

Abstract
Lecithin:cholesterol-acyl transferase (LCAT) plays a major role in cholesterol metabolism as it is the only extracellular enzyme able to esterify cholesterol. LCAT activity is required for lipoprotein remodeling and, most specifically, for the growth and maturation of HDLs. In fact, genetic alterations affecting LCAT functionality may cause a severe reduction in plasma levels of HDL-cholesterol with important clinical consequences. Although several hypotheses were formulated, the exact molecular recognition mechanism between LCAT and HDLs is still unknown. We employed a combination of structural bioinformatics procedures to deepen the insights into the HDL-LCAT interplay that promotes LCAT activation and cholesterol esterification. We have generated a data-driven model of reconstituted HDL (rHDL) and studied the dynamics of an assembled rHDL::LCAT supramolecular complex, pinpointing the conformational changes originating from the interaction between LCAT and apolipoprotein A-I (apoA-I) that are necessary for LCAT activation. Specifically, we propose a mechanism in which the anchoring of LCAT lid to apoA-I helices allows the formation of a hydrophobic hood that expands the LCAT active site and shields it from the solvent, allowing the enzyme to process large hydrophobic substrates.
Keywords: Lecithin:cholesterol-acyl transferase (LCAT), HDL/Structure, Physical biochemistry, Cholesterol metabolism, Diseases/Dyslipidemias, Structural bioinformatics, LCAT deficiency, Molecular modeling
Abbreviations: apoA-I, apolipoprotein A-I; CE, cholesteryl ester; CV, collective variable; FC, free cholesterol; FLD, familial LCAT deficiency; LCAT, lecithin:cholesterol-acyl transferase; MD, molecular dynamics; PDB, Protein Data Bank; rHDL, reconstituted HDL; RMSD, root-mean-square deviation; RMSF, root-mean-square fluctuation; wtMTD, well-tempered metadynamics
Lecithin:cholesterol-acyl transferase (LCAT) is a 65 kDa plasmatic protein of the α/β-hydrolase family synthesized mainly in the liver and in lower amounts also in the brain, testes, and kidneys. It circulates in plasma reversibly bound to lipoproteins, where it catalyzes the esterification of free cholesterol (FC) through a two-step reaction mechanism that involves the hydrolysis of a phospholipid sn-1 or sn-2 alkyl chain and its transfer to FC. Cholesteryl esters (CEs) are then removed from the surface and accumulate within the lipoproteins core, allowing the particle to store larger amounts of cholesterol. Being the only extracellular enzyme able to catalyze cholesterol esterification, LCAT plays a crucial role in the maturation, remodeling, and function of lipoproteins (1, 2). Although LCAT is also active on (V)LDLs, HDLs, the primary effectors of reverse cholesterol transport, are its preferential substrate and their principal protein constituent, apolipoprotein A-I (apoA-I), seems to be its strongest activator. Indeed, LCAT is required for the maturation and functionality of discoidal nascent pre-β-HDLs, which grow into spherical, mature, HDLs as they incorporate CEs produced by LCAT: while FC diffuses from the membranes of peripheral cells (including arterial wall macrophages) to nascent HDLs through the membrane ABCA1 transporter, LCAT preserves the concentration gradient by removing FC from the HDL surface (3).
Although the role of HDL in the prevention of coronary heart diseases is still debated, elucidating the role of LCAT in HDL-mediated reverse cholesterol transport remains a central issue that could aid in the treatment of cardiovascular diseases (3). Mutations in the LCAT gene lead to 2 rare recessive syndromes, fish-eye disease and familial LCAT deficiency (FLD), characterized by decreasing levels in LCAT residual activity. Clinical manifestations of the disease include corneal opacity, anemia, hypoalphalipoproteinemia, and alterations in blood lipids and CE levels (4, 5). In the case of FLD, alterations in HDL maturation may result in the formation of abnormal multilamellar lipoproteins, LpX, which can cause nephropathy leading to life-threatening renal failure (6).
Although the three-dimensional structure of LCAT has been elucidated via X-ray crystallography (7, 8, 9), the complete lipid-bound structure of apoA-I is still unresolved, although several models backed up by experimental data have been proposed. Since the work of Koppaka et al. (10), the so-called double belt arrangement (opposed to the picket-fence model) has been established almost unambiguously. Further work by Segrest et al. (11) has then proposed the antiparallel LL (left-to-left) orientation of apoA-I chains with a 5/5 registry, based on computational calculations; their findings were then supported by the cross-linking/MS experiments of Silva et al. (12). Since then, other computational models have been proposed, showing significant differences albeit sharing the common configuration of double-belt in the LL/5 registry. Notably, the solar flare model proposed by Wu et al. (13), which identified, via hydrogen-deuterium exchange experiments, solvent-exposed protruding bulges in apoA-I chains (corresponding to residues 159–180) that were proposed to activate LCAT; the belt buckle model, from Bhat et al. (14), where cross-linking/MS data were collected on 145 POPC reconstituted HDL (rHDL) particles, showing that the N and C termini folded back onto apoA-I helices; the looped belt model, by Martin et al. (15), generated by performing electron paramagnetic resonance and Förster resonance energy transfer experiments on 100 POPC rHDLs, suggesting the presence of a central loop region comprised by residues 133–146. Another model was proposed by Wu et al. (16) in accordance with Förster resonance energy transfer, ESR, and cross-linking/MS data obtained by others; this model displayed apoA-I helices spiraling around a 100 POPC cylindrical core as a double superhelix; however, the reliability of this model was challenged by Jones et al. (17) who tested the thermodynamic and kinetic stability of the double superhelix via extensive coarse-grained and simulated-annealing molecular dynamics (MD) simulations.
In the present paper, we employed a combination of computational approaches to model a rHDL and analyze the structure-function relationships underlying the LCAT reaction mechanism and activation mediated by apoA-I.
