

Abstract
We previously reported that Hedgehog (Hh) signal was enhanced in gallbladder cancer (GBC) and was involved in the induction of malignant phenotype of GBC. In recent years, therapeutics that target Hh signaling have focused on molecules downstream of smoothened (SMO). The three transcription factors in the Hh signal pathway, glioma-associated oncogene homolog 1 (GLI1), GLI2, and GLI3, function downstream of SMO, but their biological role in GBC remains unclear. In the present study, the biological significance of GLI1, GLI2, and GLI3 were analyzed with the aim of developing novel treatments for GBC. It was revealed that GLI2, but not GLI1 or GLI3, was involved in the cell cycle-mediated proliferative capacity in GBC and that GLI2, but not GLI1 or GLI3, was involved in the enhanced invasive capacity through epithelial-mesenchymal transition. Further analyses revealed that GLI2 may function in mediating gemcitabine sensitivity and that GLI2 was involved in the promotion of fibrosis in a mouse xenograft model. Immunohistochemical staining of 66 surgically resected GBC tissues revealed that GLI2-high expression patients had fewer numbers of CD3+ and CD8+ tumor-infiltrating lymphocytes (TILs) and increased programmed cell death ligand 1 (PD-L1) expression in cancer cells. These results suggest that GLI2, but not GLI1 or GLI3, is involved in proliferation, invasion, fibrosis, PD-L1 expression, and TILs in GBC and could be a novel therapeutic target. The results of this study provide a significant contribution to the development of a new treatment for refractory GBC, which has few therapeutic options.
Keywords: glioma-associated oncogene homolog 2, hedgehog signal, gallbladder cancer, cell cycle, epithelial-mesenchymal transition, tumor microenvironment, tumor infiltrating lymphocyte, programmed cell death ligand 1, hypoxia
Introduction
Gallbladder cancer (GBC) is the seventh most common gastrointestinal carcinoma and accounts for 1.2% of all cancer cases and 1.7% of all cancer-related deaths (1). GBC develops from metaplasia to dysplasia to carcinoma in situ and then to invasive carcinoma over 5–15 years (2). During this time, GBC exhibits few characteristic symptoms, and numerous cases have already developed into locally advanced or metastasized cancer by the time of diagnosis. Gemcitabine (GEM), cisplatin (CDDP), and 5-fluorouracil (5-FU) are used as single agents or in combination as chemotherapy for GBC. However, the 5-year survival rate of patients with GBC remains low (at <10%) (3), and thus the development of new treatment strategies is required.
The Hedgehog (Hh) signal is a morphogenesis signaling pathway that is crucial for growth and patterning during the embryonic period (4,5). The Hh signal is activated by the binding of its ligand sonic hedgehog (SHH) to patched (PTCH), which activates smoothened (SMO), promoting the nuclear translocation of three downstream transcription factors, glioma-associated oncogene homolog 1 (GLI1), GLI2, and GLI3 (6–8). Recent studies have revealed that this signal is reactivated and implicated in the development of various types of cancer (9–18). We previously reported that the Hh signal is enhanced and involved in the induction of malignant phenotype in GBC and that the Hh signal could be a therapeutic target for GBC (19).
Several SMO inhibitors have been developed to target Hh signaling such as cyclopamine (20–22) and vismodegib (23–25). Clinical trials using vismodegib has been conducted for various types of cancer, but vismodegib has not exhibited significant therapeutic efficacy in trials (26). Causes for the lack of efficacy in clinical trials include mutation of SMO and cross-talks with other signaling pathways downstream of the signal. From this perspective, molecules downstream of SMO have been investigated as potential therapeutic targets. While the GLI1, GLI2, and GLI3 transcription factors have been revealed to function in Hh signaling in mammals, the role of each transcription factor in GBC has not been fully elucidated. In the present study, the biological significance of the Hh signal transcription factors GLI1, GLI2, and GLI3 were analyzed for the development of novel therapies for GBC.
Materials and methods
Cell lines
The three human GBC cell lines NOZ, TGBC2TKB (27), and TYGBK-1 (28) were used. NOZ and TYGBK-1 cells were purchased from the Japanese Collection of Research Bioresources (JCRB) bank. The TGBC2TKB cell line was purchased from Riken Cell Bank (Tsukuba, Japan). All cell lines were cultured according to the supplier's specifications. Absence of mycoplasma contamination in the cell lines was confirmed using a mycoplasma detection kit (Lonza Group, Ltd.). For normoxic conditions, cells were cultured in 5% CO2 and 95% air; for hypoxic conditions, cells were cultured in 1% O2, 5% CO2, and 94% N2 in a multi-gas incubator (Sanyo).
RNA interference
ON-TARGETplus™ SMARTpool small interfering (si)RNA targeting siRNA against GLI1 (cat. no. L-003896), GLI2 (cat. no. L-006468), GLI3 (cat. no. L-011043), and hypoxia-inducible factor 1α (HIF-1α) (cat. no. L-004018) and negative control siRNA (ON-TARGETplus™ Control non-targeting siRNA, cat. no. D-001810) were purchased from Dharmacon; Horizon Discovery Ltd. Cells were seeded in a 6-well plate (2.0×105 cells per well) and transfected with 20 µM of each construct using Lipofectamine® RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 48 h according to the manufacturer's protocol. The knockdown efficiency of siRNAs for each target gene in each GBC cell line was evaluated by real time RT-qPCR analysis (Fig. S1). The time course result of knockdown efficiency of GLI2 siRNA by real time RT-qPCR analysis is presented in Fig. S2.
Real time reverse transcription-quantitative PCR (real time RT-qPCR)
Total RNA of each GBC cell line was extracted using a High Pure RNA Isolation kit (Roche Diagnostics) and quantified by spectrophotometry (NanoDrop 1000; Thermo Fisher Scientific, Inc.). RNA (1.0 µg) was reverse transcribed to cDNA with the Verso cDNA Synthesis Kit (Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol. Reactions were run on a 7500 Real-Time PCR System™ (Applied Biosystems; Thermo Fisher Scientific, Inc.) using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.). The thermocycling parameters were as follows: Initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 15 sec and annealing/elongation at 60°C for 1 min. The primer sequences used were as follows: Gli1 forward, 5′-TACATCAACTCCGGCCAATAGG-3′ and reverse, 5′-CGGCGGCTGACAGTATAGGCA-3′; Gli2 forward, 5′-CGAGAAACCCTACATCTGCAAGA-3′ and reverse, 5′-GTGGACCGTTTTCACATGCTT-3′; Gli3 forward, 5′-AAACCCCAATCATGGACTCAAC-3′ and reverse, 5′-TACGTGCTCCATCCATTTGGT-3′; HIF-1α forward, 5′-GAAGTGTACCCTAACTAGCCGAGG-3′ and reverse, 5′-TTTCTTATACCCACACTGAGGTTGG-3′; and β-actin forward, 5′-TTGCCGACAGGATGCAGAAGGA-3′ and reverse, 5′-AGGTGGACAGCGAGGCCAGGAT-3′. The amount of each target gene in a given sample was normalized to the level of β-actin. Relative fold expression of the target genes was calculated according to the 2−∆∆Cq method (29).
