

Abstract
Optimal innate immune response to infection includes eradication of potential pathogens, resolution of associated inflammation, and restitution of homeostasis. Phagocytosing human polymorphonuclear leukocytes (hPMN) undergo accelerated apoptosis, a process referred to as phagocytosis-induced cell death (PICD) and an early step in their clearance from inflammatory sites. Among human pathogens that modulate hPMN apoptosis, Neisseria gonorrhoeae delays PICD, which may contribute to the exuberant neutrophilic inflammation that characterizes gonorrhea. To elucidate the mechanisms underlying delayed PICD, we compared features of hPMN cell death that followed phagocytosis of N. gonorrhoeae FA1090 wild-type (GC) or serum-opsonized zymosan (OPZ), a prototypical stimulus of PICD. Phosphatidylserine externalization required NADPH oxidase activity after ingestion of GC or OPZ, and Annexin V staining and DNA fragmentation were less after phagocytosis of GC compared to OPZ. Caspase 3/7 and caspase 9 activities after phagocytosis of GC were less than after ingestion of OPZ, but caspase 8 was the same after ingestion of GC or OPZ. When hPMN sequentially ingested GC followed by OPZ, both caspase 3/7 and 9 activities were less than that seen after OPZ alone, and the inhibition was dose-dependent for GC, suggesting that ingestion of GC actively inhibited PICD. Sequential phagocytosis did not block caspase 8 activity, mitochondrial depolarization, or Annexin V/propidium iodide staining compared to responses of hPMN fed OPZ alone, despite inhibition of caspases 3/7 and 9. Taken together, these data suggest that active inhibition of the intrinsic pathway of apoptosis contributes to the delay in PICD after hPMN ingestion of N. gonorrhoeae.
Keywords: caspase 3, caspase 8, caspase 9, mitochondrial depolarization, intrinsic pathway, apoptosis
Summary Sentence
Neisseria gonorrhoeae modifies caspase activation and mitochondrial depolarization in human neutrophils to modulate phagocytosis-induced cell death
Introduction
Polymorphonuclear leukocytes (hPMN) are the most abundant circulating leukocytes in human blood and the first responders to invading pathogens [1]. Recruited to sites of nascent infection, hPMN ingest and sequester invading microbes into phagosomes, where an array of antimicrobial agents attack [2]. Coincident with phagocytosis, hPMN undergo a burst of transcriptional activity and express many genes directly involved with cell death pathways [3]. The accelerated apoptosis that accompanies receptor-mediated ingestion of many different types of bacteria [4], a response referred to as phagocytosis-induced cell death or PICD (reviewed in [5]), readies spent hPMN for ingestion by macrophages by efferocytosis (reviewed in [6, 7]). The removal of effete hPMN in this way promotes resolution of the inflammatory response and restitution of homeostasis. Conversely, failure of prompt efferocytosis of apoptotic hPMN can produce disease [8, 9], and some microbes disrupt PICD as a tactic to be pathogenic.
Neisseria gonorrhoeae is an obligate human pathogen that causes gonorrhea, a sexually transmitted infection that characteristically provokes a robust neutrophilic response [10], persistent infection, and prolonged inflammation, all of which can lead to pelvic inflammatory disease in females and result in significant morbidity including infertility [11]. Despite the exuberant inflammation elicited during infection, N. gonorrhoeae not only survives inside hPMN but also delays PICD of hPMN that have ingested N. gonorrhoeae [12–14]. The mechanisms underlying how N. gonorrhoeae delays hPMN PICD have not been fully elucidated.
Using for comparison serum-opsonized zymosan (OPZ) as the prototypical PICD-inducing target for phagocytosis [3, 14], we examined the fate of primary hPMN fed N. gonorrhoeae to determine how ingestion of N. gonorrhoeae undermines PICD.
Materials and Methods
Reagents:
All reagents were purchased from Fisher Scientific (Pittsburgh, PA) unless otherwise indicated. Gonococcal medium base was from BD Difco™ (Pittsburgh, PA) and IsoVitaleX was from BD BBL™ (Pittsburgh, PA). Proteose Peptone No. 3 was from BD Bacto™ (Pittsburgh, PA). Heparin was purchased from Fresenius Kabi USA LLC (Lake Zurich, IL), clinical grade dextran T500 from Pharmacosmos (Holbaek, Denmark), Ficoll-Hypaque PLUS from GE Healthcare (Piscataway, NJ). Sterile endotoxin-free water and 0.9% sterile endotoxin-free sodium chloride were obtained from Baxter (Deerfield, IL). Zymosan A from Saccharomyces cerevisiae was from MPBio (Santa Ana, CA). Zymosan A (S. cerevisiae) BioParticles™-Alexa Fluor 488 conjugate was from Invitrogen (Grand Island, NY). MitoProbe™ JC-1 Assay Kit was from Invitrogen (Grand Island, NY) and Annexin V-FITC Apoptosis Kit was from BioVision (Milpitas, CA). Caspase-Glo 3/7, 8 and 9 assay systems were from Promega (Madison, WI). APO-BRDU™ kit was from BD Bioscience (San Jose, CA). Gentamicin sulfate solution was from IBI Scientific (Dubuque, IA). Chloramphenicol, DPI, luminol, ferricytochrome C, PMA and SOD were purchased from Sigma (St. Louis, MO).
Bacteria and culture conditions:
N. gonorrhoeae FA1090 wild-type (GC) or GC with green fluorescent protein (GC-GFP) were used in this study. Bacteria were reconstituted from frozen stock cultures and grown on gonococcal agar base plate supplemented with 1% IsoVitaleX at 37 °C with 5% CO2 overnight. Bacteria were diluted to an OD600 of 0.1 in gonococcal broth (15 g/L Proteose Peptone No.3, 4 g/L K2HPO4, 1 g/L KH2PO4, 5 g/L NaCl, supplemented with 10 ml/L IsoVitaleX) and incubated at 37 °C and 180–200 RPM until mid-logarithmic growth. For GC-GFP, chloramphenicol (5 μg/ml) was added to the gonococcal agar base plate and broth. Gentamicin-killed GC (kGC) was prepared by adding gentamicin (1mg/ml) to GC culture at mid-logarithmic growth, incubating at 37°C for 30 minutes. Bacteria were centrifuged and bacterial pellet was washed with HH++ (Hanks’ Balanced Salt Solution with calcium and magnesium with 20 mM HEPES). Gentamicin-killed GC was resuspended in HH++.
Human PMN isolation:
hPMN were isolated from venous blood collected from healthy volunteers, as previously described [15]. A written consent was obtained from each volunteer in accordance with a protocol approved by the Institutional Review Board for Human Subjects at the University of Iowa. Briefly, hPMN were isolated from heparinized venous blood using dextran sedimentation followed by a density gradient separation on Ficoll-Hypaque PLUS. After hypotonic lysis of erythrocytes, hPMN were resuspended in HH−− (Hanks’ Balanced Salt Solution without calcium or magnesium) to 2.0 × 107 cells/ml.
Phagocytosis:
Zymosan particles were opsonized with 10% pooled human serum in HH++ for 20 minutes at 37 °C while tumbling and then washed with HH++. hPMN in suspension were tumbled with OPZ at 10:1 MOI or GC at different MOI (10:1, 25:1 or 50:1) for 30 minutes at 37 °C. For experiments to study the consequences of sequential phagocytosis, hPMN in suspension were tumbled with GC at different MOI (10:1, 25:1 or 50:1) for 20 minutes at 37 °C, then OPZ at 10:1 MOI were added, and the final sample was tumbled for 10 minutes at 37 °C. Unbound particles or bacteria were removed by centrifugation of hPMN (300 × g, 5 minutes at room temperature) and aspiration of the supernatant. The cell pellet was resuspended in HH++ and GC/OPZ-laden hPMN were tumbled at 37 °C for indicated time and analyzed.
For measuring phagocytosis, hPMN were incubated with FITC-labelled OPZ (FITC-OPZ) or GC-GFP as described above. Phagocytosis was measured using flow cytometry (Accuri™ C6, BD Biosciences) and the fraction of hPMN that ingested fluorescent targets was expressed as a percent of the total hPMN population. The geometric mean of MFI reflected the relative number of fluorescent targets per hPMN and was calculated using FlowJo (FlowJo, LLC).
Phosphatidylserine (PS) externalization and plasma membrane permeability:
After phagocytosis, GC/OPZ-laden hPMN were tumbled at 37 °C for 2 and 6 hours. PS externalization and plasma membrane permeabilization were measured using Annexin V-FITC Apoptosis Kit according to the manufacturer’s protocol. In summary, at each time point 5 × 105 cells were q.s. to 500 μl with Annexin V binding buffer. 5 μl of Annexin V-FITC and propidium iodide (PI)-PE were added and the samples were incubated at room temperature for 5 minutes in the dark. Fluorescence was measured using flow cytometry (Accuri™ C6, BD Biosciences). Externalization of PS and loss of plasma membrane integrity were expressed as percentages of Annexin V-FITC and PI-PE positive cells, respectively.
To inhibit nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, diphenyleneiodonium (DPI; 10 μM) was added to hPMN suspensions during and after phagocytosis. Otherwise, phagocytosis and Annexin V-FITC and PI-PE staining were performed as described above.
DNA fragmentation:
GC/OPZ-laden hPMN were tumbled at 37 °C for 6 hours. DNA fragmentation was measured with a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay using APO-BRDU™ kit. At 6 hours, 5 × 106 cells were fixed with 4% paraformaldehyde in PBS without calcium and magnesium at 4 °C for 1 hour at 2.5 × 106 cells/ml. Cells were permeabilized in 70% ethanol at −20 °C for at least 12 hours. DNA fragments were labeled and stained with the kit’s reagents according to the manufacturer’s protocol and analyzed by flow cytometry (Accuri™ C6, BD Biosciences). DNA fragmentation was expressed as the percent of anti-5’-bromo-2’-deoxyuridine (BrdU)-FITC positive cells.