Materials and methods
Computational procedures
Structure preparation
The LCAT structures 5BV7 and 5TXF were prepared using the “Structure preparation and refinement” panel of the Maestro program (Maestro, Schrödinger, LLC, New York, NY, 2018). Missing residues 236–242 of 5BV7 and 240–141 of 5TXF (lid loop) were modeled using an ab initio method available in the module Prime of the same suite; since LCAT N and C termini were proven to be necessary for LCAT activity (18, 19, 20), residues 1–20 and 399–416 of both structures were also modeled. Despite the high number of residues to be modeled ab initio, we do not expect that inaccuracies in the predicted structure of these residues could affect our MD simulations. The Protein Data Bank (PDB) codes of utilized apoA-I structures are 3R2P (21), 1AV1 (22), and CNS (23).
rHDL modeling
To generate the apoA-I all-atom model we merged structural fragments of existing templates into a chimeric structure. Template patches were selected considering the following properties: i) residues are in an alpha-helix secondary structure, ii) suitable orientation of amphipathic helices, iii) ability to confer a circular shape to the model. We used 3R2P as the main template; the first 36 residues of the missing C-terminal helices (residues 183–218) were reconstructed from the corresponding residues of the 1AV1 structure, whereas residues 219–243 were shaped on 3R2P central helices (residues 156–182). Residues 1–37 of the 3R2P folded N terminus (residues 1–79) were remodeled using residues from: 3R2P central helices (residues 156–182), a small fragment of the consensus model (CNS), and residues 44–79 of 1AV1. Structure alignment algorithms were used to correctly position residue patches using 2–5 overlapping residues as a guide. Energy-based homology modeling (Prime, Schrödinger, LLC, New York, NY, 2018) was used as a tool to model N and C termini last residues onto 3R2P residues 156–182 and to seal the final patched model into a continuous chain. A single apoA-I chain was modeled in this way; to model the homodimeric supramolecular assembly, a duplicated image of the generated chain was aligned to residues 80–182 of the antiparallel chain of 3R2P. The lipid core of the rHDL was built from an equilibrated POPC bilayer extracted from a validated MD simulation. The lipids bilayer was trimmed to a circular disk of 164 POPC molecules with a diameter of 9.2 nm and symmetrically positioned at the center of the chimeric model by aligning the coordinates of reference centers of mass. Residues within 3 Å from the phospholipidic core were minimized to avoid steric clashes (Polak-Ribiere Conjugate Gradient with convergence on RMS gradient with a threshold of 0.5).
Molecular dynamics simulations
All MD simulations were performed with the Desmond Molecular Dynamics System (D. E. Shaw Research, New York, NY, 2018; Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018) with the following base setting: timestep for close and far interactions, 300 K Nose-Hoover thermostat, Martina-Tobias-Klein barostat with isotropic coupling and a 9.0 Å cutoff for Coulombic interactions.
Folding of the apoA-I chains around the lipid core was achieved with a 100 ns restrained MD simulation to ensure the reproducibility of the simulation and consistency toward experimental structural data. Selected atom pairs were pulled together by flat-bottomed distance restraints with a force constant of 0.15 kcal mol−1 Å−1 and a distance threshold of 7.5 (± 7.5) Å. Atom pair sets were chosen considering data from cross-linking experiments (12, 16, 24, 25) (Figs. 2 and 3A). POPC residues were restrained on the Z axis with a force constant of 5 kcal mol−1 Å−1 to prevent the formation of a lipid drop that would have hindered hydrophobic interactions with apoA-I amphipathic helices. All restraints were removed after 100 ns and the MD was extended up to 300 ns.
Fig. 2.
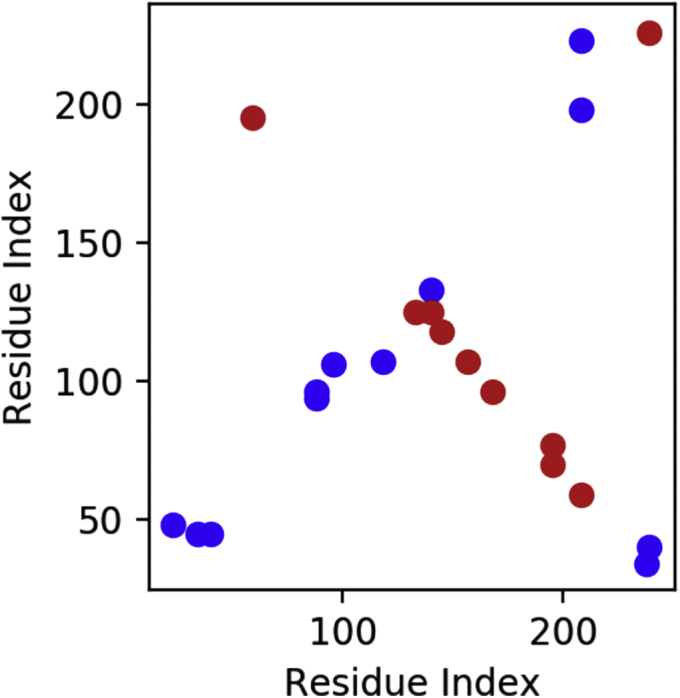
apoA-I cross link map. Inter-chain cross-links (red), intra-chain cross-links (blue).
Fig. 3.

apoA-I folding. A: 3D structure of raw apoA-I model at t = 0 ns, spheres and dashed lines represent cross-linking atoms. B: RMSD plot. C: Radius of gyration. D: RMSF plot. E: apoA-I model at t = 100 ns (before the removal of distance restraints), spheres and dashed lines represent cross-linking atoms. F: Restrained atom pairs mean distance after constraints removal. G: Model at t = 300 ns, ribbon representation is color-coded on RMSF (D).
Simulations of LCAT, LCAT::Fab, and rHDL::LCAT complexes were all carried out with default settings.
Metadynamics
A well-tempered metadynamics (wtMTD) simulation was set up to explore the energy landscape of the LCAT structure as a function of lid positioning. Two collective variables (CVs) were chosen to track the lid loop movements: 1) the dihedral angle formed by the centers of mass of the following groups: β6/β7 (214–303) and β7/αE (319–344) domains; the catalytic triad (181, 345, 377); the whole structure except the lid; the tip of the lid domain (232–238); 2) the distance between Cα of residues Met234 and Gly119. The height of the gaussian potential was set to 0.04 kcal mol−1, with a kT of 5 kcal mol−1 and a deposition rate of 0.2 ps; a σ of 2.5° and 0.25 Å was set for the 2 CVs, respectively. A 100.0 kcal mol−1 positional constraint was also applied to all Cα atoms, except for the lid loop (225–250) and the β3/αA domain (32–119), to prevent the biasing potential to affect every atom included in the CVs definition, thus ensuring values of the CVs to depend on lid positioning only. The system was prepared using the LCAT open 5TXF structure.
