

Abstract
Background
Long-acting injectable cabotegravir (CAB LA) is a novel integrase inhibitor in advanced clinical development for HIV treatment and HIV prevention. We investigated the terminal pharmacokinetics and safety of CAB LA after final injection as part of a Phase 2 study.
Methods
We performed a randomized, double-blind placebo-controlled trial at 8 sites in the United States (US), sub-Saharan Africa, and Brazil. Participants, age 18–65, HIV-uninfected and at low-risk for HIV, were randomized 3:1 to CAB LA or placebo. Participants received CAB LA at one of two doses/intervals (800 mg as a split 2 × 2mL injection or 600 mg as a single 3mL injection), which were aggregated for this analysis. The current analysis includes participants who received at least one injection and examines the tail phase (time from final injection to 52–76 weeks post-final injection). The outcomes were safety and pharmacokinetics. Safety was reported as incidence rates of grade 2 or higher adverse events. The apparent terminal phase half-life (t1/2app) and time to decay of CAB drug concentration to below the lower limit of quantification (LLOQ) were estimated using non-compartmental methods. The trial was registered with ClinicalTrials.gov, number NCT02178800. This trial is completed.
Findings
Overall, 177 (133 CAB, 44 PBO) participants received at least one injection. Median age was 31 years, 117 (66·1%) were female, 123 (69·5%) were non-white, 93 (52·5%) were from the US. Rates of grade 2 or higher adverse events on CAB were higher in the injection phase compared to the tail phase. At one time point from 52–60 weeks and at 76 weeks after final injection, 9 (22·5%) and 4 (13·3%) of males, respectively, had detectable CAB concentrations; 52 (63·4%) and 27 (42·2%) of females had detectable CAB concentrations at those time points. The estimated median time from last injection to CAB concentration below the LLOQ was 43·7 (range 20·4–152·5) weeks for males and 67·3 (range 17·7–225·5) weeks for females. Terminal phase half-life was 32% longer for female sex at birth participants compared to male participants, and 33% longer for participants with body mass index (BMI) above vs. below the study median of 27·2 for females and 25·0 for males.
Interpretation
The clinical importance of the long pharmacokinetic tail, significantly longer for females compared to males, and those with higher BMI, will need to be addressed in pivotal trials of treatment and prevention.
Trial Registration: ClinicalTrials NCT02178800
Research in Context
Evidence before this study
We searched ClinicalTrials.gov and PubMed.gov from inception to 03/27/2020 using the search terms ((((long-acting) AND cabotegravir) AND pharmacokinetics) OR pharmacology).
Long-acting injectable cabotegravir (CAB LA) is a novel integrase inhibitor in advanced clinical development for HIV treatment (in combination with long-acting injectable rilpivirine [RPV LA]) and HIV prevention as monotherapy. A potential limitation of use of long-acting injectable antiretroviral drugs for HIV treatment is the prolonged terminal decay of drug concentrations after product discontinuation. This could create a period of vulnerability during which viral replication could rebound, allowing for selection of drug-resistant variants. Similarly, when these drugs are used for HIV prevention, the pharmacokinetic tail could create a period of vulnerability in persons who acquire HIV infection, allowing for emergence of drug-resistant strains. In the Phase 2 ECLAIR study, 17% of US male participants had detectable cabotegravir concentrations 52 weeks after the final injection of cabotegravir injected intramuscularly every 12 weeks. Duration of detectable cabotegravir concentrations in women has not been described.
Added value of this study
In this secondary analysis of a Phase 2 safety, tolerability, and pharmacokinetic study of long-acting injectable cabotegravir, we evaluated the terminal safety and pharmacokinetics in a multi-national population of males and females. We identified no new adverse events during the tail phase and the active injection phase, with lower rates of adverse events in the tail phase. We found approximately 30% prolongation of terminal pharmacokinetics (t1/2app and time to lower-limit of quantitation) for females compared to males, as well as for those with higher vs. lower body mass index.
Implications of all the available evidence
The clinical implications of the prolonged pharmacokinetic tail are unclear. They may represent a prolonged interval of protection in the prevention context and, possibly, a prolonged period of risk for selection of resistant viral species, in those who acquire HIV infection. Phase 3 clinical trials and anticipated subsequent clinical use will be required to define the clinical importance of the pharmacokinetic tail for long acting injectable antiretrovirals.
Background
Oral pre-exposure prophylaxis (PrEP) with tenofovir disoproxil fumarate (TDF)-based regimens has been shown to be highly effective for HIV prevention in diverse populations and routes of exposure, but requires rigorous adherence to prescribed dosing especially to prevent vaginal transmission.1–5 Candidate agents are being evaluated that offer alternatives to a daily oral PrEP regimen (e.g., by using products that require less frequent dosing and using alternate methods for drug delivery). A potential limitation of using long-acting antiretroviral (ARV) drugs for PrEP is that the prolonged clearance of these agents after dosing cessation at sub-inhibitory concentrations may increase the risk of HIV infection and lead to emergence of drug-resistant HIV in those who become infected despite prophylaxis.
Cabotegravir (CAB) is a novel HIV-1 strand transfer integrase inhibitor in advanced development, both for HIV prevention as monotherapy and for HIV treatment maintenance, in combination with rilpivirine (RPV) as a long-acting (LA) injectable suspension for intramuscular delivery at 1–2-month intervals (CAB LA).
The multi-national HIV Prevention Trials Network 077 study (HPTN 077) was a Phase 2a, double-blind, randomized placebo-controlled trial of CAB LA. The study was conducted in Brazil, South Africa, Malawi, and the US. CAB LA was administered at two doses and administration schedules in HIV-uninfected males and females; the primary objectives of the study were to determine the safety, tolerability, and pharmacokinetics of CAB LA in this population The study was designed to help select a CAB LA dosing regimen for evaluation in Phase 3 studies of CAB LA for HIV prevention. The primary results of HPTN 077 corroborated pharmacokinetic modeling studies evaluating 600 mg of CAB LA administered as an intramuscular (IM) gluteal injection every 8-weeks after an initial 4-week interval between injections; this regimen resulted in CAB concentrations that achieved targets shown to be effective for prevention of simian-human immunodeficiency virus (SHIV) infection in non-human primate (NHP) challenge models.6 This dose/interval achieved target concentrations for both males and females.
An important characteristic of ARV-based PrEP regimens is how long the drug persists after final administration (i.e., the tail phase of the drug). The tail phase of CAB LA was first evaluated in the ECLAIR study, a Phase 2 study of CAB LA 800 mg IM every 12 weeks among HIV-uninfected men in the United States (US).7 In that study, the terminal phase half-life was 40·0 days (95%CI: 35·1–45·7) and 17% of men still had quantifiable CAB concentrations (above the lower limit of quantification [LLOQ]; >25 ng/mL) in plasma 52 weeks after final injection.7 In this report, we describe the safety and pharmacokinetics of CAB LA during the tail phase data of HPTN 077, which included male and female participants in US and non-US settings.
