

Abstract

Mass spectrometry imaging (MSI) can analyze the spatial distribution of hundreds of different molecules directly from tissue sections usually placed on conductive glass slides to provide conductivity on the sample surface. Additional experiments are often required for molecular identification using consecutive sections on membrane slides compatible with laser capture microdissection (LMD). In this work, we demonstrate for the first time the use of a single conductive slide for both matrix-assisted laser desorption ionization (MALDI)-MSI and direct proteomics. In this workflow, regions of interest can be directly ablated with LMD while preserving protein integrity. These results offer an alternative for MSI-based multimodal spatial-omics.
Mass spectrometry imaging (MSI) offers unlabeled in-depth detection of molecules directly from tissue sections while maintaining their spatial information. Different sample preparation protocols allow the analysis of a wide range of molecular classes, from small metabolites to large proteins.1 Although subsequent molecular identification can be done on the same tissue section using tandem mass spectrometry (MS/MS), this direct identification remains limited to the most abundant molecules, especially for intact proteins. Hence, increasing the number of identified molecules often requires separate experiments, with additional dimensions of separation such as liquid chromatography (LC) using, for example, tissue homogenates. The major drawback of this approach is however the loss of spatial information.
In this regard, laser capture microdissection (LMD) enables the selection of specific tissue areas allowing subsequent molecular identification.2 A first study by Banks et al. explored the use of an infrared laser beam to retrieve selected cells by activating a transfer film placed in contact with the tissue section, followed by protein analysis.3 Alternatively, UV lasers have been used in the past for negative sample selection or more recently using polyethylene napthalate (PEN), polyethylene tetraphthalate (PET), or polyphenylene sulfide (PPS) membrane slides. Target cells are then harvested through the action of the UV laser that cuts around the selected areas and tissue elements are subsequently collected by gravity or catapulting. These systems were used in more recent studies coupled with MSI, showing the potential of MSI-guided LMD.4−8 In these studies, the spatial molecular information obtained from MSI is used for region of interest (ROI) selection in consecutive sections, acquiring more in-depth molecular information and improving the overall molecular identification. Additionally, the necessity of a proper co-registration between histology, MSI, and LMD was described.6 However, using a consecutive section might introduce issues due to section-to-section variability especially now that the MSI field is moving to higher spatial resolution and single-cell imaging.9
MSI analysis coupled to LMD typically use nonconductive membrane slides for molecular identification. A main disadvantage, however, is that these slides are not compatible with all MSI instruments. Most ToF-based instruments need an electrically conductive surface such as an indium tin oxide (ITO)-coated glass slide or IntelliSlides and their implementation in the LMD workflow would be beneficial as it avoids the necessity for additional tissue sections. It will also allow the direct acquisition of imaging and “omics” data from exactly the same sample.
In the present study, we demonstrate the compatibility of conductive slides for matrix-assisted laser desorption/ionization (MALDI)-MSI followed by an LMD proteomics workflow.
Experimental Section
Chemicals and Solvents
All solvents (ULC grade) were purchased from Biosolve Chimie SARL (Dieuze, France) unless stated otherwise. 9-aminoacridine (9AA), ammonium bicarbonate (ABC), citric acid, dithiothreitol (DTT), eosin-Y (Avantor), formic acid (FA, ULC grade), Gill’s hematoxylin, iodoacetamide (IAM), norharmane, trifluoroacetic acid (TFA, ULC grade), and xylene were purchased from Sigma-Aldrich (Zwijndrecht, The Netherlands). RapiGest SF was purchased from Waters (Milford, USA). Trypsin (modified porcine, Sequencing Grade) was purchased from Promega (Madison, USA). PEN microdissection membrane slides and 0.2 mL tubes were purchased from Leica Microsystems (Wetzlar, Germany). ITO glass slides were obtained from Delta Technologies (Loveland, USA) and conductive IntelliSlides from Bruker Daltonics GmbH (Bremen, Germany).
Tissue Samples
All animal experiments were approved by the Institutional Animal Care and User Committee of Maastricht University, and they were performed adhering to the Dutch law.
Residual Wistar Han rat cardiac tissue was provided by the Department of General Surgery, Maastricht University Medical Center, Maastricht, The Netherlands. Rat cardiac tissue was flash-frozen after organ removal. Using a cryotome (Leica Microsystems), 10 μm-thick sections were cut at −20 °C and thaw-mounted onto either a PEN membrane, an ITO slide, or an IntelliSlide. The slides were stored at −80 °C until further analysis.
Residual mouse cardiac tissue was provided by the Department of Physiology, Maastricht University, Maastricht, The Netherlands. After removal, the tissue was fixed in 4% paraformaldehyde for 48 h, embedded in paraffin, and stored at room temperature until sectioning. From this formalin-fixed paraffin-embedded (FFPE) tissue, sections of 4 μm thickness were cut with a rotary microtome (Microm GmbH HM 355, Waldorf, Germany) and placed on either a PEN membrane, an ITO slide, or an IntelliSlide. The slides were stored at +4 °C until further analysis.
