

Abstract
Although Huntington’s disease is a late-manifesting neurodegenerative disorder, both mouse studies and neuroimaging studies of presymptomatic mutation carriers suggest that Huntington’s disease might affect neurodevelopment. To determine whether this is actually the case, we examined tissue from human fetuses (13 weeks gestation) that carried the Huntington’s disease mutation. These tissues showed clear abnormalities in the developing cortex, including mislocalization of mutant huntingtin and junctional complex proteins, defects in neuroprogenitor cell polarity and differentiation, abnormal ciliogenesis, and changes in mitosis and cell cycle progression. We observed the same phenomena in Huntington’s disease mouse embryos, where we linked these abnormalities to defects in interkinetic nuclear migration of progenitor cells. Huntington’s disease thus has a neurodevelopmental component and is not solely a degenerative disease.
Huntington’s disease (HD) is a neurodegenerative disease that is part of the larger family of “proteopathies,” which includes the polyglutamine diseases, amyotrophic lateral sclerosis, and Alzheimer’s and Parkinson’s diseases. These diverse disorders share a delayed onset in mid-adulthood or later despite the expression, at least in hereditary cases, of the disease-driving protein from the first days of life. This raises the question of whether early events might set the stage for later disease. For example, huntingtin (HTT), the protein mutated in HD, is essential for development, at least in mice (1–3). The mutant HTT (mHTT) impairs neural progenitor cell division and neuronal migration and maturation (4–6), giving HD mice a thinner cortex (7). The fact that expression of either mHTT or hypomorphic HTT solely during early life is sufficient to produce HD features in adult mice strongly suggests that there is a developmental component to the disease (8, 9).
In support of this notion, human neuroimaging studies have revealed smaller intracranial volume in HD mutation carriers as young as 7 years of age (10, 11). Loss of cortical volume takes place long before any symptoms appear, and defects in the corticostriatal network lead to striatal dysfunction and degeneration (12–15). Studies in neurons derived from HD human induced pluripotent stem cells (iPSCs) have identified changes in gene expression that support an altered developmental program (16, 17), and mHTT alters neuronal identity in cortical populations of HD brain organoids (18). But does mHTT affect early human development? And if so, how early? To answer these questions, we recruited HD mutation carriers who sought prenatal testing in order to determine whether the fetus carried an HD-causing mutation.
Mutant huntingtin mislocalizes in human and mouse embryos
We were able to procure rare intact cortical tissues from four HD mutation carrier fetuses and four healthy controls at gestation week 13 (GW13) (table S1). At this developmental stage, the cortical neurons that project to the striatum and later deteriorate in HD are arising from the division of progenitor cells at the ventricular zone. These apical progenitors extend processes toward both the apical and basal surfaces of the neuroepithelial wall, and their nuclei move back and forth between surfaces in concert with cell cycle progression in a process known as interkinetic nuclear migration. This process, common to all developing pseudostratified neuroepithelia (19, 20), maintains the balance between progenitor renewal and differentiation by controlling when apical progenitor nuclei are exposed to proliferative versus neurogenic signals, and in what proportions.
To examine the expression pattern of HTT at the ventricular zone of the GW13 cortex, we used an antibody that recognizes both HTT and mHTT (4C8; Fig. 1, A and B, and fig. S1A). In wild-type tissues, HTT staining demarcated the apical surface of the ventricular zone and spread diffusely throughout the basal region. In cortical tissue of HD mutation carriers, however, HTT staining concentrated at the apical endfeet (the apical surface of the processes).
Fig. 1. Huntingtin and junctional complex proteins mislocalize in the ventricular zone of human fetuses carrying HD-causing mutations.