Cell proliferation assay
All GBC cell lines were transfected with GLI2 siRNA or control siRNA and seeded onto 96-well plates (5.0×103 cells/well). Cells were then incubated with or without 1.0 µg/ml recombinant Human Sonic Hedgehog/SHH (R&D SYSTEMS, Inc.) at 37°C for 24 and 48 h. In cytotoxic assays, siRNA-transfected cells were incubated with or without GEM or CDDP (0–100 µg/ml) at 37°C for 48 h. Cell Count Reagent SF (Nacalai Tesque, Inc.) at the original concentration was then added to the cells and incubated at 37°C for 1 h. Cell proliferation was assessed by absorbance at 492 nm using a plate reader (Biotrak visible plate reader; Amersham Biosciences; Cytiva) with a reference wavelength 620 nm.
Cell invasion assay
The invasive capacity of the GBC cell lines was assessed by Matrigel invasion assay as previously described (19). Briefly, siRNA-transfected cells (2.0×105) were placed in the upper chamber of a Transwell chamber with or without recombinant human SHH and incubated for 18 h. The cells that invaded to the lower side of the filter were fixed and stained with Diff-Quik reagent (Sysmex Corporation). Diff-Quik Fixative, Diff-Quik Solution I, and then Diff-Quik Solution II were used in this order at their original concentrations for 10 min at room temperature. The stained cells were counted at an ×200 magnification under a light microscope (Nikon Eclipse TE 300; Nikon Corporation).
Western blot analysis
Western blotting was performed as previously described (30). The protein-transferred membranes were incubated overnight at 4°C with primary antibodies for GLI1 (1:500; cat. no. ab151796; Abcam), GLI2 (1:500; cat. no. ab187386; Abcam), GLI3 (1:200; cat. no. sc-6154; Santa Cruz Biotechnology, Inc.), SMO (1:200; cat. no. 20787-1-AP; ProteinTech Group, Inc.), SHH (1:200; cat. no. sc-365112; Santa Cruz Biotechnology, Inc.), cyclin D1 (1:200; cat. no. sc-246; Santa Cruz Biotechnology, Inc.), cyclin B1 (1:200; cat. no. sc-245; Santa Cruz Biotechnology, Inc.), Ki-67 (1:200; cat. no. sc-15402; Santa Cruz Biotechnology, Inc.), E-cadherin (1:200; cat. no. sc-7870; Santa Cruz Biotechnology, Inc.), vimentin (1:1,000; cat. no. ab92547; Abcam), SNAI1 (1:200, cat. no. sc-271977; Santa Cruz Biotechnology, Inc.), Slug (1:200; cat. no. sc-15391; Santa Cruz Biotechnology, Inc.), transforming growth factor-β1 (TGFβ1) (1:200; cat. no. sc-146; Santa Cruz Biotechnology, Inc.), programmed cell death ligand 1 (PD-L1) (Clone; 29E.22A3) (1:400; cat. no. B7-H1; BioLegend, Inc.), or HIF-1α (1:200; cat. no. ab51608; Abcam). The membranes were then incubated for at least 1 h at room temperature with horseradish peroxidase-linked anti-mouse antibody (1:10,000; cat. no. NA931; Amersham Biosciences; Cytiva), horseradish peroxidase-linked anti-rabbit antibody (1:10,000; cat. no. NA934; Amersham Biosciences; Cytiva), or horseradish peroxidase-linked anti-goat antibody (1:10,000; cat. no. sc-2020; Santa Cruz Biotechnology, Inc.). α-Tubulin (1:1,000; cat. no. T6199; Sigma-Aldrich; Merck KGaA) was used as a protein loading control.
Cell cycle analysis
Cells (2.0×105 cells/well) were treated for 48 h with GLI2 siRNA. Cells were harvested by trypsinization and fixed in ice-cold 75% ethanol for at least 1 h. Cell pellets were incubated and stained for 30 min at room temperature in 1 ml PBS containing 50 µg propidium iodide (Sigma-Aldrich; Merck KGaA), 0.1% Triton X-100, 1 mM/l EDTA, and 0.5 mg ribonuclease A (Sigma-Aldrich; Merck KGaA). After staining, samples were analyzed using FACScan (BD Biosciences) and BD CellQuest™ Pro software 6.0 (BD Biosciences) at 20,000 events per sample. The percentages of cells in G0/G1, S and G2/M phases were calculated for each sample.
In vivo xenograft tumor model
Five-week-old female athymic nude mice (BALB/c nu/nu; weight, 16–18 g; n=8) were purchased from Charles River Laboratories Japan and acclimated for 2 weeks. All experimental procedures were approved by the Animal Care and Use Committee of Kyushu University (permit no. A30-340-0). All experiments were performed in strict accordance with the Guidelines for Proper Conduct of Animal Experiments (Science Council of Japan). Briefly, all mice were housed and maintained in a specific pathogen-free animal facility at Kyushu University in housing conditions at a temperature of 26–28°C, humidity of 40–70% and lighting time of 12 h from 8 a.m. to 8 p.m.. Food and water were provided freely. All efforts were made to minimize the number of animals used and their suffering. The humane endpoints for euthanasia were defined as a tumor diameter exceeding 15 mm or conditions with long-lasting pain. A mouse with a skin ulceration at the tumor implantation site was euthanized as an indication for a humane endpoint. At humane and experimental endpoints, mice were euthanized by overdoses of inhaled anesthetics with sevoflurane, and the mortality of the mice was verified by cardio-respiratory arrest and loss of pupillary light reflex. NOZ cells transfected with GLI2 siRNA or control siRNA were subcutaneously implanted into the flank of nude mice (1.0×106 cells in Matrigel per mouse; n=4 in each group). Tumor size was measured twice a week, and tumor volume was calculated as follows: A × B2 × 0.5, where A is the longest diameter and B is the smaller of the two perpendicular diameters of the tumor.
