

Abstract

Tetracyanoquinodimethane (TCNQ) is known to react with various amines to generate substituted TCNQ derivatives with remarkable optical and nonlinear optical characteristics. The choice of amine plays a crucial role in the outcome of molecular material attributes. Especially, mono/di-substituted TCNQ’s possessing strong fluorescence in solutions than solids are deficient. Furthermore, cation recognition in the solid-state TCNQ derivatives is yet undetermined. In this article, we present solution-enhanced fluorescence and exclusive solid-state recognition of K+ ion achieved through the selection of 4-(4-aminophenyl)morpholin-3-one (APM) having considerable π-conjugation and carbonyl (C=O) functionality, particularly in the ring. TCNQ when reacted with APM, in a single-step reaction, resulted in two well-defined distinct compounds, namely, 7,7-bis(4-(4-aminophenyl)morpholin-3-ono)dicyanoquinodimethane (BAPMDQ [1], yellow) and 7,7,8-(4-(4-aminophenyl)morpholin-3-ono)tricyanoquinodimethane (APMTQ [2], red), with increased fluorescence intensity in solutions than their solids. Crystal structure investigation revealed extensive C–H−π interactions and strong H-bonding in [1], whereas moderate to weak interactions in [2]. Surprisingly, simple mechanical grinding during KBr pellet preparation with [1, 2] triggered unidentified cation recognition with a profound color change (in ∼1 min) detected by the naked eye, accompanied by a drastic enhancement of fluorescence, proposed due to the presence of carbonyl functionality, noncovalent intermolecular interactions, and molecular assemblies in [1, 2] solids. Cation recognition was also noted with various other salts as well (KCl, KI, KSCN, NH4Cl, NH4Br, etc.). Currently, the recognition mechanism of K+ ion in [1, 2] is demonstrated by the strong electrostatic interaction of K+ ion with CO and simultaneously cation−π interaction of K+ with the phenyl ring of APM, supported by experimental and computational studies. Computational analysis also revealed that a strong cation−π interaction occurred between the K+ ion and the phenyl ring (APM) in [2] than in [1] (ΔGbinding calculated as ∼16.3 and ∼25.2 kcal mol–1 for [1] and [2], respectively) providing additional binding free energy. Thus, both electrostatic and cation−π interactions lead to the recognition. Scanning electron microscopy of drop-cast films showed microcrystalline “roses” in [1] and micro/nano “aggregates” in [2]. Optical band gap (∼3.565 eV) indicated [1, 2] as wide-band-gap materials. The current study demonstrates fascinating novel products obtained by single-pot reaction, resulting in contrasting optical properties in solutions and experiencing cation recognition capability exclusively in the solid state.
1. Introduction
Small organic fluorophores always attract a wide range of intensive applications not only in the field of materials science like organic light-emitting diodes (OLED), optoelectronic devices, and solar cells, but also in bio-imaging, sensing, etc.1−5 The key aspect of tetracyanoquinodimethane (TCNQ) derivatives is their sensitivity to even minor structural changes. The subtle changes in the choice of amines can fine-tune physical attributes and optical responses, leading to a profound impact on molecular material properties. This captivating feature inspires the design and development of new TCNQ derivatives with marked optical properties in solutions and solids. Diaminodicyanoquinodimethane derivatives (DADQs) possess high dipole moments5 and are known to exhibit stronger fluorescence in the solid state than solutions. Very recently, on benzene substitution, high-fluorescence diaminodicyanoquinones with 90% quantum yields were reported.6 Originally, TCNQ derivatives were exploited as potential candidates for second-order nonlinear optical (NLO) materials5,7,8 due to their large hyperpolarizabilities (β).8−10 Recently, a substantial study on the optical property of TCNQ derivatives has been conducted. Specifically, mono/di-substituted TCNQ (DADQ) fluorophores developed by the reaction of TCNQ with various heterocyclic,11−15 aliphatic,16,17 and countable (∼5) aromatic amines,18−22 precisely with primary and secondary amines, resulted in a divergent phenomenon like aggregation-enhanced solid-state emission,11,15,23 colloids,20 nanoparticles,24−26 phase change attributes,27−29 and second-harmonic generation (SHG).30−33 Reasonable fluorescence in TCNQ derivatives as solutions was due to noncovalent interactions, strong hydrogen bonding, and self-assembly.6 However, enhanced fluorescence in solids was observed due to aggregation,10,34 strong intermolecular attractions,8,10−15 restricted geometric relaxation in excited states,12,13,21,22,35 and decrease in intermolecular interactions like π–π/CH−π,35−37 in comparison to solutions. Currently, the inadequacy of TCNQ derivatives with