Methods
Study Design and Participants
Enrollment criteria for HPTN 077 were described previously.6 Briefly, participants were 18–65 years old, HIV uninfected at screening, and at low risk for HIV infection. Participants were generally in good health, with acceptable laboratory parameters, and negative tests for hepatitis B and C virus infection. Females who were of reproductive potential were required to use an effective contraception method. Individuals were not eligible for enrollment if they had significant cardiovascular disease; had used post-exposure prophylaxis or PrEP in the 90 days prior to study entry; had known liver disease, coagulopathy, or seizures; were pregnant or breastfeeding; or had a score of ≥8 on the Alcohol Use Disorders Identification Test (AUDIT).8 Two sequential cohorts of participants were enrolled, with different dosing regimens. The study design for the second cohort was informed by data from the ECLAIR study and data from the CAB LA treatment program. Written informed consent was obtained from all participants.
Procedures
Original study randomization and power considerations were previously described.6 Briefly, upon study enrollment, participants were randomly assigned 3:1 by a computer-generated algorithm stratified by sex at birth and region (US or non-US) to CAB or placebo (PBO). CAB LA and 0·9% saline PBO injections were further masked with semi-opaque syringe overlays. For both cohorts, the primary endpoint was evaluated at week 41 (at completion of the 3rd injection cycle for Cohort 1 and the fifth injection cycle for Cohort 2. Blinding remained in place until all participants completed the week 41 study visit and all data for the primary endpoint (i.e., all data from the week 41 visit) had been collected.
Participants in both cohorts first received four weeks of oral CAB (or a matching oral PBO tablet) in a lead-in phase to establish safety using a short-acting product prior to first injection. Participants in Cohort 1 received 800 mg of CAB or a 0·9% saline PBO as a split 2 × 2mL injection at three study visits at 12-week intervals. In Cohort 2, participants received CAB LA 600 mg (or a 0·9% saline PBO), administered as a single 3mL injection initially, once at a 4-week interval; and then at 8-week intervals for a total of five injections. Injection guidelines recommended use of a 2-inch needle for IM injection for participants with a body mass index (BMI) of 30 or greater, and a 1½ -inch needle for BMI under 30.
Participants were followed for 52 or 76 weeks after the last injection, depending on timing of study enrollment. The original protocol specified 52 weeks of follow-up after terminal injection. When the tail phase results from the ECLAIR trial became available, the HPTN 077 protocol was amended to offer 76 weeks of follow-up after the last injection to participants who had not already completed the study. Plasma for CAB concentrations was collected 1 week, 4–6 weeks, 8–12 weeks, and every 12 weeks (excepting the ultimate assessment, scheduled 4–8 weeks after the penultimate assessment) after the final injection.
All visits included clinical and laboratory assessments for adverse events (AEs), HIV testing (using an FDA-approved rapid test and instrumented antigen/antibody testing at all visits), vital signs, clinical examination, and review of concomitant medications. AEs were graded according to the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2·0.9 Testing for sexually transmitted infections (STIs) included urine and rectal Neisseria gonorrhoeae and Chlamydia trachomatis nucleic acid amplification testing and serologic syphilis testing at approximately 6-month intervals throughout the study. For females of reproductive potential, pregnancy testing was performed at Week 41 and quarterly at follow-up visits. CAB concentrations in plasma were determined via liquid chromatographic-tandem mass spectrometric (LC-MS/MS) analysis by the Clinical Pharmacology Analytical Laboratory at the Johns Hopkins University School of Medicine. The assay was validated in accordance with FDA, Guidance for Industry: Bioanalytical Method Validation recommendations, and was reviewed by the DAIDS-sponsored Clinical Pharmacology Quality Assurance (CPQA) program10–11. Briefly, CAB was isolated from 0.025 mL K2DTA via protein precipitation and was subsequently subjected to chromatographic separation on a Waters UPLC HSS C18 column (1.8 μm, 2.1 × 50 mm, Waters Corporation, Milford, MA). CAB was quantified on an API 4000 (SCIEX, Redwood City, CA) operated in positive ionization and selective reaction monitoring (SRM) modes. CAB peak area ratios were normalized to an isotopically labeled-internal standard, and study specimens were quantified from a calibration curve generated using linear regression with a 1/x2 weighting. The primary linearity of the assay was 25–25,000 ng/mL. Any results below the lower limit of quantitation (LLOQ) were reported as below the limit of quantitation. Safety was determined by the proportion of participants experiencing any Grade 2 or higher clinical AE or any Grade 2 or higher laboratory abnormality occurring from the date of the terminal injection to the end of protocol-defined study follow-up.
Outcomes
The primary outcomes of the HPTN 077 study were the number of participants experiencing any grade 2 or higher clinical AE or laboratory abnormality from initial injection to Week 41; the number of injectable study product discontinuations by participants due to toxicity, tolerability, of acceptability between initials injection and week 41; and to determine the geometric mean and 90% prediction intervals of CAB LA by sex at time points between initial injection and week 41. Results of the primary outcomes have been previously reported.6
Statistical Analysis
This secondary analysis evaluated the safety and pharmacokinetics of CAB LA among participants who received at least one injection. Per protocol, once the primary endpoint at Week 41 had been reached for all participants, treatment assignments were unblinded and participants in the placebo arm were discontinued from follow-up; this created an unequal follow-up of placebo and CAB participants in the tail phase.
The rate of Grade 2 or higher AEs among the CAB LA recipients was compared in the injection and tail phases of the study. The period assessed in the injection phase started at the first injection and extended either 12 or 8 weeks after the last injection (in Cohorts 1 and 2, respectively). The period assessed in the tail phase started at 12 or 8 weeks after the last injection (in Cohorts 1 and 2, respectively) and ended at the last study visit. The incidence rate of AEs was compared using Poisson regression to account for the correlations within participants between the injection and tail-phase portions of the study. Data from Cohort 1 and Cohort 2 were combined for all non-PK analyses. Pharmacokinetic analysis encompassed all timepoints subsequent to the terminal injection for CAB LA receiving participants.
To contextualize the risk profile of 077 participants, STI incidence rates and 95% confidence intervals were estimated based on an exact Poisson distribution for all participants including PBO-treated participants; incidence was assessed separately for the tail phase and the injection phase. STI incidence rates in these two phases were compared using Poisson regression.