Lipid Mass Spectrometry Imaging on Frozen Tissue
Frozen rat cardiac tissue sections were deposited on ITO slides or IntelliSlides and covered with 15 layers of 7 mg/mL norharmane in 2:1 chloroform/methanol using a Suncollect pneumatic sprayer (SunChrom GmbH, Germany) with increasing flow rates of 10, 20, 30, and 40 μL/min for the first four layers and 40 μL/min for the remaining layers. The sections were imaged at 75 μm raster size on a rapifleX tissueTyper (Bruker Daltonics GmbH) in the positive or negative ion reflector mode in a m/z range of 400–2000, summing 500 laser shots per pixel position with 70% of laser power and attenuator at 85%. The instrument was calibrated using red phosphorus. Following MSI, the slides were stored at −80 °C until LMD.
Metabolite Mass Spectrometry Imaging on FFPE Tissue
FFPE mouse cardiac tissues underwent deparaffinization with two 8 min xylene washes, as described previously,10 followed by the application of 11 layers of 10 mg/mL 9AA in 70% methanol using a Suncollect pneumatic sprayer with increasing flow rates of 10, 20, 30, and 40 μL/min for the first four layers and 40 μL/min for the remaining layers. All sections were imaged at 75 μm raster size on a rapifleX tissueTyper (Bruker Daltonics GmbH) in the negative ion reflector mode at a m/z range of 40–1000, summing 500 laser shots per position with 60% of laser power and attenuator at 85%. Instrument calibration was performed using red phosphorus. After MSI, the slides were stored at +4 °C until LMD.
Laser Capture Microdissection
LMD was performed using a Leica LMD 7000 instrument (Leica Microsystems). The experimental workflow is depicted in Figure 1, showing the different dissection methods used for the PEN membrane and conductive, nonmembrane slides. For FFPE tissues, the paraffin was removed by 2 h of heating at 60 °C followed by two 5 min washes with xylene and two 2 min washes with isopropanol.7 Before LMD, the tissue sections were dried in a desiccator. A total of 0.1, 0.2, 0.5, or 1.0 mm2 dissected material was collected in triplicate, from FFPE and frozen material, before and after hematoxylin and eosin (H&E) staining. The areas were dissected using the following laser settings: wavelength 349 nm, power 40, aperture 30, speed 5, specimen balance 0, line spacing 5, head current 60%, and pulse frequency 501 Hz (later referred to as settings A). A second set of laser parameters was also used for ITO and IntelliSlides: wavelength 349 nm, power 50, aperture 38, speed 17, specimen balance 0, line spacing 5, head current 60%, and pulse frequency 310 Hz (referred to as settings B). For PEN membrane slides, “draw and cut” was used, and “draw and scan” was used for ITO slides and IntelliSlides.
Figure 1.
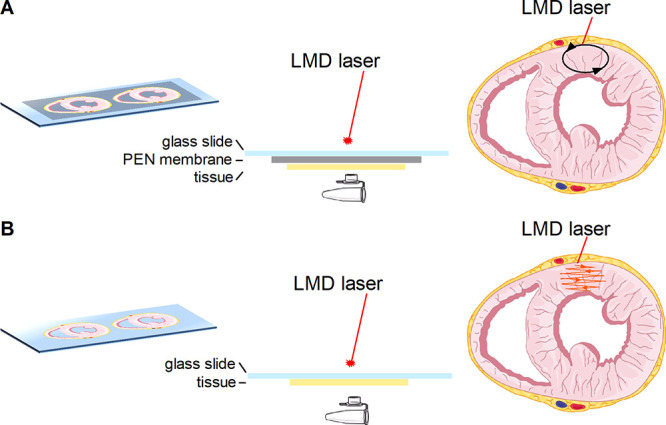
Schematic representation of the dissection method used for LMD. The selected region is dissected from a PEN membrane slide [(A) black oval] or ablated from a conductive, nonmembrane slide [(B) orange pattern]. Graphical elements were adapted from Servier Medical Art.
Dissected areas were collected in the caps of 0.2 mL centrifuge tubes, prefilled with 20 μL of buffer (50 mM ABC for frozen, 50 mM citric acid for FFPE samples), and stored at −20 °C until further processing for LC–MS/MS.
LMD after MALDI-MSI was performed on ITO slides and IntelliSlides after matrix removal with 70% ethanol, as shown in Figure 2. An ROI was selected based on segmentation data and co-registered with the LMD using an in-house-build MATLAB script.6 Areas of 0.5 mm2 were ablated from the ITO slide or IntelliSlide using laser settings B, as described above, collected in 20 μL of buffer (50 mM ABC for frozen, 50 mM citric acid for FFPE samples) and stored at −20 °C until further processing for LC–MS/MS.
Figure 2.
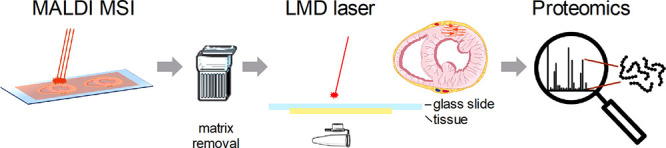
Schematic representation of the workflow coupling MALDI-MSI and LMD. After MALDI-MSI experiments, the matrix was removed followed by the ablation of the selected region from the ITO slide or IntelliSlide (orange pattern). The collected material was then processed for proteomics. Graphical elements were adapted from Servier Medical Art or created by Fredrik Edfors from the Noun project.