NADPH Oxidase activity:
Activity of the phagocyte NADPH oxidase was assessed in two ways, using luminol-enhanced chemiluminescence (LUM-CL) [13, 16, 17] to monitor relative intracellular oxidant production and superoxide dismutase (SOD)-inhibitable reduction of ferricytochrome C [18] to quantitate extracellular superoxide anion production. For the former, hPMN (5 × 105 cells) were added to a 96-well white-walled plate (Costar 3912). OPZ (10:1 MOI), GC (10:1 or 50:1 MOI) or phorbol myristate acetate (PMA; 100 ng/ml) were added as stimuli. DPI (10 μM), NADPH oxidase inhibitor, was added to selected wells. Finally, luminol (50 μM) was added to all the wells and luminescence was measured in Polarstar Omega (BMG). The plate was read every 20 seconds for 1 hour, and triplicate measurements were averaged. Maximum relative light unit (RLU) and time to reach maximum RLU were compared.
Extracellular superoxide generated by hPMN was measured by SOD-inhibitable reduction of ferricytochrome C [18]. In summary, hPMN (1 × 106 cells) were incubated with ferricytochrome C (100 nM) and one of the following stimuli: GC (50:1 MOI), OPZ (10:1 MOI), or PMA (100 ng/ml; positive control). Duplicate samples with SOD (50 ng/ml) were also prepared. The samples were tumbled at 37 °C for 30 minutes, then centrifuged (300 × g, 5 minutes at room temperature) to pellet the cells and collect supernatant. For sequential phagocytosis, hPMN were initially tumbled with GC for 20 minutes then OPZ was added; the final sample was tumbled at 37 °C for 10 minutes then centrifuged to collect supernatant. A550nm of the supernatant was read in a spectrophotometer (PerkinElmer UV/Vis Lambda 35) and the amount of extracellular superoxide anion (nmoles produced/106 cells), defined as SOD-inhibitable reduction of ferricytochrome C, was calculated using the millimolar extinction coefficient at 550 nm (21.1 mM−1 cm−1).
Caspase activity:
GC/OPZ-laden hPMN were tumbled at 37 °C for the indicated time (5–6 hours). Caspase activity was measured using Caspase-Glo assay system according to the manufacturer’s protocol. The assay measures luminescence produced by cleaved substrates by corresponding caspases. Caspase 3/7, 8, and 9 cleave DEVD, LETD and LEHD, respectively. In summary, 1 × 104 cells were q.s. to 100 μl with HH++ in a white-walled 96-well plate in triplicates. 100 μl of Caspase-Glo reagent was added to each well. The plate was incubated at room temperature for 45 minutes in the dark. Luminescence was measured with NOVOstar (BMG Labtech). Relative light units (RLU) were calculated by subtracting luminescence of HH++ alone from that of the sample. Finally, RLU of each condition was expressed as percent relative to RLU of hPMN fed OPZ (set as 100 %).
In experiments to evaluate the role of the NADPH oxidase in caspase 3/7 activity, hPMN in the absence or presence of DPI (10 μM) were compared. DPI was present during and after phagocytosis. Otherwise, phagocytosis and caspase activity measurement were performed as described above.
To determine if soluble factors released by GC mediate the observed changes in caspase 3/7 activity, a conditioned buffer (cBuffer) was prepared. GC at mid-logarithmic growth was suspended in HH++ for 20 minutes at 37 °C, bacteria removed by centrifugation, and the supernatant collected as cBuffer. hPMN were tumbled with either GC or cBuffer for 20 minutes at 37 °C and then OPZ added as described above for sequential phagocytosis. Caspase 3/7 activity of hPMN was assessed as described above.
Mitochondrial depolarization:
GC/OPZ-laden hPMN were tumbled at 37 °C for 1 hour. Mitochondrial depolarization was measured using MitoProbe™ JC-1 Assay Kit according to the manufacturer’s protocol. In brief, 1 × 106 cells from each sample were resuspended in 1 mL of PBS with calcium and magnesium and 2 μM of JC-1 stain was added. The sample was incubated for 15 minutes at 37 °C in the dark. The cells were washed twice with 1 mL PBS at 4°C and resuspended in 500 μl of PBS. Fluorescence was measured with flow cytometry (Accuri™ C6, BD Biosciences). Mitochondrial depolarization was calculated by measuring the shift in fluorescence emission from red (590 nm) to green (530 nm), then normalizing against that of cells treated with CCCP.
Statistical analysis:
Statistical comparisons were performed using either one-way or two-way ANOVA followed by Turkey’s posttests. All analyses used GraphPad Prism software. P values < 0.05 were considered statistically significant.
Results
Phagocytosis-induced cell death:
To assess how phagocytosis of GC activates the programmed cell death pathways in hPMN, we compared the externalization of plasma membrane PS, changes in plasma membrane permeability, and DNA fragmentation of hPMN that ingested GC or OPZ, an inert particle that served as a control for PICD [19]. We used flow cytometry to quantitate Annexin V-FITC binding and PI-PE staining, measures of PS expression and plasma membrane permeability, respectively (Figure 1A). Based on prior studies of markers of programmed cell death [19], we defined operationally the early and late phases of PICD as Annexin+/PI− and Annexin+/PI+ cells, respectively. The ingestion of OPZ triggered a higher percent of Annexin V+/PI− hPMN than did GC at both 2 and 6 hours. Similarly, the percent of cells with increased plasma membrane permeability (Annexin V+/PI+) was less in hPMN fed GC than those that ingested OPZ. Viability of ingested GC was not required, as gentamicin-killed GC likewise elicited less Annexin V/PI staining than did OPZ (Supplement Figure 1).
Figure 1. Delayed PICD of hPMN in suspension by GC.
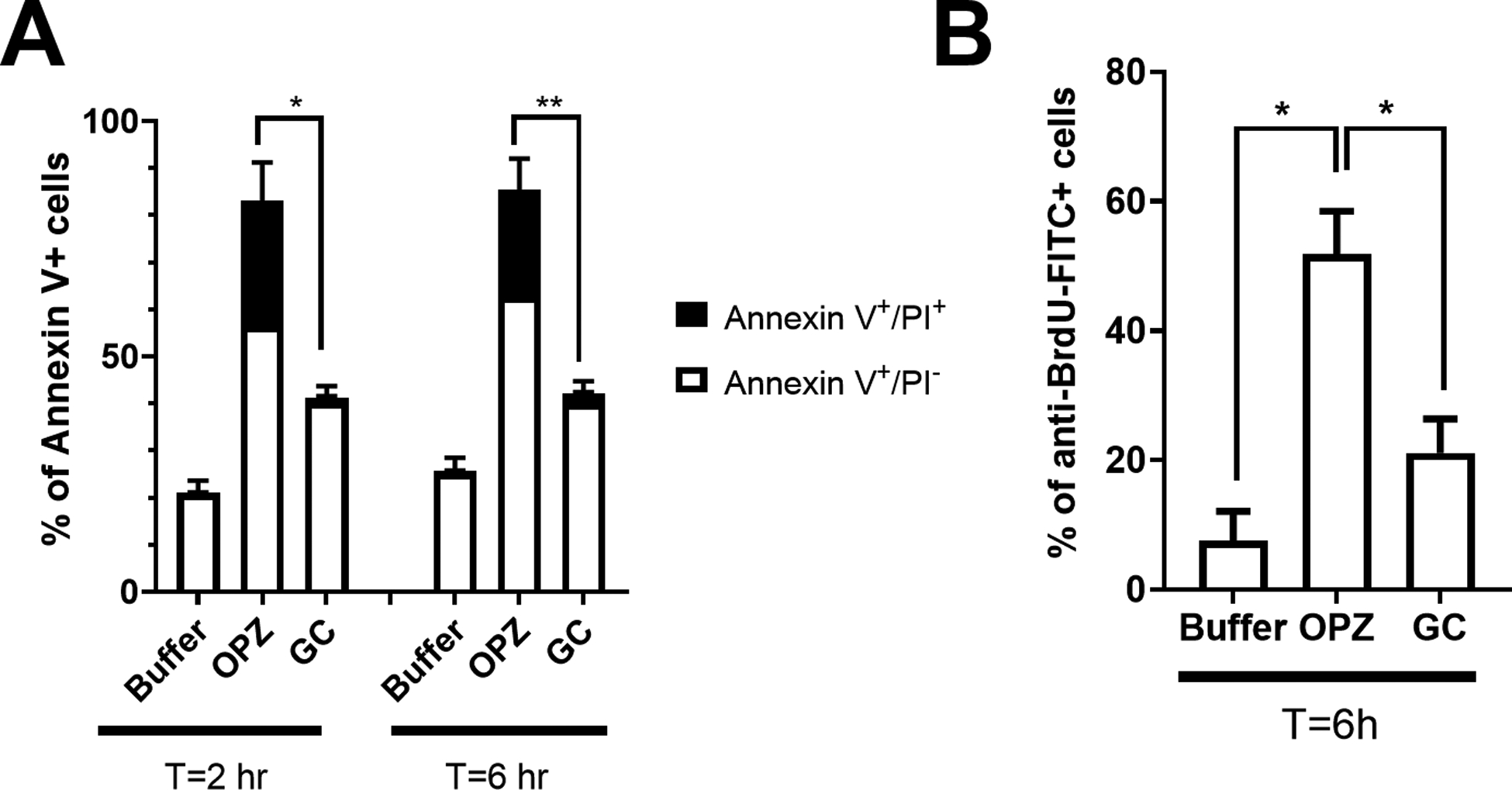
(A) PS exposure and plasma membrane integrity loss, markers of PICD, were quantitated with Annexin V-FITC and PI-PE staining, respectively. The increase in the percent of apoptotic hPMN (Annexin V+/PI−) after phagocytosis of GC (10:1 MOI) was significantly less than that after ingestion of OPZ (10:1 MOI), both at 2 and 6 hours, as was the percent of Annexin V+/PI+ hPMN. (B) DNA fragmentation, another marker of PICD, was decreased in hPMN fed GC (10:1 MOI) compared to that of OPZ (10:1 MOI) at 6 hours. Bars represent the average ± SEM from at least three independent experiments. p-values were determined using a two-way ANOVA and Turkey’s posttest (** p < 0.005; * p < 0.05).