Abduction of a phospholipid from the surface of the rHDL toward the LCAT active site was carried out using standard MTD; settings: wall 20 Å, deposition rate 1 ps, σ of 0.1 Å, and height of the gaussian potential 0.3 kcal mol−1. Other default MD settings were kept as reported in the Molecular Dynamics paragraph.
Protein::protein docking
The PIPER FFT-based protein::protein docking program (licensed by Schrödinger, New York, NY, 2018) was used to dock LCAT on the final rHDL model. The last frame (300 ns) of the rHDL model from the MD simulation was minimized and set as the receptor, while the open- and closed-lid conformations of LCAT, obtained from the largest wtMTD minima, were set as ligands. Constraints were added to the docking protocol on the basis of experimental cross-linking and mutagenesis data: interactions with apoA-I residues 143–187 were favored by weighting the internal scoring function; at least one pair of residues observed in experimental cross-linking (26) were forced to stay within 12 Å. Poses were then analyzed and accepted or rejected on the basis of geometric considerations: i) proximity and orientation of LCAT active site toward phospholipid head groups; ii) contacts between LCAT β3/αA domain and lipids.
Results and discussion
rHDL model
Since LCAT is active mainly on the surface of HDLs, where apoA-I is the main activator, understanding the interactions between LCAT, apoA-I, and a lipid interface is crucial to picture the LCAT activation and reaction mechanism. We chose to model an rHDL as this type of lipoprotein is the best simplified system to maintain general HDL properties, allowing the cross-validation between our models and experimental results.
As experimentally resolved lipid-bound apoA-I structures are currently unavailable, we relied on lipid-free apoA-I crystallographic data (21, 22). We aligned residue patches of 3 structures, 1AV1, 3R2P, and CNS, to reconstruct a full-atom model of a lipid-free apoA-I poised to wrap around a lipidic core (Fig. 1). A mixed protocol of restrained and free MD was then used to drive the folding of the modeled apoA-I chains around the lipid core (see Methods section) (Fig. 3).
Fig. 1.

apoA-I chimeric model. A: Ribbon representation of the patched apoA-I chimeric model; residues not used in the model generation are shown in light gray. B: Color-coded apoA-I primary structure. Color code: 3R2P green, homology model blue, 1AV1 orange, CNS pink.
During the course of the simulation, apoA-I chains rearranged to better adapt to the size of the lipid core by assuming a hybrid zig-zag conformation and exposing hydrophobic residues toward the lipid core (Fig. 4A) while maintaining an 80% α-helicity content. The computed tryptophan solvent accessible surface hindrance (fa = 80%) is also consistent with results by Guerini Rocco et al. (27). This measure is readily comparable with experimental data that measure, via fluorescent quenching methods, the amount of tryptophan surface area that is not exposed to the solvent. Indeed, this accordion-like conformation would allow apoA-I helices to stretch in order to accommodate larger amounts of lipids during HDL maturation and growth.
Fig. 4.

apoA-I structural features. A: Split view of apoA-I inner hydrophobic surfaces (red scale), viewed from the inside of the lipoprotein, central helices (above), termini (below). B: Van der Waal spheres view of the H5 tunnel located between residues 121 and 143 of apoA-I.
ApoA-I N and C termini wrapped around the POPC bilayer within the first 20 ns of the simulation, as shown by the initial drop in the radius of gyration, which eventually stabilizes at 4.5 nm, consistently with the literature data (27, 28) (Fig. 3B). The root-mean-square deviation (RMSD) profile of apoA-I residues indicates that the rHDL model reached a stable conformation within the first 100 ns (Fig. 3C); after the removal of distance restraints at 100 ns, more conformations become available for the system, which converges toward a new stable conformation between 200 ns and up to the end of the simulation (Fig. 3G); nonetheless, distances between restrained atoms are preserved ( = 14.4 Å, SD = 3.2 Å) (Fig. 3F).
According to root-mean-square fluctuation (RMSF) measures, the most mobile apoA-I residues are within the extremities of the N and C termini of both chains; the C-terminal mobility is comparable with a common end effect, whereas the increased N-terminal RMSF seems a characteristic feature also described by Jones et al. (29). However, residues 22–43 and 225–237 folded in a globular structure stabilized by strong electrostatic interactions: hydrogen bonds Arg27-Glu235/Glu234 (existing for 95.6% of the simulation time), Gly26-Ser231 (58.9%), Asn43-Gly39 (53.1%), salt bridges Glu34-Lys238/Lys239 (78.8% and 76.8%), Glu234-Lys23/Arg27 (78.6% and 74.0%). (Fig. 3D). The RMSF analysis also highlighted other mobile domains in the apoA-I helical structure: mobile residues 102–111 (H4), 161–169 (H6/7), and 205–221 (H8–H10) formed solvent-exposed bulges with reduced interactions with the lipid core; the elevated mobility of these regions has been previously established by limited proteolysis experiments (30), which have identified cleavage sites to be in positions compatible with our findings. Moreover, as proposed by Wu et al. (13), the mobility of these regions may be relevant for LCAT recognition. The poorly folded secondary structure encompassing these regions is compliant with observations from Sevugan Chetty et al. (31), who measured a low stability for the central amphipathic helices, indicating that these residues may be in a dynamic unfolding and refolding state. Another notable structural feature of this model is the formation of a tunnel in apoA-I chains in correspondence with residues 121–143 (H5) (Fig. 4B). This particular feature has been already observed in a previous computational experiment performed by Jones et al. (32), where the authors suggest that this tunnel may be involved in the presentation of substrates to LCAT. A computational study from Rocco et al. (33) compared the alignment of adjacent residues (143–165, H6) in natural apoA-I mutants apoA-IMilano and apoA-IParis, showing that the misalignment of helices 5 and 6 due to a disulfide bridge (Cys173) in apoA-IMilano mutant significantly reduces LCAT activation compared with apoA-IParis, where the antiparallel 5/5 organization is preserved, but the suboptimal LCAT activation of this latter mutant is likely due to Cys151 disulfide bond falling within a probable LCAT recognition site.
Assessing LCAT lid dynamics
We then moved on the evaluation of published LCAT structures resolved by X-ray crystallography and chose to consider 5TXF and 5BV7 as the 2 most representative conformations: indeed, these 2 crystals mainly differ by the arrangement of the lid loop (residues 225–248), a key domain that regulates substrate active site accessibility (9).