considerable fluorescence in solution and in solid state is a severe limitation; furthermore, cation recognition by mono/di-substituted TCNQ derivatives has not been discovered yet. In this article, we introduce enhanced fluorescence exhibited in solutions (especially in mono/di-substituted TCNQs) than solids, and cation recognition identified in novel TCNQ derivatives. As per our knowledge, only five mono/di-TCNQ derivatives with aromatic amines, viz., 4-haloanilines and 2-methyl-4-chloroaniline, as substituents are reported, a related analogue is with anisidine, obtained as brown-green needles of α-(p-anisyliamino)-α,α′,α′-tricyano-p-xylene, yet only synthesis and spectroscopic characterization are described. Recently, stimuli-responsive TCNQ derivatives with reversible crystalline–amorphous transformation have been noted.27,33 One among these was 7,7-bis(N-benzoylpiperazine)-8,8-dicyanoquinodimethane (BBPDQ)27 obtained by reacting TCNQ with 1-benzoylpiperazine; notably, 1-benzoylpiperazine possesses an amide functionality attached to a phenyl ring (amide attached to an aromatic ring), and BBPDQ manifested phosphorescence property. In particular, the carbonyl group of BBPDQ was involved in intermolecular hydrogen bonding with CH2 of piperazine. Nevertheless, the reaction of TCNQ with an amide within a saturated cyclic ring such as 4-(4-aminophenyl)morpholin-3-one (APM) was not attempted. APM is a class of lactams that plays a crucial role in various alkaloids and building blocks in biologically active compounds, also behaves as a platelet inhibitor,38 as well as a key precursor in the preparation of an inhibitor of coagulation factor Xa used for prophylaxis and treatment of thromboembolic disorders. Xa inhibitors have also been used in the clinical and preclinical research39 as anticoagulating agents. In our case, as APM has considerable π-electron conjugation from the amine group to the carbonyl group, the product obtained from the reaction of APM with TCNQ exhibited interesting molecular material properties with optical characteristics. Therefore, we designed and synthesized two new derivatives by reacting TCNQ with APM through a single-pot reaction, resulting in yellow and red emissive molecular materials, namely, 7,7-bis(4-(4-aminophenyl)morpholin-3-ono)dicyanoquinodimethane (BAPMDQ [1]) and 7,7,8-(4-(4-aminophenyl)morpholin-3-ono)tricyanoquinodimethane (APMTQ [2]). Interestingly, the single-pot reaction generated [1] as orange-red crystals, showing yellow-green fluorescence in the solid state and yellow in solution, and [2] as brown-red crystals with red fluorescence in solid as well as solution. Solutions of [1, 2] demonstrated increased fluorescence intensity than solids. Lifetime measurements were performed for solutions besides solids to understand the decay patterns. Compounds [1, 2] showed an optical band gap (∼3.565 eV), indicative of wide-band-gap materials. More interestingly, current state of the art lies on the naked eye detection of an unexpected color change (in ∼1 min) observed in [1, 2] during KBr pellets preparation for IR spectra characterization. This further guided us to explore the possible interaction of KBr with [1] and [2]. At present, detection/recognition of ions in solutions gained significant research interest, especially with fluorescent organic molecules.40,41 In general, mechanical-force-induced solid-state reactions are mostly known for inorganic complexes;42,43 in this view, solvent-free reaction or solvent-free detection of ions43 has attracted researchers beyond solution state due to environmental issues. Organic phosphonium salts were prepared by the ball milling technique with a solvent-free process, and such conventional organic synthesis is helpful to avoid side reactions and topological problems with green procedures.44 We exploited the two new derivatives by simple hand grinding (for ∼1 min) using a mortar and pestle, resulting in solid-state recognition of various cations. Intriguingly, there are no reports to date on the interaction of any TCNQ derivatives with KX (X = Br, Cl, I)/other salts exclusively in the solid state. Moreover, contrasting results were obtained from [1] and [2] compared to reported mono/di-substituted TCNQ derivatives: (i) increase in the fluorescence intensity in solutions than solids; (ii) mixture of [1] and [2] obtained through a single-step reaction, each being a major product, respectively, at instances, depending on the amount of APM and workup conditions; (iii) [1] presented as yellow and [2] as red in solutions following large Stokes shift (ca. 100–140 nm) and appreciable quantum yields; (iv) grinding [1] and [2] with KX (X = Br, Cl, I)/other salts leads to an unexpected visible color change with stimuli-responsive cation recognition, particularly in solids boosting fluorescence, accompanied by 110–150 nm Stokes shift.