Immunohistochemistry
Tissue samples were obtained from 66 patients with GBC who underwent resection at the Department of Surgery and Oncology, Kyushu University Hospitals, Fukuoka, Japan between January 2001 and December 2012. Patients who had received other anticancer treatment prior to surgery were excluded from this study. The median age of the patients was 69.5 years (range 53–91 years), and 56.1% were females. Approval for the use of tissues was obtained from patients in accordance with the Ethical Committees for Clinical Study at Kyushu University (reference no. 30–230). Tissue samples obtained from mice implanted with NOZ calls transfected with GLI2 siRNA or control siRNA were also analyzed. Immunohistochemical staining was performed using 4-µm-thick formalin-fixed, paraffin-embedded tissue sections and primary antibodies for GLI2 (1:500; product code ab187386, Abcam), α-smooth muscle actin (αSMA) (1:100; product code ab5694, Abcam), Ki-67 (1:50; cat. no. sc-15402; Santa Cruz Biotechnology, Inc.), vascular endothelial growth factor (VEGF) (1:50; cat. no. sc-152; Santa Cruz Biotechnology, Inc.), CD3 (1:100; product code ab5690; Abcam), CD8 (1:50; product code ab17147; Abcam), forkhead box P3 (FOXP3) (1:100; product code ab10563; Abcam), or PD-L1 (1:100; cat. no. B7-H1; BioLegend, Inc.). Endogenous peroxidase activity was blocked using 3% hydrogen peroxide solution for 5 min. Antigen retrieval was conducted by a high-pressure method for 10 min with Target Retrieval Solution, pH 9.0 (Agilent Technologies, Inc.). The slides were incubated with primary antibodies overnight at 4°C, followed by incubation with Histofine Simple Stain MAX-PO (M) (4 µg/ml; cat. no. 424131) or Histofine Simple Stain MAX-PO (R) (4 µg/ml; cat. no. 424141; both from Nichirei Biosciences, Inc.) for 40 min at room temperature. The labeled antigens were visualized using diaminobenzidine (DAB). Counterstaining was performed with hematoxylin for 3 min at room temperature. Negative controls were obtained in all cases by omitting the first antibodies (31). For Masson's trichrome staining, the slides were treated with Bouin's solution (Sigma-Aldrich; Merck KGaA) for 4 h at room temperature. The samples were stained using working Weigert's iron hematoxylin solution (Muto Pure Chemicals Co., Ltd.) for 10 min at room temperature and then in 0.9% Biebrich scarlet-acid fuchsin (Waldeck GmbH & Co. KG), 0.1% acid fuchsin (Sigma-Aldrich; Merck KGaA), and 1% acetic acid (Kanto Chemical Co., Inc.) for 20 min at room temperature. Subsequently, the slides were treated with working phosphotungstic/phosphomolybdic acid solution for 5 min at room temperature and placed in Aniline Blue solution (Muto Pure Chemicals Co., Ltd) for 20 min at room temperature. GLI2 expression was evaluated using the Allred score (32). Total score (TS) ≥3 was used as a cut-off value (33,34) and the samples were divided into two groups: GLI2-high expression, with TS ≥3, and GLI2-low expression, with TS <3. PD-L1 expression was considered as positive when samples contained ≥5% PD-L1-positive tumor cells (35). For the evaluation of CD3-, CD8-, and FOXP3-positive cell counts, five microscopic fields were observed at a magnification of ×400 (35). For the evaluation of αSMA expression, Ki-67 expression, and Masson's Trichrome staining of mouse xenograft tumors, we observed five microscopic fields at a magnification of ×200. Positive-stained areas were quantified using ImageJ 2.0.0-rc-69/1.52p (Fiji software) (36).
Statistical analysis
All data are expressed as the mean ± standard deviation (SD). The unpaired Student's t-test was used for comparison of mean values between two groups. One-way ANOVA followed by Tukey's post hoc test was performed when more than two groups were compared. The χ2 test was used to compare GLI2 and PD-L1 expression in human tissue specimens and to analyze the relationship between clinicopathological parameters in patients. Survival curves were plotted using the Kaplan-Meier method and analyzed using Wilcoxon tests. Cox proportional hazards model was used for multivariate analysis. The factors identified as significant in univariate analysis were included for the multivariate Cox analysis (37). Hazard ratios (HRs) were shown as relative risks, with corresponding 95% confidence intervals (CIs). Calculations were carried out using JMP 14.0 software (SAS Institute) or Microsoft Excel software (Microsoft). A P-value of <0.05 was considered to be statistically significant.
Results
GLI2 is involved in cell cycle-mediated proliferation in GBC
Protein expression of the transcription factors GLI1, GLI2, and GLI3 and Hh signal-related molecules was confirmed in three GBC cell lines (NOZ, TYGBK-1, and TGBC2TKB) (Fig. 1A). The role of each transcription factor on the proliferative capacity of GBC cells was analyzed. siRNA-mediated GLI2 knockdown significantly reduced the proliferative capacity of the cell lines (Fig. 1B). In contrast, suppression of GLI1 and GLI3 expression did not affect cell proliferation (Fig. 1C and D). These results indicated that GLI2, but not GLI1 or GLI3, is involved in the proliferative capacity of GBC. Addition of SHH, a ligand for the Hh signal, significantly enhanced the proliferative capacity of the GBC cell lines. However, SHH had no effect in GBC cells with suppression of GLI2 (Fig. 1E). In the cell cycle analysis, GLI2 suppression increased the numbers of cells in the G0/G1 phase and decreased cells in the S phase (Fig. 1F). Western blot analysis revealed a decrease in the expression of cyclin D1 and Ki-67, whereas cyclin B1 expression was not altered (Fig. 1G). Collectively, these results indicated that GLI2, but not GLI1 or GLI3, was involved in cell proliferation by regulating the cell cycle in GBC.
Figure 1.

GLI2 is involved in cell proliferation in GBC via regulation of the cell cycle. (A) Expression levels of GLI3, GLI2, GLI1, and Hh signal-related molecules (SMO and SHH) were examined in three GBC cell lines by western blot analysis. (B-D) Proliferation assays in GBC cell lines treated with (B) GLI2 siRNA or control siRNA, (C) GLI1 siRNA or control siRNA, and (D) GLI3 siRNA or control siRNA for 24 and 48 h. (E) Proliferation assays in GBC cell lines treated with GLI2 siRNA or control siRNA and then incubated with or without rhSHH for 24 and 48 h. (F) Cell cycle analysis of GBC cell lines treated with GLI2 siRNA or control siRNA. Percentages of cells in the G0/G1, S, and G2/M phase were calculated. Histograms and the bar graphs are presented. (G) Expression levels of GLI2 and cell cycle-related molecules (cyclin D1, Ki-67, and cyclin B1) were examined in GBC cell lines treated with GLI2 siRNA or control siRNA. *P<0.05, **P<0.01 and ***P<0.001. Bar, mean ± SD. GLI2, glioma-associated oncogene homolog 2; GBC, gallbladder cancer; SMO, smoothened; SHH, sonic hedgehog; siRNA or si, small interfering RNA; rhSHH, recombinant human SHH; siCont, control siRNA; siGLI2, GLI2 siRNA; siGLI1, GLI1 siRNA; siGLI3, GLI3 siRNA; n.s, not significant.
GLI2 enhances the invasive capacity of GBC cells through epithelial-mesenchymal transition (EMT)
Next, the effect of each transcription factor on the invasive capacity of GBC was examined. Suppression of GLI2 expression significantly reduced the invasive capacity of GBC cell lines (Fig. 2A). In contrast, GLI1 and GLI3 suppression did not affect the invasive capacity of GBC cells (Fig. 2B and C). These results indicated that GLI2, but not GLI1 or GLI3, enhanced the invasive capacity of GBC. Addition of SHH significantly enhanced the invasive capacity of the GBC cell lines. However, SHH had no impact on invasion in GBC cell lines in which GLI2 was suppressed (Fig. 2D). When GLI2 expression was suppressed, protein expression of the EMT-related molecule E-cadherin was increased, and vimentin was suppressed with GLI2 knockdown, while SNAI1, Slug and TGF-β1 were not altered (Fig. 2E). Collectively, these findings indicated that GLI2, but not GLI1 or GLI3, was involved in EMT-mediated enhancement of invasive capacity in GBC.
Figure 2.