Although these results suggest that the ingestion of GC delayed the appearance of features associated with the accelerated apoptosis that typically follows phagocytosis, we sought to confirm these changes by an independent means to assess apoptotic cell death and used the TUNEL assay to measure DNA fragmentation, another parameter of PICD (Figure 1B). At 6 hours after phagocytosis, the percentage of apoptotic hPMN, quantified as a percentage of DNA binding to BrdU, was nearly three times higher in cells that ingested OPZ than that in hPMN fed GC. Taken together, independent measures of cell death demonstrated that phenotypic changes associated with PICD in hPMN in suspension appeared later after ingestion of GC than after phagocytosis of OPZ. These findings extend prior observations that ingestion of N. gonorrhoeae delayed apoptosis of adherent hPMN [12–14].
Respiratory burst of hPMN:
Reactive oxygen species produced by the NADPH oxidase of stimulated hPMN have been implicated as stimuli that initiate or accelerate apoptosis [20, 21]. With that in mind, we used LUM-CL to determine if the relative oxidase activity in hPMN fed GC or OPZ contributed to the observed differences in cell death. As a positive control for activation of the NADPH oxidase, we used the soluble stimulus phorbol myristate acetate (PMA) in parallel with OPZ and GC. Both the peak and the maximum chemiluminescence of OPZ-fed hPMN were greater than responses in buffer alone but less than that of PMA-stimulated hPMN (Figure 2A). In response to GC, hPMN produced ~1.5 to 2-fold greater chemiluminescence than did PMA-stimulated hPMN, and the response was dose-dependent, with more RLU generated at 50:1 MOI than at 10:1 (Figure 2A). Stimulation in the presence of DPI decreased NADPH oxidase activity, as anticipated (Supplement Figure 2), and resulted in a decrease in the percentage of Annexin V+/PI− and Annexin V+/PI+ hPMN fed OPZ (Figure 2B). DPI inhibition of NADPH oxidase activity decreased also the percent of Annexin V+/PI+ in GC-laden hPMN (Figure 2B). Although the decrease in Annexin V staining was not statistically significant at lower MOI (10:1) of GC, the inhibitory effect of DPI at higher MOI (50:1) of GC was significant. Taken together, these data suggest that NADPH oxidase activity was necessary for externalization of PS on the surface of hPMN fed OPZ or GC.
Figure 2. NADPH oxidase activity and hPMN cell death.
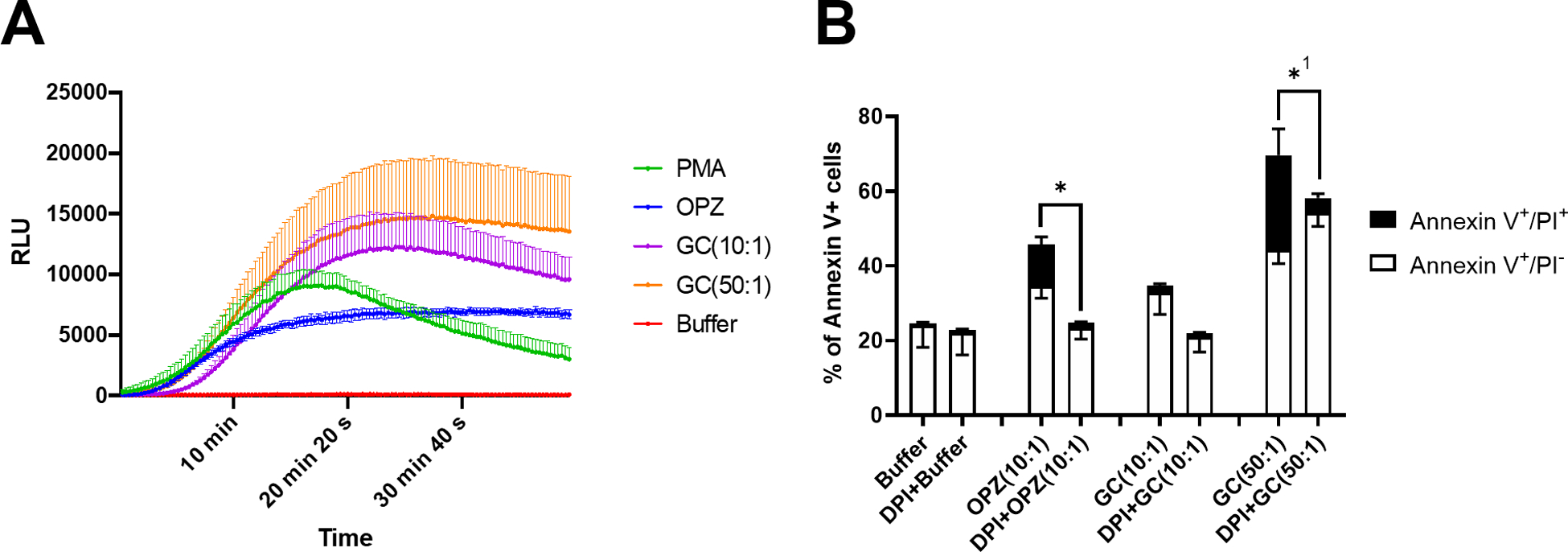
(A) The NADPH oxidase activity of hPMN was assessed by monitoring luminol-enhanced chemiluminescence of stimulated cells. The soluble agonist PMA was used as a positive control for oxidase activity. Both OPZ (10:1 MOI) and GC triggered oxidant production and the response to GC was dose dependent. (B) At 2 hours after phagocytosis in DPI-treated hPMN, the percent of Annexin V+/PI− and Annexin V+/PI+ cells in OPZ-fed hPMN were less than those fed OPZ without DPI treatment. Although DPI treatment did not significantly reduce Annexin or PI positivity of hPMN fed GC at 10:1 MOI, reduction in Annexin V+/PI+ cells was statistically significant at 50:1 MOI GC. Bars represent the average ± SEM from three independent experiments. p-values were determined using a two-way ANOVA and Turkey’s posttest (* p < 0.05). 1 denotes statistical significance in Annexin V+/PI+ population, and not in Annexin V+/PI−.
Caspase 3/7 Activity:
Apoptosis in hPMN culminates in activation of caspase 3, the executioner caspase [9]. To determine the contribution of caspase 3 to the differential fates of hPMN after phagocytosis of GC versus OPZ, we used a luminescence-based assay to quantify cleavage of DEVD as a measure of caspase 3/7 activity (Figure 3). Phagocytosis of OPZ stimulated caspase 3/7 activity in hPMN that was > 2-fold that in hPMN incubated in buffer alone for 6 hours (Figure 3A). In contrast, hPMN fed GC at the same MOI as used with OPZ challenge (10:1) exhibited less caspase 3/7 activity than did OPZ-laden hPMN or hPMN in buffer alone (Figure 3A).
Figure 3. Caspase 3/7 activity in hPMN after phagocytosis.
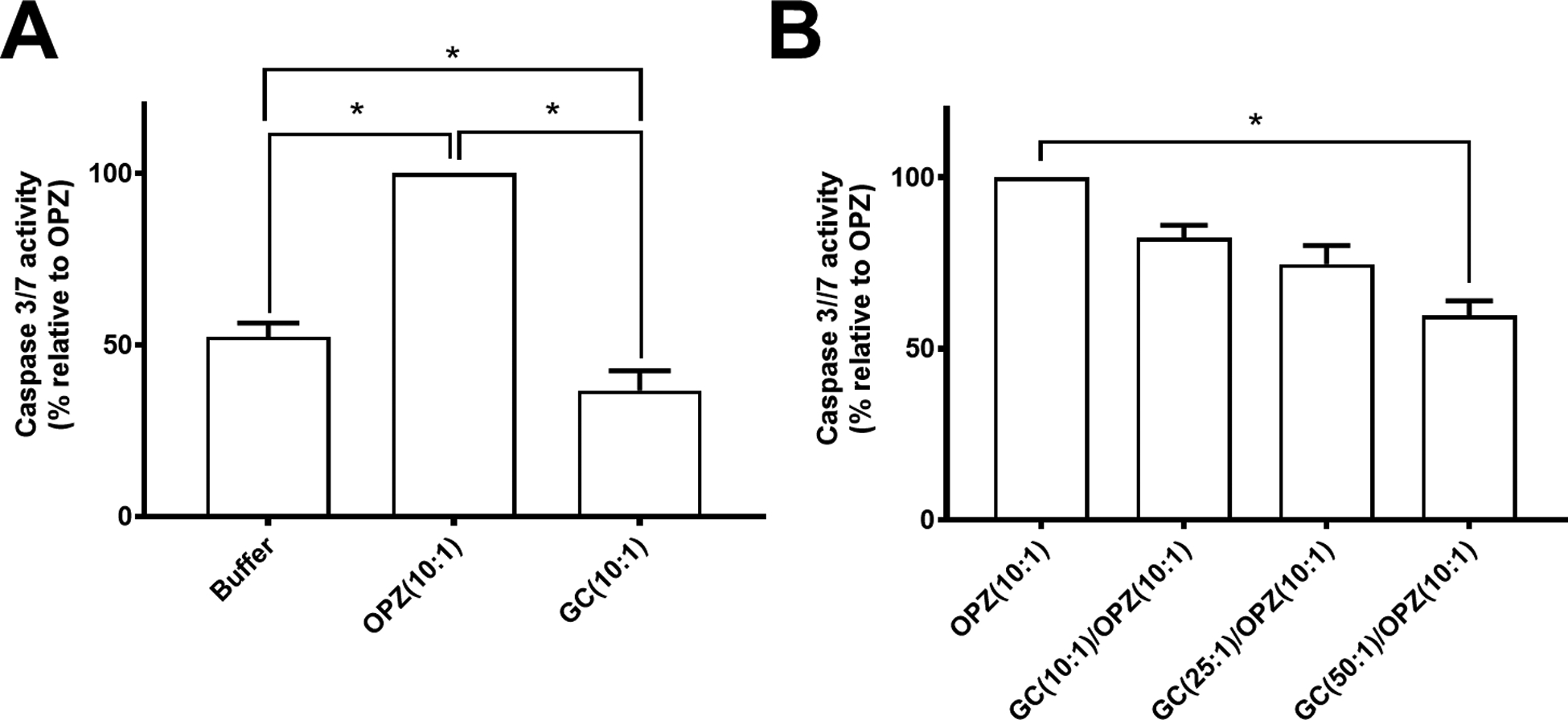
(A) At 6 hours after phagocytosis, the activity of caspase 3/7 in hPMN fed GC was decreased compared to that of hPMN fed OPZ as well as to hPMN in buffer alone. (B) hPMN sequentially fed GC followed by OPZ had a GC dose-dependent decrease in caspase 3/7 activity compared to that of hPMN fed OPZ alone. Bars represent the average ± SEM from three independent experiments. p-values were determined using a one-way ANOVA and Turkey’s posttest (* p < 0.05).
