

Abstract
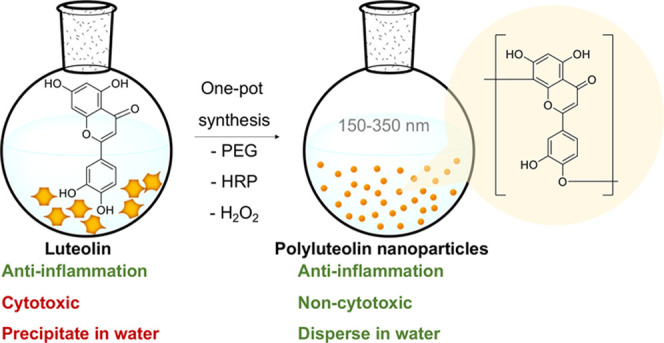
Luteolin is an anti-inflammatory flavonoid commonly found in many edible plants. The compound is popularly consumed as a supplement regardless of its poor water solubility (27.8 μg/mL at 25 °C) and low bioavailability. Here, mild one-pot polymerization of luteolin into water-dispersible nanospheres, with an average dry size of 234.8 ± 101.6 nm, an aqueous size distribution of 379.1 ± 220.5 nm (PDI = 0.338), an average ζ-potential of −36.2 ± 0.2 mV, and an 89.3 ± 4.8% yield, is described. The nanospheres consist of polymerized luteolin (polyluteolin) with a weight-average molecular mass of around 410000 Da. The chemical structure of polyluteolin is identified through 1H–1H correlated spectroscopy (COSY), 1H–13C heteronuclear single-quantum coherence (HSQC), and 1H–13C heteronuclear multiple-bond correlation (HMBC) NMR spectroscopic analyses of the oligomers, and a polymerization mechanism is proposed. Unlike luteolin that showed both dose-dependent anti-inflammatory activity and cytotoxicity when tested in lipopolysaccharide-stimulated macrophages, the polyluteolin nanoparticles possess dose-dependent anti-inflammatory activity without causing cell death even at high concentrations.
1. Introduction
Inflammation is an immune response that the body produces to inhibit infection and heal the damaged tissue. However, chronic inflammation can develop into diseases such as arthritis, chronic obstructive pulmonary disease, and cancer.1−3 Fruits and vegetables contain secondary metabolites that possess anti-inflammatory activities; therefore, consuming enough of them can reduce the risk of those diseases.4−6
Researchers from various places have reported that luteolin (3′,4′,5,7-tetrahydroxyflavone), a flavonoid found in many fruits, vegetables, and medicinal plants including onion leaves, celery, broccoli, and carrot (luteolin contents of 391.0, 80.5, 74.5, and 37.5 mg/kg of dry weight, respectively),7 possesses strong anti-inflammatory activity.8−11 Although the compound has been sold as a health supplement or part of health supplement formulations for various claims, the low bioavailability of this compound has been reported. A pharmacokinetic study on oral administration of extracts containing luteolin in beagle dogs at 7.6 mg/kg showed a peak concentration of luteolin in plasma (Cmax) of 0.6 μg/mL and the total luteolin in plasma (area of luteolin concentration peak over time or AUC0→∞) of 6.3 μg h/mL. The report agrees with the low luteolin absorption in mini-pigs (luteolin dose of 6.8 mg/kg, Cmax of 0.5 μg/mL, and AUC0→∞ of 4.9 μg h/mL) and humans (luteolin dose of 1.5 mg/kg, Cmax of 0.3 μg/mL, and AUC0→∞ of 2.0 μg h/mL).12 Improvement of luteolin bioavailability could be achieved using nanostructured lipid carriers (NLCs) and microemulsions (MEs). When rats were given luteolin at a dose of 3.5 mg/kg, the Cmax changed from 0.6 μg/mL for the original luteolin to 0.5 μg/mL for luteolin in NLC and 1.6 μg/mL for luteolin in ME, and the AUC0→∞ increased from 1.2 μg h/mL for the original luteolin to 10.7 μg h/mL luteolin in NLC and 16.2 μg h/mL for luteolin in ME. Nevertheless, the luteolin loading content in both the NLC and ME was only 0.15%, meaning that large amounts of auxiliary materials were administered to animals when NLC or ME was used to deliver luteolin.13
Another approach to solving the low-bioavailability problem is by encapsulating luteolin into various polymeric particles. Researchers have reported the use of methoxy poly(ethylene glycol) (PEG)–polylactide-co-glycolide, methoxy poly(ethylene glycol)–polylactide, methoxy poly(ethylene glycol)–polycaprolactone, and polylactic acid–poly(ethylene glycol) ether for the encapsulation of luteolin.14−16 These encapsulations have resulted in aqueous formulations of luteolin with better biological activity. Nevertheless, less than 4.0% of luteolin loading content could be achieved; therefore, large amounts of auxiliary polymeric materials are parts of formulations.
Autopolymerization of flavonoid compounds in harvested plants have been extensively studied.17,18 Improved anticancer and antihypertriglyceridemia activities were observed for the small oligomeric products such as theasinensin, theaflavin, thearubigin, and procyanidin, obtained from natural catechin polymerization, which automatically takes place in tea leaves during storage and fermentation.19−22 Some of the studies on natural autopolymerization of flavonoids indicated that only water and heat are needed to allow the reaction to take place in situ.23,24 However, some reports stated the importance of oxidative enzymes such as polyphenol oxidase (PPO), peroxidase (POD), and catalase, of which PPO was reported as the most important oxidative enzyme in polymerization of polyphenols in tea leaves.25−31 These reports inspired us to explore the possibility to polymerize other plant extracts that possess chemical structures related to catechin.
Here, we show one-pot synthesis of water-dispersible polyluteolin nanoparticles from luteolin via an enzymatic template polymerization using hydrogen peroxide as a reagent, PPO as a catalyst, and poly(ethylene glycol) polymer (PEG) as a template. In addition to reaction optimization, this work also involves product characterizations regarding chemical structure, morphology, anti-inflammatory activity, and cytotoxicity.
2. Results and Discussion
Natural fermentation of catechin can take place with the peroxidase enzyme present naturally in tea leaves and oxygen in the atmosphere.32,33 Because of the related chemical structure between luteolin and catechin, here, we have explored the possibility to polymerize luteolin by mimicking the natural environment in tea leaves. We explored the roles of oxidative enzyme horseradish peroxidase (HRP), oxidizing agent H2O2, and the PEG template. The biocompatible and bioinert PEG template was used to mimic macromolecular substrates in tea leaves, which may act as seeds or templates of the polymerization reaction.