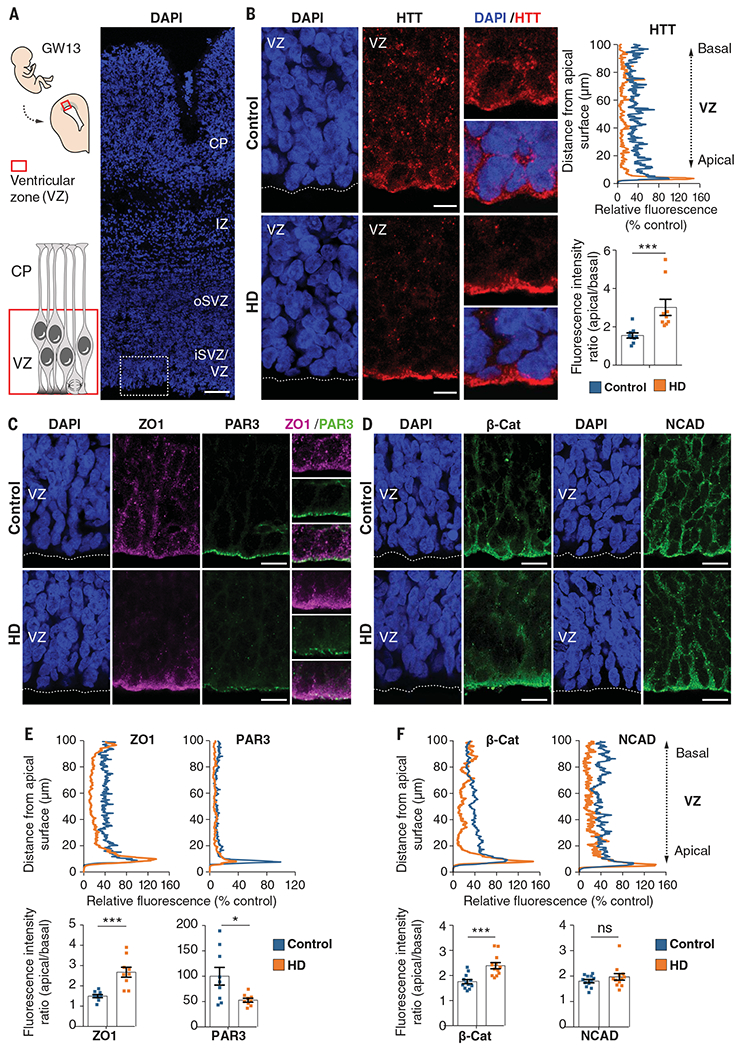
(A) Left: Diagram showing the position of the fetal ventricular zone relative to the cortical plate (CP). Right: Coronal brain sections of GW13 control human cortex were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). The dotted square shows the region imaged in (B). Scale bar, 100 μm. (B) Left: Coronal GW13 brain sections from control fetus and fetus carrying HD-causing mutation were immunostained for HTT. Scale bars, 10 μm. Right: Representative line-scan analysis (relative fluorescence intensity) of HTT immunostaining and quantification of apical/basal human HTT fluorescence intensity in the ventricular zone. For each condition, n = 3 fetuses from different mothers; ***P = 0.0044 (unpaired t test). (C and D) Coronal GW13 fetal brain sections were immunostained for ZO1 and PAR3 (C) and β-catenin and NCAD (D). Scale bars, 15 μm. (E and F) Representative line-scan analysis (relative fluorescence intensity) of indicated immunostainings (top) and quantification of indicated fluorescence intensities in the ventricular zone (bottom graphs). For each condition, n = 3 fetuses from different mothers. ZO1: ***P = 0.0003 (unpaired t test); PAR3: *P = 0.0177 (unpaired t test); β-cat: ***P = 0.0003 (unpaired t test); NCAD: P = 0.4682 (Mann-Whitney U test), ns (not significant). Results are means ± SEM. VZ, ventricular zone; iSVZ, inner subventricular zone; oSVZ, outer subventricular zone; IZ, intermediate zone; CP, cortical plate. Nuclei were counterstained with DAPI.
Given the preciousness of the human tissue, we turned to mice to further investigate these observations, using embryonic day 13.5 (E13.5) mouse embryos, which correspond to GW13 in human neurodevelopment. We studied an HD knock-in mouse model in which the first exon of the HTT gene is replaced by human exon 1 carrying 111 CAG repeats (HdhQ111/Q111) (21). Immunostaining coronal sections revealed a pattern of HTT expression that paralleled our observations in human fetuses (fig. S1B). To determine the distribution of mHTT specifically, we used another HD knock-in mouse model in which Flag tags are inserted in the N terminus of wild-type HTT (HdhF7Q/+) or mutant HTT carrying 140 CAG repeats (HdhF140Q/+) (22) (fig. S1C). The Flag labeling showed that mHTT localized to the apical surface and was decreased in the basal region.
Mutant huntingtin impairs endosome secretion and recycling
Apical progenitors maintain their polarity through endocytosis and the trafficking of proteins from the trans-Golgi network to the plasma membrane at the apical endfeet (19). In HD, both endocytosis and Golgi-membrane trafficking are dysregulated (23). Because one of HTT’s main functions is to transport vesicles, we used markers of the endosomal pathway to map the subcellular localization of HTT in HD and to gauge whether transport is affected this early in HD.
We stained for calnexin (a marker of the endoplasmic reticulum), GRASP65 (Golgi assembly stacking protein of 65 kDa to mark the cis-Golgi network), TGN38 (trans-Golgi network integral membrane protein 38), EEA1 (early endosome antigen 1), and transferrin receptor (recycling endosomes). In control samples, HTT colocalized partially with these markers (figs. S2 and S3). In both human and mouse HD samples, however, HTT strongly colocalized with TGN38, EEA1, and transferrin receptor, and to a lesser extent with calnexin and GRASP65. These results suggest that mHTT hinders endosomal trafficking in apical progenitors, even at this very early stage of development.