GLI2 is required for cell invasion via augmentation of EMT in GBC. Invasion assay of GBC cell lines treated with (A) GLI2 siRNA or control siRNA, (B) GLI1 siRNA or control siRNA, and (C) GLI3 siRNA or control siRNA for 18 h. (D) Invasion assay of GBC cell lines treated with GLI2 siRNA or control siRNA and then incubated with or without rhSHH for 18 h. (E) Expression levels of GLI2 and EMT-related molecules (E-cadherin, vimentin, SNAI1, Slug, and TGF-β1) in GBC cell lines treated with GLI2 siRNA or control siRNA. Scale bar, 100 µm. *P<0.05, **P<0.01 and ***P<0.001. Bar, mean ± SD. GLI2, glioma-associated oncogene homolog 2; EMT, epithelial-mesenchymal transition; GBC, gallbladder cancer; siRNA or si, small interfering RNA; siCont, control siRNA; siGLI2, GLI2 siRNA; siGLI1, GLI1 siRNA; siGLI3, GLI3 siRNA; rhSHH, recombinant human SHH; TGF-β1, transforming growth factor β1; n.s, not significant.
GLI2 influences the sensitivity of GBC cells to GEM
The results indicated that GLI2 may have a central role in the induction of a malignant phenotype in GBC, and therefore GLI2 was focused on in further investigations. First, the effect of GLI2 was examined on the sensitivity of GBC cells to GEM, a key drug in GBC chemotherapy (38,39). Suppression of GLI2 significantly increased the relative viability of GBC cell lines treated with GEM (Fig. 3A). In experiments using CDDP, there was no significant difference in the relative viability between GLI2 knockdown and controls nor a concentration-dependent decrease in viability in both groups (Fig. 3B). These results indicated that GLI2 influenced GEM sensitivity of GBC cells in vitro.
Figure 3.

GLI2 suppression increases the relative survival of GBC cells treated with GEM. Relative absorbance ratio of GBC cell lines transfected with GLI2 siRNA or control siRNA and then treated with (A) GEM and (B) CDDP at 0–100 µg/ml for 48 h. *P<0.05, **P<0.01, and ***P<0.001. GLI2, glioma-associated oncogene homolog 2; GBC, gallbladder cancer; GEM, gemcitabine; CDDP, cisplatin; siRNA or si, small interfering RNA; siCont, control siRNA; siGLI2, GLI2 siRNA.
GLI2 enhances GBC cell-derived tumor proliferation in vivo
Next, the role of GLI2 in the tumor proliferative capacity of GBC was investigated in vivo. NOZ cells transfected with control siRNA or GLI2 siRNA were subcutaneously injected into the flank of BALB/c nude mice (Fig. 4A). GLI2 suppression in cells was confirmed by western blot analysis (Fig. 4B). The tumor volume of the GLI2 siRNA group was significantly reduced compared with the tumors in the control siRNA group (Fig. 4C). Immunohistochemical staining revealed a significant decrease in αSMA expression in tumor tissue of the GLI2 siRNA group, but no difference was observed between GLI2 siRNA and control tumors in Masson's Trichrome staining. Consistent with the in vitro results, Ki-67 expression was significantly downregulated in the GLI2 siRNA group compared with controls. VEGF was weakly expressed in both groups, but no significant difference was observed (Fig. 4D). These results indicated that GLI2 enhanced the proliferative capacity of cancer cells in vivo and that GLI2 also promoted tumor fibrosis.
Figure 4.

Inhibition of GLI2 suppresses GBC-cell derived tumor proliferation in vivo. (A) BALB/c nude mice were implanted with control siRNA- or GLI2 siRNA-transfected NOZ cells. (B) GLI2 expression in implanted NOZ cells. (C) Representative images of mice implanted with NOZ cells transfected with control siRNA (left) or GLI2 siRNA (right), and tumor volumes in the control siRNA group and GLI2 siRNA group. (D) Immunohistochemical staining of αSMA, MT, Ki-67, and VEGF in resected xenograft tumors. Original magnification is ×200. Positively-stained areas were quantified using ImageJ. Scale bar, 100 µm. *P<0.05, **P<0.01 and ***P<0.001. Bar, mean ± SD. GLI2, glioma-associated oncogene homolog 2; GBC, gallbladder cancer; siRNA or si, small interfering RNA; siCont, control siRNA; siGLI2, GLI2 siRNA; αSMA, α-smooth muscle actin; MT, Masson's Trichrome staining; VEGF, vascular endothelial growth factor.
GLI2 expression may be a biomarker for prognosis of GBC patients
Immunohistochemical staining was performed on 66 surgically resected specimens of GBC and GLI2 expression was evaluated using the Allred score (32). TS ≥3 was used as a cut-off value (33,34) and the cases were divided into the GLI2-high expression group (TS ≥3) and GLI2-low expression group (TS <3). Representative examples of the intensity score (0–3) and proportion score (0–4) are presented in Fig. 5A. No specimen with a proportion score of 5 was observed in this study. The clinicopathological characteristics (age, sex, depth, lymph node metastasis, lymphatic invasion, venous invasion, and Union for International Cancer Control (UICC) TNM stage of the patients are summarized in Table I. The 5-year survival rate of the patients was 54.55% (n=36/66) (Fig. S3). No significant difference in characteristics was observed between the two groups. GLI2 expression was significantly enhanced in tumor tissues compared with adjacent normal epithelial tissues (Fig. 5B). This result indicated that GLI2 expression was enhanced in GBC tissues, demonstrating its specificity as a therapeutic target. A significant difference was not observed in the overall survival between the GLI2-high expression group and GLI2-low expression group (Fig. 5C). As the results in Fig. 3A indicated that GLI2 influenced GEM sensitivity, a relationship between GLI2 expression and survival in the GEM-treated patient population was analyzed. Among the patients for whom adjuvant chemotherapy could be confirmed in their medical records, 30.43% (n=14/46) received GEM. The data regarding the chemotherapy background of the 14 patients who received adjuvant GEM treatment are presented in Table II. Notably, a significant higher survival rate in the GLI2-high expression group was observed (Fig. 5D). The univariate analysis results for the overall survival are presented in Table III. Samples were divided into two groups according to PD-L1 expression; samples with ≥5% of PD-L1-positive tumor cells were considered positive for PD-L1. For age, CD3-positive cells, CD8-positive cells, and FOXP3-positive cells, samples were divided into two groups using the median split method. The median value for age was 69.5, CD3-positive cells was 25.3, CD3-positive cells was 12.6, and FOXP3-positive cells was 10.7. UICC T category, UICC N category, lymphatic permeation, venous invasion, and stage classification were positively correlated with overall survival of GBC, and PD-L1 expression was negatively correlated with overall survival of GBC. The multivariate analysis of factors identified as significant in the univariate analysis are presented in Table IV. There was no independent prognostic factor in the present study. Even when other important prognostic factors for gallbladder cancer were included, an independent prognostic factor was still not detected (Tables SI–SIII).
Figure 5.

