

Abstract
Background
Acute myeloid leukemia (AML) is biologically heterogeneous diseases with adverse prognosis. This study was conducted to find prognostic biomarkers that could effectively classify AML patients and provide guidance for treatment decision making.
Methods
Weighted gene co-expression network analysis was applied to detect co-expression modules and analyze their relationship with clinicopathologic characteristics using RNA sequencing data from The Cancer Genome Atlas database. The associations of gene expression with patients’ mortality were investigated by a variety of statistical methods and validated in an independent dataset of 405 AML patients. A risk score formula was created based on a linear combination of five gene expression levels.
Results
The weighted gene co-expression network analysis detected 63 co-expression modules. The pink and darkred modules were negatively significantly correlated with overall survival of AML patients. High expression of FNDC3B, VSTM1 and CALR was associated with favourable overall survival, while high expression of PLA2G4A was associated with adverse overall survival. Hierarchical clustering analysis of FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR uncovered four subgroups of AML patients. The cluster1 AML patients showed younger age, lower cytogenetics risk, higher frequency of NPM1 mutations and more favourable overall survival than cluster3 patients. The risk score was demonstrated to be an indicator of adverse prognosis in AML patients
Conclusions
The FNDC3B, VSTM1, PLA2G4A, GOLGA3, CALR and risk score may serve as key prognostic biomarkers for the stratification and ultimately guide rational treatment of AML patients.
Keywords: The cancer genome atlas database, Acute myeloid leukemia, Weighted gene co-expression network analysis, Risk score, Overall survival
Background
Acute myeloid leukemia (AML) is biologically heterogeneous diseases with a relatively adverse survival rate [1]. The Surveillance, Epidemiology, and End Results Program [2], reports an incidence rate of 4.3 per 100,000 persons and mortality rate of 2.8 per 100,000 persons annually. AML patients show a relatively poor 5-year survival rate of 27.4%. The 2017 European Leukemia Net (ELN) guidelines are well established tools for the assessment of risk of resistance and prognosis for AML patients. The ELN 2017 could effectively classify AML patients into three subgroups, including favorable, intermediate and poor subgroups, according to leukemia cell genetic abnormalities and mutations in driver genes [3]. For instance, some cytogenetic abnormalities are related to favorable clinical outcome, such as inv(16)(p13.1q22) and t(8;21)(q22;q22.1). While, others are indicative of poor overall survival in AML patients, such as t(6;9)(p23;q34.1), inv(3)(q21.3q26.2) [3]. RUNX1-RUNX1T1 or MYH11-CBFB fusions are indicators of good clinical outcomes in AML patients who underwent chemotherapy based consolidation regimens [4, 5]. While, a large proportion of AML genomes are lack of structural abnormalities [6, 7]. In addition to cytogenetic abnormalities, the 2017 ELN also includes mutations in several genes for risk stratification. The TP53 mutation is one of the known adverse factors and frequently associated with complex cytogenetics. NPM1 and CEBPA mutations are indicative of favorable prognosis regardless of cytogenetic abnormalities. A FLT3 internal tandem duplication (ITD) with the ratio of mutated to normal alleles > 0.5 is associated with poor prognosis [3]. DNMT3A, NPM1 mutations and MLL translocations have been shown to ameliorate risk classification for patients showing normal karyotype [8]. However, these genes are not applied to those AML patients who didn’t have DNMT3A, NPM1 mutations and MLL translocations [8]. Therefore, none of the current markers is entirely accurate, novel biomarkers are required to improve prognostic classification.
The weighted gene co-expression network analysis (WGCNA) package identifies co-expression modules in which the expression of a set of genes is highly correlated and seeks for associations between interested co-expression modules and clinical characteristics. The analysis enables researchers to detect co-expression networks related to certain phenotypic trait [9]. In this study, we applied the WGCNA algorithm to a genome-wide study of 18,366 genes using RNA-seq expression data of 173 AML patients from The Cancer Genome Atlas (TCGA) database. The WGCNA analysis revealed two co-expression modules which were significantly associated with patients’ overall survival (OS). Further analysis of the two mortality-associated modules identified a gene panel of FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR. Hierarchical clustering analysis of the five genes enabled the identification of a subgroup of AML patients with favourable OS. The co-expression modules and gene panel may be of importance in evaluating the prognosis of AML patients.