The observed difference in caspase 3/7 activity after phagocytosis of GC or OPZ could reflect a failure of GC ingestion to activate the cell death pathways to the same extent as did OPZ or suggest that the uptake of GC actively inhibited the phagocytosis-induced signaling that culminates in accelerated apoptosis. To explore which of these alternative mechanisms was more likely, we measured caspase 3/7 activity of hPMN fed OPZ alone or after sequential ingestion of GC and OPZ (Figure 3B), controlling for the total duration of phagocytosis (i.e. 30 min). Caspase 3/7 activity of hPMN that underwent sequential phagocytosis of GC followed by OPZ was less than that of hPMN fed OPZ alone (Figure 3B). Furthermore, as the MOI of GC increased, there was a dose-dependent decrease in the caspase 3/7 activity in hPMN subsequently fed OPZ. Increasing the MOI of GC fed alone to hPMN did not have similar dose-dependent effect on caspase 3/7 activity (Supplement Figure 3A), despite increased ingestion at higher MOI (Supplement Figure 3B). Buffer conditioned by GC (cBuffer) did not alter OPZ-triggered caspase 3/7 activity (Supplement Figure 3C) or affect hPMN phagocytosis (Supplement Figure 3D), thus excluding contributions from soluble factors released by hPMN fed GC. Taken together, these data suggest that GC modulated hPMN PICD by inhibiting the activation of caspase 3/7 that typically accompanies phagocytosis and accelerates apoptosis.
Caspase 8 and 9 activity:
As the executioner caspase, caspase 3 is downstream of caspases 8 and 9, intermediate effector proteins in the extrinsic and intrinsic pathways, respectively [9]. To determine if phagocytosis of GC altered one or more signaling pathways upstream of caspase 3, we measured activities of caspase 8 and 9 in hPMN fed OPZ, GC or both presented sequentially (Figure 4). Phagocytosis of OPZ, GC or both in a sequential manner increased caspase 8 activity in hPMN compared to that in hPMN in buffer alone (Figure 4A). In contrast to caspase 3/7 activity (Figure 3B), caspase 8 activity did not decrease after sequential phagocytosis of GC followed by OPZ (Figure 4A). Activity of caspase 9, however, mirrored that of caspase 3/7 under the same experimental conditions. Phagocytosis of OPZ stimulated activity of caspase 9 that was ~2-fold that of hPMN in buffer alone (Figure 4B), whereas hPMN that had ingested GC had less caspase 9 activity than did control hPMN or hPMN fed OPZ (Figure 4B). Furthermore, sequential ingestion of GC before OPZ triggered less caspase 9 activity than did OPZ alone, paralleling the effect of sequential phagocytosis on caspase 3/7 activity (Figure 3B). Taken together, these data suggest that GC acted on the intrinsic pathway for apoptosis, inhibiting caspase 9 and the downstream activation of caspase 3/7 and thereby delaying PICD.
Figure 4. Caspase 8 and 9 activities in hPMN after phagocytosis.
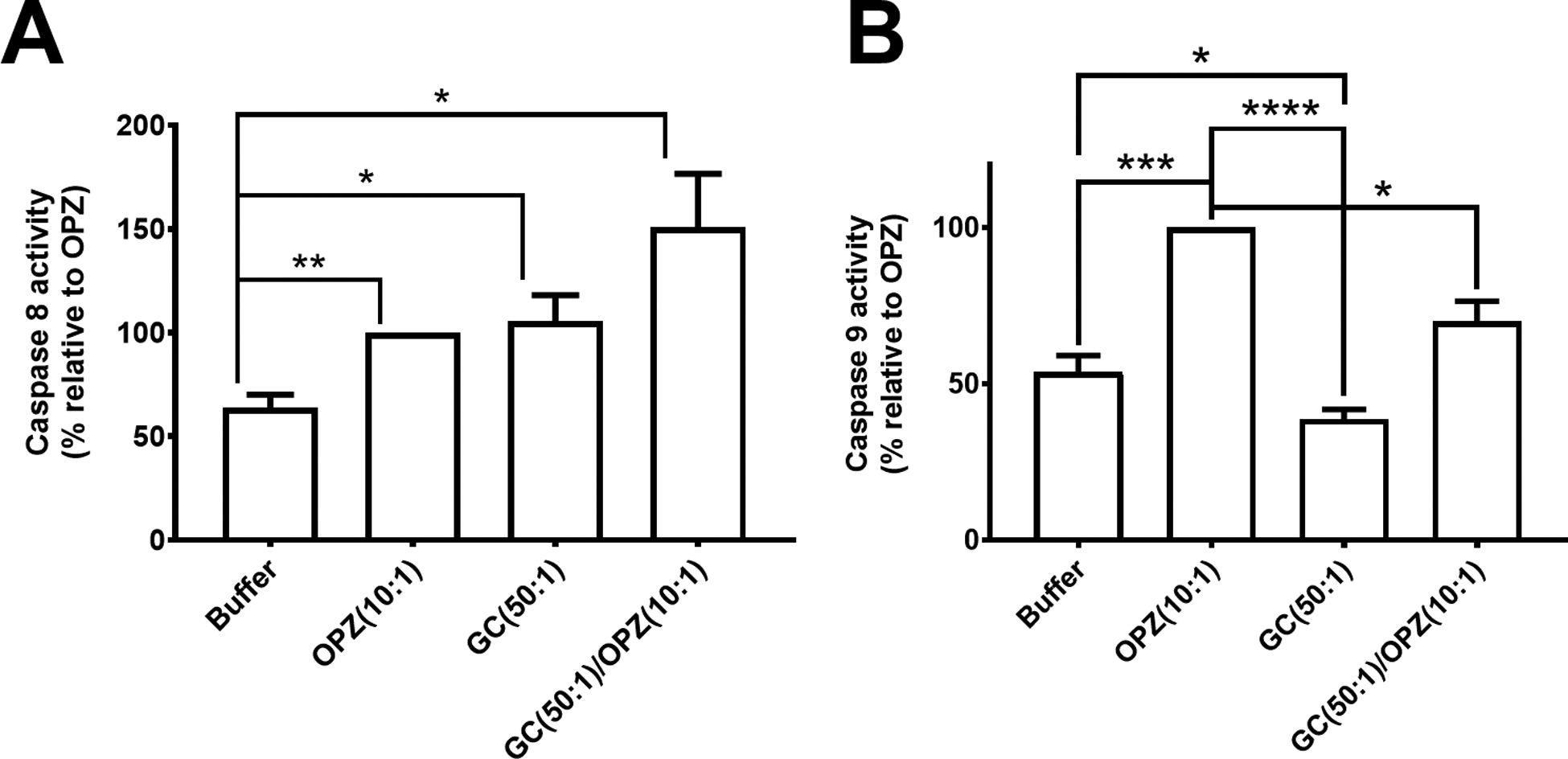
(A) Caspase 8 activity in hPMN fed OPZ, GC or both (sequentially, GC followed by OPZ) was greater than that of hPMN in buffer alone. (B) In contrast, activity of caspase 9 in hPMN fed GC was lower than that of OPZ-laden hPMN or hPMN in buffer alone. Sequential ingestion of GC followed by OPZ triggered less caspase 9 activity in hPMN than did ingestion of OPZ alone. Bars represent the average ± SEM from three independent experiments. p-values were determined using a one-way ANOVA and Turkey’s posttest (**** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05).