Immunohistochemical analysis of GLI2 in primary GBC specimens. GLI2 expression was evaluated with the Allred Score. (A) Representative examples of the intensity score (0–3) and proportion score (0–4). Original magnification is ×200. (B) Representative examples of GLI2-positive tumor tissue (upper image) and GLI2-negative normal tissue (lower image), and GLI2-positive ratio in normal tissue and tumor tissue. Original magnification is ×200. (C) Kaplan-Meier survival curves of all GBC patients of all stages according to GLI2 expression. (D) Kaplan-Meier survival curves of 14 patients who received adjuvant GEM treatments according to GLI2 expression. Scale bar, 100 µm. *P<0.05 and ***P<0.001. GLI2, glioma-associated oncogene homolog 2; GBC, gallbladder cancer; GEM, gemcitabine.
Table I.
Associations between clinicopathological characteristics and GLI2 expression.
| Variable | GLI2 low expression (n=37) | GLI2 high expression (n=29) | P-value |
|---|---|---|---|
| Age (median=69.5) | 0.4569 | ||
| <69.5 | 17 | 16 | |
| ≧69.5 | 20 | 13 | |
| Sex | 0.7106 | ||
| Male | 17 | 12 | |
| Female | 20 | 17 | |
| UICC T category | 0.4666 | ||
| Tis | 4 | 2 | |
| T1a/T1b | 7 | 5 | |
| T2a/T2b | 24 | 17 | |
| T3a/T3b | 2 | 5 | |
| UICC N category | 0.9938 | ||
| N0 | 23 | 18 | |
| N1 | 14 | 11 | |
| Lymphatic permeation | 0.5297 | ||
| (−) | 22 | 15 | |
| (+) | 15 | 14 | |
| Venous invasion | 0.1136 | ||
| (−) | 25 | 14 | |
| (+) | 12 | 15 | |
| Stage classification | 0.9304 | ||
| 0 | 4 | 2 | |
| IA/IB | 7 | 5 | |
| IIA/IIB | 12 | 11 | |
| IIIA/IIIB | 14 | 11 | |
| Stage classification | 0.6115 | ||
| 0/IA/IB | 11 | 7 | |
| IIA/IIB/IIIA/IIIB | 26 | 22 |
GLI2, glioma-associated oncogene homolog 2; UICC, Union for International Cancer Control.
Table II.
Chemotherapy background of the 14 patients who received adjuvant GEM treatments.
| Age | Sex | Stage | GLI2 | Surgery | Recurrence | RT (months) | SP (months) |
|---|---|---|---|---|---|---|---|
| 59 | M | 0 | Low | C | R-LN | 30 | 44 |
| 61 | M | II | High | C | Liver | 51 | 60 |
| 63 | M | II | High | C | None | – | 60 |
| 53 | F | II | Low | C | Peritoneum | 7 | 20 |
| 53 | F | II | High | C | None | – | 60 |
| 65 | M | II | Low | C | None | – | 60 |
| 67 | F | IIIB | High | C | None | – | 60 |
| 65 | M | II | High | C | None | – | 60 |
| 73 | F | II | Low | C | Liver | 24 | 50 |
| 58 | M | IIIB | Low | N | – | – | 31 |
| 71 | F | IIIB | Low | C | PA-LN | 12 | 35 |
| 65 | M | IIIB | Low | N | – | – | 60 |
| 65 | F | IIIB | Low | N | – | – | 49 |
| 60 | M | II | Low | C | PA-LN | 48 | 60 |
GEM, gemcitabine; GLI2, glioma-associated oncogene homolog 2; GLI2, GLI2 expression; C, curative; N, non-curative; R-LN, regional lymph node; PA-LN, para-aortic lymph node; RT, duration to recurrence; SP, survival period.
Table III.
Univariate analysis using 66 GBC patients of overall survival.
| Variable | Category (Number of cases) | P-value |
|---|---|---|
| GLI2 | Low expression (37)/High expression (29) | 0.6974 |
| Age | <69.5 (33)/≧69.5 (33) | 0.2736 |
| Sex | Female (37)/Male (29) | 0.2091 |
| UICC T category | Tis (6)/T1a and T1b (12)/T2a and T2b (23)/T3a and T3b (25) | 0.0017a |
| UICC N category | N0 (41)/N1 (25) | <0.0001a |
| Lymphatic permeation | ly(−) (37)/ly(+) (29) | <0.0001a |
| Venous invasion | v(−) (39)/v(+) (27) | 0.0001a |
| Stage classification | 0 (6)/IA and IB (12)/IIA and IIB (23)/IIIA and IIIB (25) | <0.0001a |
| PD-L1 expression | Negative (51)/Positive (15) | 0.0301a |
| CD3-positive cells | <25.3 (33)/>25.3 (33) | 0.1585 |
| CD8-positive cells | <11.7 (33)/>11.7 (33) | 0.0882 |
| FOXP3-positive cells | <9.8 (33)/>9.8 (33) | 0.2562 |
P<0.05. GBC, gallbladder cancer; GLI2, glioma-associated oncogene homolog 2; UICC, Union for International Cancer Control; PD-L1, programmed cell death ligand 1; FOXP3, forkhead box P3.
Table IV.
Multivariate analysis using 66 GBC patients of overall survival.
| Variable | HR (95% CI) | P-value |
|---|---|---|
| UICC T category | ||
| Tis/T1a and T1b/T2a and T2b/T3a and T3b | 1.4441 (0.5954–3.4837) | 0.4106 |
| UICC N category | ||
| N0/N1 | 2.1063 (0.8352–5.6633) | 0.1236 |
| Lymphatic permeation | ||
| Ly(−)/ly(+) | 1.9603 (0.817–5.1008) | 0.1463 |
| Venous invasion | ||
| v(−)/v(+) | 1.5576 (0.6083–4.1683) | 0.3631 |
| PD-L1 expression | ||
| Negative/Positive | 1.5179 (0.6617–3.4817) | 0.3245 |
GBC, gallbladder cancer; HR, hazard ratio; CI, confidence interval; UICC, Union for International Cancer Control; PD-L1, programmed cell death ligand 1.
GLI2 is involved in suppressing intratumoral infiltration of cytotoxic T lymphocytes (CTLs) and increasing PD-L1 expression
In recent years, the intratumoral infiltration of immune cells has attracted increasing attention as an important factor of the tumor microenvironment (TME) (40,41). Immunohistochemical staining was used to analyze the correlation between GLI2 expression and tumor infiltrating lymphocytes (TILs) in GBC specimens. The number of intratumoral CD3-positive cells was significantly higher in the GLI2-low expression group (Fig. 6A), and the number of intratumoral CD8-positive cells was also significantly higher in the GLI2-low expression group (Fig. 6B). When comparing the ratio of intratumoral CD8-positive cells and FOXP3-positive cells between the two groups, the CD8(+)/FOXP3(+) cell ratio was significantly higher in the GLI2-low expression group (Fig. 6C). To evaluate the anti-tumor effect of immune cells, it is also important to examine the influence of regulatory T cells (Tregs), which negatively regulate immunity. The number of intratumoral cells positive for FOXP3, a master gene of Tregs, was not significantly different in the two groups (Fig. 6D). A correlation between TILs and PD-L1 expression in cancer has been reported (42). The number of PD-L1-positive cancer cells was significantly lower in the GLI2-low expression group (Fig. 6E). Decreased expression of PD-L1 protein was also observed in cells with GLI2 suppression in western blot analysis (Fig. 6F). We previously reported that hypoxia, one of the important factors in the TME, activates the Hh signaling pathway in pancreatic cancer (43,44). We therefore analyzed GLI2 expression in GBC under hypoxia. The results revealed that GLI2 expression was enhanced in GBC cells cultured in hypoxic environments (Fig. 6G). Suppression of HIF-1α, a key transcription factor in the hypoxic environment, resulted in reduced GLI2 expression in hypoxic conditions (Fig. 6H).