Methods
Data acquisition and processing
In total, normalized read counts (RNA-seq) data of 20,531 genes of 173 AML patients and their clinical data were acquired from the TCGA database [10]. Genes without expression values in 90% AML patients were removed. Totally, 18,366 genes met the inclusion criterion of the WGCNA analysis. The FAB subtypes consisted of 8 subtypes, including minimal maturation AML (M0), no maturation AML (M1), maturation AML (M2), acute promyelocytic (M3), myelomonocytic (M4), monoblastic or monocytic (M5), erythroid (M6), megakaryoblastic (M7) leukemia and others. Cytogenetic risk comprised favorable, intermediate and poor prognosis categories. Gene expression and clinical characteristics of AML patients (n = 405) were obtained from the Oregon Health & Science University (OHSU) database for validation analysis [11].
The weighted gene co-expression network analysis in AML
Co-expression networks were built by the R package of WGCNA using normalized read count data of 18,366 genes of 173 AML patients in R3.2.0. The parameter of soft thresholding was set to 7, the minimum number of genes was set to 30, other parameters were used with the default values. Heatmap tools package was painted to analyze the strength of the interactions. The constructed modules were ranged by the number of genes and genetic information was extracted from each module. In order to identify co-expression modules which showed significant correlation with phenotypes, associations between modules and clinical traits were investigated by analyzing the correlation of the co-expression module eigengenes with clinical traits.
Functional enrichment analysis
We utilized the Gene ontology (GO) [12] and Search Tool for the Retrieval of Interacting Genes/Proteins (STRING)[13] to analyze the potential functional importance for the genes in the co-expressed modules. The enrichment of GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were regarded to be statistically significant based on the cutoff values of adjusted P value and false discovery rate (FDR) < 0.05 respectively.
Survival analyses
We followed the methods of Lai et al. and Sha et al. to perform the survival analyses [14, 15]. In brief, AML patients were grouped into two subgroups, including high and low expression groups, according to the cutoff values determined by the pROC package [16]. The difference in overall survival rates was compared between the two groups of AML patients using the Kaplan–Meier survival analysis. The prognostic importance of genes were further evaluated by the logistic regression model [17, 18]. Survival-related genes were further divided into risk genes (odd ration [OR] > 1) and protective genes (0 < OR < 1). The five genes FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR were used to build the risk score model. Risk score = expression of gene 1 × β1 + expression of gene 2 × β2 + ⋯ + expression of gene n × βn. The β values were coefficients generated by the logistic regression model. The relation between risk scores and OS was investigated by Kaplan–Meier survival analysis and logistic regression model analysis. P < 0.05 was considered statistically significant.
Unsupervised hierarchical clustering analysis
Unsupervised hierarchical clustering of CALR, VSTM1, PLA2G4A, GOLGA3 and FNDC3B was conducted with the R package of pheatmap [19]. Difference in quantitative clinical factors was analyzed by analysis of variance test among four clusters of patients. For between-group comparison, the Wilcoxon sum rank test was used. Difference in qualitative variables was investigated by fisher exact test. To characterize the prognostic probabilities of clusters of AML patients, we plotted Kaplan–Meier curves and compared overall survival rate differences using the log-rank test [20]. P < 0.05 was considered statistically significant.
Results
General characteristics of 173 AML patients
The mean age was 55.28 (range 18–88 years old). The mean percent of bone marrow blast cells was 38.85 at diagnosis. The AML patients comprised 16 M0, 42 M1, 39 M2, 16 M3, 35 M4, 18 M5, 2 M6, 3 M7 and 2 unclassified samples. 32, 103 and 36 patients were predicted to have favorable, intermediate and poor prognosis by the ELN guidelines respectively. The number of AML patients with IDH1, IDH2, DNMT3A, NPM1, FLT3 and CEBPA mutations was 16, 17, 43, 48, 48 and 13 respectively. 45 AML patients received neoadjuvant treatment. 114 AML patients were dead, 59 were alive and 10 patients were lost to contact. The average follow-up time was 563.61 days (range 0–2861 days).