NADPH oxidase activity and caspase 3/7 activity:
Given the published evidence [22] and our data (Figure 2) linking oxidant production with changes in cell death signaling pathways of hPMN fed GC, we explored how the NADPH oxidase contributes to changes in caspase activity after phagocytosis. Since sequential phagocytosis of GC followed by OPZ demonstrated decreased caspase 3/7 and caspase 9 activity compared to that after ingestion of OPZ alone (Figure 3 and 4B, respectively), we measured NADPH oxidase activity under the same conditions. We quantitated extracellular superoxide anion production as a measure of NADPH oxidase activity because the addition of OPZ to hPMN fed GC quenched LUM-CL under our experimental conditions (data not shown). Extracellular superoxide anion generated by GC-fed hPMN, even at as high as 50:1 MOI, was less than that of hPMN stimulated by OPZ at 10:1 MOI (Figure 5). Sequential phagocytosis of GC followed by OPZ did not significantly decrease extracellular generation of superoxide anion by hPMN compared to that by hPMN fed OPZ alone (Figure 5). These data demonstrate GC did not inhibit subsequent OPZ-triggered extracellular superoxide anion generation by hPMN.
Figure 5. Extracellular superoxide anion generation in hPMN after phagocytosis.
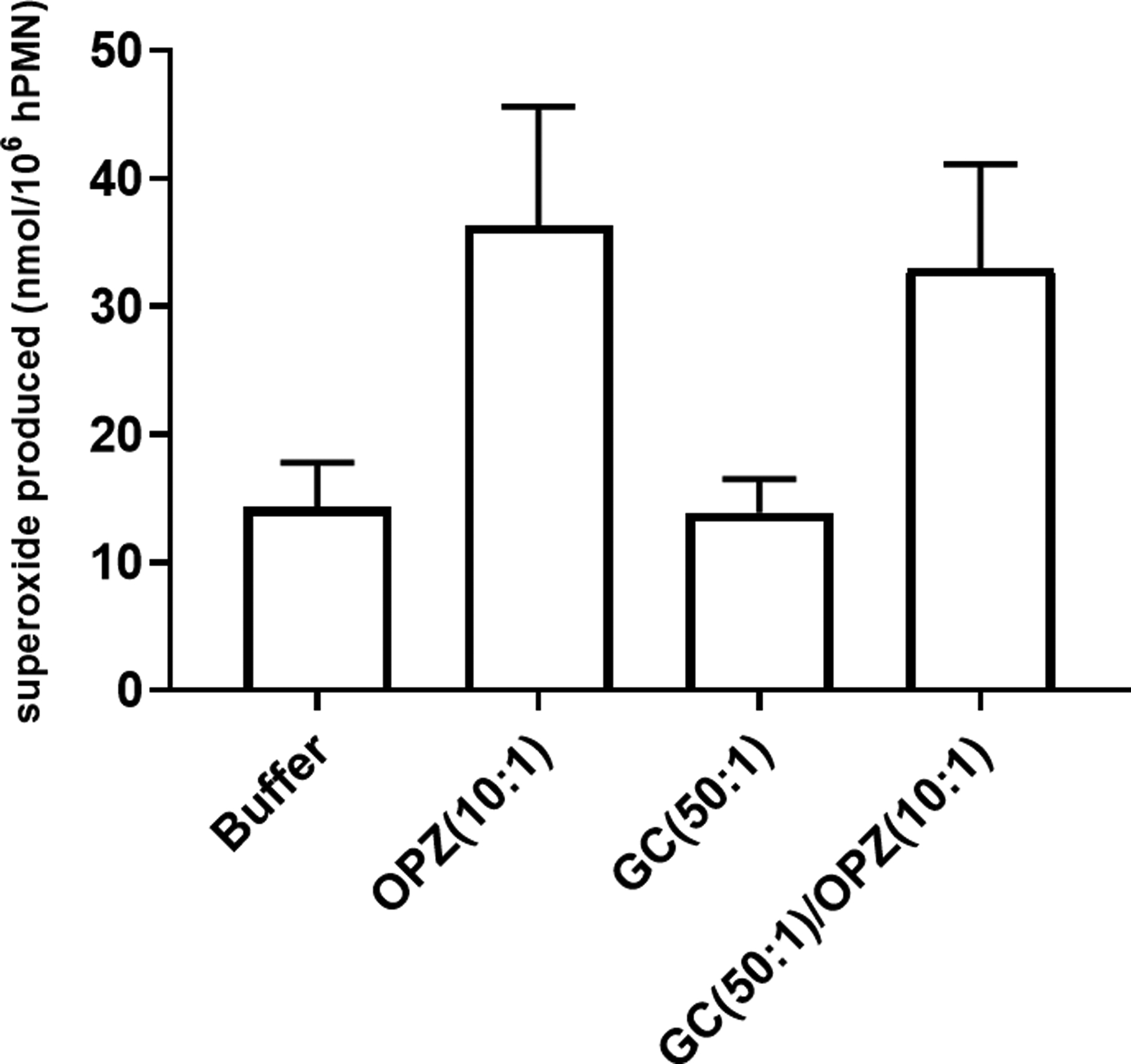
Extracellular superoxide anion generated by hPMN was measured as SOD-inhibitable reduction of ferricytochrome C. GC (50:1 MOI) triggered little extracellular superoxide production by hPMN, and sequential phagocytosis of GC then OPZ did not decrease the amount of extracellular superoxide anion produced by hPMN. Bars represent the average ± SEM from three independent experiments.
To determine if oxidant production after phagocytosis was linked to caspase 3/7 activity, we measured caspase 3/7 after phagocytosis of GC, OPZ, or both sequentially by hPMN in the presence of DPI, a flavoprotein inhibitor that blocks NADPH oxidase activity [23]. With the exception of hPMN fed GC at 50:1 MOI, the caspase 3/7 activity was significantly greater (1.4- to 2.6-fold) in hPMN in the presence of DPI (Figure 6A). Furthermore, the relative decrease in caspase 3/7 activity in hPMN fed GC and OPZ sequentially compared to that in hPMN fed OPZ alone was the same in the absence or presence of DPI (Figure 6B). Taken together, these data demonstrate that the capacity of GC to inhibit OPZ-driven caspase 3/7 activity was independent of the NADPH oxidase.
Figure 6. Effect of NADPH oxidase on caspase 3/7 activity in hPMN.
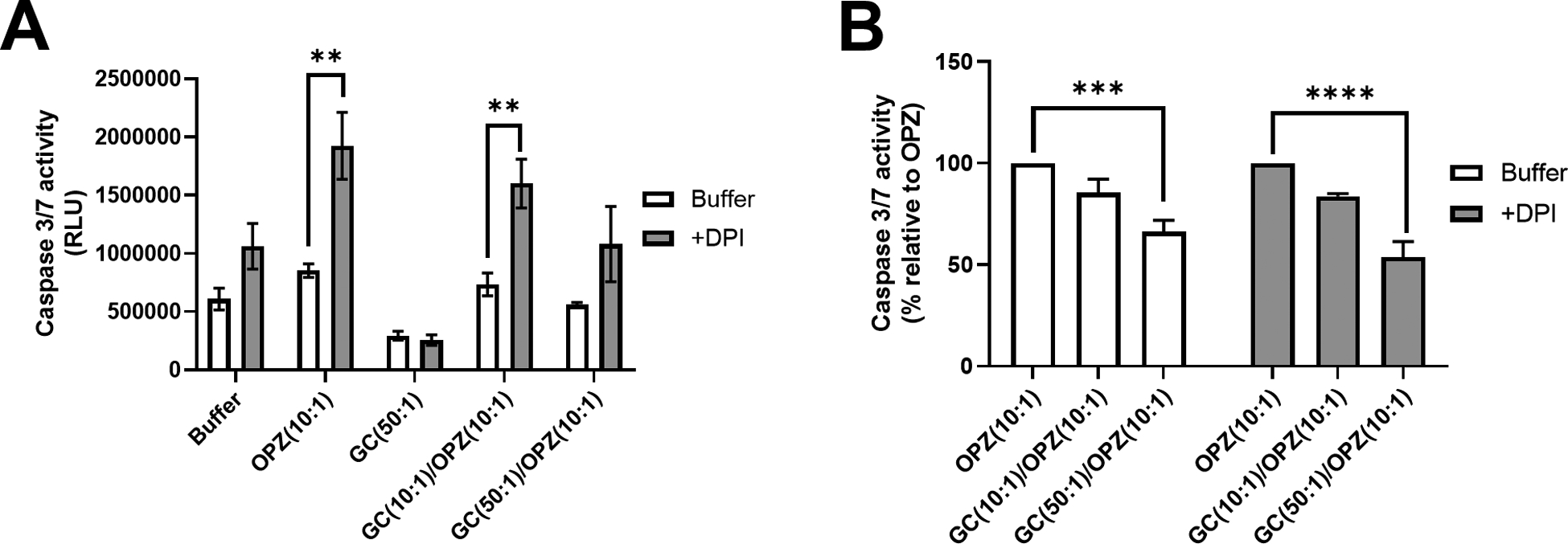
hPMN in the absence or presence of DPI were stimulated with GC, OPZ or GC followed by OPZ, and caspase 3/7 activity was measured 6 hours after phagocytosis. (A) Inhibition of NADPH oxidase activity with DPI increased caspase 3/7 activity in hPMN independent of the stimulus and even in buffer alone, with the exception of hPMN fed GC at 50:1 MOI. (B) hPMN fed sequentially GC and OPZ had less caspase 3/7 activity than did hPMN fed OPZ alone. The inhibition was in a GC dose-dependent manner and the same in the absence or presence of DPI. Bars represent the average ± SEM from three independent experiments. p-values were determined using a two-way ANOVA and Sidak’s posttest (**** p < 0.0001; *** p < 0.001; ** p < 0.01).