Figure 6.

GLI2 expression is inversely correlated to the number of tumor-infiltrating CD8-positive T lymphocytes and correlated to PD-L1 expression. Comparison of the number of tumor infiltrating (A) CD3-positive cells and (B) CD8-positive cells in GLI2-high and GLI2-low expression groups. Original magnification is ×400. (C) Comparison of tumor-infiltrating CD8-positive/FOXP3-positive cell ratio in the two groups. (D) Comparison of the number of tumor-infiltrating FOXP3-positive cells. Original magnification is ×400. (E) Comparison of the PD-L1-positive ratio in the two groups. (F) GLI2 and PD-L1 expression in GBC cell lines treated with GLI2 siRNA or control siRNA. (G) GLI2 expression of GBC cell lines incubated in normoxic or hypoxic condition for 48 h. (H) HIF-1α and GLI2 expression in GBC cell lines treated with HIF-1α siRNA or control siRNA in hypoxic conditions for 48 h. Scale bar, 50 µm. *P<0.05, **P<0.01 and ***P<0.001. GLI2, glioma-associated oncogene homolog 2; PD-L1, programmed cell death ligand 1; GBC, gallbladder cancer; HIF-1α, hypoxia-inducible factor 1α; FOXP3, forkhead box P3; siRNA or si, small interfering RNA; siCont, control siRNA; siGLI2, GLI2 siRNA; siHIF-1α, HIF-1α siRNA.
Discussion
In the present study, it was revealed that GLI2, but not GLI1 or GLI3, regulated the proliferative and invasive capacity of GBC. The Hh signal is involved in fetal morphogenesis (4,5). Duman et al reported that this signal enhanced gene transcription of the cell cycle regulators cyclin D and cyclin E (45). Cyclin D1 is a checkpoint molecule required for transition through the G1 phase (46). Regulation of the cell cycle by GLI2 through cyclin D1 was previously reported in lung cancer (47) and hepatocellular carcinoma (48). Ki-67 is a well-known proliferation marker that is expressed at all proliferative phases in the cell cycle except the G0 phase (49). The present cell cycle analysis and cell cycle-related molecular expression analysis indicated that GLI2 exacerbated cell proliferation by enhancing G1 phase transition in GBC.
Because GEM has revealed insufficient treatment results, and resistance to GEM has become a major problem in bile duct cancer (50), the combination therapy of GEM with various anticancer agents has been investigated (26). In the present study, the potential synergistic effects of GEM and GLI2 inhibition were examined and it was revealed that suppression of GLI2 increased the relative survival of GBC cells treated with GEM. GEM irreversibly inhibited ribonucleotide reductase, which is incorporated into DNA to cause chain termination, leading to S or G2 phase arrest (51,52). As revealed in the present study, GLI2 suppression in GBC cells reduced the percentage of cells in the S phase and significantly inhibited cell proliferation. Thus, a synergistic effect of GEM administration on cytotoxicity may not have been observed because GLI2 suppression already reduced the number of S-phase cells, which are the therapeutic targets of GEM. On the other hand, Ou et al reported that aspirin, which causes G1 arrest via cyclin D1 as well as GLI2 suppression, restored GEM sensitivity in a GEM-resistant pancreatic cancer cell line (53). Although there was no synergistic effect against GEM-sensitive gallbladder cancer cell lines, GLI2-targeted therapy for GEM-resistant strains may be beneficial.
In pancreatic cancer, the Hh ligands secreted by cancer cells promote tissue fibrosis (54). In the present study it was revealed that GLI2 suppression reduced tissue αSMA expression in a mouse xenograft model. This result indicated that GLI2 may be involved in tissue fibrosis in GBC. In our past study of pancreatic cancer, we used pancreatic cancer cells and pancreatic tissue-derived cancer-associated fibroblasts (CAFs) to generate tumors in a xenograft model (55), whereas the reproduction of human pancreatic cancer tissue was not perfect because inducing fibrosis resembling the actual cancer tissue was incomplete. In addition, CAFs derived from GBC tissues has not been established. Further efforts are required to experimentally analyze the interaction between cancer cells and CAFs in GBC.
In view of the in vitro results revealing that GLI2 is involved in the induction of malignant phenotype in GBC, we hypothesized that there may be a correlation between GLI2 expression and GBC patient prognosis. However, no correlation was revealed between GLI2 expression and overall survival. The present study included only 66 resectable cases, and unresectable stage IV cases and elderly patients who were intolerable to surgery were not included. The 5-year survival rate in the patient group was 54.55% (n=36/66), which is substantially higher than the published 5-year rate of <10% (3). Thus, if GLI2 induces malignant phenotype, then the number of advanced cancer cases affected by GLI2 is likely to be lower than the actual proportion in the present study. A significant correlation between GLI2 expression and overall survival could be observed if the overall number of cases increased, including such inoperable cases. In addition, it is possible that GEM administration as adjuvant chemotherapy masks the correlation. Consistent with in vitro experiments, patients with high GLI2 expression had potential sensitivity to GEM, and the administration of GEM may have reduced the gap between the prognostic survival curves of the two groups. As revealed, in the population that received GEM as adjuvant chemotherapy, the GLI2-high expression group had a higher survival rate. Further studies need to be performed with a larger number of cases in the future.
In recent years, cancer immunotherapy has been a focus of therapeutic treatments for cancer with the development of immune checkpoint inhibitors (56,57). In addition, the TME has also attracted increased attention (58). We previously reported that inhibition of the Hh signaling pathway suppressed pancreatic cancer fibrosis and promoted intratumoral lymphocyte infiltration in pancreatic cancer (55). As our results indicated that GLI2 may promote tumor fibrosis, we analyzed the association between GLI2 expression and TILs in GBC specimens and found an inverse correlation between GLI2 expression in tumor cells and TIL counts. In addition, of the TIL fraction, significantly fewer CD8-positive cells infiltrated in the group with high GLI2 expression. We previously reported that inhibition of Hh signal with cyclopamine reduced PD-L1 expression in GBC cell lines (59). Lin et al reported a correlation between tissue PD-L1 expression and the number of tumor-infiltrating CD8-positive cells in GBC specimens (35). Consistent with their results, in the present study, a correlation was revealed between GLI2 expression and PD-L1 expression in cancer cells of GBC specimens. The finding that suppression of GLI2 reduced PD-L1 expression in vitro indicated that GLI2 may be involved in tumor immune evasion in GBC. A potential association between a hypoxic environment and GLI2 expression was also revealed, suggesting that Hh signaling, hypoxic environment, PD-L1 expression, and immune cell infiltration may influence each other to form the TME.
The schema of the results of the present study is presented in Fig. 7. The present findings indicated that GLI2 played a central biological role in GBC, and not GLI1, a transcription factor that is thought to play a central role in the Hh signaling pathway. GLI2 was involved in proliferation, invasion, fibrosis, PD-L1 expression, and TILs in GBC and could be a novel therapeutic target. The results of the present study provide a significant contribution to the development of new treatments for refractory GBC, which has few therapeutic options.