Detection of co-expression modules in AML
The WGCNA package was applied to construct the co-expression network and detect co-expression modules using normalized read counts of 18,366 genes of the 173 AML samples. The scale-free fit index was greater than 0.8 and the mean connectivity of the WGCNA network was stable at the soft-thresholding power value of six. Therefore, we used the soft-thresholding power value of seven in the WGCNA analysis (Additional file 1: Figure 1). The WGCNA analysis detected 63 co-expression modules and turquoise, blue, brown, yellow and green modules were the top 5 modules having largest number of genes (Fig. 1 and Additional file 1: Figure 2, Additional file 2: Table 1).
Fig. 1.

The associations between co-expression modules and clinical traits. Each row and column corresponded to a module eigengene and clinical trait respectively. The correlation co-efficient and P value were presented in each cell. The red-to-blue bar on the right showed the degree of correlation between co-expression modules and clinical traits
Module-trait association analysis in AML
The majority of modules (59/63) showed significant correlation with the 15 clinical traits. 14, 26, 22, 5, 3, 3, 6,23, 2,12,27 and 12 modules were significantly correlated with patients’ age, bone marrow blast cell, cytogenetic risk, gender, IDH1 mutation, IDH2 mutation, DNMT3A mutation, NPM1 mutation, CEBPA mutation, FLT3 mutation, FAB subtypes and neoadjuvant treatment respectively (P value < 0.05 for all cases, Fig. 1 and Additional file 2: Table 1). Importantly, the pink and darkred modules (hereinafter referred to as overall survival-associated module1: OSAM1, overall survival-associated module2: OSAM2 respectively) were negatively correlated with patients’ OS (P value < 0.05 for all cases, Fig. 1). Moreover, the OSAM1 module also showed significantly negative correlation with patients’ age, PBMBC, cytogenetic risk, DNMT3A mutation and NPM1 mutation. The OSAM2 module was negatively correlated with FAB subtypes (P value < 0.05 for all cases, Fig. 1, Table1).
Table 1.
The associations between clinical traits and the OSAM1 and OSAM2 in the WGCNA network
| Module | Number of genes | Correlation with clinical traits |
|---|---|---|
| OSAM1 | 429 | Age, PBMBC, Cytogenetic risk, OS, DNMT3A mutation, NP1 mutation |
| OSAM2 | 151 | OS, FAB |
PBMBC percent of bone marrow blast cells, OSAM1 overall survival-associated module1, OSAM2 overall survival-associated module2
Functional annotation of genes in the OSAM1 and OSAM2 modules
The functional values of genes in the OSAM1 and OSAM2 modules were analyzed by GO and KEGG pathway enrichment analysis. Genes in the OSAM1 module were significantly enriched in 53 GO terms (adjusted P value < 0.05), such as negative regulation of signaling (GO:0023057), regulation of cell communication (GO:0010646), negative regulation of developmental process (GO:0051093), cell differentiation (GO:0030154), negative regulation of developmental process (GO:0051093). Moreover, the genes in the OSAM1 module were over-represented in the KEGG pathway of protein processing in endoplasmic reticulum (FDR < 0.05). The OSAM2 module genes were significantly enriched in the KEGG pathway of other types of O-glycan biosynthesis (FDR = 0.001).
Identification of survival-related genes in AML
Kaplan–Meier survival analysis suggested that patients with high expression levels of 327 genes exhibited favorable clinical outcome, such as FNDC3B, VSTM1 and CALR. Whereas, patients with high expression levels of 12 genes were associated with a poor prognosis, such as PLA2G4A (P < 0.05 for all cases, log rank test, Fig. 2 and Additional file 2: Table 2). Among the clinicopathologic characteristics, cytogenetic risk and patients’ age were significantly associated with patients’ mortality (P < 0.001 for all cases, Fisher exact test or Wilcoxon sum rank test, Additional file 2: Table 3). However, the association was not observed between OS and other factors, such as gender, PBMBC, IDH1, IDH2, DNMT3A, NPM1, FLT3, CEBPA mutations and neoadjuvant treatment (P > 0.05 for all cases, Fisher exact test or Wilcoxon sum rank test, Additional file 2: Table 3). Then, logistic regression model was applied between patients’ OS and patients’ age, cytogenetic risk, 339 gene expression levels. High expression of 207 genes was associated with favorable prognosis, such as FNDC3B, VSTM1 and CALR (P < 0.05 for all cases, OR: 0.32–0.44, Additional file 2: Table 2). While high expression of 8 genes was associated with inferior overall survival, including IL15RA, ITGB1BP1, STAB1, PLA2G4A, STIM2, VCL, DCLRE1B and USP20 (P < 0.05 for all cases, OR:2.31–3.88, Additional file 2: Table 2).