Mutant huntingtin disrupts neuroepithelial junctional complexes
Apical endfeet contain junctional complexes (19, 24) composed of tight-junction and adherens-junction proteins, including ZO1, PAR3, NCAD, and β-catenin (25), that link neighboring progenitors to each other, thereby sealing the neuroepithelium. Because HTT regulates the trafficking of these proteins, which are dysregulated in HD (6, 26–30), we hypothesized that mHTT hinders the correct positioning of these junctions, which would diminish the integrity of the neuroepithelium. As predicted, HTT partially codistributed with ZO1, PAR3, β-catenin, and NCAD at the apical endfeet of human GW13 control and mouse E13.5 neuroepithelium (figs. S4 and S5A). The levels of ZO1, NCAD, and β-catenin were high at the apical surface of the human and mouse control ventricular zone and even higher in HD tissues, with a concomitant reduction in these proteins in the basal region (Fig. 1, C to F, and fig. S5, B to E). PAR3 was also misregulated in HD but in a different pattern from these other proteins: Its expression levels were down-regulated, so its demarcation of the apical surface in control samples was diminished, rather than intensified, in HD.
To better understand how junctional complexes in individual apical endfeet are affected in HD, we electroporated E13.5 control and HD mouse embryos in utero with a pCAG-GFP (green fluorescent protein) construct and performed immunohistochemistry on E15.5 coronal sections (Fig. 2, A and B). At this stage in mice and at the corresponding stage GW16 in humans (table S1), the mislocalization of HTT and junction proteins in HD apical progenitors persisted (figs. S6 and S7). Indeed, GFP-expressing knock-in HD progenitors, but not controls, showed a bright line of HTT along the apical surface (Fig. 2B). In control embryos, ZO1, NCAD, and β-catenin immunostaining marked the sides of the apical endfeet; PAR3 staining was more apical (Fig. 2C). In HdhQ111/Q111 embryos, ZO1, NCAD, and β-catenin spread throughout the apical endfeet and PAR3 staining was diminished. These observations were corroborated by immunoblotting protein extracts from HdhQ7/Q7 and HdhQ111/Q111 E15.5 cortices (Fig. 2D). The levels of NCAD and β-catenin were similar in control and HD conditions, but ZO1 and PAR3 protein levels were lower in the mutant mice. Coimmunoprecipitation showed that HTT associates with ZO1, PAR3, and β-catenin, but these interactions were disrupted in HD (Fig. 2E and fig. S8).
Fig. 2. Junctional protein complexes are disrupted in the apical endfeet of HD mouse embryos.
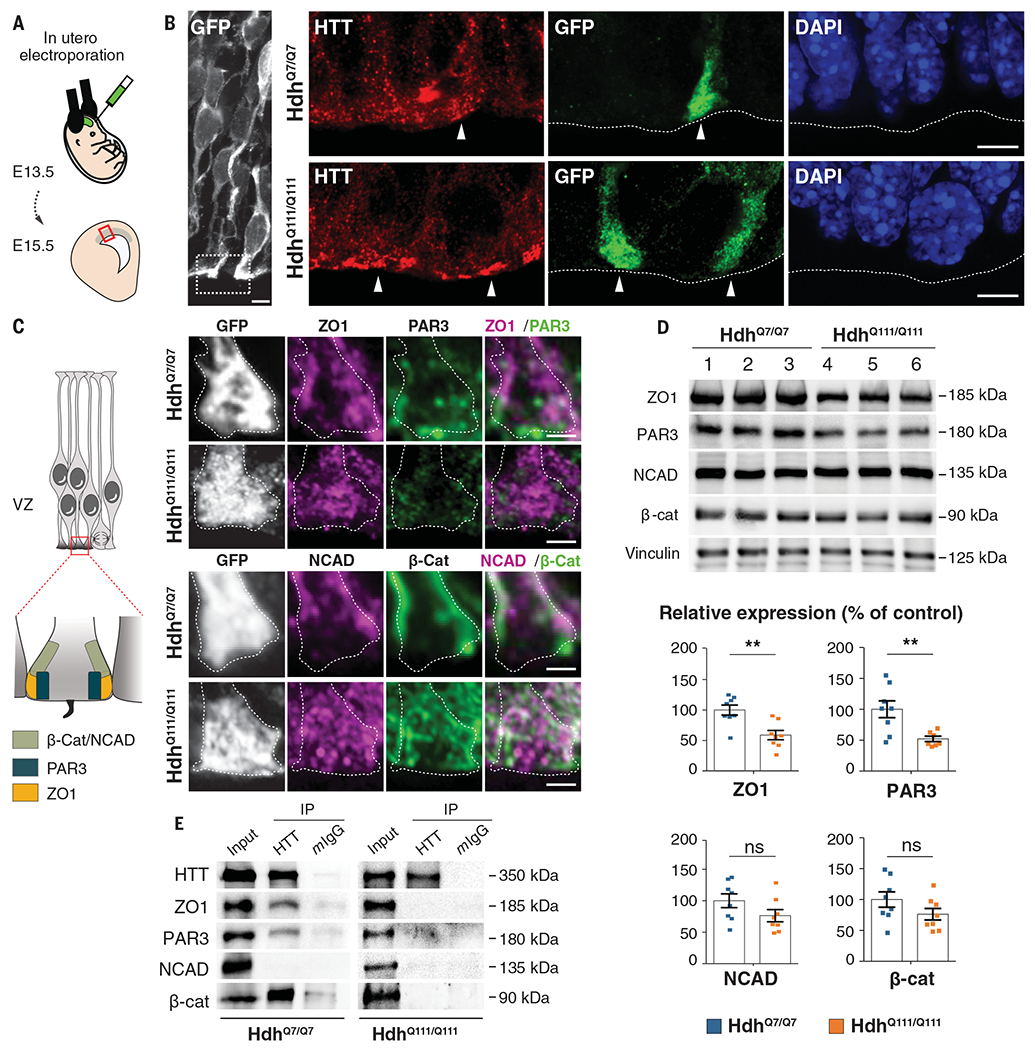