During one-pot synthesis of polyluteolin, it was noticed that with not enough HRP in the reaction mixture (Table 2, Products 1 and 2), there was no chemical transformation. The NMR, IR, and UV spectra of Products 1 and 2 (Figures 2 and S1 and S2 in the Supporting Information, SI) are similar to those of the starting luteolin. This information suggests that the obtained Products 1 and 2, which appeared as water-insoluble precipitates, are luteolin aggregates (Figures 1, 2a, and 3a). When the amount of HRP was increased to 3.3% (Table 2, Product 3), water-dispersible nanoparticles with an average dry size of 234.8 ± 101.6 nm (Figures 1 and 4a) were obtained. Product 3 possesses significant differences in the NMR, IR, and UV spectra from those of luteolin (Figures 2 and S1 and S2 in the SI). The IR spectrum of Product 3 (Figure S1 in SI) possesses a broader O–H stretching vibration (at 3265 cm–1) than that of luteolin (at 3417 cm–1). The C=C stretching vibration of luteolin appears as triple peaks at 1606, 1574, and 1500 cm–1, whereas that of Product 3 appears as a broad peak with maxima at 1612 and 1504 cm–1. The phenolic C–O stretching vibration of luteolin appears as triple peaks at 1365, 1263, and 1246 cm–1 whereas that of Product 3 appears as a broad peak with maxima at 1354 and 1257 cm–1. These changes correspond well with the speculative polymer structure of Product 3, in which the broad peaks are caused by overlapping of many slightly different frequencies of vibration resulted from geometrical irregularities and different physical environments among various connected units. Additionally, the UV absorption band of luteolin in dimethyl sulfoxide (DMSO) appears at λmax values of 271 and 353 nm (π–π* transition of benzoyl system and n−π* transition of cinnamoyl system, respectively), whereas that of Product 3 shows a broader absorption band with more absorption at a longer wavelength, indicating the increased conjugation of Product 3 as compared to the conjugation system in luteolin.
Table 2. Amount of Materials Used in the Synthesis of Polyluteolin Nanoparticles.
| Luteolin | PEG |
HRP |
30% H2O2 | PB pH 7.4 | Ethanol | ||||
|---|---|---|---|---|---|---|---|---|---|
| Product | mg | Mw | mg | mg | % relatively to luteolin | mL | mL | mL | % yield |
| 1 | 75.0 | 2050 | 7.5 | 0.0 | 0.0 | 3.6 | 26.4 | 15.0 | NRa |
| 2 | 75.0 | 2050 | 7.5 | 1.25 | 1.7 | 3.6 | 26.4 | 15.0 | NRa |
| 3 | 75.0 | 2050 | 7.5 | 2.5 | 3.3 | 3.6 | 26.4 | 15.0 | 89.3 ± 4.8 |
| 4 | 75.0 | 2050 | 7.5 | 2.5 | 3.3 | 0.0 | 26.4 | 15.0 | NRa |
| 5 | 75.0 | 0.0 | 2.5 | 3.3 | 3.6 | 26.4 | 15.0 | 67.9 ± 0.9 | |
| 6 | 75.0 | 200 | 7.5 | 2.5 | 3.3 | 3.6 | 26.4 | 15.0 | 56.7 ± 2.2 |
| 7 | 75.0 | 35000 | 7.5 | 2.5 | 3.3 | 3.6 | 26.4 | 15.0 | 69.7 ± 2.6 |
NR, no polymerization reaction.
Figure 2.
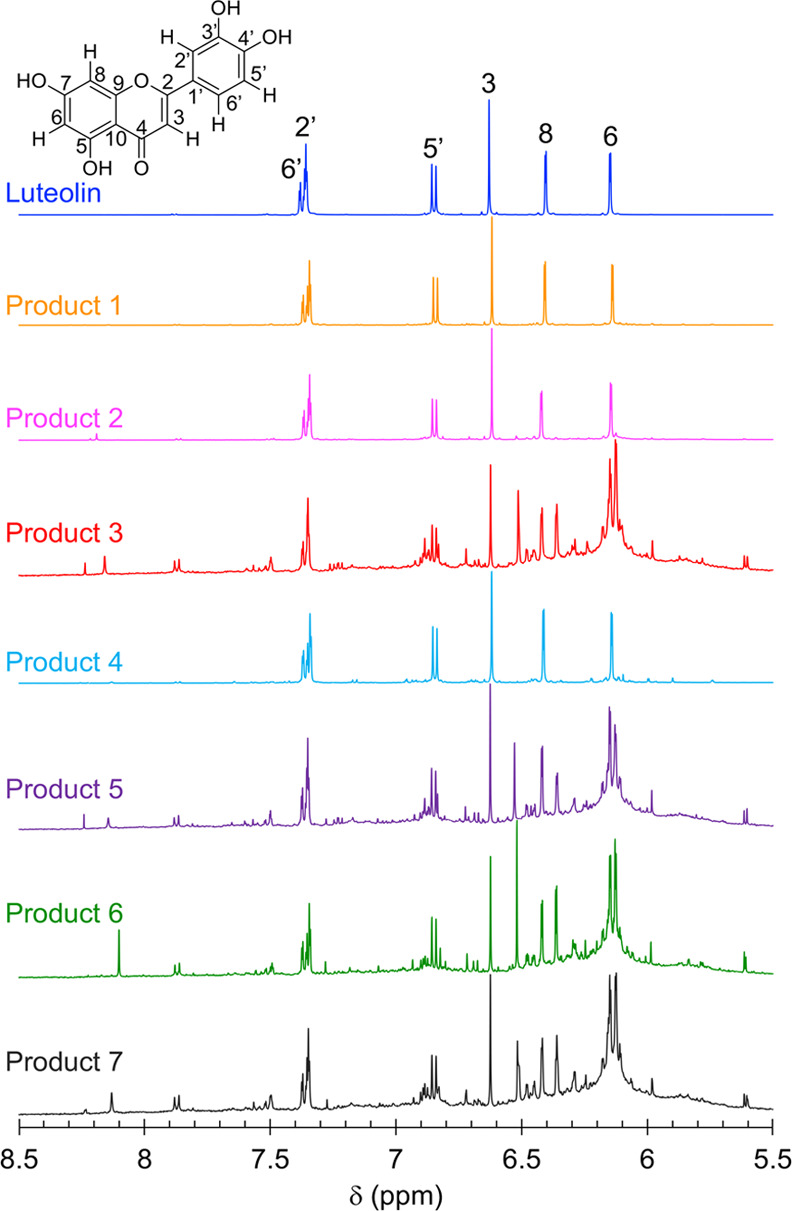
1H NMR (20 mg/mL, DMSO-d6, 500 MHz) spectra of luteolin and products prepared under different conditions: no HRP | H2O2 | PEG 2050 (Product 1), 1.7% HRP | H2O2 | PEG 2050 (Product 2), 3.3% HRP | H2O2 | PEG 2050 (Product 3), 3.3% HRP | no H2O2 | PEG 2050 (Product 4), 3.3% HRP | H2O2 | no PEG (Product 5), 3.3% HRP | H2O2 | PEG 200 (Product 6), and 3.3% HRP | H2O2 | PEG 35000 (Product 7).
Figure 1.

Scanning electron microscopy (SEM) images of luteolin (1a) and products prepared under different reaction conditions (2a–4b). Reaction conditions are denoted in parentheses. (2a) Product 1 (no HRP | H2O2 | PEG 2050), (3a) Product 2 (1.7% HRP | H2O2 | PEG 2050), (4a) Product 3 (3.3% HRP | H2O2 | PEG 2050), (1b) Product 4 (3.3% HRP | no H2O2 | PEG 2050), (2b) Product 5 (3.3% HRP | H2O2 | no PEG), (3b) Product 6 (3.3% HRP | H2O2 | PEG 200), and (4b) Product 7 (3.3% HRP | H2O2 | PEG 35000).
Figure 3.