Fig. 2.

Kaplan–Meier survival analysis of patients’ OS with VSTM1 (a), FNDC3B (b), PLA2G4A (c) and CALR (d) expression levels in 173 AML patients of the TCGA dataset. The blue and red plots are low and high expression groups respectively
Validation of survival-related genes
The clinical characteristics of AML patients in the OHSU cohort are presented in Additional file 2: Table 4. Kaplan–Meier survival analysis suggested that patients with high expression of 55 genes showed a favourable prognosis than those with low expression, such as FNDC3B, VSTM1 and CALR. While, AML patients with high IL15RA, VCL and PLA2G4A expression had a poor prognosis than those with low IL15RA, VCL and PLA2G4A expression (P < 0.05 for all cases, log rank test, Fig. 3, Additional file 2: Table 5 and Additional file 1: Figure 3). Then, logistic regression model was applied between patients’ OS and 48 gene expression levels and the survival-related features, including age, cytogenetic risk, chemotherapy, bone marrow transplant, targeted therapy. 37 genes were demonstrated to be protective genes, such as FNDC3B, VSTM1 and CALR (P < 0.05 for all cases, OR 0.29–0.34), while VCL and PLA2G4A was confirmed to be risk genes (P = 0.01, OR 1.99, P < 0.001, OR: 2.85, respectively, Additional file 2: Table 5 and Additional file 1: Figure 3).
Fig. 3.

Kaplan–Meier survival analysis of patients’ OS with VSTM1 (a), FNDC3B (b), PLA2G4A (c) and CALR (d) expression levels in 405 AML patients of the OHSU dataset
Unsupervised hierarchical clustering analysis
To build a panel of prognostic biomarkers to accurately evaluate the prognosis of AML patients, we selected the top five genes most significantly associated with patients’ OS. FNDC3B, VSTM1, GOLGA3 and CALR showed the smallest four OR values and P values among the protective genes, PLA2G4A had the largest OR and smallest P value among the risk genes in the validation cohort. Therefore, these five genes were included in the gene panel for further survival analysis. Hierarchical clustering analysis of the five genes revealed four subgroups of AML patients (Additional file 1: Figure 4). The cluster1 AML patients showed younger age, lower cytogenetics risk, higher frequency of NPM1 mutations and better OS than cluster3 patients (P values < 0.05 for all cases, Wilcoxon sum rank test, fisher exact test or log-rank test, Fig. 4 and Additional file 2: Table 6). The hierarchical clustering of the five genes also uncovered four subgroups of AML patients in the OHSU dataset (Additional file 1: Figure 5). Cluster1 tumors exhibited lower cytogenetics risk than those in cluster 3 or 4, lower frequency of NPM1 mutations than cluster2 tumors, lower frequency of FLT3-ITD mutations than cluster 2 or 4 tumors and better OS than cluster 2, 3 or 4 tumors (P values < 0.05 for all cases, Wilcoxon sum rank test, fisher exact test or log-rank test, Additional file 1: Figure 6 and Additional file 2: Table 7).
Fig. 4.