Sample Processing for Proteomics
The dissected material was further processed based on the protocol as previously described by Longuespée et al.7 In short, for FFPE samples, antigen retrieval was performed by heating to 99 °C for an hour while shaking at 800 rpm in a Thermoshaker (Eppendorf, Hamburg, Germany). For both FFPE and frozen samples, RapiGest (final concentration 0.01%) was added and incubated for 10 min at room temperature (RT = 21 °C) and the pH of FFPE samples was adjusted by the addition of ABC. All samples were reduced using DTT (final concentration 10 mM) at 56 °C for 40 min at 800 rpm and alkylated using IAM (final concentration 20 mM) at RT for 30 min at 800 rpm. DTT (final concentration 10 mM) was used to quench the excess of IAM at RT for 10 min at 800 rpm. Digestion using trypsin (final concentration 15 μg/mL) was performed overnight at 37 °C and 800 rpm. The second digestion step (trypsin final concentration 5 μg/mL) was performed in 80% ACN for 3 h at 37 °C and 800 rpm. The digestion was stopped by adding TFA (final concentration 0.5%) and an incubation step at 37 °C for 45 min and 800 rpm. After centrifugation (15,000g, 10 min at 4 °C, Thermo Scientific Heraeus Biofuge stratos, Waltham, USA), the supernatant was collected and concentrated to a final volume of approximately 30 μL using a Speedvac (Hetovac VR-1, Heto Lab Equipment, Denmark). The concentrated samples were stored at −20 °C until LC–MS/MS analysis.
LC–MS/MS Analysis
Peptide separation was performed on a Thermo Scientific (Dionex) Ultimate 3000 Rapid Separation UHPLC system equipped with a PepSep C18 analytical column (15 cm, ID 75 μm, 1,9 μm Reprosil, 120 Å). An aliquot of 10 μL of the sample was desalted using an online installed C18 trapping column, and the peptides were separated on the analytical column with a 90 min linear gradient from 5 to 35% ACN with 0.1% FA at a 300 nL/min flow rate.
The UHPLC system was coupled to a Q Exactive HF mass spectrometer (Thermo Scientific). Mass spectra were acquired in the positive ionization mode between m/z 250–1250 at a resolution of 120.000, followed by MS/MS scans of the top 15 most intense ions at a resolution of 15.000 in DDA mode.
Data Analysis
The triplicates were analyzed individually, and protein identification was performed using Proteome Discoverer 2.2 (Thermo Scientific). The search engine Sequest was used with the SwissProt Rattus norvegicus (SwissProt TaxID = 10116) or Mus musculus (SwissProt TaxID = 10090) databases from October 2017. The following settings were used for the database search: Trypsin was used as enzyme with a maximum of two missed cleavages and a minimal peptide length of six amino acids, mass tolerance of 10 ppm for precursor ions, and 0.02 Da for MS2, dynamic modifications of methionine oxidation and protein N-terminus acetylation, and static modifications of cysteine carbamidomethylation. The false discovery rate was used to estimate the certainty of the identification, and only proteins with a high protein confidence were used for further analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE11 partner repository with the dataset identifier PXD023196.
Results on the number of proteins identified per triplicates are presented as mean ± standard deviation (SD). Comparisons were performed with the t-test or one-way ANOVA and p < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism (version 5.00, GraphPad Software Inc., San Diego, CA).
Proteins commonly identified in the triplicates were used for gene ontology cellular component analysis. UniProt ID mapping was used to obtain the gene names which were then submitted to EnrichR,12 where cellular components with p-value < 0.05 were considered for further analysis. The components were categorized based on a higher level in the Gene Ontology Cellular Component tree for a more concise and structured analysis. Pathway analysis was performed for the differentiation of the clusters after MSI. EnrichR used Reactome’s cell signaling database and pathways with p-value < 0.05 were used for the analysis.
MSI data were analyzed using SCiLS lab MVS, version 2020a (Bremen, Germany) after TIC normalization. TIC normalization is commonly used in ToF-based MSI, when samples contain similar cell types.13 Segmentation by bisecting k-means with correlation distance was performed to obtain ROI information. mMass14 was used to generate a peak list (15 precision baseline correction with 25 relative offset, Savitzky–Golay smoothing with a window size of 0.2 m/z and two cycles, S/N threshold of 3.5, a relative intensity threshold of 0.5%, and a picking height of 75).
H&E Staining
For FFPE samples, the paraffin was removed after 15 min of heating at 60 °C followed by two 10 min xylene washes. This was followed by a standard H&E protocol for staining of all tissues: in short, 3 min distilled water, 3 min 0.1% Gill’s hematoxylin, 3 min running tap water, rinse with distilled water, 30 s 0.2% eosin, 10 dips 70% ethanol, two times 2 min 100% ethanol, and two times 5 min xylene. The H&E-stained tissue for LMD was not covered with a cover slip and stored at +4 °C until LMD.