For pharmacokinetic analysis, participants were included if they received at least one injection and had at least three CAB measurements above the LLOQ after the last injection (Appendix p. 2). The apparent terminal phase half-life (t1/2app) and estimated time to LLOQ were calculated using non-compartmental analysis in Phoenix WinNonlin 8·1 (Certara USA, Inc., Princeton, NJ). Terminal slope data point selection was optimized using adjusted R2, which accounts for the number of points used in the estimation of terminal slope (ka), to evaluate the goodness of fit for the log concentration decline over time in a linear regression. Then, t1/2app was estimated by log(2)/ ka. Time to LLOQ was estimated using the time from last injection to the intercept of the estimated model fit for terminal slope with the assay LLOQ (WinNonlin Therapeutic Response module). No imputations for concentrations below the LLOQ were used.
The associations of participants’ baseline characteristics (e.g., sex at birth, race, BMI, weight, smoking status), # of injections, and cohort with t1/2app were evaluated by using a log-linear model. BMI was dichotomized by ≥Median vs <Median. A different threshold (25.0 for males and 27.2 for females) was used in the analysis of assessing the association of BMI. All variables except weight were included in the multivariable log-linear regression model. Weight was excluded due to the collinearity with BMI. Smoking was included because cigarette smoke effect on UDP-glucuronosyltransferase (UGT), the primary metabolic pathway for cabotegravir. A sensitivity analysis was performed to evaluate whether the associations would be affected by those estimates of t1/2app with adjusted R2 <0·85, a stringent threshold for non-compartmental analysis. Wilcoxon rank sum tests were used to compare time to LLOQ between sexes and between BMI groups (≥ median vs. < median).
Informed Consent
Local institutional review boards or ethics committees at each participating site granted ethics and regulatory approvals of the protocol and all participants provided written informed consent.
Role of the funding source
The funders of the study had roles in study design, data collection, data analysis, data interpretation, and writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
This trial is registered with ClinicalTrials.gov, number NCT02178800.
Results
Of the 199 eligible participants randomized into HPTN 077, 177 (133 CAB, 44 PBO) received at least one injection and were included in this analysis (Table 1, Appendix p. 3). The reasons for non-receipt of injections for 22 participants have been previously reported (Appendix p. 4).6 The median age for the 177 participants included in the analysis was 31 years, 66·1% of participants were female at birth, 46·3% non-Hispanic black, 26·6% Latinx, and 30·5% non-Hispanic white; 52·5% were from the US, 28·8% from sub-Saharan Africa, and 18·6% from Brazil. Ten percent of participants reported smoking cigarettes at study entry; the median BMI was 26. One hundred thirty-five participants agreed to an extended 76 week of post-terminal injection follow-up.
Table 1:
Baseline demographics of participants who received at least one injection of study product
| Overall (n=177) | Cabotegravir (n=134) | Placebo (n=43) | Male participants (n=60) | Female participants (n=117) | |
|---|---|---|---|---|---|
| Age (years) | 31(24–39) | 29 (24–38) | 34(27–40) | 33 (29–50) | 29 (23–37) |
| Weight (kg) | 75 (62–91) | 75 (61–91) | 74(65–91) | 80 (71–99) | 72 (59–89) |
| Body-mass index | 26 (23–33) | 26 (23–33) | 26 (24–32) | 25 (23–29) | 28 (23–34) |
| Race | |||||
| Non-Hispanic white | 47 (27%) | 36 (27%) | 11 (26%) | 21 (35%) | 26 (22%) |
| Non-Hispanic black | 71(40%) | 55(41%) | 16 (37%) | 17 (28%) | 54 (46%) |
| Asian | 3(2%) | 2 (1%) | 1(2%) | 2 (3%) | 1(1%) |
| Mixed or other | 10 (6%) | 5 (4%) | 5 (12%) | 4 (7%) | 6 (5%) |
| Latino | 46 (26%) | 36 (27%) | 10 (23%) | 16 (27%) | 30 (26%) |
| Region | |||||
| USA | 93 (53%) | 69 (51%) | 24(56%) | 45 (75%) | 48(41%) |
| Sub-Saharan Africa | 51(29%) | 41(31%) | 10 (23%) | 6 (10%) | 45 (38%) |
| Brazil | 33 (19%) | 24 (18%) | 9 (21%) | 9 (15%) | 24 (21%) |
| Current smoker* | |||||
| Yes | 18 (10%) | 13 (10%) | 5 (12%) | 11 (18%) | 7(6%) |
| No | 158 (89%) | 120(90%) | 38 (88%) | 49 (82%) | 109 (93%) |
Data are median (IQR) or n (%). Some percentages do not sum to 100 because of rounding.
One female participant in the cabotegravir group did not report smoking status.
Incidence rates (95% CIs) of Grade 2 or higher clinical and laboratory AEs experienced in CAB participants during the tail phase are shown in Table 2 and are compared to the corresponding AEs experienced by these participants during the injection phase of the study; the incidence rate of grade 2 or higher AEs in the tail phase was lower than the incidence rate in the injection phase. There were only seven grade 2 AEs assessed as related to CAB by the site investigator. Twenty-five Grade 3 or higher events were observed in 21 participants during the tail phase. None were assessed by the site investigator as related to CAB. Adverse events occurring more than once in participants treated with CAB LA included unintentional weight loss, decreased creatinine clearance and increased direct bilirubin. No seizure events were reported during the tail phase. One previously reported seroconversion occurred in a participant 48 weeks after her terminal active CAB injection at a time when CAB concentrations were below the assay LLOQ; the participant’s virus had no mutations anticipated to confer resistance to CAB or other integrase inhibitors on population level or next-generation deep sequencing.6
Table 2.
Incidence rate of any grade 2 or higher clinical and laboratory AEs between tail phase and injection phase
| Tail Phase (n=133*, person-years=148·7) | Injection Phase (n=134*, person-years=85·6) | ||||
|---|---|---|---|---|---|
| Incidence Rate (per 100 PY) | 95% CI | Incidence Rate (per 100 PY) | 95% CI | P-value | |
| Any grade 2 AE† | 214·9 | (180·3, 256·1) | 750·0 | (662·3, 849·2) | <0·0001 |
| Conjunctivitis | 6 | (2·9, 12·8) | 14 | (7·5, 26·3) | 006 |
| Creatinine renal clearance decreased ‡ | 107 | (6·5, 17·6) | 134·5 | (108·1, 167·2) | <0·0001 |
| Genital candidiasis | 3·4 | (1·4, 8·2) | 7·6 | (3·4, 16·6) | 0·14 |
| Headache | 9·8 | (5·4, 17·7) | 28·8 | (18·4, 45·1) | 0·001 |
| Influenza | 5·8 | (2·7, 12·9) | 16·1 | (10·0, 25·8) | 0·02 |
| Injury | 3·8 | (1·7, 8·6) | 3·3 | (1·0, 10·3) | 0·82 |
| Musculoskeletal discomfort | 11·7 | (7·3, 18·9) | 48·3 | (34·8, 67·2) | <0·0001 |
| Nasopharyngitis | 13·8 | (8·6, 22·3) | 23·7 | (14·5, 38·7) | 0·07 |
| Rash | 8·6 | (4·9, 15·0) | 11·3 | (5·4, 23·8) | 0·51 |
| Sinusitis | 4 | (1·8, 8·6) | 10·3 | (4·6, 22·7) | 0·07 |
| Urinary tract infection | 11·1 | (5·1, 24·3) | 14·5 | (7·3, 28·8) | 0·47 |
| Upper respiratory tract infection | 33·4 | (23·5, 47·3) | 40·3 | (26·9, 60·5) | 0·38 |
One participant in the CAB arm of the injection phase did not attend study visits during the tail phase.