Differences in patients’ age (a), cytogenetic risk (b), NPM1 mutation (c), and OS (d) were compared among the four clusters of AML patients (1–4) in the TCGA dataset
Risk score is a risk factor for overall survival in AML
We established the risk score model by a linear combination of the five genes FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR using the coefficients generated from the logistic regression models. Risk score = 0.32 × expression of CALR + 0.39 × expression of VSTM1 + 3.88 × expression of PLA2G4A + 0.25 × expression of GOLGA3 + 0.44 × expression of FNDC3B in the TCGA dataset. Kaplan–Meier survival analysis exhibited the risk score was negatively associated with OS of AML patients in the TCGA dataset (P < 0.05, log rank test and Fig. 5). The logistic regression model analysis validated that risk score was significantly associated with inferior OS following adjustment of survival-related features (P < 0.05 for all cases, Table2 and Fig. 5). To validate the findings above, risk score was calculated following the formula: risk score = 0.34 × expression of CALR + 0.32 × expression of VSTM1 + 2.85 × expression of PLA2G4A + 0.35 × expression of GOLGA3 + 0.29 × expression of FNDC3B. The negative correlation between OS and risk score was confirmed in the OHSU cohort (Table 2 and Fig. 5).
Fig. 5.
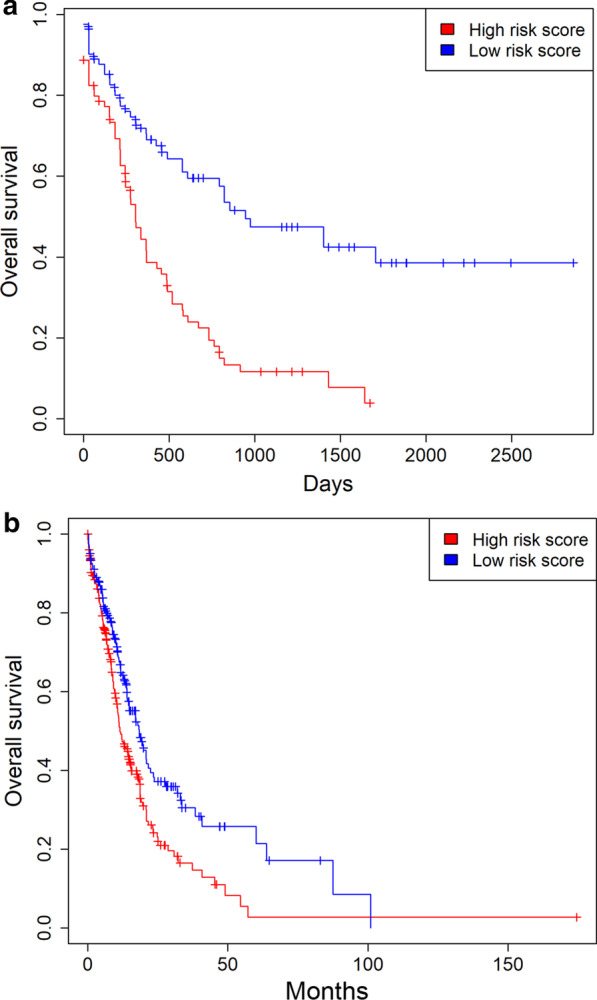
Risk score is a negative prognostic factor. a High risk score is associated with a poor prognosis in the TCGA dataset. b High risk score is associated with a poor prognosis in the OHSU dataset
Table 2.