Results
Protein Identification from the PEN Membrane and ITO Slides
The potential use of ITO slides for LMD-based proteomic applications was evaluated and compared to the conventionally used PEN membrane slides for cardiac tissue, as depicted in Figure 1. The number of identified proteins was determined for different dissected tissue areas (0.1, 0.2, 0.5, and 1.0 mm2) for both frozen and FFPE samples (images of the dissected tissue can be found in Figure S1). Figure 3 demonstrates the feasibility of LMD-based proteomic workflows directly from ITO slides. The number of proteins identified increased when bigger areas were dissected. This trend was significant for frozen tissue from both slide types (p = 0.0047 for the PEN membrane and p = 0.0168 for ITO slides), and for FFPE tissue only for PEN membrane slides, p < 0.0001. As expected, the number of identified proteins was significantly higher in PEN membrane slides for both frozen and FFPE tissue (p < 0.05 is indicated with an asterisk *). Interestingly, the number of identified proteins for frozen and FFPE tissue was similar in PEN membrane slides, while the number of identified proteins from ITO slides was lower for FFPE compared to frozen tissue.
Figure 3.

Proteins identified from PEN membrane and ITO slides for (A) frozen tissue and (B) FFPE tissue. Data are presented as mean ± SD, * indicates p < 0.05 when comparing equal areas from PEN membrane versus ITO slides.
The number of identified proteins was also assessed after staining the sections with H&E (Figure S2). Protein identification directly from ITO slides after H&E staining was still possible but with less favorable numbers compared to PEN membrane slides.
An enrichment analysis on commonly found proteins within the triplicates was performed to assess the cellular component origin and to verify whether the protein integrity was maintained after tissue dissection. Table 1 depicts the top 10 most significant cellular components and shows the preservation of cellular components from mitochondrial and secretory granule proteins for all studied samples.
Table 1. Top 10 Most Significant Cellular Components (p < 0.05) That Were Identified after LMDa.
| frozen
tissue |
FFPE
tissue |
|||
|---|---|---|---|---|
| PEN membrane | ITO | PEN membrane | ITO | |
| cell junction | × | × | × | |
| cell projection | × | |||
| cytoplasm | × | |||
| cytoplasmic vesicle | × | |||
| cytoskeleton | × | × | ||
| cytosol | × | × | ||
| mitochondrion | × | × | × | × |
| ribosome | × | × | ||
| secretory granule | × | × | × | × |
The cellular components were clustered and ordered alphabetically.
A more detailed analysis of all significant cellular components revealed that 87 and 38 cellular components were found in PEN membrane and ITO slides from frozen tissue, respectively. In comparison, analysis of FFPE tissue resulted in 72 and 11 cellular components, from PEN membrane and ITO slides, respectively (Figure S3 and Table S1). Despite this variability, the categorized analysis showed the preservation of cytoskeletal, mitochondrial, and secretory granule proteins.
Effect of the LMD Laser on Protein Identification
Next, the laser parameters were adjusted with the aim to improve the number of proteins identified, which was based on visual inspection of the residue left on the ITO slide after collection with LMD. The results shown in Figure 4 indicate an improvement in the number of identified proteins when using settings B, especially for FFPE tissue (p = 0.0119 and p = 0.0065 for 0.5 and 1.0 mm2, respectively). Cellular component analysis showed no differences for the top 10 most significant components (Table S2). Based on the improvement seen for FFPE tissue on ITO slides, settings B were used without further optimization.
Figure 4.

Comparison of two laser settings for ablation from an ITO slide. The figure represents the number of proteins from (A) frozen tissue and (B) FFPE tissue. Data are presented as mean ± SD, * indicates p < 0.05.
Proteomics after MALDI-MSI
Lipid and metabolite imaging experiments, for frozen and FFPE tissue, respectively, were performed on ITO slides and IntelliSlides (Figure 2) to evaluate the effect of a MALDI-MSI workflow prior to our LMD-proteomics approach. Figure 5 shows that proteins can still be identified from tissue sections that were previously used for lipid or metabolite MSI on ITO or IntelliSlides (marked as INT in the figure). However, comparing the number of identified proteins from frozen tissue on ITO slides before (Figure 4A, laser settings B) and after MSI showed a significant decrease in the number of identified proteins, as indicated with an asterisk (*) in Figure 5A,B. Still, the number of identified proteins after MALDI-MSI remained above one hundred. No statistically significant differences were found before (Figure 4A, laser settings B) and after MALDI-MSI for FFPE tissues (Figure 5C).
Figure 5.

Number of proteins identified after MSI from ITO and IntelliSlides (INT) followed by LMD. Lipid MSI on frozen tissue in the (A) positive ion mode and (B) negative ion mode and (C) metabolite MSI on FFPE tissue in the negative ion mode. Data are presented as mean ± SD. * indicates p < 0.05 when comparing the results from ITO slides before versus after MSI. The horizontal lines represent the mean number of proteins identified before MSI, light gray for 0.5 mm2 and dark gray for 1.0 mm2.