(A) Schematic of the in utero electroporation experiment. (B and C) Mouse embryos were electroporated at E13.5 with a pCAG-GFP construct to delineate the apical endfoot in E15.5 cortices. (B) HdhQ7/Q7 and HdhQ111/Q111 cortical sections were immunostained for GFP (left) and for HTT and GFP (right). White arrowheads point to apical endfeet. Nuclei were counterstained with DAPI. (C) Left: Diagram indicating the position of junctional complexes at the apical endfeet. Right: Cortical sections were immunostained for GFP, ZO1, and PAR3 (upper panel) and GFP, NCAD, and β-Cat (lower panel). Scale bars, 5 μm (B), 2 μm (C). (D) ZO1, PAR3, NCAD, β-catenin, and vinculin immunoblotting analyses of lysates from E15.5 HdhQ7/Q7 and HdhQ111/Q111 cortices. Bar graphs correspond to the quantitative evaluation of the indicated proteins. For each condition, n = at least 7 embryos from different mothers. ZO1: **P = 0.0026; PAR3: **P = 0.0075; NCAD: P = 0.1255; β-cat: P = 0.1476 (unpaired t tests). Results are means ± SEM. (E) HTT-associated complexes were immunoprecipitated with the 4C8 antibody from E15.5 HdhQ7/Q7 and HdhQ111/Q111 cortical extracts. Mouse IgG (mIgG) was used as a negative control.
Mutant HTT alters progression through the cell cycle
The integrity of the apical junctional complexes is essential for progression through interkinetic nuclear migration, when the nuclei of progenitor cells born at the apical surface move toward the basal side during the G1 phase of the cell cycle, enter and complete the S phase, then return to the apical surface, where they undergo division (19, 20, 31, 32). Given that the junctional complexes do not form properly with mHTT, we examined cell cycle progression in the apical progenitors.
To measure apical (G1 phase) and basal (G2 phase) movements in vivo, we used the fluorescent ubiquitination-based cell cycle indicator (FUCCI), which tracks the expression of markers of the different phases of the cell cycle (33). We electroporated wild-type E13.5 embryos with plasmids encoding CDT1 (chromatin licensing and DNA replication factor 1)–mKO2 and geminin-GFP, then carried out time-lapse imaging on acute cortical slices 2 days after in utero electroporation (Fig. 3A) so that we could distinguish cycling progenitors in G1 from neurons exiting the cell cycle and migrating away from the ventricular zone (fig. S9A and movie S1). As expected, CDT1 levels peaked during G1 and fell upon entry into S phase, whereas geminin levels were high during S phase and G2 (fig. S9B and movie S2). The velocities of nuclear movement in G1 and G2 in control cells were as previously reported (34) (Fig. 3, B, C, and E, and movies S3 to S6), but in HdhQ111/Q111 embryos, migrating nuclei moved more slowly in both G1 and G2, causing these phases to lengthen while the G1/S phase transition was shortened (Fig. 3, D and E, and movies S7 and S8). We next immunostained cortical sections of HdhQ111/Q111 embryos and GW13 HD carrier fetuses with antibody against phospho-histone 3 (PH3), a marker of mitosis, and evaluated the mitotic index (Fig. 3F and Fig. 4, A and B). HD mice and human mutation carriers had roughly half the mitotic index of controls. In HD, therefore, the pool of proliferating cells is diminished.