Multivariate analyses between OS and the risk score in the TCGA and OHSU datasets
| TCGA dataset | OHSU dataset | ||||||
|---|---|---|---|---|---|---|---|
| Clinical feature | OR | 95% CI | P value | Clinical feature | OR | 95% CI | P value |
| Age | 1.03 | 1.01–1.06 | < 0.01 | Age | 1.04 | 1.02–1.05 | < 0.001 |
| Cytogenetic risk | 1.71 | 0.97–3.1 | 0.07 | Cytogenetic risk | 0.97 | 0.74–1.28 | 0.85 |
| Risk score | 3.88 | 1.84–8.46 | < 0.001 | Chemotherapy | 0.27 | 0.01–1.47 | 0.22 |
| Transplant | 0.38 | 0.23–0.63 | < 0.001 | ||||
| Targeted therapy | 2.87 | 1.51–5.71 | < 0.002 | ||||
| Risk score | 2.37 | 1.49–3.81 | < 0.001 | ||||
OR odds ratio, CI confidence interval
Discussion
WGCNA is a common computational tool to develop co-expression network and to identify the co-expression modules. Genes in the same module were regarded as functionally relevant. Thus, the application of WGCNA analysis enables the identification of clinical trait-associated modules which might become potentially prognostic and therapeutic targets [9]. In this study, 63 co-expression modules were detected by the WGCNA method using RNA-seq expression data of 18,366 genes from 173 AML samples. We identified 59/63 co-expression modules showed significant correlation with clinical traits. The OSAM1 module showed significantly negative correlation with age, cytogenetic risk, PBMBC, DNMT3A mutation and NPM1 mutation and OS. The OSAM2 module was negatively associated with FAB subtypes and OS. GO enrichment analysis suggested that genes in the OSAM1 module were significantly enriched in 53 GO terms, such as negative regulation of signaling, regulation of cell communication, negative regulation of developmental process. Moreover, the genes in the OSAM1 module were over-represented in the KEGG pathway of protein processing in endoplasmic reticulum. Thus, we speculate that the OSAM1 and OSAM2 modules play a pivotal role in the overall survival of AML patients.FLT3-ITD frequently occurs in AML patients and indicates an inferior prognosis in AML [21]. There are 200 and 672 AML samples in the TCGA and OHSU datasets respectively, however, only 173 and 451 patients had somatic mutation and RNA-seq expression data. 173 AML patients in the TCGA cohort and 405 patients in the OHSU cohort were included in the study. Owing to the lack of FLT3-IDT information in the TCGA dataset, we analyzed the association between the FLT3 mutation and OS in the 173 patients with RNA-seq data and the 200 AML patients. However, no significant correlation was observed between the FLT3 mutation and overall survival (Additional file 2: Table 8). In the OHSU cohort, the FLT3-IDT mutation was indicative of poor prognosis in the 672 AML patients. However, the association was not statistically significant in our study (Additional file 2: Table 9). Therefore, the difference of our results and previous publications on the association of FLT3-IDT mutation with overall survival is probably caused by the selection of different cohorts of AML patients.
We analyzed the associations between 580 genes in the OSAM1 and OSAM2 modules and AML patients’ OS in the TCGA and OHSU datasets using many statistical methods and identified set of genes was significantly associated with OS in AML patients, such as FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR. The PLA2G4A gene encodes a member of the cytosolic phospholipase A2 group IV family which plays an important role the regulation of hemodynamics, inflammatory responses and other intracellular pathways [22]. The expression of PLA2G4A is up-regulated in a wide range of cancer types [23–26]. PLA2G4A depletion significantly repressed cellular proliferation in glioblastoma, lung cancer and colon cancer [23, 25, 26]. These results demonstrate PLA2G4A may play an oncogenic role in cancers. Another gene, CALR, has been involved in calcium retention and protein folding, as well as in immune responses [27]. In line with the finding in our study, CALR exposure by malignant blasts is correlated with robust anticancer immunity and superior OS in AML patients [28]. Activation of unfolded protein response, including CALR, is associated to more favorable clinical outcome and lower relapse rate [29]. These studies suggest CALR is a positive prognostic biomarker for AML patients.
High FNDC3B, VSTM1, GOLGA3 and CALR expression and low PLA2G4A expression were indicative of decreased mortality of AML patients. Among the four subgroups of AML patients identified by hierarchical clustering analysis, the cluster1 AML patients showed younger age, lower cytogenetics risk, higher frequency of NPM1 mutations and more favourable OS than cluster3 patients. Therefore, expression analysis of the gene panel might be clinically useful in the future. AML patients exhibiting low FNDC3B, VSTM1, GOLGA3 and CALR expression or high PLA2G4A expression are expected to have poor clinical outcome. Therefore, these patients may need more aggressive therapies or more frequent follow-up.