Figure 7.
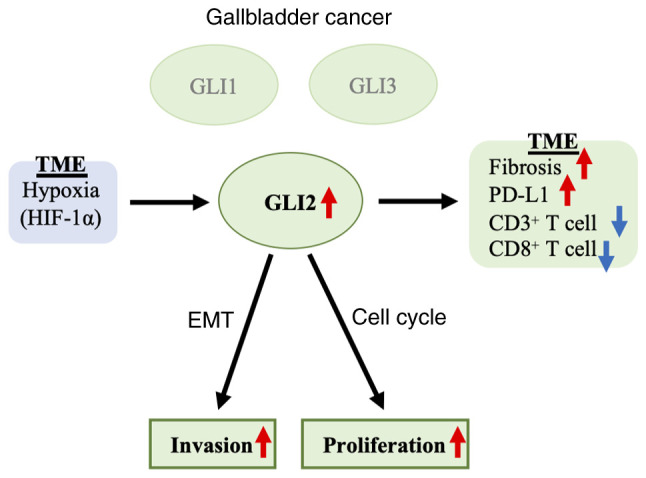
Schematic findings of the study. In GBC, GLI2 augmented the proliferative capacity and invasive capacity, and regulated tissue fibrosis and TILs in the TME. Hypoxia may be involved in the expression of GLI2. GBC, gallbladder cancer; GLI2, glioma-associated oncogene homolog 2; TILs, tumor-infiltrating lymphocytes; TEM, tumor microenvironment.
Supplementary Material
Acknowledgements
We thank Ms. Emi Onishi for her skillful technical assistance at Department of Cancer Therapy and Research, Kyushu University Graduate School of Medical Sciences, Ms. Megumi Kiyota for technical assistance at The Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences.
Funding Statement
The present study was supported by the Japan Society for the Promotion of Science KAKENHI grant no. 18K08620.
Funding
The present study was supported by the Japan Society for the Promotion of Science KAKENHI grant no. 18K08620.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
SI performed the majority of the experiments and wrote the manuscript. HO, YoO and MN made notable contributions to the design, data interpretation and the manuscript revision. SN, SK, KS, KN, AF, YaO and AI performed experiments and established experimental techniques. KS, YaO, and AI helped to design and plan the experiments. KS and YaO were involved in the validation of the data. All authors have read and approved the manuscript.
Ethics approval and consent to participate
Protocols involving the use of human tissues were approved by the Ethical Committees for Clinical Study at Kyushu University (reference no. 30-230) and conducted in accordance with the Ethical Guidelines for Human Genome/Gene Research enacted by the Japanese Government and the Helsinki Declaration. Written informed consent was obtained from all patients who agreed to the use of their samples in the present research. Protocols involving the use of animals were approved by the Animal Care and Use Committee of Kyushu University (permit no. A30-340-0).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Lazcano-Ponce EC, Miquel JF, Muñoz N, Herrero R, Ferrecio C, Wistuba II, Alonso de Ruiz P, Aristi Urista G, Nervi F. Epidemiology and molecular pathology of gallbladder cancer. CA Cancer J Clin. 2001;51:349–364. doi: 10.3322/canjclin.51.6.349. [DOI] [PubMed] [Google Scholar]
- 3.Misra S, Chaturvedi A, Misra NC, Sharma ID. Carcinoma of the gallbladder. Lancet Oncol. 2003;4:167–176. doi: 10.1016/S1470-2045(03)01021-0. [DOI] [PubMed] [Google Scholar]
- 4.Hebrok M. Hedgehog signaling in pancreas development. Mech Dev. 2003;120:45–57. doi: 10.1016/S0925-4773(02)00331-3. [DOI] [PubMed] [Google Scholar]
- 5.Cohen MM., Jr The hedgehog signaling network. Am J Med Genet A. 2003;123A:5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- 6.Ingham PW, McMahon AP. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- 7.Kalderon D. Similarities between the hedgehog and Wnt signaling pathways. Trends Cell Biol. 2002;12:523–531. doi: 10.1016/S0962-8924(02)02388-7. [DOI] [PubMed] [Google Scholar]
- 8.Ruel L, Rodriguez R, Gallet A, Lavenant-Staccini L, Therond PP. Stability and association of smoothened, costal2 and fused with cubitus interruptus are regulated by hedgehog. Nat Cell Biol. 2003;5:907–913. doi: 10.1038/ncb1052. [DOI] [PubMed] [Google Scholar]
- 9.Onishi H, Katano M. Hedgehog signaling pathway as a therapeutic target in various types of cancer. Cancer Sci. 2011;102:1756–1760. doi: 10.1111/j.1349-7006.2011.02010.x. [DOI] [PubMed] [Google Scholar]
- 10.Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O'Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–73. doi: 10.1126/science.3563490. [DOI] [PubMed] [Google Scholar]
- 11.Gailani MR, Bale AE. Developmental genes and cancer: Role of patched in basal cell carcinoma of the skin. J Natl Cancer Inst. 1997;89:1103–1109. doi: 10.1093/jnci/89.15.1103. [DOI] [PubMed] [Google Scholar]
- 12.Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- 13.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/S0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 14.Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 15.Kubo M, Nakamura M, Tasaki A, Yamanaka N, Nakashima H, Nomura M, Kuroki S, Katano M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004;64:6071–6074. doi: 10.1158/0008-5472.CAN-04-0416. [DOI] [PubMed] [Google Scholar]
- 16.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watkins DN, Berman DM, Burkholder SG, Wang B, Beachy PA, Baylin SB. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature. 2003;422:313–317. doi: 10.1038/nature01493. [DOI] [PubMed] [Google Scholar]
- 18.Berman DM, Karhadkar SS, Maitra A, Montes De Oca R, Gerstenblith MR, Briggs K, Parker AR, Shimada Y, Eshleman JR, Watkins DN, Beachy PA. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 19.Matsushita S, Onishi H, Nakano K, Nagamatsu I, Imaizumi A, Hattori M, Oda Y, Tanaka M, Katano M. Hedgehog signaling pathway is a potential therapeutic target for gallbladder cancer. Cancer Sci. 2014;105:272–280. doi: 10.1111/cas.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahmane N, Sánchez P, Gitton Y, Palma V, Sun T, Beyna M, Weiner H, Ruiz I, Altaba A. The sonic hedgehog-gli pathway regulates dorsal brain growth and tumorigenesis. Development. 2001;128:5201–5212. doi: 10.1242/dev.128.24.5201. [DOI] [PubMed] [Google Scholar]
- 21.Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by Hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- 22.Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–1172. doi: 10.1056/NEJMoa0905360. [DOI] [PubMed] [Google Scholar]
- 26.Sabol M, Trnski D, Musani V, Ozretić P, Levanat S. Role of GLI transcription factors in pathogenesis and their potential as new therapeutic targets. Int J Mol Sci. 2018;19:E2562. doi: 10.3390/ijms19092562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horiuchi H, Kawamata H, Fujimori T, Kuroda Y. A MEK inhibitor (U0126) prolongs survival in nude mice bearing human gallbladder cancer cells with K-ras mutation: Analysis in a novel orthotopic inoculation model. Int J Oncol. 2003;23:957–963. [PubMed] [Google Scholar]
- 28.Sekine S, Shimada Y, Nagata T, Moriyama M, Omura T, Yoshioka I, Hori R, Matsui K, Sawada S, Okumura T, et al. Establishment and characterization of a new human gallbladder carcinoma cell line. Anticancer Res. 2012;32:3211–3218. [PubMed] [Google Scholar]