Reported events included AE categories reported by 5 or more participants during the tail phase.
Grade 2 AEs for creatinine clearance are determined by a result of <90 to 60 ml/min or a 10% to <30% decrease from baseline. Grade 3 AEs for creatinine clearance are determined by a result of <60 to 30 ml/min or a 30% to <50% decrease from baseline. One participant in the CAB arm of the injection phase did not attend study visits during the tail phase.
Note: Models were adjusted for cohort when incidence rates and their 95% CIs were estimated and the differences between the injection and the tail phase were compared.
Four pregnancies occurred in HPTN 077 participants; two pregnancies occurred among participants receiving PBO and two among those receiving CAB LA. The first pregnancy occurring in the CAB LA arm was in a Cohort 2 participant that received five injections of 600 mg CAB LA at 8-week intervals. Pregnancy was estimated to have occurred 32 weeks after final injection of CAB LA, and although late pregnancy was complicated by pre-eclampsia, the participant delivered a near-full term healthy infant. The second pregnancy was in a Cohort 1 participant who received two injections of 800 mg CAB LA at 12-week intervals. Pregnancy occurred approximately 108 weeks after final injection and the participant delivered a healthy infant after an uncomplicated pregnancy.
Rates of STIs did not differ from injection phase to tail phase in this low-risk population, with five incident infections per 100 person-years in each phase. Eight incident STIs in six participants were diagnosed during the tail phase, compared with six STIs in five participants during the injection phase with the majority occurring in non-US based females (Table 3).
Table 3.
Rates of sexually transmitted infections experienced by HPTN 077 participants in the tail phase of the study compared to the injection phase
| Tail phase (n=175*, person-years=181·2) | Injection phase (n=177*, person-year=113·5) | p-value | |||
|---|---|---|---|---|---|
| Incidence Rate (per 100 PY) | 95% CI | Incidence Rate (per 100 PY) | 95% CI | ||
| Any STI | 4·4 | (1·9, 8·7) | 6·2 | (2·5, 12·7) | 0·52 |
| Urine | |||||
| C. trachomatis | 1·7 | (0·3, 4·8) | 1·8 | (0·2, 6·4) | |
| N. gonorrheae | 1·1 | (0·1, 4·0) | 0·9 | (0·0, 4·9) | |
| Rectal | |||||
| C. trachomatis | 0·6 | (0·0, 3·1) | 1·8 | (0·2, 6·4) | |
| N. gonorrheae | 0·6 | (0·0, 3·1) | 0·9 | (0·0, 4·9) | |
| Syphilis | |||||
| Incident | 0·6 | (0·0, 3·1) | 0·9 | (0·0, 4·9) | |
Two participants in the injection phase did not attend study visits during the tail phase.
Individual participant terminal log-linear regression curves were fitted to plasma CAB concentration selecting among concentrations from the observed Cmax to Clast following the last injection; these data are shown for males (Figure 1A) and females (Figure 1B). Relevant non-human primate (NHP) correlates of protection against rectal (Figure 1A) and vaginal (Figure 1B) challenges are overlaid for reference.12–14 The distribution of plasma CAB concentrations aggregated by study visit (approximately 12-week intervals after final injection) are shown in Figure 2 for males and females. The categories used in Figure 2 represent the NHP correlates of protection observed in the SIVmac251 vaginal challenge model, in which plasma levels >4x PA-IC90 (664 ng/mL) provided calculated protective efficacy of 87% compared to control-untreated macaques [CM]); plasma levels <4x PA-IC90 (166–664 ng/mL) were not protective compared to CM).11 Similarly, for the rectal SHIV162p3 challenge model, plasma CAB concentrations above 3x PA-IC90 (498 ng/mL) had 100% calculated protective efficacy, between 1 and 3x PA-IC90(166–498 ng/mL) provided 90% protective efficacy, and <1x provided 45% protective efficacy, respectively, compared to CM.14
Figure 1.
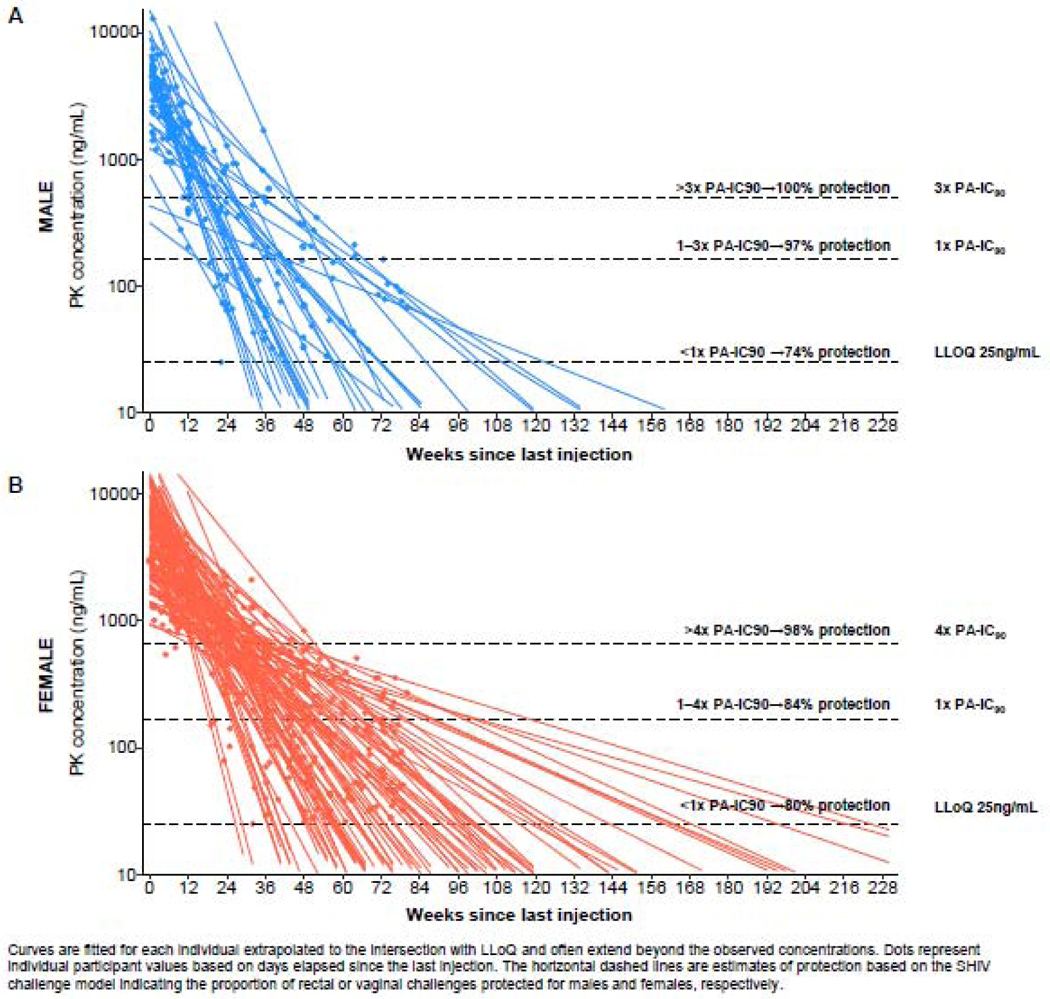
Individual participant log-linear regression curves of plasma CAB concentration over time since Cmax to Clast after the last injection by sex
Figure 2.