Fig. 3. Interkinetic nuclear migration and mitosis of cortical apical progenitors are impaired in HD mouse embryos.
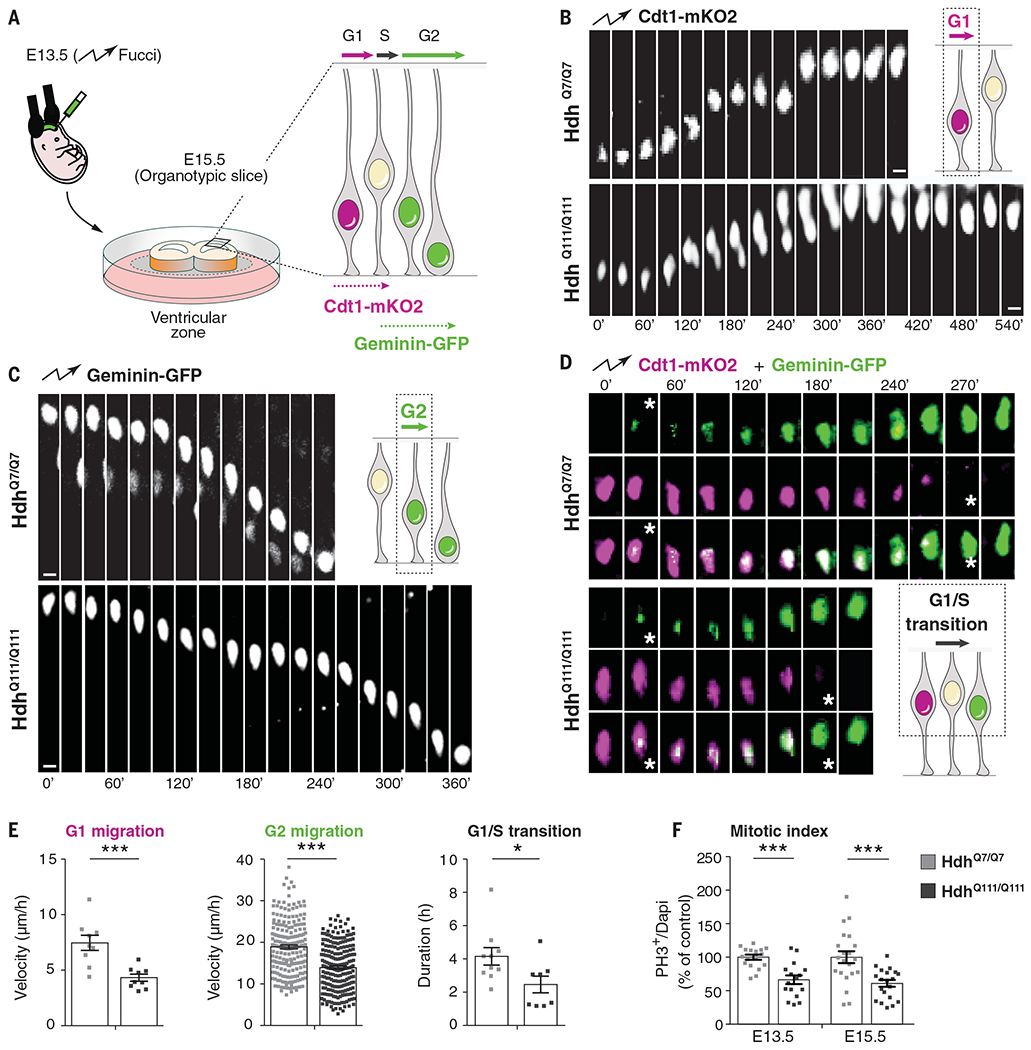
(A) Schematic of the experiment for analysis of interkinetic nuclear migration. E13.5 HdhQ7/Q7 and HdhQ111/Q111 embryos were electroporated with Cdt1-mKO2 and geminin-GFP constructs. After 48 hours, the movement of the GFP- and mKO2-labeled nuclei was followed by spinning disc microscopy, taking one image every 10 or 15 min for 10 hours. (B to D) Representative images showing the movement of nuclei in G1, G2, and G1/S transition phases as indicated. (D) Stars indicate the beginning and ending of the G1/S transition. Scale bars, 5 μm. (E) Quantitative differences in the velocity of G1-phase nuclei [for each condition, n = 9 cells from three embryos from different mothers; ***P = 0.0008 (unpaired t test)], velocity of G2-phase nuclei [for each condition, n = at least 202 cells from four embryos from different mothers; ***P < 0.0001 (Mann-Whitney U test)], and length of G1/S transition [for each condition, n = at least 8 cells from three embryos from different mothers; *P = 0.0356 (unpaired t test)]. (F) Bar graphs show the percentage of phospho-histone 3 (PH3) cells (mitotic index) of dividing progenitors [E13.5: for each condition, n = at least 2151 cells from four embryos from different mothers, ***P < 0.0001 (unpaired t test); E15.5: for each condition, n = at least 1801 cells from three embryos from different mothers, ***P = 0.0005 (unpaired t test)]. Results are means ± SEM.