Mitochondrial depolarization after sequential phagocytosis of GC and OPZ:
Mitochondrial depolarization precedes caspase 9 activation in the intrinsic pathway of apoptosis [9]. We measured the shift in fluorescence of JC-1 to assess mitochondrial depolarization in hPMN after phagocytosis of OPZ, GC, or both. At 1 hour after phagocytosis, mitochondrial depolarization of hPMN fed GC at 10:1 MOI was less than that of OPZ at 10:1 MOI but increased in a dose-dependent fashion with the MOI of GC. (Figure 7). In contrast to the effect of sequential phagocytosis on activity of caspase 3/7 (Figure 3B) and 9 (Figure 4B), ingestion of GC, even at 50:1 MOI, did not reduce mitochondrial depolarization of hPMN subsequently fed OPZ (Figure 7). These data suggest that GC delayed OPZ-triggered PICD of hPMN by reducing activity of the intrinsic pathway downstream of determinants of mitochondrial depolarization.
Figure 7. Mitochondrial depolarization in hPMN after phagocytosis.
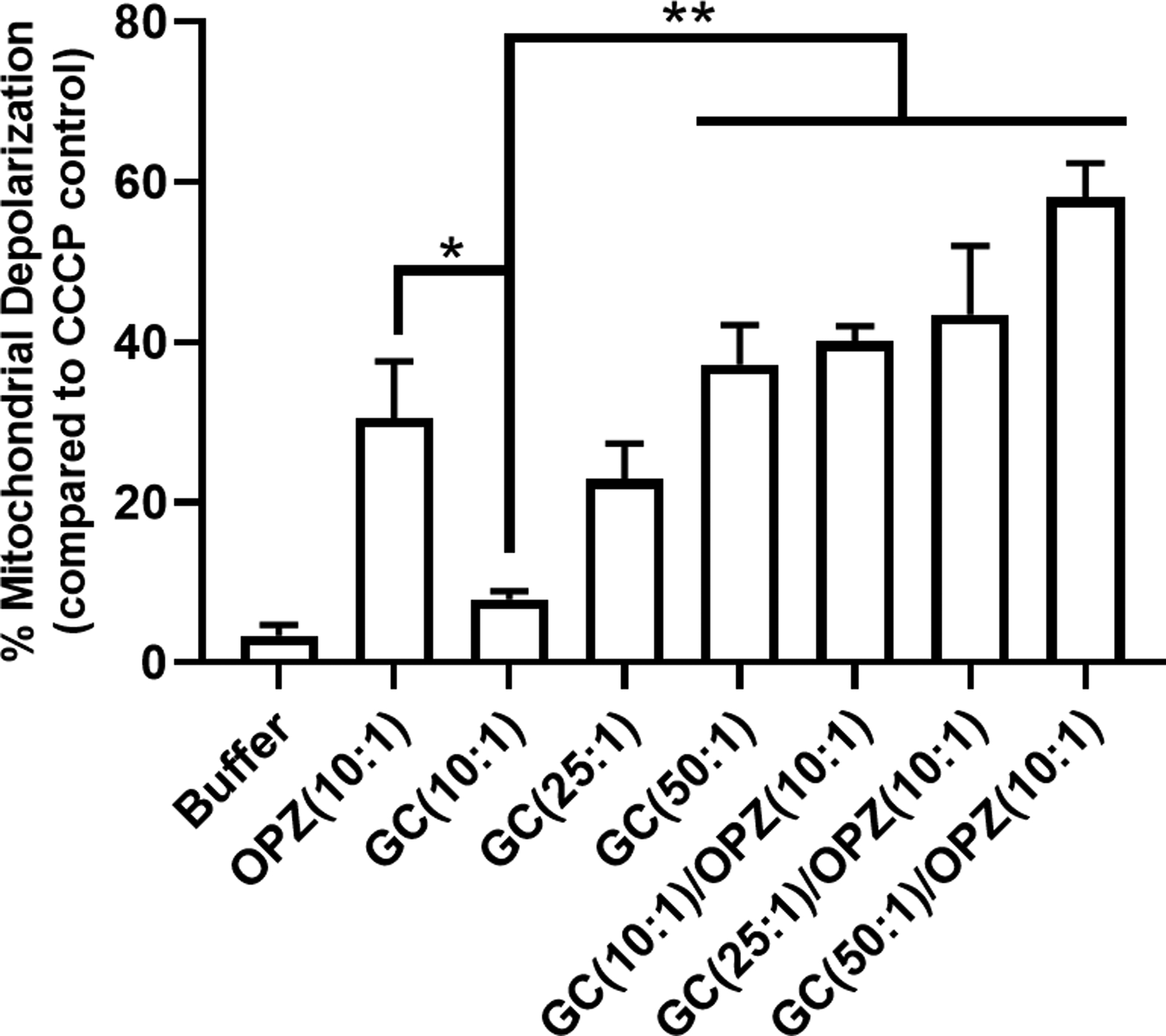
Mitochondrial depolarization was measured fluorometrically after staining with JC-1. At 1 hour after phagocytosis, mitochondrial depolarization in hPMN fed GC at 10:1 MOI was decreased compared to that of OPZ. Phagocytosis of GC at higher MOI led to increased mitochondrial depolarization in hPMN in a MOI-dependent manner and sequential phagocytosis of GC followed by OPZ did not decrease mitochondrial depolarization of hPMN compared to that in OPZ alone. Bars represent the average ± SEM from three independent experiments. p-values were determined using a one-way ANOVA and Turkey’s posttest (** p < 0.01; * p < 0.05).
Effect of sequential phagocytosis of GC and OPZ on PICD:
The ingestion of GC before phagocytosis of OPZ triggered less activity of caspases 9 and 3/7 than did uptake of OPZ alone (Figure 4B and 3B, respectively). We anticipated that GC-driven inhibition of caspases 9 and 3/7 would be manifested in overall cell fate, as measured by Annexin V-FITC/PI-PE staining (Figure 1). To test this expectation, we measured Annexin V-FITC/PI-PE of hPMN after phagocytosis of OPZ, GC, or both to assess cell fate. At 2 hours after phagocytosis, the percentage of Annexin V+/PI− cells was similar in hPMN alone and hPMN fed GC at 10:1 MOI but less than that of hPMN fed OPZ (Figure 8, inset A). Similarly, the fraction of Annexin V+/PI+ hPMN was decreased both in hPMN alone and in those fed GC at 10:1 MOI compared to hPMN fed OPZ alone (Figure 8, inset B). Despite the decreased activity of caspase 9 and 3/7, hPMN that ingested GC at a higher MOI (50:1) had more Annexin V+/PI− hPMN than did those fed GC at a lower MOI (10:1) as well as hPMN alone (Figure 8, inset A). Sequential phagocytosis of GC at 10:1 MOI and OPZ resulted in increased percent of Annexin V+/PI− and Annexin V+/PI+ cells compared to those fed GC at 10:1 MOI alone, but were similar to those fed OPZ alone (Figure 8, inset A and B). The percent of Annexin V+/PI+ cells was higher in hPMN that sequentially phagocytosed GC at 50:1 MOI and OPZ than those that ingested OPZ alone or GC alone at 50:1 MOI (Figure 8, inset B).
Figure 8. Effect of sequential phagocytosis of GC followed by OPZ on hPMN PICD.
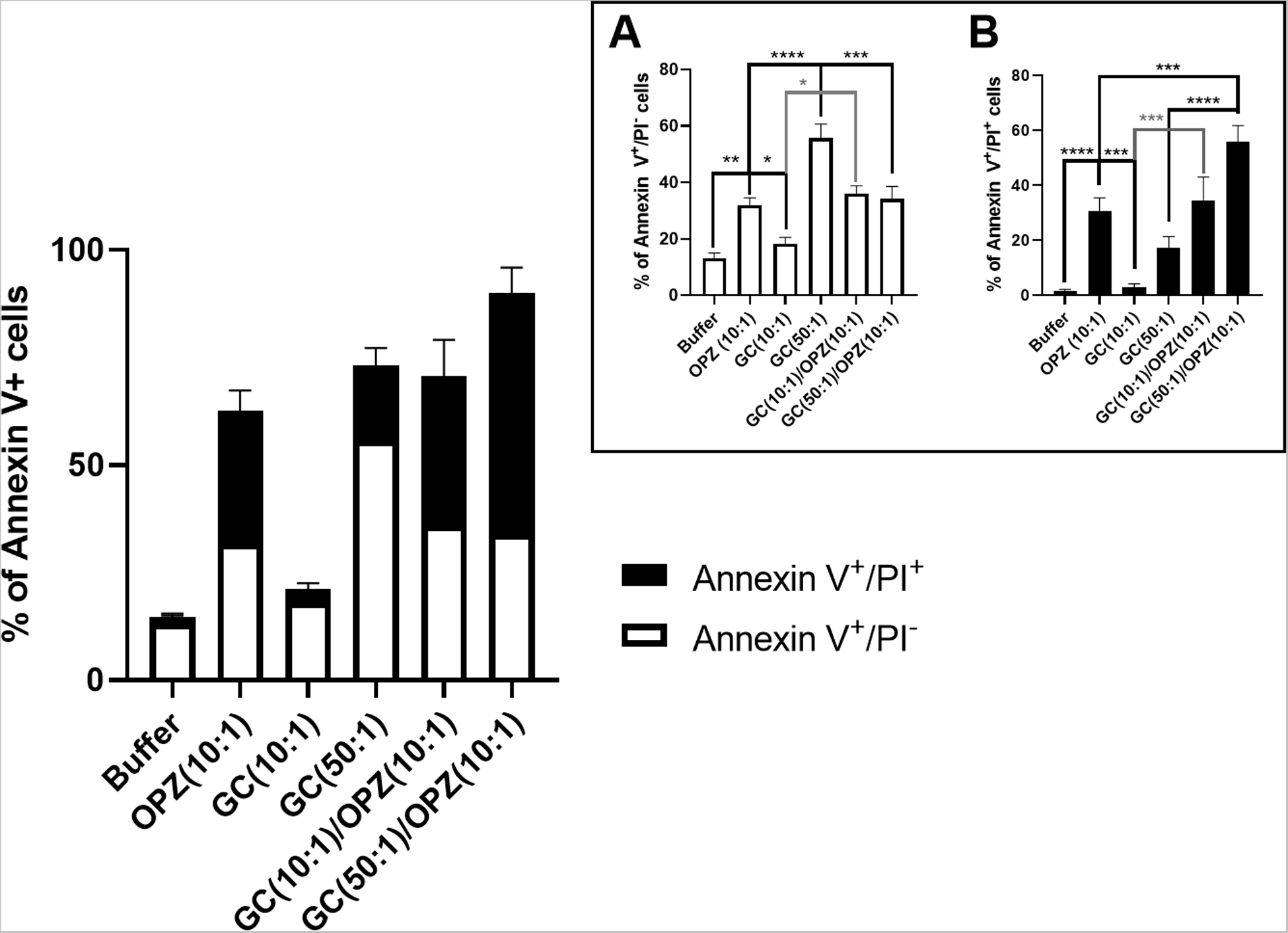
hPMN PICD 2 hours after phagocytosis was assessed as PS externalization and plasma membrane permeabilization, measured with Annexin V and PI, respectively. The percent of Annexin V+/PI− hPMN in buffer alone was similar to that after phagocytosis of GC at 10:1 MOI, but was less than that of hPMN fed GC at 50:1 MOI (inset A) Sequential ingestion of GC, either at 10:1 or 50:1 MOI, followed by OPZ led to fewer Annexin V+/PI− cells than when hPMN were fed GC at 50:1 alone but similar to after ingestion of OPZ alone (inset A). Similar to the previous data, hPMN fed OPZ had more late-stage PICD cells (Annexin V+/PI+) than did hPMN in buffer alone or hPMN fed GC at 10:1 MOI (inset B). The percent of Annexin V+/PI+ cells was greater after sequential ingestion of GC (50:1) and OPZ (10:1) than in hPMN fed OPZ or GC alone (inset B). hPMN fed GC at 10:1 MOI followed by OPZ had a percent of Annexin V+/PI+ hPMN similar to that after ingestion of OPZ alone but higher than hPMN that ingested GC alone (inset B). Bars represent the average ± SEM from at least three independent experiments. p-values were determined using a one-way ANOVA and Turkey’s posttest (**** p < 0.0001; *** p < 0.001; ** p < 0.01; * p < 0.05).