Detailed investigation revealed that [1] could detect various cations (K+, NH4+, Na), whereas [2] was selective to Na+ and K+. Since this is a breakthrough result among any TCNQ compounds known so far, a rigorous analysis is required; in this article, we have focused on and demonstrated the interaction of [1, 2] specifically with potassium salts KX (X = Br, Cl, I), while the results with other salts are given in the Supporting Information. A computational study also supports the recognition of K+ ion. Compounds [1, 2] were also found to be thermally stable. They were thoroughly investigated and characterized by IR, 1H, and 13C NMR, and high-resolution mass spectrometry (HRMS) techniques. X-ray crystallography was studied for structure–property relationship. Crystallographic studies disclosed weak to moderate intermolecular interactions in both [1] and [2]; however, strong intermolecular hydrogen bonding was noted in [1], and found to be absent in [2]. Morphological studies of drop-cast solutions in acetonitrile revealed micro “roses” in [1] and nanoaggregates in [2]. Thus, the subtle variation in the course of introducing APM has drastically refashioned the structural aspects, optical responses, and wide band gaps, as well as created solid-state cation recognition capability in [1] and [2]. Thus, we provide a comprehensive study of [1] and [2] to envisage solution and solid-state properties, beginning from the crystal structure analysis, photophysical property, and oxidation–reduction phenomenon due to the carbonyl functionality present, especially within the morpholine ring of AMP.
2. Results and Discussion
2.1. Crystallographic Investigations
Crystal structure analysis showed that [1, 2] belonged to the P21/c space group with one molecule of each in the asymmetric unit. The molecular structures of [1, 2] indicate that the central ring (C1–C6) is nearly benzenoid with average carbon–carbon bond distances of 1.404 Å [1] and 1.391 Å [2] owing to the zwitterionic nature.15,18 The diaminomethylene unit in [1] is found to be twisted with reference to the benzenoid ring plane accompanied by torsion angles τN4–C7–C3–C4 = −18.47° and τN3–C7–C3–C2 = −16.45°, with the average being −17.46°; similarly, the aminomethylene unit in [2] is twisted with respect to the benzenoid ring plane, and torsional angles are τN4–C7–C3–C2 = 16.75° and τN1–C7–C3–C4 = 19.77° with an average of 18.26°; lower twist angles imply the role of APM generating conjugation effect in [1] and [2]. The farther placement of the phenyl group of APM could also be an important factor that contributes to the lower twist angles in both [1] and [2].21 Interestingly, intramolecular interactions were not at all observed in either [1] or [2]. In particular, TCNQ derivatives having aliphatic secondary amines as substituents and intramolecular interactions play a significant role, leading to second-harmonic generation (NLO property).15,29 In this work, only moderate to weak intermolecular interactions were observed in the crystals of [1] and [2], triggering solid-state cation recognition. The molecular packing diagrams of [1] and [2] with different orientations of the molecular dipoles are shown in Figure 1. In [1], each dipole was aligned one above the other, slightly inclined in a tail–head manner along the c-axis, antiparallel arrangements of dipoles with an interplanar distance of 4.454 Å were noted along the ab plane and the dipoles were arranged in a parallel manner at a large interplanar distance of 12.415 Å through the a-axis (Figures S1a,b and S3a). The bond distances of C3–C7 and C6–C8 were 1.468 and 1.446 Å, respectively. Significant short intermolecular interactions (shorter than the sum of the van der Waals radii of atoms) were involved in [1, 2] (Figure 2). Intriguingly, in [1], these were mainly from methylene hydrogens of morpholine ring (H19A, H28B) and phenyl ring hydrogen (H11) of APM. Notably, H19A and H28B interact with phenyl ring (APM) carbons (C13, C14, C15, C17) and H11 with benzenoid ring carbons (C3, C4, C5) generating various C–H···π interactions (Table 1). Moreover, in [1], morpholine ring oxygens O1 and O3, N1 (of one of the free C≡N at the δ– end), N5, and N6 (phenyl ring of APM) did not involve in any of the intermolecular interactions. N2 (another C≡N of the δ– carbon end) undergoes intermolecular interactions with H3(N–H, APM), H2(Ar–H), C11(phenyl carbon), and N3(N of aminophenyl) of neighboring molecules. Carbonyl oxygens (O2, O4), N2, and N3 experienced weak interactions. Only one of the amino hydrogens, H4 (of APM moiety), is hydrogen-bonded to O4 (carbonyl oxygen; O4···H4–N, 2.759 Å). Benzenoid rings between donor and acceptor were found to be separated with the centroid distance of 7.175 Å (Figure S2). The phenyl ring (APM) was twisted out of the benzenoid ring plane by ≈67.49°. The benzenoid and phenyl rings were in different planes; in particular, the carbonyl group was found to be above these planes (Figure S3), probably facilitating the solid-state cation recognition. The angles between the substituted diaminomethylene units C16–N3–C7 and C26–N4–C7 were found to be 129.59 and 127.89° respectively.