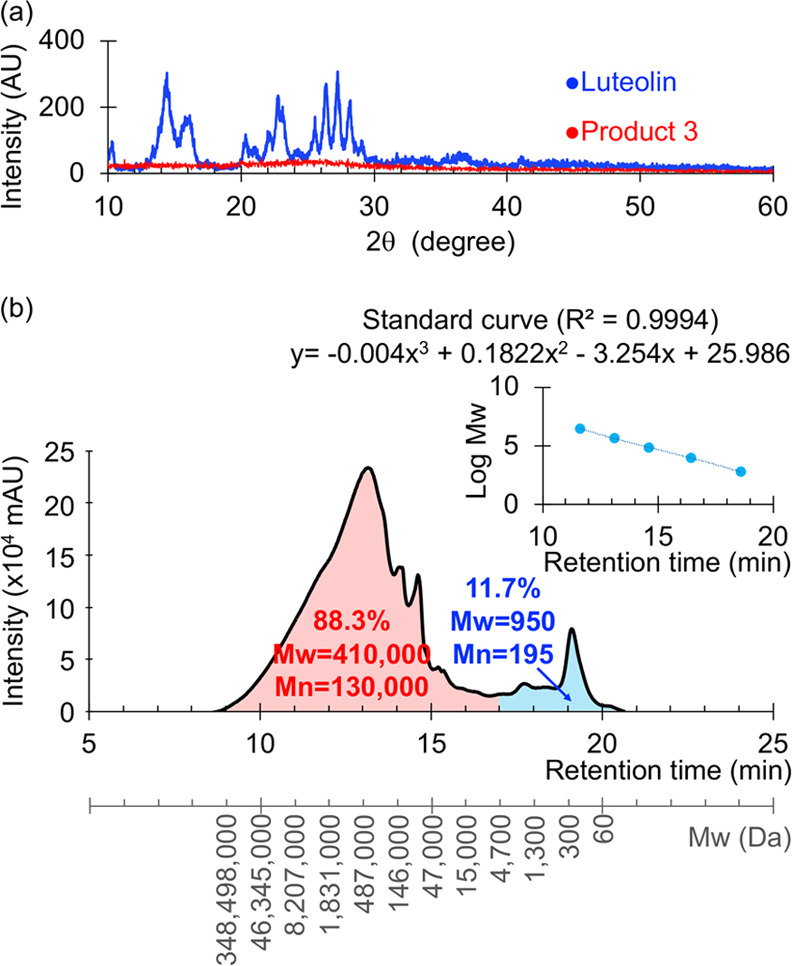
(a) X-ray diffraction (XRD) patterns of luteolin and polyluteolin nanoparticles (Product 3) and (b) gel filtration chromatography (GFC) chromatogram (10 mg/mL, N,N-dimethylformamide (DMF); PDA detector at 254 nm) of polyluteolin (Product 3). Polystyrene (Mp = 580, 9820, 67600, 466300, and 3152000 Da) standard curve is shown in the upper left.
Figure 4.

Overlay of 1H–13C heteronuclear multiple-bond correlation (HMBC) NMR correlations (MeOD-d4, 500 MHz) of luteolin and luteolin oligomers.
It was observed through the SEM images that the reaction performed without H2O2 (Table 2, Product 4) gave a product with similar morphology to the luteolin morphology (Figure 1b). The NMR and UV spectra of Product 4 (Figures 2 and S2 in the SI) are also similar to those of luteolin, indicating that the chemical transformation did not take place.
Next, the effect of molecular mass of PEG on product formation was investigated. Replacing PEG 2050 (Table 2, Product 3) with PEG 200 and PEG 35000 (Table 2, Products 6 and 7), also a reaction without PEG (Table 2, Product 5), still gave obvious chemical transformation of luteolin. The NMR, IR, and UV spectra of Products 5–7 are different from those of luteolin but similar to those of Product 3, indicating that similar polymerization reactions were taking place under these conditions. The molecular mass of PEG or the absence of PEG did not affect the chemical transformation of luteolin but rather affected the morphology of the obtained polymerized products. Product 3 (obtained with PEG 2050) is uniform spherical particles of 200–300 nm diameter, and Products 5 (obtained with no PEG) and 6 (obtained with PEG 200) are spherical particles of similar size with some obvious big aggregates (Figures 1, 2b, and 3b). A product with a bigger particle size (average dry size of 857.8 ± 235.1 nm) was observed when PEG 35000 was used (Product 7, Figures 1 and 4b).
With our goal of making water-dispersible nanoparticles for water-based formulations that could target inflammation, the morphology of Product 7 was considered too big. This is because a previous study has shown that only nanoparticles in the size range of 10–500 nm can leave the blood vessel and accumulate inside the inflamed tissue.34 Due to the small uniform size of Product 3, this product was selected for further characterization and evaluation of anti-inflammatory activity.
It should be noted here that the first intention of using PEG in the reaction was for the material to act as a template for polymerization.35 Nevertheless, the observation that polymerization could take place with no PEG (Product 5) suggests a different role of PEG. It should be noticed here that for all reactions that contained PEG (Product 1−4, 6 and 7), the amount of PEG added to the reaction was at 10% relative to the amount of luteolin. We observed that PEG plays a crucial role in particle formation of the polymerized products, e.g., uniformed particles could not be obtained without PEG (Product 5). We speculate that PEG acts as a corona of the self-assembled particles, making the obtained polymerized particles water-dispersible and nonaggregated. Without PEG, big aggregates were observed (Product 5). With too large PEG molecules (35000 Da), big aggregated particles were observed (Product 7). By fixing the 10% weight of PEG used in the reaction but increasing the molecular weight of the material, the numbers of PEG chains present in the reaction automatically decreased. It was likely that in the case of PEG 35000, there were not enough PEG molecules to surround each smaller particle; therefore, aggregation into bigger particles took place. In contrast, there were higher numbers of PEG chains to surround smaller polyluteolin particles when smaller PEG molecules (2050 Da) were used. As a result, uniform 200–300 nm polyluteolin particles were obtained from the reaction using PEG 2050 (Product 3). Nevertheless, with too small PEG (200 Da), the PEG corona was probably not long enough to prevent the aggregation of the particles; thus, aggregated particles were produced when PEG 200 was used (Product 6).
In aqueous, more than 90% of Product 3 accounts for the particles with an average diameter of 379.1 ± 220.5 nm (PDI = 0.338, Figure S3 in the SI). The high S.D. value indicates broad size distribution and corresponds well with the depicted size distribution curve (Figure S3). The broad size distribution of Product 3 might stem from the self-assembling process that took place in tandem to the growth of Product 3 chains. With an abundance of PEG in the system at the beginning of synthesis, there would be enough PEG to cover the surface of small particles. Later, with less amount of PEG, the particle size had to increase so that the particle surface would have PEG to cover. This resulted in the formation of small to large particles. It should be noted here that we limited the amount of PEG in the system based on our intention to prepare particles of polyluteolin with a minimal amount of auxiliary material.
The obtained product 3 gave a PDI of 0.338, corresponding well to the unimodal size distribution (one maximum) of the particles (Figure S3). The high negative ζ-potential of −36.2 ± 0.2 mV agrees with the observation that the aqueous suspension of Product 3 is stable, i.e., no precipitation or sedimentation was observed even when the aqueous suspension was placed unattended for 3 months.
The XRD pattern of luteolin (Figure 3a) shows sharp crystalline peaks with the 2θ of 10.3, 14.4, 16.1, 20.4, 21.2, 22.8, 23.0, 24.6, 25.5, 26.4, 27.2, 28.2, and 29.2°, indicating crystallinity of this compound. In contrast, no 2θ scattering peak can be observed for Product 3 (Figure 3a), suggesting that Product 3 nanoparticles are amorphous. The crystallinity and amorphous states of luteolin and Product 3 also agree well with the higher decomposition temperature of luteolin (379 °C) compared to that of Product 3 (188 °C).