In relation to protein preservation after MALDI-MSI, the top 10 most significant cellular components were found to be similar to those from tissue before MALDI-MSI (Table S3). Figure 6 shows all significant cellular components found in frozen tissue before and after positive lipid MSI analysis. Cytoplasmic vesicle, cytoskeleton, mitochondrion, and secretory granule proteins are the main contributors under both conditions (results for negative lipid MSI can be found in Figure S4, the number of proteins per cellular category can be found in Table S4). In comparison, also for FFPE tissue, the cellular components map to similar categories as before MSI, having cytoplasmic, cytoskeleton, mitochondrion, and secretory granule proteins as the biggest contributors (Figure S5, Table S5).
Figure 6.
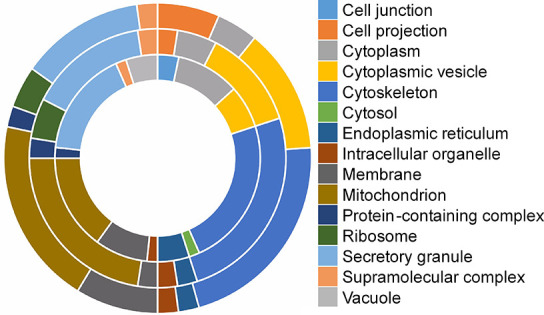
Clustered representation of all significant cellular components found from frozen tissue before (ITO; inner circle) and after positive lipid MSI (ITO; middle circle, IntelliSlide; outer circle).
Finally, segmentation data from the positive ion-mode lipid MALDI-MSI was used for the selection of two clusters, as indicated in Figure 7A, to illustrate the use of conductive slides for MSI-guided LMD. Figure 7B shows the number of identified proteins from both clusters, both approximately 0.5 mm2. Categorized analysis of cellular components showed a similar distribution (Figure S6, Table S6). Further analysis of the biological differences between clusters was performed with pathway analysis, and the detailed results can be found in Table S7. In total, 103 pathways were identified, of which 29 and 33 pathways were specific for cluster 1 and 2, respectively.
Figure 7.
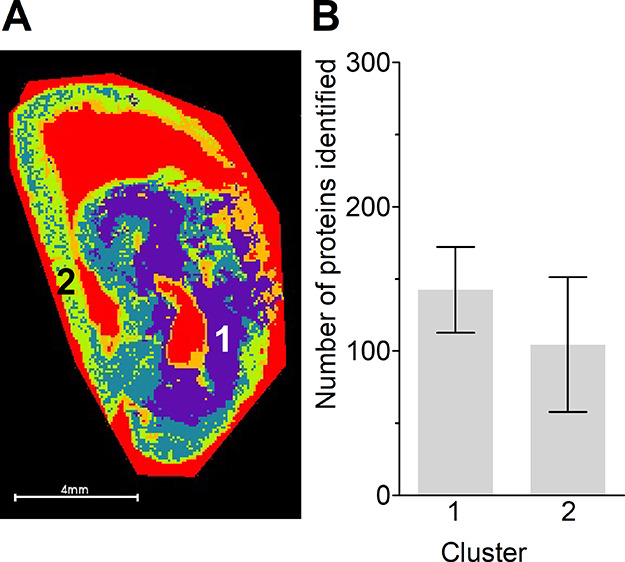
Segmentation data from positive ion-mode lipid analysis (A). The numbers indicate clusters 1 (purple) and 2 (green), and an area of 0.5 mm2 was ablated. (B) The number of proteins identified from clusters 1 and 2 Data are represented as mean ± SD.
Discussion
In this work, we demonstrate the use of conductive slides for LMD-based proteomic workflows and the possibility to identify proteins from ITO slides and IntelliSlides after MALDI-MSI. This will positively impact MSI-based spatial-omics workflows for different reasons. Our method enables the use of the same tissue section for both MSI and subsequent LC–MS/MS-based identification. This reduces the identification variability from section-to-section and it is relevant for co-registration strategies with other imaging modalities without the need of additional material. Also, the IntelliSlides contain engraved marks and a unique identifier for sample registration, which improves automation capabilities and speeds up the co-registration between staining, MSI, and LMD. And finally, with a conductive slide, it is possible to use any MSI instrument of choice for MSI-based LMD.
The influence of the LMD laser on protein integrity was evaluated in this work. The main difference between a membrane slide and a conductive slide is the dissecting method: the laser either cuts around the tissue for dissection or it scans across the tissue for ablation, respectively. In the latter case, the LMD laser is pointed directly on the tissue section, which could induce unwanted side effects such as tissue degradation. Nevertheless, our cellular component analysis showed a good preservation of proteins after ablation. It should be noted that in the present study, only two laser settings were compared, which were chosen based on the efficacy of tissue collection. A too-high LMD laser power or defocusing of the laser might cause tissue damage by inducing an increase in the temperature, as was found for nonmembrane slides.15,16 Finally, the LMD laser speed in combination with its frequency determines the time and intensity of exposure to the laser. Based on our and previous studies,4−7 there is an optimal laser setting for every tissue, sample type, and glass slide combination.