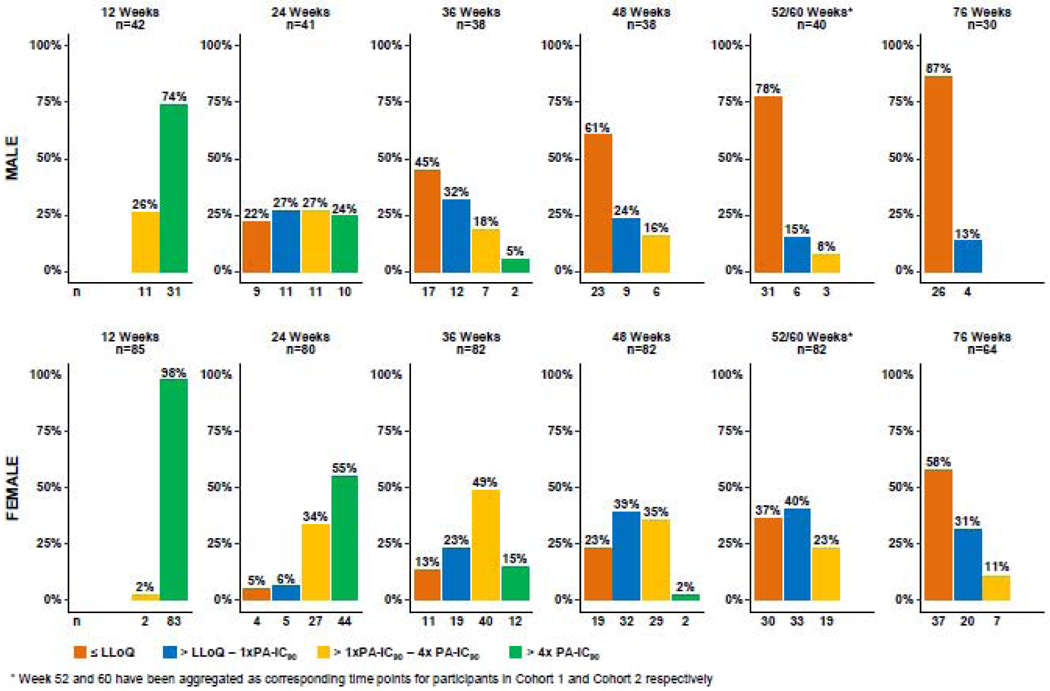
Distribution of participant plasma CAB concentrations aggregated at 12-week intervals after the final injection
Approximately 1 year after the final injection (Week 52/60), 78% of male participants had CAB concentrations below the LLOQ (<25 ng/mL); at 18 months (W76) after the final injection 87% of male participants had CAB concentrations below the LLOQ. For male participants who had detectable CAB concentrations 76 weeks after the final injection, CAB concentrations all fell between the LLOQ and ≤ 1x PA-IC90. In contrast, only 37% of female participants had CAB concentrations below the LLOQ at Week 52/60, and 58% of female participants had CAB concentrations below the LLOQ at Week 76. For Weeks 52/60 and 76, the CAB concentrations of 40% and 31% respectively for female participants with detectable plasma CAB fell between the LLOQ and ≤ 1x-PAIC90; 23% and 11% respectively fell between > 1x and 4x PA-IC90. The proportions of CAB concentrations in each category between male and female participants were significantly different at Week 60 (p=0·0001) and at Week 76 (p=0·01).
The geometric mean of the apparent terminal half-life (t1/2app) was 45·3 days (95%CI 37·6–54·5) for males and 60·4 days (95%CI 52·9–69·0) for females. In simple linear regressions, only sex at birth and BMI were significantly associated with t1/2app (Table 4). The t1/2app was 1·33 times longer in females than males (95%CI 1·06–1·68, p=0·014) and 1·31 times longer in the high BMI group (≥ median) than the low BMI group (< median) (95%CI 1·06–1·63, p=0·015). The associations of sex at birth and BMI remained significant after accounting for all relevant parameters except weight in a multivariable linear regression (1·32- fold increase in Female vs Male, p=0·035; 1·33-fold increase in BMI ≥ median vs < median, p=0·016, Table 4). Although sex at birth and BMI were significantly associated with t1/2app, only 9·4% of the variability in t1/2app was accounted for by these parameters. Notably, the number of injections administered did not influence the elimination rate. The results do not change in a meaningful way if we adjust for cohort in the baseline characteristics analysis. However, in the number of injections analysis, cohort and interaction between number of injections and cohort were included in the model to obtain the effect of number of injections in each cohort when the data were pooled from both cohorts (Table 4). In classical PK studies, the possibility for differences based on prior dosing frequency (with very similar overall dose per unit time precluding concentration-dependent clearance) is almost nonexistent; our findings are therefore reassuring in corroborating that supposition.
Table 4.