The LCAT 5TXF (closed) structure formed crystals with an homotetrameric protein organization, whereas 5BV7 (open) was crystallized in complex with an agonist antibody that enhances LCAT phospholipasic activity on soluble substrates. Although this may suggest that 5BV7 depicts LCAT in its active conformation, the reported open conformation of the lid may be artificial; in fact, a visual inspection of the surrounding crystal mates reveals that the first C-terminal residues of the adjacent structure reached into the LCAT active site, forcing a wide-open conformation of the lid, as also discussed by Manthei et al. (9).
To test the hypothesis that the lid loop conformation in structure 5BV7 depends on the presence of the agonist antibody, we set up two 50 ns MD simulations of 5BV7 LCAT, with and without the cocrystallized antibody fragment. The MD trajectory analyses pointed out that the Fab-bound LCAT simulation is the most stable, as shown by the constant RMSD profile (Fig. 5A); however, residues with the highest ΔRMSF between the 2 simulations, that is, residues that account for the highest changes in the Fab-free LCAT simulation, are located within the LCAT β3/αA domain (Fig. 5B). This is also the region recognized by the Fab, indicating that the antibody actually stabilizes an otherwise highly mobile domain of LCAT.
Fig. 5.

LCAT MD. Comparison of residues mobility between 2 LCAT (5BV7) MD simulations with (blue line) and without (orange line) the cocrystallized agonistic antibody. A: RMSD plot. B: RMSF plot, lid loop is comprised between residues 225 and 250, within this region RMSFfab − RMSFnofab = −0.075 A, indicating that there is no significant difference between the mobility of the lid loop in the 2 simulations.
These results suggest that the open arrangement of the lid observed in 5BV7 is likely not caused by the binding of the antibody fragment to the LCAT β3/αA domain; therefore, in our opinion, although not necessarily a crystallographic artefact, 5BV7 is not suitable, without further refinement, to model a physiologically active conformation of LCAT. We compared 5BV7 with another open LCAT structure (PDB ID: 6MVD) (34) in complex with a small molecule activator, even if the positioning of the lid loop does not differ significantly from the 5BV7 structure; in addition to that, the same crystallographic issues may still persist; in fact, an inspection of the orientation of LCAT proteins within the crystal reveals that the LCAT lid loop could be forced in an open position by the surrounding crystal mates. To sample other possible lid loop conformations we then performed a wtMTD simulation, a sampling technique that allows one to describe the energy of the system as a function of reference frames, named collective variables (CVs) (Fig. 6A), chosen to track a particular movement.
Fig. 6.

LCAT wtMTD. A: Collective variables definition: CV1 is the dihedral angle defined by the following atom groups: (1) center of mass of Cap domain, (2) center of mass of catalytic triad, (3) center of mass of the entire protein except the lid loop, (4) center of mass of the lid loop; CV2 is the distance between C of Met234 and Gly119. B: Free energy surface map as a function of collective variables, red dots indicate relative minima, yellow stars indicate the value of the CVs as in 5TXF and 5BV7 crystallographic structures. C: LCAT structures corresponding to the 3 relative minima indicated in (B), closed (red), intermediate (green), open (yellow).
The free energy surface (FES) generated by the wtMTD displayed 3 energy wells within 3.5 kcal mol−1 from the global minimum (Fig. 6B). The structures associated with the minima of such wells correspond to an open, an intermediate, and a closed LCAT structure (Fig. 6C). These results indicate that energy barriers exist along the path that links the open and closed conformations and suggest that external forces may be required for the open-close transition to happen (i.e., the binding to lipoproteins). Intrigued by the identification of a metastable intermediate lid loop configuration, we compared it with the LPLA2 lid loop (residues 209–231) (PDB ID: 4X90) (35) and found a remarkable similarity between the 2 structures with respect to the lid loop positioning (RMSD: 16.00 Å) albeit LCAT lid loop lacks the secondary structure element a4 alpha helix described in (35) for LPLA2 and thus exhibits a wider range of motion. It is plausible that the protrusion of the lid loop in the intermediate configuration may help in the interaction with lipoproteins, during which LCAT has to interact both with lipids and apoA-I residues and the lid loop has to stretch further to shield the catalytic site. Notably, the lid positioning of the closed conformation predicted by the wtMTD almost perfectly overlaps with the lid positioning of the 5TXF crystal structure (RMSD 5.6 Å); conversely, the lid positioning of the predicted open conformation shows a greater difference from the one observed in the 5BV7 crystal (RMSD 9.4 Å). Therefore, our predicted open LCAT structure may display a more probable and energetically favorable rearrangement of the lid loop in solution to be used as a starting point for the rHDL-LCAT interactions simulation.
LCAT binds rHDLs in different positions with varying specificity
To obtain a model of an LCAT::rHDL complex, we employed a protein::protein molecular docking procedure adapted to integrate data gathered from cross-linking experiments and apoA-I mutagenesis studies; the former highlight the residues that are in proximity between LCAT and apoA-I, the latter isolate residues that, when mutated, impaired only LCAT activation while leaving ABCA1-mediated cholesterol efflux unaffected (1, 26, 32).
We also considered the functional characterization of LCAT glycosylation (36), which showed how the removal of the N-glycan on Asn384 improved LCAT activity on apoA-I-containing proteoliposomes, suggesting that LCAT αF helix of the α/β-hydrolase domain may be directly involved in apoA-I recognition.
Both the open and closed LCAT structures determined from the wtMTD were used as ligands to be docked on the rHDL model after 300 ns MD followed by minimization (for details, see Methods section).
When an unguided docking procedure was applied, most of the generated poses showed nonspecific interactions between the LCAT β3/αA domain and the phospholipidic surface of the rHDL (data not shown). However, when docking restraints were applied, more specific protein::protein interactions between LCAT and apoA-I could be observed. Experimental data suggest that LCAT may interact with rHDLs in multiple ways; in fact, not all the constraints could be satisfied simultaneously, so different existing models of the LCAT::rHDL complex can be described. We then compared the docking results to apoA-I::LCAT cross-links reported in Manthei et al. (37), which had not been used to guide the protein::protein docking program, and selected a subset of docking poses that could satisfy multiple interaction hypotheses. Binding poses were also shortlisted on the basis of the proximity between the β3/αA region (membrane binding domain) and catalytic triad to phospholipids and of the presence of protein-protein interactions between LCAT and apoA-I. The selected binding poses (Fig. 7) were then submitted to MD simulations to assess the complex stability over time.