The changes in weight percentage of the materials as a function of temperature were measured by thermogravimetric analysis (TGA) in the differential thermal analysis mode (TG-DTA). The DTA curves of the two starting materials used in the preparation of Product 3, luteolin and PEG 2050 (Figure S4a,b in the SI), show single sharp peaks at 367.8 and 391.2 °C, respectively. Interestingly, the DTA curves of Product 3 (Figure S4c in the SI) shows five broad peaks at 92.0, 175.8, 278.9, 389.1, and 454.5 °C. It is obvious that none of these peaks matches those of luteolin or PEG 2050. This implies that neither luteolin nor PEG 2050 was left as a physical mixture in Product 3. The DSC curves of luteolin and PEG 2050 (Figure S4e in the SI) show glass transition (Tg) at −110.0 and −109.7 °C, and the two materials melt (Tm) at 171.3 and 56.6 °C, respectively, whereas Product 3 shows Tg at only −110.7 °C. The higher degradation temperature of luteolin (367.8 °C) compared to those of Product 3, and the sharp melting temperature of luteolin agree with the above-mentioned XRD result, which indicates that luteolin is crystalline. These differences in thermal properties align well with the different crystallinity properties and different chemical structures of luteolin and Product 3.
The GFC profile (Figure 3b) indicates that Product 3 is a mixture of polyluteolin of various molecular masses. Approximately 88.3% of Product 3 are polyluteolin with molecular masses of 871147500–5400 Da (Mw of 410000 Da, Mn of 130000 Da, retention time of 8.5–17.0 min) and 11.7% are polyluteolin with molecular masses of 5400–18 Da (Mw of 950 Da, Mn of 195 Da, retention time of 17.0–20.5 min). It is very likely that this latter potion of the small-Mw polyluteolin was actually the material in the ethanolic extract of Product 3 because the ethanolic extract of Product 3 coincidentally accounted for 11.2% of whole Product 3.
Attempts to directly elucidate chemical structures of Product 3 cannot be accomplished due to the severe broadening of NMR resonance peaks. The broadening of peaks was probably caused by the slower molecular motion of polymer chains, which leads to repeating units being situated in slightly different chemical environments. Therefore, to elucidate the chemical structure and propose the reaction mechanism of polymerization, the luteolin oligomer was extracted from Product 3 and characterized by 1H, 13C, 1H–1H correlated spectroscopy (COSY), 1H–13C heteronuclear single-quantum coherence (HSQC), and 1H–13C HMBC NMR spectroscopy (Table 1, Figures 4 and 5, and Table S1 and Figures S5–S7 in the SI). Compared to the spectra of original luteolin, many changes are observed in the oligomers’ spectra, e.g., a 75% decrease in the intensity of H8 resonance at 6.41 ppm from 1:1 to 0.25:1H8:H6 integral ratio and decreases in HMBC correlations between H8 and all carbons. The HMBC spectrum of the oligomers shows new correlations between H6 and a new carbon peak at 177.03 ppm. It is likely that this C8 in a new environment (designated as C8A) is the connecting point between one unit of the luteolin monomer and the next unit. With the downfield resonance of C8A, its connection to an electronegative atom is quite possible. For finding where this C8A is connected to, we have identified a small downfield shift from 149.65 of the original C4′ to 154.38 ppm. Confirming with HMBC correlations to H5′ and H2′, this carbon is C4′ in the new environment (designated as C4A). It is likely that the hydroxyl group on original C4 changes to an ether functionality to produce C4A. We propose that C8A (in flavonoid ring A) of one luteolin oligomeric unit is connected to C4′A (in flavonoid ring B) of another luteolin monomeric unit via an ether linkage. This oligomer is designated as oligomer I (see more HMBC, COSY, and HSQC of this proposed oligomer I in Figure 5).
Table 1. 1H and 13C NMR Chemical Shift of Luteolin and Luteolin Oligomer I (MeOD-d4, 500 MHz).
| Luteolin |
Luteolin oligomer I (major product) |
|||
|---|---|---|---|---|
| Position | 1H δ (ppm), J (Hz) | 13C δ (ppm) | 1H δ (ppm), J (Hz) | 13C δ (ppm) |
| 2 | 164.63 | 164.61 | ||
| 3 | 6.51 (1H, s) | 102.53 | 6.51 (1H, s) | 102.50 |
| 4 | 182.49 | 182.23 | ||
| 5 | 161.84 | 161.94 | ||
| 6 | 6.18 (1H, d, 2.3) | 98.77 | 6.17 (1H, m) | 98.74 |
| 7 | 164.96 | 165.01 | ||
| 8 | 6.41 (1H, d, 2.3) | 93.66 | 6.41 (1H, d, 2.3) | 93.60 |
| 8A | 177.03 | |||
| 9 | 158.05 | 157.40 | ||
| 10 | 103.97 | 103.66 | ||
| 1′ | 122.36 | 122.40 | ||
| 2′ | 7.35 (1H, m) | 112.85 | 7.35 (1H, m) | 112.73 |
| 3′ | 145.69 | 145.85 | ||
| 4′ | 149.65 | 149.65 | ||
| 4′A | 154.38 | |||
| 5′ | 6.88 (1H, dd, 8.9, 2.0) | 115.45 | 6.87 (1H, dd, 8.9, 2.0) | 115.38 |
| 6′ | 7.36 (1H, m) | 118.99 | 7.36 (1H, m) | 118.81 |
Figure 5.

(a) Key 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC correlations of luteolin and luteolin oligomer. (b) Proposed structures of polyluteolin.
In addition to oligomer I, a new singlet 1H resonance peak at 7.51 ppm can be observed. This new proton peak shows HMBC correlations to carbons at 150.77 ppm (C4′B) and 148.34 ppm (C3′B). Therefore, it is likely that this new singlet proton resonance peak represents H5′ in the new environment (designated as H5′B). A new carbon peak at 123.12 ppm, which shows correlation to H6, is likely C8 in the new environment (designated as C8B). The HMBC correlation of H5′B to C8B indicates the connection of C8B (in flavonoid ring A) to C6′B (in flavonoid ring B). The oligomer with this connection is designated as oligomer II (see more HMBC, COSY, and HSQC of this proposed oligomer II in Figure 5).
In addition to oligomers I and II, oligomer III structure, where C8C (from flavonoid ring A) is connected to C2′C (in flavonoid ring B), is also proposed based on COSY (see H5′C–H6′C correlation of oligomer III in Figure S5), HMBC, HSQC, and splitting pattern of the 1H NMR spectra (Figure 5 and Table S1).
It should be noted here that the three types of connections proposed here for the oligomerized luteolin products resemble the connections previously proposed for polymerized tea polyphenols.22−29 Among the three identified oligomers, oligomer I is the most abundant (the highest integral of the 1H NMR resonance peaks). Apart from the three proposed structures, some other minor unidentified structures are also present in the extracted oligomerized products used in the analyses.
Due to the oxidative nature of the three oligomers and the requirement of both H2O2 and PPO, it is likely that the oxidative polymerization proceeds via a free-radical mechanism. The ortho diphenolic moieties on the flavonoid ring B are the first oxidative points, resulting in an intermediate with diketo moieties on ring B. The electron-rich site at the 8 position in flavonoid ring A of another unit then reacts with this oxidized intermediate at three different positions, resulting in three different connection types (Figure S7 in the SI).