Association of the baseline characteristics and # of injections with t1/2app in CAB participants
| Simple Linear Regression | Multivariable Linear Regression | |||||
|---|---|---|---|---|---|---|
| fold-change | 95% CI | P-value | fold-change | 95% CI | P-value | |
| Sex at birth (female vs. male) | 1·33 | (1·06, 1·68) | 0·014 | 1·32 | (1·02, 1·69) | 0·035 |
| BMI (≥median* vs. <median*) | 1·31 | (1·06, 1·63) | 0·015 | 1·33 | (1·06, 1·67) | 0·016 |
| Age | 1·00 | (0·99, 1·01) | 0·759 | 1 | (0·99, 1·01) | 0·834 |
| Weight+ (kg) | 1·01 | (1·00, 1·01) | 0·104 | - | - | - |
| Race (non-Hispanic White as reference) | ||||||
| Hispanic | 1·11 | (0·82, 1·50) | 0·490 | 1·07 | (0·71, 1·63) | 0·742 |
| Non-Hispanic Asian | 1·11 | (0·40, 2·45) | 0·971 | 1·1 | (0·44, 2·75) | 0·846 |
| Non-Hispanic Black | 1·30 | (0·99, 1·72) | 0·059 | 1·42 | (0·95, 2·11) | 0·086 |
| Non-Hispanic Others | 0·91 | (0·50, 1·67) | 0·767 | 0·92 | (0·5, 1·68) | 0·784 |
| Region (US as reference) | ||||||
| Sub-Saharan Africa | 1·23 | (0·95, 1·59) | 0·110 | 1·08 | (0·69, 1·69) | 0·741 |
| Brazil | 1·11 | (0·83,1·50) | 0·473 | 0·81 | (0·54, 1·22) | 0·317 |
| Current Smoker (Yes vs. No) | 0·80 | (0·56,1·13) | 0·204 | 0·83 | (0·58, 1·19) | 0·317 |
| # of Injections | ||||||
| Cohort 1 | 1·09 | (0·88, 1·35) | 0·423 | 1·12 | (0·9, 1·38) | 0·316 |
| Cohort 2 | 0·88 | (0·69, 1·12) | 0·290 | 0·97 | (0·76, 1·24) | 0·785 |
| Cohort (2 vs 1) | 1·02 | (0·82, 1·28) | 0·851 | 1·61 | (0·43, 6·05) | 0·481 |
Median BMI was 27·2 for females and 25·0 for males.
Weight was not included in the multivariable linear regression due to the collinearity between weight and BMI.
To evaluate the sensitivity of these associations to goodness of fit in the terminal slope, we excluded 8 of 130 participants with adjusted R2 < 0·85. With this exclusion, the association of t1/2app with BMI remained significant in both simple and multivariable linear regressions; the association of t1/2app with sex at birth with became slightly weaker, but remained significant in the simple linear regression (p=0·051) and in the multivariable linear regression (p=0·042) (Appendix p. 5).
The median time from the last injection to the time when CAB concentration fell below the LLOQ was 43·7 weeks (IQR 31·1–66·6, range 20·4–152·5) for males and 67·3 weeks (IQR 29·1–89·6, range 17·7225·5) for females (p=0·0003) (Appendix p. 6 – 8). The median (IQR, range) of the time to the LLOQ was 65·4 (49·8–95·3, 19·7–198·2) in the higher BMI group compared with 57·7 (36·2–76·3, 17·7–225·5) in the lower BMI group (p = 0·03).
Discussion
The characterization of the pharmacokinetic tail of long-acting injectable agents is critical to evaluating the clinical consequences of their use for HIV treatment and prevention. The prolonged persistence of CAB LA after intramuscular injection is simultaneously its greatest asset and its greatest potential liability. Once injected into a large muscle, CAB LA (a crystalline suspension of the parent drug cabotegravir) is slowly absorbed into blood plasma over time, providing sustained plasma concentrations. This intramuscular depot cannot be removed once injected, and its metabolism or elimination cannot be accelerated in the event of toxicity. The ability of CAB LA to provide high plasma concentrations for prolonged periods is an attractive advantage over daily administration potentially alleviating pill fatigue and adherence challenges for those in whom daily pill-taking is difficult or impossible. Of course, failure to receive ongoing or timely injections provides a new set of challenges to preventive or therapeutic success. Presuming HIV prophylactic efficacy, there will be a plasma (and/or genital tissue) threshold for such protection. As CAB concentrations wane after a final injection and gradually fall below that threshold, protection will eventually wane, even though non-protective CAB concentrations may persist for some interval. It is during this period that there is a theoretical increased risk for HIV acquisition in the presence of persistent CAB concentrations, which may be inadequate to suppress replication but sufficient to apply selective pressure to select for existing or de novo CAB-, or integrase strand transfer inhibitor (INSTI)-resistant viral quasispecies. An understanding of the concentrations and time intervals at which protection persists post injection, and also the concentrations at which risk of selection for resistance increases will only be determined with clinical experience. Importantly, the NHP experiments in which a single dose of CAB LA was provided to macaques followed by serial rectal or vaginal SHIV/SIV challenges to determine threshold for protection, no INSTI signature mutations or other mutations causing decreased CAB susceptibility were detected in infected animals.13–15
In HPTN 077, we expanded data obtained in the ECLAIR study which demonstrated that 17% of males in the US maintained detectable plasma CAB concentrations 52 weeks after a final injection of 800 mg CAB LA administered every 12 weeks. In addition, our terminal half-life estimate for men, 45·3 days (95%CI 37·6–54·5), largely overlapped with the men in ECLAIR, 40·0 days (95%CI: 35·1–45·7). HPTN 077 evaluated the safety, tolerability, and pharmacokinetics in both females and males, and included analysis of participants in the US, South Africa, Malawi, and Brazil. The study also compared the 800 mg injection dose administered every 12 weeks to a 600 mg dose administered every 8 weeks, since the ECLAIR showed that the every 12-week dose failed to meet NHP-derived pharmacokinetic targets. We aggregated data from both Cohorts in this study to evaluate decay characteristics after the final CAB LA injection; this was done after initial analyses demonstrated no statistically significant difference between terminal pharmacokinetics with these two dosing regimens.
Importantly, we identified no new safety concerns during this tail phase; the rates and types of AEs were similar to or at lower rates than rates seen during the injection phase of the study. No new seroconversion events were reported in this low-risk population during the tail phase of the study. Two new pregnancies occurred in participants who received active CAB LA, one was conceived at a time when CAB concentrations were still detectable. Both pregnancies resulted in healthy infants without birth defects. HPTN 083, an ongoing phase 3 clinical trial enrolling women of child-bearing potential will further define the safety profile of CAB LA in this population.
A large proportion of enrollment occurred in Malawi, South Africa and Brazil, suggesting that some participants were at-risk for tuberculosis (TB) infection. Rifampin, an antibiotic that is commonly used to treat or prevent active TB, is contraindicated with cabotegravir based on an approximately 60% anticipated reduction in AUC for cabotegravir.16While the implications of waning cabotegravir levels in the setting of rifampin use may have implications for duration of protection (should such protection be demonstrated in ongoing Phase 3 trials), they are not anticipated to have implications for efficacy of TB treatment regimens. Therefore, alternative methods of HIV protection should be deployed during periods of rifampin and cabotegravir coadminatration; the time frame of this interaction losing clinical significance during the tail phase remains undefined.