Fig. 7.

rHDL::LCAT binding modes. Results of rHDL-LCAT protein-protein docking. Binding poses differ on the basis of the number of constraints satisfied simultaneously. Color code: apoA-I (gray), H4 (pink), H5 (red), H6 (pale yellow), H7 (dark yellow), LCAT a/b-hydrolase fold (orange), lid loop (purple), membrane-binding domain (cyan), cap domain (dark blue), catalytic triad (green sticks), lipids (gray hollow surface). Each figure depicts a small overview of the LCAT position with respect to the lipoprotein and a closeup view of the interaction interface. (1) maximized interactions between the LCAT a/b-hydrolase domain and apoA-I H6; (2) LCAT Lys240 close to apoA-I Lys182, the LCAT a/b-hydrolase domain is close to H6/H7; (3 and 4) LCAT Lys240 and Ser108 are close to apoA-I Lys140 and Lys118, the LCAT a/b-hydrolase domain contacts apoA-I H5/6; (5) maximized proximity between LCAT Lys240 and Ser108 and apoA-I Lys140 and Lys118 (on both chains), interaction is localized on apoA-I H5; (6) maximized interactions between LCAT and rHDL phospholipid surface, LCAT Lys240 is proximal to apoA-I Lys182 (H7); (7) closed LCAT structure, no protein-protein contacts. Cumulative RMSD between cross-linking atom pairs (Ca): 1 54.44 A, 2 73.89 A, 3 53.17 A, 4 53.59 A, 5 47.49 A, 6 70.48 A, 7 60.58 A.
In simulations 2, 3, 4, and 7, the interactions between LCAT and the rHDL are not suitable to maintain the enzyme anchored to the lipoprotein and LCAT drifts away from the complex toward the end of each simulation (Fig. 8A). In simulation 2, the cross-linking residue pairs LCAT Lys240 and apoA-I Lys182 (H7) are in proximity and the LCAT α/β-hydrolase domain is close to H6/H7; in simulations 3 and 4, the cross-linking residue pairs LCAT Lys240 and Ser108 are close to apoA-I Lys140 and Lys118 (H5/6), whereas the LCAT α/β-hydrolase domain contacts apoA-I H5/6; in simulation 7, no protein-protein interactions form between LCAT and apoA-I and the LCAT lid loop is in a closed configuration.
Fig. 8.

rHDL::LCAT MD. A: RMSD of LCAT fit on apoA-I. B: Mean LCAT::rHDL binding energy throughout the simulation.
However, in simulations 1, 5, and 6, despite some rearrangements in its positioning, LCAT remains bound to the rHDL with the active site facing the phospholipids. These simulations are characterized by the most negative mean interaction energy values throughout the simulation (Fig. 8B), indicating a higher stability of the complexes. Moreover, simulation 5 has the lowest RMSD of distances between residues that can form cross-links (37) (Fig. 7, caption). Protein::protein interactions occurring between LCAT and apoA-I are summarized in Table 1. In simulation 1, interactions between the LCAT α/β-hydrolase domain and apoA-I H6 are maximized compared with the other systems, whereas in simulation 5 LCAT binding is localized on apoA-I H5; in simulation 6, protein-protein interactions are limited to few residues in the LCAT lid loop and apoA-I H7 in favor of extensive electrostatic interactions between LCAT and the phospholipids. Despite LCAT and N and C termini having been described to affect LCAT binding to HDLs (9, 38), we could not assign to them a role in the rHDL::LCAT interaction in any of our simulations. This is probably due to the fact that in our systems initial configurations the complex is already formed and LCAT terminal regions may be required during an earlier recognition phase; or else, the simulation time scale was too short to identify LCAT termini interactions with rHDL components given the tested starting configurations.
Table 1.
rHDL::LCAT interactions: the existence of protein-protein interactions with respect to total simulation time is reported
| rHDL | LCAT | Existence % |
|---|---|---|
| Simulation 1 | ||
| Hydrogen bonds | ||
| TYR 100 | GLN 229 | 26.59 |
| LYS 118 | GLY 374 | 22.85 |
| ASP 150 | ASN 379 | 21.63 |
| LYS 118 | GLN 376 | 19.92 |
| ASP 103 | LYS 240 | 18.26 |
| ARG 151a | GLN 376 | 17.01 |
| Salt bridges | ||
| ASP 103a | LYS 240 | 48.59 |
| LYS 106 | ASP 335 | 12.39 |
| Simulation 5 | ||
| Hydrogen bonds | ||
| ARG 123 | ASP 335 | 58.92 |
| GLN 132 | ASN 228a | 29.44 |
| GLU 139 | GLY 119 | 22.14 |
| LYS 133 | LEU 70 | 22.04 |
| Salt bridges | ||
| ARG 123 | ASP 335 | 62.60 |
| ARG 116 | ASP 328 | 21.23 |
| LYS 140 | ASP 113 | 16.46 |
| Simulation 6 | ||
| Hydrogen bonds | ||
| LIP | ARG 244 | 50.70 |
| LIP | TYR 111a | 45.80 |
| LIP | GLU 241 | 33.62 |
| LIP | HIS 122 | 32.22 |
| LIP | GLN 126 | 28.42 |
| LIP | SER 114 | 27.07 |
| LIP | THR 59 | 53.54 |
| LIP | ASN 65 | 25.92 |
| LIP | TYR 111 | 24.73 |
| LIP | HIS 122 | 23.78 |
| LIP | ASN 65 | 22.73 |
| GLU 179 | LYS 240 | 36.11 |
| GLU 183 | LYS 238 | 14.09 |
| Salt bridges | ||
| GLU 179 | LYS 240 | 90.31 |
| GLU 183 | LYS 238 | 61.44 |
| GLU 179 | LYS 238 | 26.47 |
| LIP | CYS 50 | 21.78 |
Residues associated to fish-eye disease/FLD (LCAT) or that disrupt LCAT binding/activation when mutated (apoA-I).
A possible interpretation of these data is that, despite the high tendency of LCAT to form nonspecific interactions when close to a lipid interface, specific protein::protein interactions are necessary to stabilize the enzyme on the lipoprotein. Protein::lipid interactions via the LCAT β3/αA domain may be required to drive the first recognition; then, stronger, specific, protein::protein interactions between apoA-I central helices and the LCAT α/β-hydrolase domain are required to properly position LCAT on the lipoprotein and trigger the opening of LCAT lid, with the consequent exposure of the catalytic site.