- 29.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta) C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 30.Suyama K, Onishi H, Imaizumi A, Shinkai K, Umebayashi M, Kubo M, Mizuuchi Y, Oda Y, Tanaka M, Nakamura M, Katano M. CD24 suppresses malignant phenotype by downregulation of SHH transcription through STAT1 inhibition in breast cancer cells. Cancer Lett. 2016;374:44–53. doi: 10.1016/j.canlet.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 31.Kameda C, Nakamura M, Tanaka H, Yamasaki A, Kubo M, Tanaka M, Onishi H, Katano M. Oestrogen receptor-alpha contributes to the regulation of the hedgehog signalling pathway in ERalpha-positive gastric cancer. Br J Cancer. 2010;102:738–747. doi: 10.1038/sj.bjc.6605517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allred DC, Harvey JM, Berardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–168. [PubMed] [Google Scholar]
- 33.Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17:1474–1481. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 34.Mohsin SK, Weiss H, Havighurst T, Clark GM, Berardo M, Roanh le D, To TV, Qian Z, Love RR, Allred DC. Progesterone receptor by immunohistochemistry and clinical outcome in breast cancer: A validation study. Mod Pathol. 2004;17:1545–1554. doi: 10.1038/modpathol.3800229. [DOI] [PubMed] [Google Scholar]
- 35.Lin J, Long J, Wan X, Chen J, Bai Y, Wang A, Yang X, Wu Y, Robson SC, Sang X, Zhao H. Classification of gallbladder cancer by assessment of CD8+ TIL and PD-L1 expression. BMC Cancer. 2018;18:766. doi: 10.1186/s12885-018-4651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui X, Zhu S, Tao Z, Deng X, Wang Y, Gao Y, Liao Y, Ma W, Zhang Y, Ma X. Long-term outcomes and prognostic markers in gallbladder cancer. Medicine (Baltimore) 2018;97:e11396. doi: 10.1097/MD.0000000000011396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 39.Javle M, Zhao H, Abou-Alfa GK. Systemic therapy for gallbladder cancer. Chin Clin Oncol. 2019;8:44. doi: 10.21037/cco.2019.08.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu CC, Steen CB, Newman AM. Computational approaches for characterizing the tumor immune microenvironment. Immunology. 2019;158:70–84. doi: 10.1111/imm.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17:807–821. doi: 10.1038/s41423-020-0488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho YA, Yoon HJ, Lee JI, Hong SP, Hong SD. Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncol. 2011;47:1148–1153. doi: 10.1016/j.oraloncology.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T, Morisaki T, Nakashima Y, Katano M. Hypoxia activates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci. 2011;102:1144–1150. doi: 10.1111/j.1349-7006.2011.01912.x. [DOI] [PubMed] [Google Scholar]
- 44.Onishi H, Yamasaki A, Kawamoto M, Imaizumi A, Katano M. Hypoxia but not normoxia promotes smoothened transcription through upregulation of RBPJ and mastermind-like 3 in pancreatic cancer. Cancer Lett. 2016;371:143–150. doi: 10.1016/j.canlet.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 45.Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing cyclin D and cyclin E. Nature. 2002;417:299–304. doi: 10.1038/417299a. [DOI] [PubMed] [Google Scholar]
- 46.Day PJ, Cleasby A, Tickle IJ, O'Reilly M, Coyle JE, Holding FP, McMenamin RL, Yon J, Chopra R, Lengauer C, Jhoti H. Crystal structure of human CDK4 in complex with a D-type cyclin. Proc Natl Acad Sci USA. 2009;106:4166–4170. doi: 10.1073/pnas.0809645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutter M, Wang J, Huang Z, Kuliszewski M, Post M. Gli2 influences proliferation in the developing lung through regulation of cyclin expression. Am J Respir Cell Mol Biol. 2010;42:615–625. doi: 10.1165/rcmb.2008-0390OC. [DOI] [PubMed] [Google Scholar]
- 48.Zhang D, Liu J, Wang Y, Chen J, Chen T. shRNA-mediated silencing of Gli2 gene inhibits proliferation and sensitizes human hepatocellular carcinoma cells towards TRAIL-induced apoptosis. J Cell Biochem. 2011;112:3140–3150. doi: 10.1002/jcb.23240. [DOI] [PubMed] [Google Scholar]
- 49.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- 50.Kawamoto M, Umebayashi M, Tanaka H, Koya N, Nakagawa S, Kawabe K, Onishi H, Nakamura M, Morisaki T. Combined gemcitabine and metronidazole is a promising therapeutic strategy for cancer stem-like cholangiocarcinoma. Anticancer Res. 2018;38:2739–2748. doi: 10.21873/anticanres.12516. [DOI] [PubMed] [Google Scholar]
- 51.Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76:358–369. doi: 10.1111/bcp.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Montano R, Khan N, Hou H, Seigne J, Ernstoff MS, Lewis LD, Eastman A. Cell cycle perturbation induced by gemcitabine in human tumor cells in cell culture, xenografts and bladder cancer patients: Implications for clinical trial designs combining gemcitabine with a Chk1 inhibitor. Oncotarget. 2017;8:67754–67768. doi: 10.18632/oncotarget.18834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ou YQ, Zhu Wb, Li Y, Qiu PX, Huang YJ, Xie J, He SM, Zheng XK, Leng TD, Xu D, Yan GM. Aspirin inhibits proliferation of gemcitabine-resistant human pancreatic cancer cells and augments gemcitabine-induced cytotoxicity. Acta Pharmacol Sin. 2010;31:73–80. doi: 10.1038/aps.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bynigeri RR, Jakkampudi A, Jangala R, Subramanyam C, Sasikala M, Rao GV, Reddy DN, Talukdar R. Pancreatic stellate cell: Pandora's box for pancreatic disease biology. World J Gastroenterol. 2017;23:382–405. doi: 10.3748/wjg.v23.i3.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oyama Y, Onishi H, Koga S, Murahashi M, Ichimiya S, Nakayama K, Fujimura A, Kawamoto M, Imaizumi A, Umebayashi M, et al. Patched 1-interacting peptide represses fibrosis in pancreatic cancer to augment the effectiveness of immunotherapy. J Immunother. 2020;43:121–133. doi: 10.1097/CJI.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 56.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 58.Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–68. doi: 10.1016/j.canlet.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 59.Onishi H, Fujimura A, Oyama Y, Yamasaki A, Imaizumi A, Kawamoto M, Katano M, Umebayashi M, Morisaki T. Hedgehog signaling regulates PDL-1 expression in cancer cells to induce anti-tumor activity by activated lymphocytes. Cell Immunol. 2016;310:199–204. doi: 10.1016/j.cellimm.2016.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.