Consistent with data from the treatment literature for CAB LA (in combination with long-acting injectable RPV LA), we found that BMI and sex at birth were strong predictors of the CAB LA t1/2app6 and, therefore, of the estimated time to undetectable plasma levels after the last injection. T1/2app was approximately 29% longer for females than males; independently, t1/2app was approximately 35% longer for high BMI (≥ median) participants compared to low BMI (< median) participants. For each unit increase in BMI, t1/2app increased by approximately 2%. Despite this, under 10% of the variability observed was explained by these two parameters, suggesting that as yet undetermined/unmeasured factors contributed the majority of the observed variability in CAB concentrations. These might include muscle size and/or muscle fat content, host genetics, or injection intended for intramuscular delivery that was inadvertently delivered into the subcutaneous tissues or intravenously. Ongoing studies (NCT02478463) that specifically address theoretical sources of variability that will be useful to better understand the heterogeneity in terminal phase kinetics in order to optimize dosing. We had insufficient numbers of transgender men (six) and transgender women (one) in this low-risk cohort to be able to evaluate the effects of gender affirming hormone therapy.
The low number of seroconversion events in HPTN 077 was expected given the intentionally low-risk population recruited for the study. The likelihood that resistance emergence will only be clearly defined after completion of phase 3 studies and during implementation and scale up would be consistent with the experience with tenofovir disoproxil fumarate/emtricitabine (TDF/FTC) PrEP. With that intervention (1) randomized clinical trials included rare seroconversion events where resistant virus was detected, and (2) case reports describing the parameters of rare seroconversion cases in the presence of protective levels of tenofovir and emtricitabine (and their metabolites) that did not come to light until 4–5 years after initial regulatory approvals.17–22 The frequency of these events with CAB LA (compared to TDF/FTC) may be reduced by the higher resistance threshold of advanced-generation INSTIs and increased by the use of a single drug (CAB LA) in the prevention regimen.
Using these results from the post-injection tail phase of HPTN 077 we estimate that in some cases the concentration of CAB may persist above the limit of quantitation for as long as 4·3 years for females and 2·9 years for males with the longer durations occurring in individuals with higher BMI (Figure S1). This observation has implications for the risk of HIV infection, drug-drug interactions, and resistance after dosing cessation. If CAB is shown to be efficacious for HIV prevention, at-risk individuals who chose to transition off of a long-acting PrEP agent with a long pharmacokinetic tail could transition to oral TDF/FTC or oral CAB, to provide protection against HIV infection; those no longer at risk for HIV exposure would not require additional interventions. Further, a nuanced understanding of the pharmacokinetic tail is similarly relevant to the HIV treatment community and HIV treatment providers. Although the clinical significance of the pharmacokinetic “tail” remains to be defined, the potential advantages of a long-acting injectable preparation provide compelling impetus for investigation of such compounds for HIV treatment and prevention.
The analyses in this report have several limitations. These include curtailing follow-up at 76 weeks after the final injection and relying on extrapolation of earlier post-injection pharmacokinetics to model terminal decay (although model fits are generally excellent); use of an assay with a LLOQ of 25 ng/mL (limiting the ability to characterize time to complete drug washout, though demonstrating washout to levels well below concentrations associated with protection in NHP models, although absent pharmacokinetic correlates of infection, these estimates provide a useful benchmark while awaiting such data); and an incomplete population with follow-up to 76 weeks. Additionally, it was not possible to determine whether injections were delivered exclusively intramuscularly (as intended), in fat or subcutaneously; this variable may be important for understanding the heterogeneity of terminal phase pharmacokinetics and would likely be relevant to drug delivery practices encountered in actual clinical settings.
Conclusions
The tail phase of CAB LA was well tolerated. The apparent terminal plasma half-life of CAB was 33% longer (60 days) for females than for males (45 days) and was also independently influenced by BMI. Similarly, the median time from final injection to undetectable plasma CAB levels was 55% longer for females (66 weeks) than for males (43 weeks). The ongoing Phase 3 CAB LA efficacy studies, HPTN 083 and HPTN 084, may provide information about the relevance of these sex-based differences to preventive efficacy and risk of drug resistance. However, these associations may only be apparent with large-scale clinical use of CAB LA for HIV prevention.
Supplementary Material
Acknowledgments
RJL has received study medication, consulting fees and travel support from Gilead Sciences, and consulting fees and travel support from Merck, Inc. SL declares no competing interests. JJE is a consultant to Merck, Gilead Sciences, Janssen, and ViiV Healthcare; and he is an investigator on research contracts from ViiV Healthcare, Janssen, and Gilead Sciences. BG declares no competing interests. HD has received honoraria from Pfizer-South Africa, Novartis-South Africa, MSD-South Africa and Adcock Ingram for speaking engagements and has received travel support from Mylan-South Africa, Novartis-South Africa and Aspen-South Africa. AYL received research grants from NIH and has led studies in which study drug was donated by Gilead Sciences. MM, MCH, RP LC, GC, and PR declare no competing interests. MAM received grant support through the NIH and received grant support through ViiV/GSK on work external to this study. SHE reports grants from NIH. RK, AA, and DB declare no competing interests. ARR and DM are paid employees of ViiV Healthcare. MSC reports other support from Merck and Gilead.
CWH has research contracts with NIH, ViiV/GlaxoSmithKline, Gilead Sciences, and Merck through Johns Hopkins.
Funding: National Institute of Allergy and Infectious Diseases (UM1AI068619, UM1AI068613, and, UM1AI068617)
Footnotes
Data Sharing
De-identified individual participant data that underlie the results reported in this Article (text, tables, figures, and appendices) will be shared upon request. Proposals and data requests should be directed to Sue Li (sli@fredhutch.org). Those requesting de-identified data may be required to sign a data access agreement.
Declaration of Interests
MM declares no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Raphael J. Landovitz, UCLA Center for Clinical AIDS Research and Education, Division of Infectious Diseases, David Geffen School of Medicine at UCLA, 11075 Santa Monica Blvd, Suite 100, Los Angeles, CA 90025.
Sue Li, Statistical Center for HIV/AIDS Research and Prevention, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N. Mail Stop M2-C200, Seattle, WA 98109.
Joseph J. Eron, Jr., University of North Carolina, Chapel Hill, CB# 7030, Bioinformatics Building, 130 Mason Farm Road, 2nd Floor, Chapel Hill, North Carolina 27599-7030.
Beatriz Grinsztejn, Evandro Chagas National Institute of Infectious Diseases (INI), FIOCRUZ, Av. Brasil 4365, Manguinhos, 21040-360 Rio de Janeiro, Brazil.
Halima Dawood, CAPRISA, University of KwaZulu Natal, 2nd Floor, Doris Duke Medical Research Institute, 719 Umbilo Road, Durban, 4041, South Africa.
Albert Y. Liu, Bridge HIV, Population Health Division, San Francisco Department of Health, 25 Van Ness, Suite 100, San Francisco, CA 94102.