LCAT extracts phospholipids from the lipoprotein surface
We speculated that LCAT could extract a phospholipid, the first substrate required for the cholesterol trans-esterification mechanism, in the same way as LPLA2 does (35), directly from the surface of the rHDL.
Simulation 6 showed a remarkable LCAT behavior: it sits on top of the lipid core and some interactions with apoA-I H7 keep the lid loop in an open conformation. Because of the binding site shielding operated by the lid loop, according to this mechanism, substrates would transit directly from the lipid surface to the LCAT binding site without exposing their hydrophobic fatty acid chains to the solvent. To simulate the phospholipid abduction into the LCAT active site, we set up a 50 ns wtMTD starting from the last frame of simulation 6; a single cv was chosen, represented by the distance between Ser181 hydroxylic group and the carboxylic carbon of the sn-2 fatty acid chain of the closest phospholipid (Fig. 9A). As shown by the FES (Fig. 9B), there are no significant energy barriers along the reaction path, indicating that this kind of POPC transition toward the LCAT active site might be favorable.
Fig. 9.

LCAT acylation. A: Definition: distance between the Ser181 hydroxyl group and the carboxylic carbon of the sn-2 fatty acid chain of the closest phospholipid. B: FES as function of distance CV, potential grows to infinity as the atoms are pulled too close. C: Minimized structure of acylated LCAT, hydrophobicity of the binding site is shown as a red surface, salt bridge between Lys218 and phosphate head group as magenta dashed line.
To identify a putative binding mode of the POPC residue within the LCAT binding site, the frame corresponding to the lowest CV value was extracted from the wtMTD simulation, and the chemical bonds necessary to obtain the tetrahedral reaction intermediate were manually modified, the acylated enzyme structure was then optimized with the MM/GBSA method (Fig. 9C). Notably, during structural optimization of the acyl-enzyme, Lys218 formed a salt bridge with the phospholipid phosphate group; the role of this residue in stabilizing the substrate may explain why its mutation (Lys218Asn) results in FLD syndrome (39).
Conclusions
We generated an all-atom model of a rHDL making use of restrained MD by integrating experimental cross-linking and crystallographic data. The resulting model satisfied other biochemical observations, such as particle size, α-helicity content, and tryptophan solvent accessibility; high-mobility regions at the N and C termini and within apoA-I central helices were also consistent with hydrogen-deuterium exchange and limited proteolysis experiments. To our knowledge, this is the first time that a full-atom structure of lipid-bound apoA-I was generated from crystallographic data on the delipidated protein; the integration of cross-linking experiments into a restrained MD simulation was aimed at minimizing the biases related to the chosen starting configuration for the system, by allowing apoA-I to smoothly fold and rearrange itself around the lipid core instead of constraining its structure into the circular double-helix belt. This approach also allowed us to deal with apoA-I N termini without the need to guess their structure or truncate the protein sequence.
We studied and compared published LCAT crystallographic structures, and an energetically stable LCAT structure in an open (active) conformation was obtained refining the experimental data with wtMTD simulations. Notably, our LCAT MD simulations show that the lid loop arrangement is decoupled from the dynamics of the membrane binding domain, suggesting that specific interactions with apoA-I are required to stabilize an open configuration.
The generated rHDL model was used as a receptor to study the LCAT recognition and activation mechanism. We used an integrative approach to build several models of rHDL::LCAT interactions combining data from mutagenesis and cross-linking experiments through molecular docking and then tested the dynamic behavior and the stability of the generated models with MD simulations.
Our findings on the LCAT::rHDL are complementary with the model proposed by Manthei et al. (37), where the authors integrated electron microscopy and cross-link/MS analyses to conclude that LCAT preferentially binds to helices 4/6 of apoA-I on the edge of the lipoprotein. The use of MD simulations to assess the stability of several interaction models allowed us to corroborate the importance of LCAT α/β-hydrolase fold and apoA-I helices 4–7 for their molecular recognition mechanism. However, we also observed that other binding modes involving the LCAT membrane binding domain are stable and could be functional to the enzyme activity. Castelijn et al. (40) simulated the binding of LCAT to a lipid bilayer coupling their MD simulations with free-energy calculations methods; our results support their hypothesis that the main driver of the rHDL::LCAT recognition mechanism is the interaction between the LCAT membrane binding domain (β3/αA) and the lipid surface. As exemplified by simulation 6 described in this paper, we showed how less specific protein-lipid interactions indeed contribute to LCAT binding on the lipoprotein, and we further described the specific protein-protein interactions that are required to drive the opening of the LCAT lid. Elaborating further on this interaction model, we then simulated the first step of the LCAT reaction mechanism, showing that the extraction of a phospholipid from the rHDL lipid core occurs without having to cross significant energy barriers, resulting in the subsequent acylation of the enzyme.
We hope this work can help to support and pull together the accumulated literature on the subject of LCAT and (r)HDL interactions, as well as to address the focus of future work, which should be aimed at clarifying the functional role and the transition dynamics between the multiple binding modes that LCAT exhibits with respect to apoA-I-containing lipoproteins.
Data availability
The authors confirm that the data supporting the findings of this study are contained within the article. The raw models and molecular dynamics data are available upon request from the corresponding author (I. E., University of Milan, ivano.eberini@unimi.it).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgements
I. E. thanks departmental “Linea 2 – Azione A 2019” and “National FFABR 2017 (Fondo di Finanziamento per l'Attività Base di Ricerca).”
Author contributions
T. L. investigation; T. L., C. P., L. P., U. G., and I. E. methodology; T. L. and C. P. writing-original draft; C. P. and I. E. supervision; L. P. and U. G. data curation; L. P. and U. G. formal analysis; L. P. visualization; E. G., L. C., and I. E. conceptualization; E. G., L. C., and I. E. writing-review and editing; L. C. and I. E. resources; L. C. validation; I. E. funding acquisition; I. E. project administration.
Author ORCIDs
Tommaso Laurenzi https://orcid.org/0000-0002-0446-0392
Chiara Parravicini https://orcid.org/0000-0002-4115-0094
Luca Palazzolo https://orcid.org/0000-0002-5370-9588
Uliano Guerrini https://orcid.org/0000-0001-7291-5160
Elisabetta Gianazza https://orcid.org/0000-0002-1083-3714
Laura Calabresi https://orcid.org/0000-0001-5042-9532
Ivano Eberini https://orcid.org/0000-0001-5521-3829
Funding and additional information
Funding was provided in part by grants from MIUR Progetto Eccellenza.