Fig. 4. Mutant huntingtin shifts neurogenesis toward neuronal lineage.
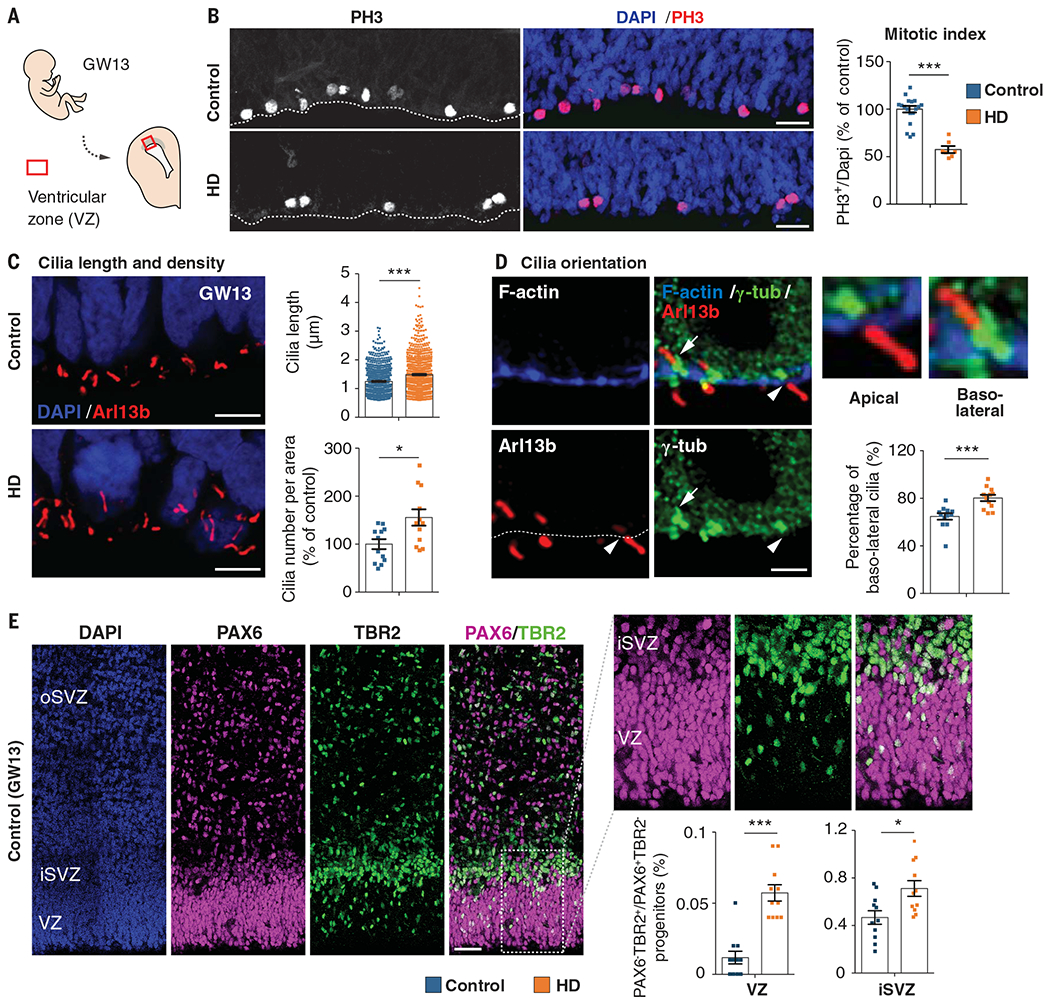
(A) Diagram showing the position of the fetal ventricular zone. (B) Cortical sections of GW13 fetuses were immunostained with antibody against phospho-histone 3 (PH3) and the mitotic index was quantified. For each condition, n = at least 1146 cells from three fetuses from different mothers; ***P < 0.0001 (Mann-Whitney U test). Scale bars, 25 μm. (C) Coronal GW13 brain sections from control fetus and fetus carrying HD-causing mutation (HD) were immunostained for the cilia marker Arl13b. Scale bars, 5 μm. Bar graphs show cilia length [for each condition, n = at least 770 cilia from four fetuses from different mothers; ***P < 0.0001 (Mann-Whitney U test)] and cilia density [for each condition, n = 4 fetuses from different mothers; *P = 0.0104 (unpaired t test)] at the apical surface. (D) Coronal brain sections of GW13 human cortex were immunostained for F-actin, γ-tubulin (γ-tub), and Arl13b. Scale bars, 2 μm. White arrowheads and white arrows show apical and basolateral cilia, respectively. Bar graph shows the percentage of basolateral cilia at the apical surface. For each condition, n = at least 260 cilia from four fetuses from different mothers; ***P = 0.0003 (Mann-Whitney U test). (E) Typical PAX6 and TBR2 staining of a GW13 human fetal sample analyzed. Scale bars, 50 μm. Bar graphs show the percentage of PAX6/TBR2-positive cells (PAX6+/TBR2+) over PAX6-positive, TBR2-negative (PAX6+/TBR2−) progenitors [for each condition, three fetuses from different mothers were analyzed; VZ, n = at least 2447 cells, ***P < 0.0001 (Mann-Whitney U test); iSVZ, n = at least 1580 cells, *P = 0.011 (unpaired t test). Results are means ± SEM. Nuclei were counterstained with DAPI.