Taken together, these data suggest that despite the decreased activities of caspase 9 and 3/7 after hPMN ingested GC before OPZ, sequential phagocytosis of GC and OPZ did not block PICD, as defined as Annexin V+/PI− staining, compared to responses of hPMN fed OPZ alone. Furthermore, sequential ingestion of GC at 50:1 MOI and OPZ led to more Annexin V+/PI+ hPMN compared to the uptake of OPZ alone, despite inhibition of caspase 9 and 3/7 by GC as shown previously. Collectively, these data suggest that ingestion of GC did not decrease all signaling pathways relevant to the changes associated with cell death and that cellular responses other than caspase activity contributed to Annexin V/PI staining under these conditions.
Discussion
Gonococcal infection typically elicits a robust influx of neutrophils, and the complicated interplay between gonococci and hPMN has been the subject of extensive investigation. Seminal studies have elucidated many of the tactics that N. gonorrhoeae employs to thwart the antimicrobial activities of hPMN, including interruption of phagosome maturation, thereby limiting recruitment of antimicrobial proteins housed in azurophilic granules, and blocking the translocation of cytosolic oxidase subunits p47phox, p67phox, and p40phox and assembly of a functional NADPH oxidase [reviewed in [12, 24]]. Less explored however have been the effects of ingested N. gonorrhoeae on the cell death pathways of hPMN.
Using chemically induced HL-60 cells, a human promyelocytic cell line, as a model of neutrophil-like cells, Chen and Seifert assessed how opa− N. gonorrhoeae influenced spontaneous, staurosporine-, and TRAIL-induced apoptosis [22]. The single set of experiments in that report using hPMN demonstrates that N. gonorrhoeae (50:1 MOI) inhibits spontaneous DNA fragmentation but not that due to staurosporine, which triggers apoptosis through the intrinsic pathway. However, N. gonorrhoeae inhibits both spontaneous and staurosporine-triggered caspase 3 activity. Taken together, these data indicate that opa− N. gonorrhoeae inhibits the intrinsic pathway of caspase 3 activation by staurosporine without altering DNA fragmentation. Previous studies that focused on hPMN alone examined adherent cells [13, 14, 25, 26] and demonstrated that N. gonorrhoeae remains viable after phagocytosis and replicates intracellularly despite stimulation of oxidant generation by hPMN. When compared with changes seen following ingestion of OPZ, N. gonorrhoeae-stimulated adherent hPMN display delayed DNA fragmentation; depressed activity of caspases 3, 7, and 9; and sustained activity of inhibitors of apoptosis, cIAP-2 and X-IAP [14].
We explored how the ingestion of N. gonorrhoeae by hPMN in suspension might derail PICD, the accelerated apoptosis triggered by phagocytosis. Because several functional parameters of adherent and suspended hPMN differ [27, 28], we compared the fate of hPMN in suspension that were fed GC or OPZ, monitoring PS externalization, plasma membrane permeability, and DNA fragmentation as indicators of PICD (Figure 1). By all three independent measures of apoptosis, ingestion of GC delayed PICD of hPMN in suspension, thus validating our use of the experimental model to determine how GC delays hPMN PICD.
Focusing our attention first on caspase 3, the executioner caspase of apoptosis [29], we found that. caspase 3/7 activity in hPMN fed GC was less than that of hPMN fed OPZ and similar to the baseline caspase 3/7 activity of resting hPMN in buffer alone (Figure 3A). Although higher MOI of GC increased both the percent of hPMN that ingested GC and the number of GC per hPMN, caspase 3/7 activity remained the same (Supplement Figure 3), indicating that inadequate activation by GC did not fully account for the observed delay in PICD and that GC instead might exert an active inhibition of PICD. Consistent with the presence of active inhibition of PICD by GC, the phagocytosis of GC muted subsequent OPZ-triggered activation of caspase 3/7 and did so in a dose-dependent fashion (Figure 3B). The inhibition of OPZ-dependent caspase 3/7 activity did not reflect negative effects of GC on the uptake of OPZ, since ingestion of GC did not compromise subsequent phagocytosis of OPZ (Supplement Figure 4) and buffer conditioned by viable GC had no effect on OPZ-triggered caspase 3/7 activity (Supplement Figure 3). Taken together with observations that N. gonorrhoeae decreases spontaneous and staurosporine-induced caspase 3 activity in hPMN [22], these data suggest that binding or phagocytosis of GC likewise can inhibit caspase 3/7 activity in PICD..
To determine whether GC modified cell death pathways upstream of caspase 3/7, we measured the activities of caspase 8 and 9 in hPMN fed either GC alone or GC followed by OPZ. Caspase 9 activity in hPMN fed GC was less than that in hPMN suspended in buffer alone or after phagocytosis of OPZ (Figure 4B). In contrast, the effects of GC and OPZ on caspase 8 activity appeared to be additive (Figure 4A). Taken together, these data suggest that the target for inhibitory effects of GC was in the intrinsic pathway, affecting both caspase 9 and 3/7.
Substantial data link NADPH oxidase activity and signaling of apoptosis and cell death pathways in hPMN, with reactive oxygen species promoting apoptosis in most experimental settings, including PICD [reviewed in [30]]. Pharmacologic inhibition of the NADPH oxidase with DPI blocks the apoptosis induced by β2-integrin-dependent phagocytosis, and neutrophils from patients with CGD, which lack a functional oxidase, likewise fail to exhibit an apoptotic morphology or DNA fragmentation after β2-integrin-dependent phagocytosis [31]. Although caspase activation accompanies the phenotypic changes of apoptosis, including Annexin V staining and DNA fragmentation, its relationship with oxidant generation is the opposite; oxidants generated by the NADPH oxidase reduce caspase activity. For example, caspase activity in hPMN stimulated with PMA can be detected only when NADPH oxidase activity is inhibited by DPI [20]. Caspases are cysteine proteases and consequently have in their active site critical cysteine residues, which are highly susceptible to oxidation [reviewed in [32]], particularly when oxidative stress is high. In a sense, the redox state of hPMN, whether at rest or after stimulation, dictates in large part what signaling pathways contribute to cell death, and, when oxidase activity is high, oxidants produced by the phagocyte oxidase can mediate oxidative inactivation of caspases.
Our data demonstrate that DPI-inhibition of NADPH oxidase activity reduced Annexin V/PI staining triggered by OPZ or GC (Figure 2B), suggesting that externalization of PS after phagocytosis required a functional NADPH oxidase, consistent with earlier studies of β2-integrin-dependent phagocytosis [31]. DPI treatment of hPMN increased caspase 3/7 activity in all but one of the experimental conditions (i.e. GC at 50:1 MOI), even in hPMN without agonist exposure and undergoing spontaneous apoptosis (Figure 6A). Furthermore, the relative caspase 3/7 activity in hPMN fed GC followed by OPZ was less than that in hPMN fed OPZ alone, in the absence or presence of DPI (Figure 6B), thus indicating that the GC-mediated inhibition of caspase 3/7 activity in OPZ-stimulated hPMN was independent of effects on NADPH oxidase activity.