Anti-inflammatory activity of luteolin and Product 3 was determined in RAW 264.7 macrophage cells by measuring nitrite accumulation in the culture supernatant via the Griess assay after triggering the cells with LPS.36 As shown in Figure 6a, RAW 264.7 macrophages (positive control) produced low levels of nitrite (3.76 ± 1.92%, relative to negative control). Nitrite production increased after cells were activated with LPS for 24 h (100.72 ± 12.23%, negative control). Luteolin strongly suppressed nitrite production in a dose-dependent manner. To investigate that the observed anti-inflammatory activity was not the result of cell death, cell viability at various concentrations of the tested materials was acquired by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay.37 Luteolin shows cytotoxicity in a dose-dependent manner with more than 20, 60, and 80% cell death at 12.5, 25, and 50 μg/mL, respectively (Figure 6b). Therefore, the high anti-inflammatory activities of luteolin at concentrations of 12.5 μg/mL and higher indicated from the reduced cellular nitrite production assay are, in fact, a result of cell death but not the true anti-inflammatory activity.
Figure 6.

In vitro anti-inflammatory activity of luteolin and polyluteolin nanoparticles (Product 3): (a) % nitrite production and (b) % cell viability. Aqueous suspensions of (c) luteolin and (d) Product 3 at various concentrations. Significant differences between the tested groups were statistically analyzed using the t-test and were denoted with * for a p value of <0.05.
Although not as strongly as luteolin, the t-test analysis confirms that Product 3 significantly inhibits the nitrite production in a dose-dependent manner, with almost complete suppression at 100 μg/mL (Figure 6a). Interestingly, unlike luteolin that is cytotoxic in a dose-dependent manner, Product 3 shows no cytotoxicity even at the highest concentration tested (100 μg/mL) (Figure 6b). The t-test analysis indicates no significant difference (at a p value of 0.05) among cell viabilities of negative control and Product 3 of various concentrations (Figure 6b). These results confirm that the anti-inflammatory activity observed for Product 3 was not a result of cytotoxicity of the material but the real anti-inflammatory action of the compound. As a result, we conclude that Product 3 is noncytotoxic even at high concentrations and possesses dose-dependent anti-inflammatory activity.
As mentioned earlier that there might be some PEG in Product 3; therefore, to make sure that the anti-inflammatory activity observed for Product 3 was not caused by PEG, we tested the anti-inflammatory activity of PEG at the highest possible concentrations of PEG in each sample (assuming that no PEG was eliminated during dialysis). The result (Figure S8 in the SI) confirms no anti-inflammatory activity of PEG.
3. Conclusions
This paper demonstrates the conversion of luteolin into a water-dispersible polymerized product. The polymerization reaction, induced by a catalytic amount of HRP and H2O2, is coupled to the product self-assembly induced by a small amount of PEG to produce polyluteolin nanoparticles at 89.3 ± 4.8% yield. When prepared at an optimized condition, the spherical polyluteolin nanoparticles are amorphous with a dry size of 234.8 ± 101.6 nm and an aqueous size distribution of 379.1 ± 220.5 nm (PDI = 0.338) and contains polyluteolin with Mw of 410000 Da and Mn of 130000 Da. Analyses of 1H–1H COSY, 1H–13C HSQC, and 1H–13C HMBC NMR spectra and splitting patterns of 1H reveal three different connections of luteolin monomeric units in polyluteolin: (1) the bonding of carbon 8 in flavonoid ring A of one monomeric unit via an ether bond to C4′A in flavonoid ring B of the next unit, (2) C–C bonding between C8B in ring A of one monomeric unit and C6′B of ring B of the next unit, and (3) the C–C connection of C8C of ring A of one monomeric unit to C2′C of ring B of the next unit. Unlike luteolin that is cytotoxic at high doses, polyluteolin nanoparticles (Product 3) possess dose-dependent anti-inflammatory activity with no cytotoxicity at high doses. We anticipate that these water-dispersible anti-inflammatory polyluteolin nanoparticles could be further developed into an anti-inflammatory agent for therapeutic purposes.
4. Experimental Section
4.1. Synthesis of Polyluteolin Nanoparticles
To synthesize the polyluteolin nanoparticles, 75 mg of luteolin (98% w/w, Shaanxi Kingsci Biotechnology, China) was dissolved in 15 mL of ethanol and then 7.5 mg of poly(ethylene glycol) (PEG, Sigma-Aldrich, Germany) in 2 mL of 0.01 M phosphate buffer pH 7.4 (PB pH 7.4) was added while stirring (200 rpm, C-MAG HS 7, IKA, Germany) at room temperature. After that, horseradish peroxidase (HRP, Health Biochem Technology, China) in 2 mL of PB pH 7.4 and 3.6 mL of 30% H2O2 were added, respectively. The effects of HRP concentration, presence of H2O2, and molecular mass of PEG were investigated (see the amount in Table 2). Next, phosphate buffer pH 7.4 was added dropwise to the mixture (to 45 mL of total volume) and then the mixture was stirred at room temperature for 24 h. H2O2 and ethanol were eliminated by dialysis (dialysis tubing cellulose membrane molecular cutoff 14000, Sigma-Aldrich).
The resulting aqueous suspension of the product was collected and subjected to scanning electron microscopy analyses (SEM, JSM-IT100 JEOL, Japan). Dry products were obtained by freeze-drying the suspension (FreeZone 6 Liter-50C Console Freeze Dryer, Labconco Corporation) and subjected to 1H NMR spectrophotometric (20 mg/mL in dimethyl sulfoxide-d6 (DMSO-d6), JNM-ECZR 500 MHz, JEOL, Japan), UV–vis spectrophotometric (10 μg/mL in DMSO, Optizen POP QX, K LAB, Korea), and attenuated total reflectance Fourier transform infrared (ATR FT-IR) spectrophotometric analyses (Nicolet 6700, Thermo Electron Corporation). The particle size distribution and ζ-potential of the polyluteolin nanoparticles in aqueous suspension were determined by the dynamic light scattering (DLS) technique using a Zetasizer instrument (RI = 1.441, Zetasizer Nano ZS, Malvern Instruments, Worcestershire, U.K.). The polyluteolin nanoparticle was analyzed by powder X-ray diffraction (XRD, scanning rate 10–60° in the 2θ range, scan speed 5°/min in 0.02° step size, voltage 40 kV, electronic current 30 mA, X-ray diffractometer DMAX 2200/Ultima, Rigaku, Japan), thermogravimetric analysis (TGA, scanned from 50 to 850 °C at a heating rate of 20 °C/min under nitrogen gas, PerkinElmer Pyris 1 TGA), and differential scanning calorimetry (DSC, scanned from −130 to 250 °C (for luteolin and PEG) and −130–250 °C (for Product 3) at a heating rate of 10 °C/min under nitrogen gas, NETZSCH DSC 204F1 Phoenix, Germany). Weight-average molecular mass (Mw) of polyluteolin or luteolin oligomers was investigated by gel filtration chromatography (GFC with a photodiode array detector set at 254 nm, 10 mg/mL sample concentration, 50 μL injection volume, 1 mL/min flow rate, Shimadzu Prominence HPLC series equipped with Shodex SB-803 HQ and SB-806 HQ columns) using N,N-dimethylformamide (DMF) as a solvent and polystyrenes (Mp = 580, 9820, 67600, 466300, and 3152000 Da) as standards. The percent yield is defined as follows
The percent yield is reported as the average and standard deviation of three independent samples.