Figure 1.

Molecular packing diagrams exhibiting different orientations of molecular dipoles in (a) BAPMDQ [1] and (b) APMTQ [2].
Figure 2.

Prominent hydrogen bonding and significant weak intermolecular interactions (in yellow) in (a) [1]; C–H···π, N–H···O, C–H···O, N–H···N, CN···H and (b) [2]; C–H···O, O–C, CN···H, CN···C, C–C.
Table 1. Significant Noncovalent Weak Intermolecular Interactions (Å) Detected in [1] and [2] with C–H···π and Hydrogen-Bond Type Ranging from Weak, Moderate, to Strong Leading to Different Orientations of Molecular Dipoles Consequently with Contrasting Optical Properties in the Solid State.
| C–H···π | N–H···O | C–H···O | π···π | N–H···N | O···C | C≡N–H | C–H···C | C≡N···C | C···C | |
|---|---|---|---|---|---|---|---|---|---|---|
| [1] | 2.894, 2.724 | 1.917 | 2.643 | 7.175 | 2.142 | 2.600 | 2.860 | 3.211 | ||
| 2.814, 2.767 | 2.759 | 2.675 | 2.836 | |||||||
| 2.819, 2.788 | 2.588 | |||||||||
| 2.809, 2.836 | ||||||||||
| 2.384 | ||||||||||
| [2] | 2.576 | 7.190 | 3.200 | 2.674 | 3.184 | 3.380 | ||||
| 2.136 | 3.138 | 2.746 | 3.141 | 3.397 | ||||||
| 2.488 | 3.163 | 2.596 | 3.049 | |||||||
| 2.411 | 3.006 | 3.215 |
Compound [2] is envisaged as a tautomer of a regular mono-substituted TCNQ product (referred to as aminotricyanoquinodimethane) with a H atom of the amino group being transferred to the dicyanomethylene carbon atom (H8), accommodated by the enhanced stability of benzenoid structure over the quinonoid (Scheme 1, APMTQ). The acidic hydrogen (H8) was involved in weak hydrogen bonding with O2 (carbonyl oxygen) alone and no N (Figure S4a), contrasting with one of such previously reported18 analogues, wherein the acidic hydrogen at dicyanomethylene carbon was involved in bifurcated hydrogen bonds with N atoms of unreacted cyano groups, of neighboring molecules. In [2], along the c-axis, alternate stacks of parallel dimers were found to be sandwiched between the single layers arranged in antiparallel orientations (Figure S4b), a different manner noted compared to [1] because [1] had certain strong intermolecular hydrogen bonds, which were absent in [2]. Head–tail arrangements placed one above the other in [2] were observed along the b-axis. Likewise in [1], weak π–π stacking was observed between the benzenoid rings (centroid distance, 7.190 Å; Figure S4c) in [2] as well; the angle between the phenyl ring planes of neighboring molecules was 69°. The phenyl ring (APM) was twisted out of the benzenoid ring plane by ≈55.39° (Figure S4d). Moreover, C8, H8, and N2 at the dicyanomethylene end in [2] were arranged at a tetrahedral angle (θC8–H8···N2 = 108.24°; θC8–H8···N2 = 108.61°; θN2–C8···N3 = 109.90°), emanating the stability to the carbon acid. Amidst overall five nitrogens in [2], only N1 (unsubstituted free C≡N at δ+ carbon) and N2 (free C≡N at δ– carbon) are involved in intermolecular hydrogen bonding, and N3 (other free C≡N at δ–), N4 (aminophenyl ring), and N5 (morpholine ring) did not participate in any of the interactions. Remarkably, C3–C7 and C6–C8 were 1.480 and 1.535 Å. In [2], short intermolecular interactions (shorter than the sum of the van der Waals radii of atoms involved) were mainly due to carbonyl oxygen (O2) and N from both the free C≡N at δ– and δ+ carbon (Table 1). Due to antiparallel dipole orientations, significantly weak interactions also occurred from N1, N2 (unsubstituted C≡N groups), morpholine ring oxygen (O1), and carbonyl oxygen (O2) between the neighboring molecules. Consequently, several short contacts resulted in supramolecular self-assemblies in both [1] and [2], leading to considerable fluorescence in solids. Although supramolecular self-assembly was noted in [1, 2], [1] showed dominance in extensive C–H···π, strong H-bonding (NH···O (1.917 Å), NH···N (2.142 Å)), and moderate interactions, leading to