Manya Magnus, Milken Institute School of Public Health at The George Washington University, Department of Epidemiology, 950 New Hampshire Avenue, NW, Suite 507, Washington, District of Columbia 20052.
Mina C. Hosseinipour, UNC Project-Malawi, Tidziwe Centre Private Bag A-104, Lilongwe Malawi.
Ravindre Panchia, Perinatal HIV Research Unit, Chris Hani Baragwanath Hospital, Diepkloof, 1864, Soweto, South Africa.
Leslie Cottle, Statistical Center for HIV/AIDS Research and Prevention, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N. Mail Stop M2-C200, Seattle, WA 98109.
Gordon Chau, Statistical Center for HIV/AIDS Research and Prevention, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave. N. Mail Stop M2-C200, Seattle, WA 98109.
Paul Richardson, School of Medicine, Johns Hopkins University, 733 N Broadway, Baltimore, MD 21205.
Mark A. Marzinke, School of Medicine, Johns Hopkins University, 1800 Orleans St., Baltimore, MD 21287.
Susan H. Eshleman, School of Medicine, Johns Hopkins University, Ross 646, 720 Rutland Ave., Baltimore, MD 21205.
Ryan Kofron, UCLA Center for Clinical AIDS Research & Education, 11075 Santa Monica Blvd, Suite 100, Los Angeles, CA 90025.
Adeola Adeyeye, DAIDS/NIAID/NIH, 5601 Fishers Lane, Rm 8B36, Rockville, MD 20852-9831.
David Burns, DAIDS/NIAID/NIH, 5601 Fishers Ln, Rm. 8B40, Rockville MD 20852.
Alex R. Rinehart, ViiV Healthcare, 5 Moore Drive, Durham, NC 27713.
David Margolis, ViiV Healthcare, 5 Moore Drive, Durham, NC 27713.
Myron S. Cohen, University of North Carolina, Chapel Hill, CB# 7030, Bioinformatics Building, 130 Mason Farm Road, 2nd Floor, Chapel Hill, North Carolina 27599-7030.
Marybeth McCauley, FHI360, 1825 Connecticut Avenue NW, Washington, DC 20009.
Craig W. Hendrix, School of Medicine, Johns Hopkins University, Blalock 569, 600 N. Wolfe Street, Baltimore, MD 21287.
References
- 1.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010; 363(27): 2587–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med. 2012; 367(5): 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med. 2012; 367(5): 423–34. [DOI] [PubMed] [Google Scholar]
- 4.Molina J-M, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med. 2015; 373(23): 2237–46. [DOI] [PubMed] [Google Scholar]
- 5.McCormack S, Dunn DT, Desai M, et al. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet. 2016; 387(10013): 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landovitz RJ, Li S, Grinsztejn B, et al. Safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in low-risk HIV-uninfected individuals: HPTN 077, a phase 2a randomized controlled trial. PLoS medicine 2018; 15(11): e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markowitz M, Frank I, Grant RM, et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV 2017; 4(8): e331–e40. [DOI] [PubMed] [Google Scholar]
- 8.Babor TF, Higgins-Biddle JC, Saunders JB, Monteiro MG. The alcohol use disorders identification test Guidelines for use in primary health care Geneva: World Health Organization; 1992. [Google Scholar]
- 9.Health UDo, Services H. National Institute of Allergy and Infectious Diseases, Division of AIDS. Division of AIDS (DAIDS) Table for Grading the Severity of Adult and Pediatric Adverse Events, Version 2.0.[November 2014]. 2017.
- 10.Center for Drug Evaluation and Research, Center for Veterinary Medicine. Guidance for Industry: Bioanalytical Method Validation. US FDA, Rockville, MD, USA: (2001). [Google Scholar]
- 11.DiFrancesco R, Tooley K, Rosenkranz SL, Siminski S, Taylor CR, Pande P, Morse GD. Clinical Pharmacology Quality Assurance for HIV and Related Infectious Diseases Research. Clin Pharmacol Ther. 2013; 93 (6): 479–82. PMCID: PMC3970711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Radzio J, Spreen W, Yueh Y, et al. The long-acting integrase inhibitor GSK744 protects macaques from repeated intravaginal SHIV challenge. Sci Transl Med. 2015; 7 (270): 270ra5. [DOI] [PubMed] [Google Scholar]
- 13.Spreen W, Lowry A, Pal R, et al. Correlation of in vivo cabotegravir concentration and prevention of SIV in macaques [Abstract 966LB]. 22nd Conference on Retroviruses and Opportunistic Infections (CROI); 23–26 February 2015; Seattle, Washington. [Google Scholar]
- 14.Andrews CD, Spreen WR, Mohri H, et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science. 2014; 343(6175): 1151–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andrews CD, Yueh YL, Spreen WR, et al. A long-acting integrase inhibitor protects female macaques from repeated high-dose intravaginal SHIV challenge. Sci Transl Med. 2015; 7(270): 270ra4-ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ford SL, Sutton K, Lou Y, Zhang Z, Tenorio A, Trezza C, Patel P, Spreen W. Effect of rifampin on the single-dose pharmacokinetics of oral cabotegravir in healthy subjects. Antimicrobial agents and chemotherapy. 2017. October 1;61(10):e00487–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knox DC, Anderson PL, Harrigan PR, Tan DH. Multidrug-resistant HIV-1 infection despite preexposure prophylaxis. N Engl J Med. 2017; 376(5): 501–2. [DOI] [PubMed] [Google Scholar]
- 18.Markowitz M, Grossman H, Anderson PL, et al. Newly acquired infection with multidrug-resistant HIV-1 in a patient adherent to preexposure prophylaxis. J Acquir Immune Defic Syndr. 2017; 76(4): e104–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoornenborg E, Prins M, Achterbergh RC, et al. Acquisition of wild-type HIV-1 infection in a patient on pre-exposure prophylaxis with high intracellular concentrations of tenofovir diphosphate: a case report. Lancet HIV. 2017; 4(11): e522–e8. [DOI] [PubMed] [Google Scholar]
- 20.Thaden JT, Gandhi M, Okochi H, Hurt CB, McKellar MS. Seroconversion on preexposure prophylaxis: a case report with segmental hair analysis for timed adherence determination. AIDS. 2018; 32(9): F1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colby DJ, Kroon E, Sacdalan C, et al. Acquisition of multidrug-resistant human immunodeficiency virus type 1 infection in a patient taking preexposure prophylaxis. Clin. Infect. Dis. 2018; 67(6): 962–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen SE, Sachdev D, Lee SA, et al. Acquisition of tenofovir-susceptible, emtricitabine-resistant HIV despite high adherence to daily pre-exposure prophylaxis: a case report. Lancet HIV. 2019; 6(1): e43–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.