4.2. Preparation of Luteolin Oligomers
Product 3 (Table 2) was prepared as described above. Small oligomeric products or luteolin oligomers (soluble in ethanol) were extracted by dissolving Product 3 in ethanol, the obtained ethanolic suspension was filtered through a 0.22 μm syringe filter, and the filtered solution was dried under vacuum. The obtained product was subjected to 1H, 13C, COSY, HSQC, and HMBC NMR analyses in methanol-d4 (500 MHz NMR spectrometer, JNM-ECZR 500 MHz, JEOL, Japan), and UV–visible absorption analysis in acetone (Optizen POP QX, K LAB, Korea).
Luteolin: light yellow powder; decomposition temperature of 379 °C; 1H NMR (DMSO-d6, 500 MHz): δ 7.36 (2H, m, H2′ and H6′), 6.84 (1H, d, J = 8.5 Hz, H5′), 6.63 (1H, s, H3), 6.40 (1H, d, J = 2.0 Hz, H8), and 6.15 (1H, d, J = 2.0 Hz, H6); 13C NMR (DMSO-d6, 500 MHz): δ 181.66 (s, C4), 164.33 (s, C7), 163.88 (s, C2), 161.49 (s, C5), 157.32 (s, C9), 149.79 (s, C4′), 145.77 (s, C3′), 121.45 (s, C1′), 119.02 (s, C6′), 116.02 (s, C5′), 113.33 (s, C2′), 103.64 (s, C10), 102.84 (s, C3), 98.88 (s, C6), and 93.88 (s, C8); ATR FT-IR (cm–1): 3417 (brd, O–H stretching), 1653 (str, C=O stretching of the aromatic ketone), 1606, 1574, 1500 (str, C=C stretching of aromatic ring), 1365, 1263, 1246 (str, C–O stretching of phenol), 1164, 1030 (str, C–O stretching of ether in the aromatic ring); UV–visible absorption (λmax in DMSO): 271 nm (π–π* of the benzoyl system) and 353 nm (n−π* of the cinnamoyl system), and UV–visible absorption (λmax in acetone): 340 nm (n−π* of the cinnamoyl system).
Product 1: light yellow powder; decomposition temperature of 312 °C; 1H NMR (DMSO-d6, 500 MHz): δ 7.36 (2H, m, H2′ and H6′), 6.84 (1H, d, J = 8.5 Hz, H5′), 6.63 (1H, s, H3), 6.40 (1H, d, J = 2.0 Hz, H8), and 6.15 (1H, d, J = 2.0 Hz, H6); ATR FT-IR (cm–1): 3408 (brd, O–H stretching), 1651 (str, C=O stretching of the aromatic ketone), 1602, 1574, 1498 (str, C=C stretching of the aromatic ring), 1365, 1261, 1246 (str, C–O stretching of phenol), 1165, 1030 (str, C–O stretching of ether in aromatic ring); UV–visible absorption (λmax in DMSO): 271 (π–π*) and 353 nm (n−π*).
Product 2: dark yellow powder; decomposition temperature of 212 °C; 1H NMR (DMSO-d6, 500 MHz): δ 7.36 (2H, m, H2′and H6′), 6.84 (1H, d, J = 8.5 Hz, H5′), 6.63 (1H, s, H3), 6.40 (1H, d, J = 2.0 Hz, H8), and 6.15 (1H, d, J = 2.0 Hz, H6); ATR FT-IR (cm–1): 3207 (brd, O–H stretching), 2908 (brd, sp3 C–H stretching), 1623 (str, C=O stretching of aromatic ketone), 1604, 1504 (str, C=C stretching of aromatic ring), 1352, 1255 (str, C–O stretching of phenol), 1162, 1028 (str, C–O stretching of ether in aromatic ring); UV–visible absorption (λmax in DMSO): 269 (π–π*) and 353 nm (n−π*).
Product 3: light brown powder; decomposition temperature of 188 °C; ATR FT-IR (cm–1): 3265 (brd, O–H stretching), 2914 (brd, sp3 C–H stretching), 1630 (str, C=O stretching of aromatic ketone), 1612, 1504 (str, C=C stretching of aromatic ring), 1354, 1257 (str, C–O stretching of phenol), 1163, 1028 (str, C–O stretching of ether in aromatic ring); UV–visible absorption (λmax in DMSO): 262 (π–π*) and 318 nm (n−π*).
Product 4: light brown powder; decomposition temperature 315 °C; 1H NMR (DMSO-d6, 500 MHz): δ 7.36 (2H, m, H2′and H6′), 6.84 (1H, d, J = 8.5 Hz, H5′), 6.63 (1H, s, H3), 6.40 (1H, d, J = 2.0 Hz, H8), and 6.15 (d, 1H, J = 2.0 Hz, H6); ATR FT-IR (cm–1): 3145 (brd, O–H stretching), 1647 (str, C=O stretching of aromatic ketone), 1597, 1576, 1506 (str, C=C stretching of aromatic ring), 1363, 1246 (str, C–O stretching of phenol), 1159, 1026 (str, C–O stretching of ether in aromatic ring); UV–visible absorption (λmax in DMSO): 271 (π–π*) and 353 nm (n−π*).
Product 5: light brown powder; decomposition temperature of 264 °C; ATR FT-IR (cm–1): 3400, 3197 (brd, O–H stretching), 1651 (str, C=O stretching of aromatic ketone), 1610, 1558, 1500 (str, C=C stretching of the aromatic ring), 1354, 1257 (str, C–O stretching of phenol), 1165, 1030 (str, C–O stretching of ether in the aromatic ring); UV–visible absorption (λmax in DMSO): 265 (π–π*) and 340 nm (n−π*).
Product 6: light brown powder; decomposition temperature of 187 °C; ATR FT-IR (cm–1): 3209 (brd, O–H stretching), 2953 (brd, sp3 C–H stretching), 1651 (str, C=O stretching of the aromatic ketone), 1608, 1506 (str, C=C stretching of the aromatic ring), 1354, 1257 (str, C–O stretching of phenol), 1163, 1022 (str, C–O stretching of ether in the aromatic ring); UV–visible absorption (λmax in DMSO): 262 (π–π*) and 293 nm (n−π*).
Product 7: light brown powder; decomposition temperature of 212 °C; ATR FT-IR (cm–1): 3348 (brd, O–H stretching), 2920 (brd, sp3 C–H stretching), 1637 (str, C=O stretching of the aromatic ketone), 1614, 1504 (str, C=C stretching of the aromatic ring), 1352, 1259 (str, C–O stretching of phenol), 1165, 1024 (str, C–O stretching of ether in the aromatic ring); UV–visible absorption (λmax in DMSO): 274 (π–π*) and 340 nm (n−π*).
Luteolin oligomer: dark yellow powder; Yield 11.2% weight of Product 3; dark yellow powder; UV–visible absorption (λmax in acetone): 332 nm (n−π*). See its NMR spectra in the Results and Discussion section.
4.3. In Vitro Anti-inflammatory Activity
The in vitro anti-inflammatory activity of polyluteolin nanoparticles and luteolin was determined through the nitrite production level of RAW 264.7 macrophage cells (ATCC, Manassas) using the Griess assay. The samples include the polyluteolin nanoparticle (Product 3) and luteolin at the concentrations of 6.25, 12.5, 25, 50, and 100 μg/mL and PEG 2050 at the concentrations of 0.625, 1.25, 2.5, 5, and 10 μg/mL. Effects of samples on RAW 264.7 cell viability were also acquired using the MTT assay to confirm that the obtained anti-inflammatory activity was not related to the cytotoxicity of the samples.