Histological staining provides additional spatial and morphological information to MSI data. Our results with ITO slides showed a reduction in the number of proteins identified after H&E-staining. Even though it was still possible to identify the majority of the proteins, this decrease might introduce challenges when investigating low-abundant proteins or minor differences in protein expression. As the observed loss of proteins might be due to the different washing steps in the H&E protocol, previous studies investigated truncated H&E protocols.17 A study by Craven et al. illustrated using 2D-PAGE that staining mainly affected the relative abundance of several proteins.18 On the other hand, with the microproteomics approach for PEN membrane slides as used by Dewez et al., a robust number of proteins were identified from H&E-stained tissue.6 Further optimization of the LMD laser settings as described above and adjustment of the staining protocol might also improve the number or proteins identified after staining.
In our goal to explore the use of conductive, nonmembrane glass slides in a workflow coupling MALDI-MSI, LMD, and proteomics, different MALDI-MSI sample preparation protocols were used, based on the following considerations. First, frozen tissue was investigated with a frequently used lipid MSI protocol. FFPE tissue had limited options due to formalin induced protein cross-linking and the possible modification of the molecular content during paraffin removal. Therefore, a previously established metabolite MSI protocol was used for FFPE tissue.10 Next, the MALDI matrices and organic solvents might affect the tissue, as does the removal of the matrix afterwards. When focusing on the number of proteins identified after metabolite MSI, an increase was observed, although this trend was not statistically significant. Perhaps, here, the formalin has a protective effect on protein preservation. In addition, the washing steps and the matrix might also facilitate the extraction of low-molecular-weight analytes and further reduce the presence of interfering compounds. In a study by Dilillo et al., an increase in the number of identified proteins after MSI was observed, and there it was presumed to be due to the acidic matrix solution.5 After lipid MSI, on the contrary, we observed a decrease in proteins identified. Here, we hypothesize that the mixture of chloroform and methanol used as a matrix solvent might disrupt the cell membranes and wash away the majority of highly hydrophobic proteins.19
The combination of MSI, LMD, and proteomics is described in previous studies but with membrane slides and other tissue types. Using a 1D LC–MS/MS proteomics protocol, Longuespée et al.7 identified approximately 700 proteins from approximately 0.4 mm2 dissected FFPE tumor tissue from a PEN membrane slide (estimated to contain 2700 ± 245 cells). In comparison, our 0.5 mm2 dissected frozen and FFPE cardiac tissue contained approximately 1100 and 1400 cells, respectively. This resulted in an average of 424 identified proteins for frozen tissue and 347 identified proteins for FFPE tissue, from PEN membrane slides. This indicates that for a proper comparison between dissected areas, the number of cells should be carefully considered as this varies per tissue type. Longuespée et al. further optimized the protocol by including a 2D LC–MS/MS method which doubled the number of proteins identified.4,7 A more recent study used a timsTOF flex, and identified on average 2500 proteins (approximately 2000 cells) from PEN membrane slides.8 These studies indicate that with the addition of another separation method, the number of identified proteins could further improve our results. Other studies have used a nanoPOTS sample-processing platform.20,21 This workflow did not include MSI but maintained the spatial information as proteins were identified per tissue. In contrast to all automated methods, Quanico et al. described a manual dissection approach combining MSI with a multi-omics approach on the same tissue section, resulting in metabolite and protein identifications.22 Their study shows additional omics possibilities for the dissected tissue. Incorporation of a multi-omics approach in our workflow would add another level of data obtained from a single tissue section.
So far, the direct identification of proteins by MALDI-MSI is limited to the highly abundant proteins.23,24 The incorporation of ion mobility separation helps on the separation of compounds with a similar m/z.25 However, it is still limited to a low number of proteins compared to our approach.23 Although the methods used in both studies maintain the spatial information, the on-tissue identification remains limited due to abundant species and the lack of chromatographic separation. In contrast, the workflow as proposed in our study provides the spatial mapping of compounds and additional in-depth protein identification. Our workflow could be compatible with other imaging-LMD-guided methods such as secondary ion mass spectrometry, liquid extraction surface analysis, or desorption electrospray ionization.
In summary, our results show that a single tissue section placed on a conductive slide can be used for multidimensional spatial-omics. Future adjustments of the method for different applications will further impact the MS(I) community.
Conclusions
Here, we demonstrate for the first time, the use of a single conductive nonmembrane slide in a MALDI-MSI-LMD workflow. Proteins were identified with a bottom-up proteomics approach after LMD from conductive slides with good preservation of the protein integrity. Therefore, this MSI coupled to LMD workflow in combination with omics approaches facilitates the identification of multiple compounds from a single tissue section and potentially aids in answering biological questions.
Acknowledgments
The research described in this paper was part of the research program of M4i and received funding through the LINK program of the Dutch Province of Limburg. The authors are thankful to the Department of General Surgery for providing the rat samples, M. van Bilsen and A. Kuhn for providing the mouse samples, F. Dewez for his help with the LMD, and R. Mohren for setting up the LC–MS/MS analysis. S.T.P.M., R.M.A.H., and O.B. received funding from the Maastricht University Medical Center. A.M.A.M. received funding from the Dutch government, ZonMW/NWO (09150161810155).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c04572.