Mutant HTT biases neurogenesis toward the neuronal lineage
The cell cycle correlates with the assembly (during G0) and disassembly (at the onset of M phase) of the primary cilium at the apical progenitor endfeet (19, 32). Immunostaining with the cilia marker Arl13b, a member of the adenosine diphosphate ribosylation factor-like family, revealed that both the length and density of the cilia were greater at the apical area of the developing cortex in HD human and mouse samples than in controls (Fig. 4C and fig. S10A), which confirms that the cells were not progressing through the cell cycle properly (20). Because a longer G1 phase and a shorter G1/S transition characterize progenitors committed toward the neuronal lineage (32, 35), we asked whether mHTT favors the production of apical over basal progenitors.
We evaluated cilia orientation by labeling brain sections with F-actin (to delineate the apical surface) and Arl13b and γ-tubulin (to label the basal body) (Fig. 4D and fig. S10B). The proportion of basolateral cilia, which signal the generation of basal progenitors (36), was greater in HD human and mouse samples than in controls. To discriminate between apical progenitors and basal progenitors, which are more engaged in the neuronal lineage (37), we labeled for the transcription factors PAX6 and TBR2, respectively. HD human and mouse samples showed a greater proportion of basal progenitors at the ventricular zone, subventricular zone, and inner subventricular zone than did controls (Fig. 4E and fig. S10C).
Discussion
Our data show that mHTT mislocalizes at junctional complexes, disrupts the polarity of human and mouse neuroepithelium, and interferes with the cell cycle of apical progenitors, leading to fewer proliferating cells and more neural progenitors prematurely entering lineage specification. This is consistent with previous evidence that HTT regulates cellular adhesion, polarity, and epithelial organization (27). In the presence of mHTT, the epithelial-mesenchymal transition is accelerated (28). It is possible that mHTT contributes to cellular disorganization through other means as well, such as by interfering with the orientation of the mitotic spindle (7). Given that HTT also establishes apical polarity in the mammary epithelium, where it forms a complex with PAR3, aPKC, and RAB11A and ensures the apical translocation of PAR3-aPKC through RAB11A (29), we speculate that HTT may act to maintain epithelial cell polarity throughout the body.
A recent neuroimaging study found that the posterior Sylvian fissure, normally asymmetrical between the right and left hemispheres, lacks asymmetry in the HD population studied (38). Because the Sylvian fissure appears early in utero, the authors concluded that this abnormal symmetry arises during fetal development. Our results show that mHTT does alter very early stages of brain development in human HD, even though the samples we analyzed were from mutation carriers with small pathological expansions (39, 40, and 42 repeats) that would typically cause later manifestations of HD. The defects we observed likely render the corticostriatal circuitry more vulnerable to the later dysfunctions characteristic of HD (23), as proposed for another polyglutamine disease, spinocerebellar ataxia type 1 (39). The path to degeneration is complex, however, and weaves together both pathogenic and compensatory mechanisms. For example, a recent study found that HD mutation carriers as young as 6 years of age show compensatory hyperconnectivity between the striatum and cerebellum; this initially enlarges the striatum but the metabolic load soon overwhelms it, the connections are rapidly lost, and the striatum atrophies well before the onset of motor symptoms (40).
