPA1 receptors mediate stimulation, whereas LPA2 receptors mediate inhibition, of migration of pancreatic cancer cells in response to lysophosphatidic acid and malignant ascites. Carcinogenesis 2009;30:457-65. 10.1093/carcin/bgp011 [DOI] [PubMed] [Google Scholar]
- 26.Fukushima K, Otagaki S, Takahashi K, et al. Promotion of cell-invasive activity through the induction of LPA receptor-1 in pancreatic cancer cells. J Recept Signal Transduct Res 2018;38:367-71. 10.1080/10799893.2018.1531889 [DOI] [PubMed] [Google Scholar]
- 27.Zhao S, Ammanamanchi S, Brattain M, et al. Smad4-dependent TGF-beta signaling suppresses RON receptor tyrosine kinase-dependent motility and invasion of pancreatic cancer cells. J Biol Chem 2008;283:11293-301. 10.1074/jbc.M800154200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burkhalter RJ, Westfall SD, Liu Y, et al. Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces beta-Catenin-mediated Transcription in Epithelial Ovarian Carcinoma. J Biol Chem 2015;290:22143-54. 10.1074/jbc.M115.641092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badri L, Lama VN. Lysophosphatidic acid induces migration of human lung-resident mesenchymal stem cells through the beta-catenin pathway. Stem Cells 2012;30:2010-9. 10.1002/stem.1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamada T, Sato K, Komachi M, et al. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem 2004;279:6595-605. 10.1074/jbc.M308133200 [DOI] [PubMed] [Google Scholar]
- 31.Nan L, Wei J, Jacko AM, et al. Cross-talk between lysophosphatidic acid receptor 1 and tropomyosin receptor kinase A promotes lung epithelial cell migration. Biochim Biophys Acta 2016;1863:229-35. 10.1016/j.bbamcr.2015.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Xiao Y, Fan S, et al. RACK1 overexpression associates with pancreatic ductal adenocarcinoma growth and poor prognosis. Exp Mol Pathol 2016;101:176-86. 10.1016/j.yexmp.2016.08.001 [DOI] [PubMed] [Google Scholar]
- 33.He Y, Hu H, Wang Y, et al. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell Physiol Biochem 2018;48:838-46. 10.1159/000491915 [DOI] [PubMed] [Google Scholar]
- 34.Zhang M, Yan L, Wang GJ, et al. Resistin effects on pancreatic cancer progression and chemoresistance are mediated through its receptors CAP1 and TLR4. J Cell Physiol 2019;234:9457-66. 10.1002/jcp.27631 [DOI] [PubMed] [Google Scholar]
- 35.Kim YJ, Jeong SH, Kim EK, et al. Ursodeoxycholic acid suppresses epithelial-mesenchymal transition and cancer stem cell formation by reducing the levels of peroxiredoxin II and reactive oxygen species in pancreatic cancer cells. Oncol Rep 2017;38:3632-8. 10.3892/or.2017.6045 [DOI] [PubMed] [Google Scholar]
- 36.Lin FT, Lai YJ, Makarova N, et al. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem 2007;282:37759-69. 10.1074/jbc.M705025200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li D, Jin C, Yin C, et al. An alternative splice form of CMTM8 induces apoptosis. Int J Biochem Cell Biol 2007;39:2107-19. 10.1016/j.biocel.2007.06.002 [DOI] [PubMed] [Google Scholar]
- 38.Park SY, Jeong KJ, Panupinthu N, et al. Lysophosphatidic acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011;30:1351-9. 10.1038/onc.2010.517 [DOI] [PubMed] [Google Scholar]
- 39.Marshall JC, Collins JW, Nakayama J, et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J Natl Cancer Inst 2012;104:1306-19. 10.1093/jnci/djs319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang W, Mendoza MC, Pei X, et al. Down-regulation of CMTM8 induces epithelial-to-mesenchymal transition-like changes via c-MET/extracellular signal-regulated kinase (ERK) signaling. J Biol Chem 2012;287:11850-8. 10.1074/jbc.M111.258236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sano M, Driscoll DR, DeJesus-Monge WE, et al. Activation of WNT/beta-Catenin Signaling Enhances Pancreatic Cancer Development and the Malignant Potential Via Up-regulation of Cyr61. Neoplasia 2016;18:785-94. 10.1016/j.neo.2016.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tveteraas IH, Aasrum M, Brusevold IJ, et al. Lysophosphatidic acid induces both EGFR-dependent and EGFR-independent effects on DNA synthesis and migration in pancreatic and colorectal carcinoma cells. Tumour Biol 2016;37:2519-26. 10.1007/s13277-015-4010-1 [DOI] [PubMed] [Google Scholar]
- 43.Harper K, Lavoie RR, Charbonneau M, et al. The Hypoxic Tumor Microenvironment Promotes Invadopodia Formation and Metastasis through LPA1 Receptor and EGFR Cooperation. Mol Cancer Res 2018;16:1601-13. 10.1158/1541-7786.MCR-17-0649 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as