Furthermore, we developed a risk score based on the linear combination of the five gene expression values. The risk score effectively stratifies AML patients with two distinct risk groups with significant different prognosis. Recent studies have reported a four-gene LincRNA expression signature (LINC4) and a 17-gene stemness score (LSC17) to predict risk in AML patients [30, 31]. Our risk score, LINC4 and LSC17, have all been tested on the TCGA dataset. Though the prognostic difference of subgroups of AML patients stratified by the three prognostication scores all were statistically significant, our risk showed higher OR value (3.88) than the OR values of LINC4 (2.22) and LSC17 (2.62), suggesting the risk score might have more predictive power for overall survival in AML. Moreover, the LSC17 score requires quantification of expression of 17 genes. Therefore, implementing the LSC17 risk classification might cause more experimental workload and higher cost than the LINC4 and our risk score. Lastly, the five genes may become druggable targets for AML patients. For instance, depletion of PLA2G4A caused significant decrease in cellular proliferation in glioblastoma, lung cancer and colon cancer cells [23, 25, 26].
Conclusion
In conclusion, the OSAM1 and OSAM2 modules were the most critical modules in the OS of AML patients. The five gene panel comprising FNDC3B, VSTM1, PLA2G4A, GOLGA3 and CALR and risk score may function as potential prognostic biomarkers for AML, which also needs much further research.
Supplementary Information
Additional file 1: The supplementary figures which support the findings of this study.
Additional file 2: The supplementary tables which support the findings of this study.
Acknowledgements
None.
Abbreviations
- AML
Acute myeloid leukemia
- TCGA
The Cancer Genome Altas
- OHSU
The Oregon Health & Science University
- OS
Overall survival
- GO
Gene ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- WGCNA
Weighted correlation network analysis
- FAB
French-American-British
- PBMBC
Percent of bone marrow blast cells
- FDR
False discovery rate
- AUC
Area under curve
- OR
Odds ratio
- OSAM1
Overall survival-associated module 1
- OSAM2
Overall survival-associated module 2
Authors’ contributions
Conception and design: MZ. Administrative support: LXS. Provision of study materials or patients: YLL and YLZ. Collection and assembly of data: GFOY and BBL. Data analysis and interpretation: YLL, GFOY, LXS, BBL and YLZ. Manuscript writing: All authors. All authors read and approved the final manuscript.
Funding
The study was supported by the traditional Chinese medicine administration of Zhejiang Province (Grant No. 2015ZZ018), the National Science Foundation of Zhejiang Province (Grant No. LY17H160005). The funders provide us with financial support on data analysis and interpretation, language editing service and administrative work.
Availability of data and materials
Raw data of the TCGA cohort are the normalized read counts (RNA-seq) data of 18379 genes of 173 AML patients and their clinical data were publicly available at https://figshare.com/s/7c683384c6e2add08262 (figshare ID: 13585235). The gene expression and clinical data of 405 AML patients (downloaded from the OHSU dataset) used for the validation of survival analysis in our study were publicly available at https://figshare.com/s/7c683384c6e2add08262 (figshare ID: 13585235).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare there is no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-00888-0.
References
- 1.Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–1907. doi: 10.1016/S0140-6736(06)69780-8. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Wei L, Liu J, Li S, Wang Q. Comparison of cancer incidence between China and the USA. Cancer Biol Med. 2012;9:128–132. doi: 10.3969/j.issn.2095-3941.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Breems DA, Van Putten WLJ, De Greef GE, Van Zelderen-Bhola SL, Gerssen-Schoorl KBJ, Mellink CHM, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26:4791–4797. doi: 10.1200/JCO.2008.16.0259. [DOI] [PubMed] [Google Scholar]
- 5.Byrd JC, Mro K, Dodge RK, Carroll AJ, Edwards CG, Arthur DC, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B ( CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 6.Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106:12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bullinger L, Krönke J, Schön C, Radtke I, Urlbauer K, Botzenhardt U, et al. Identification of acquired copy number alterations and uniparental disomies in cytogenetically normal acute myeloid leukemia using high-resolution single-nucleotide polymorphism analysis. Leukemia. 2010;24:438–449. doi: 10.1038/leu.2009.263. [DOI] [PubMed] [Google Scholar]
- 8.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–531. doi: 10.1038/s41586-018-0623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Gene Ontolo Consort Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2016;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai B, Lai Y, Zhang Y, Zhou M, Sheng L, OuYang G. The solute carrier family 2 genes are potential prognostic biomarkers in acute myeloid leukemia. Technol Cancer Res Treat. 2020;19:1–9. doi: 10.1177/1533033819894308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sha K, Lu Y, Zhang P, Pei R, Shi X, Fan Z, et al. Identifying a novel 5-gene signature predicting clinical outcomes in acute myeloid leukemia. Clin Transl Oncol. 2020 doi: 10.1007/s12094-020-02460-1. [DOI] [PubMed] [Google Scholar]
- 16.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Therneau T. Survival analysis. Cran. 2016 doi: 10.1007/978-1-4419-6646-9. [DOI] [Google Scholar]
- 18.Fox J. Cox proportional-hazards regression for survival data the cox proportional-hazards model. Most. 2002;2008:1–18. doi: 10.1016/j.carbon.2010.02.029. [DOI] [Google Scholar]
- 19.Warnes G, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A, et al. gplots: Various R programming tools for plotting data. 2005.