4.3.1. Cell Culture and Treatment
RAW 264.7 cells were cultured in complete media of Dulbecco’s modified Eagle’s medium (DMEM, Hyclone), 10% fetal bovine serum (FBS, Hyclone), 1% 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid (HEPES, Hyclone), 1% sodium pyruvate (Hyclone), and 1% gentamicin (Hyclone) at 37 °C in an atmosphere of 5% CO2 for 72 h. After that, the cells were seeded in a 96-well plate (10000 cell/well) overnight. Then, the supernatant was removed and replaced by the sample (50 μL/well), and the plate was incubated at 37 °C in an atmosphere of 5% CO2 for 1 h. After that, complete media containing LPS (200 ng/mL, Sigma-Aldrich) and recombinant IFN-γ (20 ng/mL, BioLegend) was added (50 μL/well). The cells were incubated at 37 °C in an atmosphere of 5% CO2 for 24 h. Culture supernatants were collected from the treated cells and subjected for the nitric oxide assay (Section 4.3.2) while cells were subjected for viability by the MTT assay (Section 4.3.3).
4.3.2. Nitrite Production
Nitrite production was measured by nitrite accumulation in the culture supernatant using a colorimetric reaction. For analysis, the supernatant (50 μL) of the treated cells from Section 4.3.1 was mixed with Griess reagent (50 μL/well 0.1% N-1-napthylethylenediamine dihydrochloride (NED, Sigma-Aldrich) and 50 μL/well 1% sulfanilamide (Merck, Germany) in 5% phosphoric acid). The absorbance was measured at 540 nm (microplate reader, Multiskan FC, Thermo Scientific), and the nitrite concentration was determined with the aid of the standard curve constructed from standard sodium nitrite solutions prepared in complete media. Cells treated with 1% water, IFN-γ, and LPS in complete media were used as a negative control, and cells treated with 1% water in complete media were used as a positive control. Nitrite production is defined as follows
The nitrite production is reported as the average and standard deviation of results from three independent experiments.
4.3.3. Cell Viability
Cell metabolism was measured by the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) to purple formazan using a colorimetric assay. All supernatants of the treated cells from Section 4.3.1 were removed and replaced with complete media (100 μL/well) and MTT solution (5 mg/mL in phosphate-buffered saline pH 7.4, 10 μL/well). After the treated cells were incubated at 37 °C in an atmosphere of 5% CO2 for 4 h, DMSO was added (200 μL/well) to dissolve the formazan crystals. Then, the absorbance was measured at 540 nm. Cells treated with 1% water, IFN-γ, and LPS in complete media were used as a negative control, cells treated with 1% water in complete media were used as a positive control, and complete media was used as a blank. Cell viability is defined as follows
The cell viability is reported as the average and standard deviation of results from three independent experiments.
4.3.4. Statistical Analysis
The data were reported as the average and standard deviation of three independent experiments. Statistical differences were analyzed by t-test analysis with the significance level at α = 0.05 for all tests. Significant differences between the tested groups are denoted with * for p value <0.05.
Acknowledgments
This work was funded by the Center of Excellence on Petrochemical and Materials Technology (PETROMAT), Chulalongkorn University, the Center of Excellence in Advanced Materials and Biointerfaces, Chulalongkorn University, and the National Nanotechnology Center (NANOTEC), NSTDA, Ministry of Science and Technology, Thailand, through its program of Research Network NANOTEC (RNN).
Glossary
Abbreviations
- PEG
poly(ethylene glycol)
- PB pH 7.4
phosphate buffer pH 7.4
- HRP
horseradish peroxidase
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- GFC
gel filtration chromatography
- LPS
lipopolysaccharides
- IFN-γ
interferon γ
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05142.
FT-IR spectra and UV spectra of products, particle size distribution graph, TG-DTA curves and DSC curves of polyluteolin nanoparticles (Product 3), overlay of 1H–1H COSY and 1H–13C HSQC NMR correlations, and proposed structure and reaction mechanism of the luteolin oligomer (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Perwez Hussain S.; Harris C. C. Inflammation and Cancer: an Ancient Link with Novel Potentials. Int. J. Cancer 2007, 121, 2373–2380. 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- Lu H.; Ouyang W.; Huang C. Inflammation, a Key Event in Cancer Development. Mol. Cancer Res. 2006, 4, 221–233. 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- Greten F. R.; Grivennikov S. I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Tsao R. Dietary Polyphenols, Oxidative Stress and Antioxidant and Anti-Inflammatory Effects. Curr. Opin. Food Sci. 2016, 8, 33–42. 10.1016/j.cofs.2016.02.002. [DOI] [Google Scholar]
- Wu X.; Schauss A. G. Mitigation of Inflammation with Foods. J. Agric. Food Chem. 2012, 60, 6703–6717. 10.1021/jf3007008. [DOI] [PubMed] [Google Scholar]
- Benavente-García O.; Castillo J. Update on Uses and Properties of Citrus Flavonoids: New Findings in Anticancer, Cardiovascular, and Anti-Inflammatory Activity. J. Agric. Food Chem. 2008, 56, 6185–6205. 10.1021/jf8006568. [DOI] [PubMed] [Google Scholar]
- Miean K. H.; Mohamed S. Flavonoid (Myricetin, Quercetin, Kaempferol, Luteolin, and Apigenin) Content of Edible Tropical Plants. J. Agric. Food Chem. 2001, 49, 3106–3112. 10.1021/jf000892m. [DOI] [PubMed] [Google Scholar]
- Al-Megrin W. A.; Alkhuriji A. F.; Yousef A. O. S.; Metwally D. M.; Habotta O. A.; Kassab R. B.; Abdel Moneim A. E.; El-Khadragy M. F. Antagonistic Efficacy of Luteolin Against Lead Acetate Exposure-Associated with Hepatotoxicity is Mediated via Antioxidant, Anti-Inflammatory, and Anti-Apoptotic Activities. Antioxidants 2020, 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz N.; Kim M. Y.; Cho J. Y. Anti-Inflammatory Effects of Luteolin: A Review of In Vitro, In Vivo, and in Silico Studies. J. Ethnopharmacol. 2018, 225, 342–358. 10.1016/j.jep.2018.05.019. [DOI] [PubMed] [Google Scholar]
- Hu C.; Kitts D. D. Luteolin and Luteolin-7-O-glucoside from Dandelion Flower Suppress iNOS and COX-2 in RAW264. 7 Cells. Mol. Cell. Biochem. 2004, 265, 107–113. 10.1023/B:MCBI.0000044364.73144.fe. [DOI] [PubMed] [Google Scholar]
- Ueda H.; Yamazaki C.; Yamazaki M. Luteolin as an Anti-Inflammatory and Anti-Allergic Constituent of Perilla frutescens. Biol. Pharm. Bull. 2002, 25, 1197–1202. 10.1248/bpb.25.1197. [DOI] [PubMed] [Google Scholar]