It is now beyond doubt that neurodegenerative diseases can have a developmental component. For HD, this discovery opens the door for future studies to identify molecular treatments. For example, the HD iPSC Consortium characterized isoxazole-9 after finding that it reverts abnormal neuronal differentiation in HD-derived pluripotent stem cells (17). It may be that treatment should be given very early in life; it remains to be seen whether reducing mHTT levels in adulthood, even in the prodromal stage, would be sufficient to forestall symptom progression, because the brain circuitry is already altered.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the patients and their families. We thank L. Benammar and M. Biet for their logistical help; midwife O. Philippon for her support; C. Benstaali and E. Martin for help with experiments; staff of the animal facility and of the Photonic Imaging Center (part of the ISdV core facility and certified by the IBiSA label) of GIN for technical help; F. Saudou, members of the Humbert laboratory, and V. L. Brandt for helpful discussions; and the Association Huntington France (AHF) for continuous support.
Funding: Supported by grants from Agence Nationale pour la Recherche (ANR-15-IDEX-02 NeuroCoG, S.H.; Network of centers of excellence in neurodegeneration COEN, A.D. and S.H.); Fondation pour la Recherche Médicale (DEQ 20170336752, S.H.); Fondation pour la Recherche sur le Cerveau (S.H.); and the AGEMED program from INSERM (S.H.). M.B. and S.H. are INSERM investigators. E.A. was supported by a CIFRE (2012-0401) doctoral fellowship. M.C. is supported by a La ligue contre le cancer post-doctoral fellowship. J.-P.L. and S.O.Z. are supported by NIH NS077926.
Footnotes
Competing interests: None declared.
Data and materials availability: All data are available in the manuscript or the supplementary materials.
REFERENCES AND NOTES
- 1.Duyao MP et al. , Science 269, 407–410 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A, Nat. Genet 11, 155–163 (1995). [DOI] [PubMed] [Google Scholar]
- 3.Reiner A, Dragatsis I, Zeitlin S, Goldowitz D, Mol. Neurobiol 28, 259–276 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Godin JD et al. , Neuron 67, 392–406 (2010). [DOI] [PubMed] [Google Scholar]
- 5.McKinstry SU et al. , J. Neurosci 34, 9455–9472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnat M, Le Friec J, Benstaali C, Humbert S, Neuron 93, 99–114 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Molina-Calavita M et al. , J. Neurosci 34, 10034–10040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arteaga-Bracho EE et al. , Neurobiol. Dis 96, 144–155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molero AE et al. , Proc. Natl. Acad. Sci. U.S.A. 113, 5736–5741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JK et al. , Neurology 79, 668–674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nopoulos PC et al. , Brain 134, 137–142 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabrizi SJ et al. , Lancet Neurol. 10, 31–42 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Tang CC et al. , J. Clin. Invest 123, 4076–4088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Virlogeux A et al. , Cell Rep. 22, 110–122 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Zhao X et al. , Proc. Natl. Acad. Sci. U.S.A. 113, E5655–E5664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ring KL et al. , Stem Cell Rep. 5, 1023–1038 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.HD iPSC Consortium, Nat. Neurosci. 20, 648–660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conforti P et al. , Proc. Natl. Acad. Sci. U.S.A. 115, E762–E771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aral Y, Taverna E, Front. Cell. Neurosci. 11, 384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyata T, Okamoto M, Shinoda T, Kawaguchi A, Front Cell. Neurosci. 8, 473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler VC et al. , Hum. Mol. Genet 11, 633–640 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Zheng S, Ghitani N, Blackburn JS, Liu JP, Zeitlin SO, Mol. Brain 5, 28 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saudou F, Humbert S, Neuron 89, 910–926 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Chou FS, Li R, Wang PS, Cell. Mol. Life Sci 75, 1027–1041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bultje RS et al. , Neuron 63, 189–202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Godin JD, Poizat G, Hickey MA, Maschat F, Humbert S, EMBO J. 29, 2433–2445 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lo Sardo V et al. , Nat Neurosci. 15, 713–721 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Moreira Sousa C et al. , EMBO Mol. Med. 5, 309–325 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elias S, McGuire JR, Yu H, Humbert S, PLOS Biol. 13, e1002142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thion MS et al. , J. Natl. Cancer Inst. 107, djv208 (2015).26293574 [Google Scholar]
- 31.Taverna E, Götz M, Huttner WB, Annu. Rev. Cell Dev. Biol 30, 465–502 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Norden C, J. Cell Sci 130, 1859–1863 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Sakaue-Sawano A et al. , Cell 132, 487–498 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Kosodo Y et al. , EMBO J. 30, 1690–1704 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arai Y et al. , Nat Commun. 2, 154–165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilsch-Bräuninger M, Peters J, Paridaen JT, Huttner WB, Development 139, 95–105 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Manuel MN, Mi D, Mason JO, Price DJ, Front Cell. Neurosci 9, 70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mangin JF et al. , Neuroimage Clin. 26, 102211 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edamakanti CR, Do J, Didonna A, Martina M, Opal P, J. Clin. Invest 128, 2252–2265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tereshchenko AV et al. , Neurology 94, e1908–e1915 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.