- 20.Therneau T. Survival Analysis. Cran. 2016.
- 21.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML : review of current knowledge and evidence. 2019; 299–312. [DOI] [PMC free article] [PubMed]
- 22.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35 Database:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Zhang H. Expression of cytosolic phospholipase A2 alpha in glioblastoma is associated with resistance to chemotherapy. Am J Med Sci. 2018;356:391–398. doi: 10.1016/j.amjms.2018.06.019. [DOI] [PubMed] [Google Scholar]
- 24.Runarsson G, Feltenmark S, Forsell PKA, Sjöberg J, Björkholm M, Claesson H-E. The expression of cytosolic phospholipase A2 and biosynthesis of leukotriene B4 in acute myeloid leukemia cells. Eur J Haematol. 2007;79:468–476. doi: 10.1111/j.1600-0609.2007.00967.x. [DOI] [PubMed] [Google Scholar]
- 25.Sundarraj S, Kannan S, Thangam R, Gunasekaran P. Effects of the inhibition of cytosolic phospholipase A2α in non-small cell lung cancer cells. J Cancer Res Clin Oncol. 2012;138:827–835. doi: 10.1007/s00432-012-1157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parhamifar L, Jeppsson B, Sjölander A. Activation of cPLA2 is required for leukotriene D4-induced proliferation in colon cancer cells. Carcinogenesis. 2005;26:1988–1998. doi: 10.1093/carcin/bgi159. [DOI] [PubMed] [Google Scholar]
- 27.Cahu X, Constantinescu SN. Oncogenic drivers in myeloproliferative neoplasms: from JAK2 to calreticulin mutations. Curr Hematol Malig Rep. 2015;10:335–343. doi: 10.1007/s11899-015-0278-x. [DOI] [PubMed] [Google Scholar]
- 28.Fucikova J, Truxova I, Hensler M, Becht E, Kasikova L, Moserova I, et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood. 2016;128:3113–3124. doi: 10.1182/blood-2016-08-731737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schardt JA, Weber D, Eyholzer M, Mueller BU, Pabst T. Activation of the unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin Cancer Res. 2009;15:3834–3841. doi: 10.1158/1078-0432.CCR-08-2870. [DOI] [PubMed] [Google Scholar]
- 30.Beck D, Thoms JAI, Palu C, Herold T, Shah A, Olivier J, et al. A four-gene LincRNA expression signature predicts risk in multiple cohorts of acute myeloid leukemia patients. Leukemia. 2018;32:263–272. doi: 10.1038/leu.2017.210. [DOI] [PubMed] [Google Scholar]
- 31.Ng SWK, Mitchell A, Kennedy JA, Chen WC, McLeod J, Ibrahimova N, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540:433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: The supplementary figures which support the findings of this study.
Additional file 2: The supplementary tables which support the findings of this study.
Data Availability Statement
Raw data of the TCGA cohort are the normalized read counts (RNA-seq) data of 18379 genes of 173 AML patients and their clinical data were publicly available at https://figshare.com/s/7c683384c6e2add08262 (figshare ID: 13585235). The gene expression and clinical data of 405 AML patients (downloaded from the OHSU dataset) used for the validation of survival analysis in our study were publicly available at https://figshare.com/s/7c683384c6e2add08262 (figshare ID: 13585235).