The discordant effects of sequential phagocytosis of GC and OPZ on caspase 9 and 3/7 activation on the one hand, and changes in mitochondrial depolarization and Annexin V/PI staining on the other, underscore the complexity and interactive nature of signaling in cell death pathways in hPMN [33]. Despite GC-dependent inhibition of OPZ-triggered caspase 9 and caspase 3/7 activation, hPMN fed GC and OPZ sequentially increased Annexin V binding to the same extent as did hPMN that phagocytosed only OPZ (Figure 8). Although it is likely that the large stimulation imposed by ingestion of both GC and OPZ would prompt extensive degranulation and possibly promote increased phospholipid flipping and surface exposure of PS independent of engaging cell death pathways, as described for fMLF-stimulated neutrophils [34], such plasma membrane remodeling would not explain the increased mitochondrial depolarization seen with increased ingestion of GC (Figure 7). Chen and Seifert likewise found that the effects of N. gonorrhoeae on DNA fragmentation and staurosporine-induced caspase 3 activity, a reflection of intrinsic pathway engagement, diverge; caspase 3 activity reduces whereas DNA fragmentation does not [22].
The impact of phagocytosis of N. gonorrhoeae on aspects of PICD in hPMN was dose-dependent, as has been previously reported in other systems [35, 36]. Whereas challenge with low MOI of GC before ingestion of OPZ failed to activate caspase 3/7, depolarize mitochondria, or elicit increased binding of Annexin V, the higher MOI of GC promoted caspase 8 activity, binding of Annexin V, and mitochondrial changes consistent with PICD despite the absence of active caspase 3/7. We interpret these findings as evidence that at low MOI, GC inhibited the intrinsic pathway distal to mitochondrial permeabilization but upstream of caspases 9 and 3/7. At high MOI, GC activated sufficient caspase 8 to override any inhibitory influence on the intrinsic pathway and to drive PICD, perhaps through activation of caspase 6, which like caspases 3 and 7 serves as executioner caspase [37, 38].
In summary, PICD represents an important step in the cascade of cellular responses that culminate in resolution of the inflammatory response, and microbial sabotage of PICD causes sustained proinflammatory events that promote tissue damage in certain infections, including those due to N. gonorrhoeae. Along with published reports, these data demonstrate that N. gonorrhoeae does not influence all apoptotic signaling pathways equally but succeeds in inhibiting activation of the intrinsic pathway of apoptosis and delaying PICD of hPMN. Many questions remain unanswered: what components of N. gonorrhoeae mediate the observed changes in hPMN fate? Do host sensors detect bacterial elements released in the phagosome or do microbial components or secreted products need to access cytoplasmic sensors? Additional study is needed to dissect in greater detail the interplay between the individual cell death pathways engaged following phagocytosis, and how some microbes, such as N. gonorrhoeae, derail resolution of the inflammatory response and thus exacerbate the clinical sequela of infection.
Supplementary Material
Acknowledgements
This work was supported by National Institute of Health grants AI116546 (WMN), AI132335 (WMN), AU996343 (CC) and Physician-Scientist Training Pathway at University of Iowa Carver College of Medicine (CC).
Abbreviations
- BrdU
5’-bromo-2’-deoxyuridine
- DPI
diphenylene iodonium
- dUTP
deoxyuridine triphosphate
- FITC
fluorescein isothiocyanate
- FITC-OPZ
FITC labelled OPZ
- GC
Neisseria gonorrhoeae FA1090 wild-type (opa+)
- GFP
green fluorescent protein
- GC-GFP
GC with GFP
- HH++
Hanks’ Balanced Salt Solution with calcium and magnesium with 20mM HEPES
- HH−−
Hanks’ Balanced Salt Solution without calcium and magnesium
- hPMN
human polymorphonuclear leukocytes
- kGC
gentamicin-killed GC
- kGC-GFP
kGC with GFP
- LUM-CL
luminol-enhanced chemiluminescence
- MFI
mean fluorescence intensity
- MOI
multiplicity of infection
- NADPH
nicotinamide adenine dinucleotide phosphate
- OPZ
pooled human serum opsonized zymosan
- PBS
Phosphate Buffered Saline
- PE
phycoerythrin
- PI
propidium iodide
- PICD
phagocytosis-induced cell death
- PMA
phorbol myristate acetate
- PS
phosphatidylserine
- RLU
relative light unit
- SOD
superoxide dismutase
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests
References
- 1.DeLeo FR and Nauseef WM, Granulocytic Phagocytes, in Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 2020, Elsevier; p. 83–98. [Google Scholar]
- 2.Nauseef WM, How human neutrophils kill and degrade microbes: an integrated view. Immunol Rev, 2007. 219: p. 88–102. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi SD, et al. , Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc Natl Acad Sci U S A, 2002. 99(10): p. 6901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi SD, et al. , Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci U S A, 2003. 100(19): p. 10948–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kobayashi SD and DeLeo FR, Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip Rev Syst Biol Med, 2009. 1(3): p. 309–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy AD and DeLeo FR, Neutrophil apoptosis and the resolution of infection. Immunol Res, 2009. 43(1–3): p. 25–61. [DOI] [PubMed] [Google Scholar]
- 7.Doran AC, Yurdagul A Jr., and Tabas I, Efferocytosis in health and disease. Nat Rev Immunol, 2020. 20(4): p. 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amulic B, et al. , Neutrophil function: from mechanisms to disease. Annu Rev Immunol, 2012. 30: p. 459–89. [DOI] [PubMed] [Google Scholar]
- 9.McCracken JM and Allen LA, Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death, 2014. 7: p. 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apicella MA, et al. , Modification by sialic acid of Neisseria gonorrhoeae lipooligosaccharide epitope expression in human urethral exudates: an immunoelectron microscopic analysis. J Infect Dis, 1990. 162(2): p. 506–12. [DOI] [PubMed] [Google Scholar]
- 11.Unemo M and Shafer WM, Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev, 2014. 27(3): p. 587–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palmer A and Criss AK, Gonococcal Defenses against Antimicrobial Activities of Neutrophils. Trends Microbiol, 2018. 26(12): p. 1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simons MP, Nauseef WM, and Apicella MA, Interactions of Neisseria gonorrhoeae with adherent polymorphonuclear leukocytes. Infect Immun, 2005. 73(4): p. 1971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simons MP, et al. , Neisseria gonorrhoeae delays the onset of apoptosis in polymorphonuclear leukocytes. Cell Microbiol, 2006. 8(11): p. 1780–90. [DOI] [PubMed] [Google Scholar]
- 15.Kremserova S and Nauseef WM, Frontline Science: Staphylococcus aureus promotes receptor-interacting protein kinase 3- and protease-dependent production of IL-1beta in human neutrophils. J Leukoc Biol, 2019. 105(3): p. 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlgren C and Stendahl O, Role of myeloperoxidase in luminol-dependent chemiluminescence of polymorphonuclear leukocytes. Infect Immun, 1983. 39(2): p. 736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stevens P, Winston DJ, and Van Dyke K, In vitro evaluation of opsonic and cellular granulocyte function by luminol-dependent chemiluminescence: utility in patients with severe neutropenia and cellular deficiency states. Infect Immun, 1978. 22(1): p. 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Babior BM, Kipnes RS, and Curnutte JT, Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest, 1973. 52(3): p. 741–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fox S, et al. , Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun, 2010. 2(3): p. 216–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fadeel B, et al. , Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood, 1998. 92(12): p. 4808–18. [PubMed] [Google Scholar]
- 21.Gardai S, et al. , Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J Biol Chem, 2002. 277(7): p. 5236–46. [DOI] [PubMed] [Google Scholar]
- 22.Chen A and Seifert HS, Neisseria gonorrhoeae-mediated inhibition of apoptotic signalling in polymorphonuclear leukocytes. Infect Immun, 2011. 79(11): p. 4447–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cross AR and Jones OT, The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J, 1986. 237(1): p. 111–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Criss AK and Seifert HS, A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol, 2012. 10(3): p. 178–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Criss AK, Katz BZ, and Seifert HS, Resistance of Neisseria gonorrhoeae to non-oxidative killing by adherent human polymorphonuclear leucocytes. Cell Microbiol, 2009. 11(7): p. 1074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Criss AK and Seifert HS, Neisseria gonorrhoeae suppresses the oxidative burst of human polymorphonuclear leukocytes. Cell Microbiol, 2008. 10(11): p. 2257–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greenlee-Wacker MC, et al. , Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol, 2014. 192(10): p. 4709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu T, et al. , Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J Innate Immun, 2014. 6(5): p. 639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McIlwain DR, Berger T, and Mak TW, Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol, 2013. 5(4): p. a008656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zeng MY, et al. , The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol, 2019. 34(2): p. 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coxon A, et al. , A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity, 1996. 5(6): p. 653–66. [DOI] [PubMed] [Google Scholar]
- 32.Poole LB, The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med, 2015. 80: p. 148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geering B and Simon HU, Peculiarities of cell death mechanisms in neutrophils. Cell Death & Differentiation, 2011. 18(9): p. 1457–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frasch SC, et al. , Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated met-leu-phe-stimulated neutrophils. Journal of Biological Chemistry, 2004. 279: p. 17625–17633. [DOI] [PubMed] [Google Scholar]
- 35.Watson RW, et al. , Neutrophils undergo apoptosis following ingestion of Escherichia coli. J Immunol, 1996. 156(10): p. 3986–92. [PubMed] [Google Scholar]
- 36.Zhang B, et al. , Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis. Journal of Biological Chemistry, 2003. 278: p. 28443–28454. [DOI] [PubMed] [Google Scholar]
- 37.Shalini S, et al. , Old, new and emerging functions of caspases. Cell Death Differ, 2015. 22(4): p. 526–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiewchengchol D, et al. , The protective effect of GM-CSF on serum-induced neutrophil apoptosis in juvenile systemic lupus erythematosus patients. Clin Rheumatol, 2015. 34(1): p. 85–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.