Figures S1: Images of the dissected tissue after LMD; Figure S2: Proteins identified from PEN membrane and ITO slides after H&E staining; Figure S3: Categorized representation of the cellular components found after LMD on both frozen and FFPE tissue; Figure S4: Cellular component analysis found in frozen tissue before and after negative lipid MALDI-MSI; Figure S5: All cellular component found in FFPE tissue before and after metabolite MALDI-MSI; Figure S6: Cellular component analysis of the significant components found in the two clusters dissected after positive lipid MALDI-MSI; Table S1: Number of cellular components and proteins after LMD on both frozen and FFPE tissue; Table S2: Top 10 cellular components for frozen and FFPE tissue comparing laser settings; Table S3: Top 10 cellular components from conductive slide before and after MALDI-MSI; Table S4: Number of cellular components and proteins from frozen tissue before and after (positive or negative) lipid MALDI-MSI; Table S5: Number of cellular components and proteins from FFPE tissue before and after metabolite MALDI-MSI; Table S6: Number of cellular components and proteins from cluster 1 and 2 after positive lipid MALDI-MSI; Table S7: Pathway analysis from clusters 1 and 2 after positive lipid MALDI-MSI (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chughtai K.; Heeren R. M. A. Mass spectrometric imaging for biomedical tissue analysis. Chem. Rev. 2010, 110, 3237–3277. 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.; Malhotra L.; Dickerson R.; Chaffee S.; Sen C. K.; Roy S. Laser capture microdissection: Big data from small samples. Histol. Histopathol. 2015, 30, 1255–1269. 10.14670/HH-11-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks R. E.; Dunn M. J.; Forbes M. A.; Stanley A.; Pappin D.; Naven T.; Gough M.; Harnden P.; Selby P. J. The potential use of laser capture microdissection to selectively obtain distinct populations of cells for proteomics analysis - Preliminary findings. Electrophoresis 1999, 20, 689.. [DOI] [PubMed] [Google Scholar]
- Alberts D.; Pottier C.; Smargiasso N.; Baiwir D.; Mazzucchelli G.; Delvenne P.; Kriegsmann M.; Kazdal D.; Warth A.; De Pauw E.; Longuespee R. MALDI Imaging-Guided Microproteomic Analyses of Heterogeneous Breast Tumors-A Pilot Study. Proteomics: Clin. Appl. 2018, 12, 1700062. 10.1002/prca.201700062. [DOI] [PubMed] [Google Scholar]
- Dilillo M.; Pellegrini D.; Ait-Belkacem R.; de Graaf E. L.; Caleo M.; McDonnell L. A. Mass Spectrometry Imaging, Laser Capture Microdissection, and LC-MS/MS of the Same Tissue Section. J. Proteome Res. 2017, 16, 2993–3001. 10.1021/acs.jproteome.7b00284. [DOI] [PubMed] [Google Scholar]
- Dewez F.; Martin-Lorenzo M.; Herfs M.; Baiwir D.; Mazzucchelli G.; De Pauw E.; Heeren R. M. A.; Balluff B. Precise co-registration of mass spectrometry imaging, histology, and laser microdissection-based omics. Anal. Bioanal. Chem. 2019, 411, 5647–5653. 10.1007/s00216-019-01983-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuespée R.; Alberts D.; Pottier C.; Smargiasso N.; Mazzucchelli G.; Baiwir D.; Kriegsmann M.; Herfs M.; Kriegsmann J.; Delvenne P.; De Pauw E. A laser microdissection-based workflow for FFPE tissue microproteomics: Important considerations for small sample processing. Methods 2016, 104, 154–162. 10.1016/j.ymeth.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Dewez F.; Oejten J.; Henkel C.; Hebeler R.; Neuweger H.; De Pauw E.; Heeren R. M. A.; Balluff B. MS Imaging-Guided Microproteomics for Spatial Omics on a Single Instrument. Proteomics 2020, 20, 1900369. 10.1002/pmic.201900369. [DOI] [PubMed] [Google Scholar]
- Ščupáková K.; Dewez F.; Walch A. K.; Heeren R. M. A.; Balluff B. Morphometric Cell Classification for Single-Cell MALDI-Mass Spectrometry Imaging. Angew. Chem., Int. Ed. Engl. 2020, 132, 17600. 10.1002/ange.202007315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly A.; Buck A.; Balluff B.; Sun N.; Gorzolka K.; Feuchtinger A.; Janssen K.-P.; Kuppen P. J. K.; van de Velde C. J. H.; Weirich G.; Erlmeier F.; Langer R.; Aubele M.; Zitzelsberger H.; McDonnell L.; Aichler M.; Walch A. High-mass-resolution MALDI mass spectrometry imaging of metabolites from formalin-fixed paraffin-embedded tissue. Nat. Protoc. 2016, 11, 1428–1443. 10.1038/nprot.2016.081. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; Pérez E.; Uszkoreit J.; Pfeuffer J.; Sachsenberg T.; Yılmaz Ş.; Tiwary S.; Cox J.; Audain E.; Walzer M.; Jarnuczak A. F.; Ternent T.; Brazma A.; Vizcaíno J. A. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov M. V.; Jones M. R.; Rouillard A. D.; Fernandez N. F.; Duan Q.; Wang Z.; Koplev S.; Jenkins S. L.; Jagodnik K. M.; Lachmann A.; McDermott M. G.; Monteiro C. D.; Gundersen G. W.; Ma’ayan A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deininger S.-O.; Cornett D. S.; Paape R.; Becker M.; Pineau C.; Rauser S.; Walch A.; Wolski E. Normalization in MALDI-TOF imaging datasets of proteins: practical considerations. Anal. Bioanal. Chem. 2011, 401, 167–181. 10.1007/s00216-011-4929-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohalm M.; Hassman M.; Košata B.; Kodíček M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun. Mass Spectrom. 2008, 22, 905–908. 10.1002/rcm.3444. [DOI] [PubMed] [Google Scholar]
- Vogel A.; Horneffer V.; Lorenz K.; Linz N.; Hüttmann G.; Gebert A. Chapter 5: Principles of Laser Microdissection and Catapulting of Histologic Specimens and Live Cells. Methods Cell Biol. 2007, 82, 153. 10.1016/S0091-679X(06)82005-4. [DOI] [PubMed] [Google Scholar]
- Schütze K.; Niyaz Y.; Stich M.; Buchstaller A. Chapter 23: Noncontact Laser Microdissection and Catapulting for Pure Sample Capture. Methods Cell Biol. 2007, 82, 649. 10.1016/S0091-679X(06)82023-6. [DOI] [PubMed] [Google Scholar]
- Kuhn D. E.; Roy S.; Radtke J.; Khanna S.; Sen C. K. Laser microdissection and capture of pure cardiomyocytes and fibroblasts from infarcted heart regions: perceived hyperoxia induces p21 in peri-infarct myocytes. Am. J. Physiol.: Heart Circ. Physiol. 2007, 292, H1245–H1253. 10.1152/ajpheart.01069.2006. [DOI] [PubMed] [Google Scholar]
- Craven R. A.; Totty N.; Harnden P.; Selby P. J.; Banks R. E. Laser capture microdissection and two-dimensional polyacrylamide gel electrophoresis: evaluation of tissue preparation and sample limitations. Am. J. Pathol. 2002, 160, 815–822. 10.1016/s0002-9440(10)64904-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertommen A.; Panis B.; Swennen R.; Carpentier S. C. Evaluation of chloroform/methanol extraction to facilitate the study of membrane proteins of non-model plants. Planta 2010, 231, 1113–1125. 10.1007/s00425-010-1121-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehowski P. D.; Zhu Y.; Bramer L. M.; Stratton K. G.; Zhao R.; Orton D. J.; Moore R. J.; Yuan J.; Mitchell H. D.; Gao Y.; Webb-Robertson B. M.; Dey S. K.; Kelly R. T.; Burnum-Johnson K. E. Automated mass spectrometry imaging of over 2000 proteins from tissue sections at 100-mum spatial resolution. Nat. Commun. 2020, 11, 8. 10.1038/s41467-019-13858-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Dou M.; Piehowski P. D.; Liang Y.; Wang F.; Chu R. K.; Chrisler W. B.; Smith J. N.; Schwarz K. C.; Shen Y.; Shukla A. K.; Moore R. J.; Smith R. D.; Qian W.-J.; Kelly R. T. Spatially Resolved Proteome Mapping of Laser Capture Microdissected Tissue with Automated Sample Transfer to Nanodroplets. Mol. Cell. Proteomics 2018, 17, 1864–1874. 10.1074/mcp.tir118.000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quanico J.; Franck J.; Wisztorski M.; Salzet M.; Fournier I. Integrated mass spectrometry imaging and omics workflows on the same tissue section using grid-aided, parafilm-assisted microdissection. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 1702–1714. 10.1016/j.bbagen.2017.03.006. [DOI] [PubMed] [Google Scholar]
- Groseclose M. R.; Andersson M.; Hardesty W. M.; Caprioli R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass Spectrom. 2007, 42, 254–262. 10.1002/jms.1177. [DOI] [PubMed] [Google Scholar]
- Cillero-Pastor B.; Eijkel G. B.; Kiss A.; Blanco F. J.; Heeren R. M. A. Matrix-assisted laser desorption ionization-imaging mass spectrometry: a new methodology to study human osteoarthritic cartilage. Arthritis Rheum. 2013, 65, 710–720. 10.1002/art.37799. [DOI] [PubMed] [Google Scholar]
- Stauber J.; MacAleese L.; Franck J.; Claude E.; Snel M.; Kaletas B. K.; Wiel I. M. V. D.; Wisztorski M.; Fournier I.; Heeren R. M. A. On-tissue protein identification and imaging by MALDI-ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 338–347. 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.