- Li L.; Wu X.; Chen Z.; Sun S.; Ye J.; Zeng S.; Jiang H. Interspecies Difference of Luteolin and Apigenin after Oral Administration of Chrysanthemum Morifolium Extract and Prediction of Human Pharmacokinetics. Die Pharm. 2013, 68, 195–200. [PubMed] [Google Scholar]
- Liu Y.; Wang L.; Zhao Y.; He M.; Zhang X.; Niu M.; Feng N. Nanostructured Lipid Carriers versus Microemulsions for Delivery of the Poorly Water-Soluble Drug Luteolin. Int. J. Pharm. 2014, 476, 169–177. 10.1016/j.ijpharm.2014.09.052. [DOI] [PubMed] [Google Scholar]
- Qing W.; Wang Y.; Li H.; Ma F.; Zhu J.; Liu X. Preparation and Characterization of Copolymer Micelles for the Solubilization and In Vitro Release of Luteolin and Luteoloside. APPS PharmSciTech 2017, 18, 2095–2101. 10.1208/s12249-016-0692-y. [DOI] [PubMed] [Google Scholar]
- Majumdar D.; Jung K. H.; Zhang H.; Nannapaneni S.; Wang X.; Amin A. R.; Chen Z.; Shin D. M. Luteolin Nanoparticle in Chemoprevention: In Vitro and In Vivo Anticancer Activity. Cancer Prev. Res. 2014, 7, 65–73. 10.1158/1940-6207.CAPR-13-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J. F.; Gao X.; Wang B. L.; Wei X. W.; Gou M. L.; Men K.; Liu X. Y.; Guo G.; Qian Z. Y.; Huang M. J. Preparation and Characterization of Monomethoxy poly (ethylene glycol)-poly (ε-caprolactone) Micelles for the Solubilization and In Vivo Delivery of Luteolin. Int. J. Nanomed. 2013, 8, 3061–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T.; Matsuo Y.; Kouno I. Chemistry of Secondary Polyphenols Produced During Processing of Tea and Selected Foods. Int. J. Mol. Sci. 2010, 11, 14–40. 10.3390/ijms11010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsume M.; Osakabe N.; Yamagishi M.; Takizawa T.; Nakamura T.; Miyatake H.; Hatano T.; Yoshida T. Analyses of Polyphenols in Cacao Liquor, Cocoa, and Chocolate by Normal-Phase and Reversed-Phase HPLC. Biosci., Biotechnol., Biochem. 2000, 64, 2581–2587. 10.1271/bbb.64.2581. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Rankin G. O.; Tu Y.; Chen Y. C. Inhibitory Effects of the Four Main Theaflavin Derivatives Found in Black Tea on Ovarian Cancer Cells. Anticancer Res. 2016, 36, 643–651. [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M.; Ichitani M.; Suzuki Y.; Unno T.; Sugawara T.; Yamahira T.; Kato M.; Takihara T.; Sagesaka Y.; Kakuda T. Black-Tea Polyphenols Suppress Postprandial Hypertriacylglycerolemia by Suppressing Lymphatic Transport of Dietary Fat in Rats. J. Agric. Food Chem. 2009, 57, 7131–7136. 10.1021/jf900855v. [DOI] [PubMed] [Google Scholar]
- Pan M. H.; Liang Y. C.; Lin S. Y.; Zhu N. Q.; Ho C. T.; Lin J. K. Induction of Apoptosis by the Oolong Tea Polyphenol Theasinensin A through Cytochrome C Release and Activation of Caspase-9 and Caspase-3 in Human U937 Cells. J. Agric. Food Chem. 2000, 48, 6337–6346. 10.1021/jf000777b. [DOI] [PubMed] [Google Scholar]
- Gali H.; Perchellet E.; Gao X.; Karchesy J.; Perchellet J. Comparison of the Inhibitory Effects of Monomeric, Dimeric, and Trimeric Procyanidins on the Biochemical Markers of Skin Tumor Promotion in Mouse Epidermis In Vivo. Planta Med. 1994, 60, 235–239. 10.1055/s-2006-959466. [DOI] [PubMed] [Google Scholar]
- Fan F. Y.; Shi M.; Nie Y.; Zhao Y.; Ye J. H.; Liang Y. R. Differential Behaviors of Tea Catechins Under Thermal Processing: Formation of Non-Enzymatic Oligomers. Food Chem. 2016, 196, 347–354. 10.1016/j.foodchem.2015.09.056. [DOI] [PubMed] [Google Scholar]
- He F.; Pan Q. H.; Shi Y.; Zhang X. T.; Duan C. Q. Identification of Autoxidation Oligomers of Flavan-3-ols in Model Solutions by HPLC-MS/MS. J. Mass Spectrom. 2009, 44, 633–640. 10.1002/jms.1536. [DOI] [PubMed] [Google Scholar]
- Davies A. P.; Goodsall C.; Cai Y.; Davis A. L.; Lewis J. R.; Wilkins J.; Wan X.; Clifford N.; Powell C.; Parry A.. Black Tea Dimeric and Oligomeric Pigments—Structures and Formation. In Plant Polyphenols 2, 3rd ed.; Gross G. G.; Hemingway R. W.; Yoshida T., Eds.; Springer: New York,1999; pp 697–724. [Google Scholar]
- Drynan J. W.; Clifford M. N.; Obuchowicz J.; Kuhnert N. The Chemistry of Low Molecular Weight Black Tea Polyphenols. Nat. Prod. Rep. 2010, 27, 417–462. 10.1039/b912523j. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Yang J.; Cui D.; Zhao D.; Benedito V. A.; Zhao J. Genome-Wide Analysis and Metabolic Profiling Unveil the Role of Peroxidase CsGPX3 in Theaflavin Production in Black Tea Processing. Food Res. Int. 2020, 137, 109677 10.1016/j.foodres.2020.109677. [DOI] [PubMed] [Google Scholar]
- Subramanian N.; Venkatesh P.; Ganguli S.; Sinkar P. Role of Polyphenol Oxidase and Peroxidase in The Generation of Black Tea Theaflavins. J. Agric. Food Chem. 1999, 47, 2571–2578. 10.1021/jf981042y. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Higashimura H. Oxidative Polymerization of Phenols Revisited. Prog. Polym. Sci. 2003, 28, 1015–1048. 10.1016/S0079-6700(03)00014-5. [DOI] [Google Scholar]
- De Regil R.; Sandoval G. Biocatalysis for Biobased Chemicals. Biomolecules 2013, 3, 812–847. 10.3390/biom3040812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourcel L.; Routaboul J. M.; Cheynier V.; Lepiniec L.; Debeaujon I. Flavonoid Oxidation in Plants: from Biochemical Properties to Physiological Functions. Trends Plant Sci. 2007, 12, 29–36. 10.1016/j.tplants.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Verloop A. J.; Vincken J. P.; Gruppen H. Peroxidase Can Perform the Hydroxylation Step in the “Oxidative Cascade” During Oxidation of Tea Catechins. J. Agric. Food Chem. 2016, 64, 8002–8009. 10.1021/acs.jafc.6b03029. [DOI] [PubMed] [Google Scholar]
- Finger A. In-Vitro Studies on the Effect of Polyphenol Oxidase and Peroxidase on the Formation of Polyphenolic Black Tea Constituents. J. Sci. Food Agric. 1994, 66, 293–305. 10.1002/jsfa.2740660306. [DOI] [Google Scholar]
- Shaji J.; Lal M. Nanocarriers for Targeting in Inflammation. Asian J. Pharm. Clin. Res. 2013, 6, 3–12. [Google Scholar]
- Kim Y. J.; Uyama H.; Kobayashi S. Peroxidase-Catalyzed Oxidative Polymerization of Phenol with a Nonionic Polymer Surfactant Template in Water. Macromol. Biosci. 2004, 4, 497–502. 10.1002/mabi.200300078. [DOI] [PubMed] [Google Scholar]
- Green L. C.; Wagner D. A.; Glogowski J.; Skipper P. L.; Wishnok J. S.; Tannenbaum S. R. Analysis of Nitrate, Nitrite, and [15N] Nitrate in Biological Fluids. Anal. Biochem. 1982, 126, 131–138. 10.1016/0003-2697(82)90118-X. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.