References
- 1.Jonas A. Lecithin cholesterol acyltransferase. Biochim. Biophys. Acta. 2000;1529(1–3):245–256. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- 2.Asztalos B.F., Schaefer E.J., Horvath K.V., Yamashita S., Miller M., Franceschini G., Calabresi L. Role of LCAT in HDL remodeling: investigation of LCAT deficiency states. J. Lipid Res. 2007;48:592–599. doi: 10.1194/jlr.M600403-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Ossoli A., Pavanello C., Calabresi L., Paoletti C.E.G. High-Density Lipoprotein, Lecithin: Cholesterol Acyltransferase, and Atherosclerosis. Endocrinol. Metab. 2016;31:223–229. doi: 10.3803/EnM.2016.31.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calabresi L., Gomaraschi M., Villa B., Omoboni L., Dmitrieff C., Franceschini G. Elevated soluble cellular adhesion molecules in subjects with low HDL-cholesterol. Arterioscler. Thromb. Vasc. Biol. 2002;22:656–661. doi: 10.1161/hq0402.105901. [DOI] [PubMed] [Google Scholar]
- 5.Saeedi R., Li M., Frohlich J. A review on lecithin:cholesterol acyltransferase deficiency. Clin. Biochem. 2015;48(7–8):472–475. doi: 10.1016/j.clinbiochem.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Ossoli A., Neufeld E.B., Thacker S.G., Vaisman B., Pryor M., Freeman L.A., Brantner C.A., Baranova I., Francone N.O., Demosky S.J., Vitali C., Locatelli M., Abbate M., Zoja C., Franceschini G. Lipoprotein X causes renal disease in LCAT deficiency. PLoS One. 2016;11 doi: 10.1371/journal.pone.0150083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piper D.E., Romanow W.G., Gunawardane R.N., Fordstrom P., Masterman S., Pan O., Thibault S.T., Zhang R., Meininger D., Schwarz M., Wang Z., King C., Zhou M., Walker N.P.C. The high-resolution crystal structure of human LCAT. J. Lipid Res. 2015;56:1711–1719. doi: 10.1194/jlr.M059873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunawardane R.N., Fordstrom P., Piper D.E., Masterman S., Siu S., Liu D., Brown M., Lu M., Tang J., Zhang R., Cheng J., Gates A., Meininger D., Chan J., Carlson T. Agonistic human antibodies binding to lecithin-cholesterol acyltransferase modulate high density lipoprotein metabolism. J. Biol. Chem. 2016;291:2799–2811. doi: 10.1074/jbc.M115.672790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manthei K.A., Ahn J., Glukhova A., Yuan W., Larkin C., Manett T.D., Chang L., Shayman J.A., Axley M.J., Schwendeman A., Tesmer J.J.G. A retractable lid in lecithin:cholesterol acyltransferase provides a structural mechanism for activation by apolipoprotein A-I. J. Biol. Chem. 2017;292:20313–20327. doi: 10.1074/jbc.M117.802736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koppaka V., Silvestro L., Engler J.A., Brouillette C.G., Axelsen P.H. The Structure of Human Lipoprotein A-I. J. Biol. Chem. 1999;274:14541–14544. doi: 10.1074/jbc.274.21.14541. [DOI] [PubMed] [Google Scholar]
- 11.Segrest J.P., Jones M.K., Klon A.E., Sheldahl C.J., Hellinger M., De Loof H., Harvey S.C. A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J. Biol. Chem. 1999;274:31755–31758. doi: 10.1074/jbc.274.45.31755. [DOI] [PubMed] [Google Scholar]
- 12.Silva R.A.G.D., Hilliard G.M., Li L., Segrest J.P., Davidson W.S. A mass spectrometric determination of the conformation of dimeric apolipoprotein A-I in discoidal high density lipoproteins. Biochemistry. 2005;44:8600–8607. doi: 10.1021/bi050421z. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z., Wagner M.A., Zheng L., Parks J.S., Shy J.M., Smith J.D., Gogonea V., Hazen S.L. The refined structure of nascent HDL reveals a key functional domain for particle maturation and dysfunction. Nat. Struct. Mol. Biol. 2007;14:861–868. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 14.Bhat S., Sorci-thomas M.G., Tuladhar R., Samuel M.P., Thomas J. NIH Public Access. 2008;46:7811–7821. [Google Scholar]
- 15.Martin D.D.O., Budamagunta M.S., Ryan R.O., Voss J.C., Oda M.N. Apolipoprotein A-I assumes a “looped belt” conformation on reconstituted high density lipoprotein. J. Biol. Chem. 2006;281:20418–20426. doi: 10.1074/jbc.M602077200. [DOI] [PubMed] [Google Scholar]
- 16.Wu Z., Gogonea V., Lee X., Wagner M.A., Li X.M., Huang Y., Undurti A., May R.P., Haertlein M., Moulin M., Gutsche I., Zaccai G., Didonato J.A., Hazen S.L. Double superhelix model of high density lipoprotein. J. Biol. Chem. 2009;284:36605–36619. doi: 10.1074/jbc.M109.039537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones M.K., Zhang L., Catte A., Li L., Oda M.N., Ren G., Segrest J.P. Assessment of the validity of the double superhelix model for reconstituted high density lipoproteins: A combined computational-experimental approach. J. Biol. Chem. 2010;285:41161–41171. doi: 10.1074/jbc.M110.187799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vickaryous N.K., Teh E.M., Stewart B., Dolphin P.J., Too C.K.L., McLeod R.S. Deletion of N-terminal amino acids from human lecithin:cholesterol acyltransferase differentially affects enzyme activity toward alpha- and beta-substrate lipoproteins. Biochim. Biophys. Acta. 2003;1646:164–172. doi: 10.1016/s1570-9639(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 19.Lee Y.-P., Adimoolam S., Liu M., Subbaiah P.V., Glenn K., Jonas A. Analysis of human lecithin–cholesterol acyltransferase activity by carboxyl-terminal truncation. Biochim. Biophys. Acta. 1997;1344:250–261. doi: 10.1016/s0005-2760(96)00149-x. [DOI] [PubMed] [Google Scholar]
- 20.Francone O.L., Evangelista L., Fielding C.J. Effects of carboxy-terminal truncation on human lecithin:cholesterol acyltransferase activity. J. Lipid Res. 1996;37:1609–1615. [PubMed] [Google Scholar]
- 21.Mei X., Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of High Density Lipoprotein (HDL) by dimerization. J. Biol. Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borhani D.W., Rogers D.P., Engler J.A., Brouillette C.G. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melchior J.T., Walker R.G., Cooke A.L., Morris J., Castleberry M., Thompson T.B., Jones M.K., Song H.D., Rye K.A., Oda M.N., Sorci-Thomas M.G., Thomas M.J., Heinecke J.W., Mei X., Atkinson D. A consensus model of human apolipoprotein A-I in its monomeric and lipid-free state. Nat. Struct. Mol. Biol. 2017;24:1093–1099. doi: 10.1038/nsmb.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pourmousa M., Song H.D., He Y., Heinecke J.W., Segrest J.P., Pastor R.W. Tertiary structure of apolipoprotein A-I in nascent high-density lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2018;115:5163–5168. doi: 10.1073/pnas.1721181115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang R., Silva R.A.G.D., Jerome W.G., Kontush A., Chapman M.J., Curtiss L.K., Hodges T.J., Davidson W.S. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat. Struct. Mol. Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooke A.L., Morris J., Melchior J.T., Street S.E., Jerome W.G., Huang R., Herr A.B., Smith L.E., Segrest J.P., Remaley A.T., Shah A.S., Thompson T.B., Davidson W.S. A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL. J. Lipid Res. 2018;59:1244–1255. doi: 10.1194/jlr.M085332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rocco A.G., Gianazza E., Calabresi L., Sensi C., Franceschini G., Sirtori C.R., Eberini I. Structural features and dynamics properties of human apolipoprotein A-I in a model of synthetic HDL. J. Mol. Graph. Model. 2009;28:305–312. doi: 10.1016/j.jmgm.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 28.Calabresi L., Meng Q.H., Castro G.R., Marcel Y.L. Apolipoprotein A-I conformation in discoidal particles: Evidence for alternate structures. Biochemistry. 1993;32:6477–6484. doi: 10.1021/bi00076a023. [DOI] [PubMed] [Google Scholar]
- 29.Jones M.K., Gu F., Catte A., Li L., Segrest J.P. “Sticky” and “promiscuous”, the yin and yang of apolipoprotein A-I termini in discoidal high-density lipoproteins: A combined computational-experimental approach. Biochemistry. 2011;50:2249–2263. doi: 10.1021/bi101301g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calabresi L., Tedeschi G., Treu C., Ronchi S., Galbiati D., Airoldi S., Sirtori C.R., Marcel Y., Franceschini G. Limited proteolysis of a disulfide-linked apoA-I dimer in reconstituted HDL. J. Lipid Res. 2001;42:935–942. [PubMed] [Google Scholar]
- 31.Sevugan Chetty P., Mayne L., Kan Z.-Y., Lund-Katz S., Englander S.W., Phillips M.C. Apolipoprotein A-I helical structure and stability in discoidal high-density lipoprotein (HDL) particles by hydrogen exchange and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2012;109:11687–11692. doi: 10.1073/pnas.1209305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones M.K., Catte A., Li L., Segrest J.P. Dynamics of activation of lecithin:cholesterol acyltransferase by apolipoprotein A-I. Biochemistry. 2009;48:11196–11210. doi: 10.1021/bi901242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocco A.G., Sensi C., Gianazza E., Calabresi L., Franceschini G., Sirtori C.R., Eberini I. Structural and dynamic features of apolipoprotein A-I cysteine mutants, Milano and Paris, in synthetic HDL. J. Mol. Graph. Model. 2010;29:406–414. doi: 10.1016/j.jmgm.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 34.Manthei K.A., Yang S., Baljinnyam B., Chang L., Glukhova A. Molecular basis for activation of lecithin: cholesterol acyltransferase by a compound that increases HDL cholesterol. 2018;2:1–61. doi: 10.7554/eLife.41604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glukhova A., Hinkovska-Galcheva V., Kelly R., Abe A., Shayman J.A., Tesmer J.J.G. Structure and function of lysosomal phospholipase A2 and lecithin:cholesterol acyltransferase. Nat. Commun. 2015;6:6250. doi: 10.1038/ncomms7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O K., Hill J.S., Wang X., McLeod R., Pritchard P.H. Lecithin:cholesterol acyltransferase: role of N-linked glycosylation in enzyme function. Biochem. J. 1993;294:879–884. doi: 10.1042/bj2940879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manthei K.A., Patra D., Wilson C.J., Fawaz M.V., Piersimoni L., Shenkar J.C., Yuan W., Andrews P.C., Engen J.R., Schwendeman A., Ohi M.D., Tesmer J.J.G. Structural analysis of lecithin:cholesterol acyltransferase bound to high density lipoprotein particles. Commun. Biol. 2020;3:28. doi: 10.1038/s42003-019-0749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vickaryous N.K., Teh E.M., Stewart B., Dolphin P.J., Too C.K.L., McLeod R.S. Deletion of N-terminal amino acids from human lecithin:cholesterol acyltransferase differentially affects enzyme activity toward α- and β-substrate lipoproteins. Biochim. Biophys. Acta. 2003;1646(1–2):164–172. doi: 10.1016/s1570-9639(03)00005-0. [DOI] [PubMed] [Google Scholar]
- 39.Calabresi L., Pisciotta L., Costantin A., Frigerio I., Eberini I., Alessandrini P., Arca M., Bon G.B., Boscutti G., Busnach G., Frascà G., Gesualdo L., Gigante M., Lupattelli G., Montali A. The molecular basis of lecithin: cholesterol acyltransferase deficiency syndromes: A comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler. Thromb. Vasc. Biol. 2005;25:1972–1978. doi: 10.1161/01.ATV.0000175751.30616.13. [DOI] [PubMed] [Google Scholar]
- 40.Casteleijn M.G., Parkkila P., Viitala T., Koivuniemi A. Interaction of lecithin-cholesterol acyltransferase with lipid surfaces and apolipoprotein A-I derived peptides. J. Lipid Res. 2018;59 doi: 10.1194/jlr.M082685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are contained within the article. The raw models and molecular dynamics data are available upon request from the corresponding author (I. E., University of Milan, ivano.eberini@unimi.it).