aggregation, whereas in [2], C–H···π and strong H-bonding were absent, only moderate (C≡N–H ∼ 2.672 Å, C–H···O ∼ 2.403) and weak interactions (O–C ∼ 3.127 Å, C≡N ∼ 3.147 Å) were noted (Table 1), hindering the dipoles from forming robust aggregates; thus, aggregation-enhanced fluorescence22,34 is noted in [1] compared to [2]. Also, since [1] contains slightly excess heteroatoms due to bis-substituted product, it could readily participate in strong intermolecular hydrogen bonding (Figure 2a); each molecule of [1] formed four H-bonds with four neighboring molecules: 1.917 Å (O4···H–N3, N4–H···O4) and 2.142 Å (N3–H···N2, N2···H–N3), whereas the same is found to be totally absent in [2]. Overall, crystal structure investigation revealed interesting and compact contrasting features between [1] and [2]. On the other hand, increased fluorescence intensity was observed in [1, 2] solutions compared to [1, 2] solids. A greater insight into the fluorescence of solution and solids is presented in the Section 2.2. The importance of CH−π interactions associated with biomolecules,45 conformational selectivity,46 and molecular recognition47−49 has been described in the literature. To our surprise, when [1] and [2] were prepared as KBr pellets for infrared spectra characterization, unexpected color change was identified by the naked eye within ∼1 min of mechanical grinding, due to a significant number of weak to moderate interactions identified in both [1] and [2]. Thus, external stimuli on weak noncovalent interactions resulting from the molecular assemblies and the choice of AMP with carbonyl functionality in the morpholine ring has induced solid-state cation recognition capability in [1, 2], supported by a detailed investigation carried out through analytical and microscopic techniques such as compositional analysis (X-ray fluorescence, XRF), Fourier transform infrared (FT-IR) spectroscopy, morphology of ground samples (scanning electron microscopy (SEM)), etc. A computational study also showed that [1, 2] exhibit cation recognition capability by electrostatic and cation−π interactions. We further exploited this sequel with various KX (X = Cl, I) and other salts, discussed in detail in the cation binding study in the Section 2.2.
Scheme 1. BAPMDQ [1] and APMTQ [2] Obtained by the Reaction of TCNQ with APM.

2.2. Photophysical Property
Interesting optical properties were recognized in the donor−π–acceptor scaffolds of BAPMDQ [1] and APMTQ [2]. Absorption spectra in solvents with increasing polarity did not show any significant solvatochromism in [1, 2] (Figure S5a,b). Plots of Stokes shifts vs ET(30) (solvent polarity scale) and Stokes shifts vs Δf (orientational polarizability) also showed a nonlinear characteristic (Figure S5c,d) supporting no considerable solvatochromism.50 A slight bathochromic shift in polar aprotic solvents (Tables S1 and S2) shows that [1, 2] are more stabilized in dimethylformamide (DMF) and dimethyl sulfoxide (DMSO) in the excited states by decreasing the S0–S1 gap. The lowest energy of absorptions in [1, 2] is a characteristic feature of amine-substituted TCNQ derivatives9,19,34 likely due to the intramolecular charge-transfer transition. λmax absorption and λmax emission in DMF solutions were found to be at 450 and 564 nm for [1] and at 500 and 634 nm for [2], respectively (Figure 3). The redshift occurred in [2] compared to [1] due to the extended π-electron conjugation. Th λmax emission values in [1] and [2] were found to be almost similar in solvents considered here, indicating that the emission is from an indistinguishable locally excited state.51 A minimal blueshift (∼12 nm) in the λmax emission (Table S2) of [2] in DMSO compared to CH3CN could be due to the restricted nonradiative decay of the excited states in DMSO owing to stabilization. In a previous report, when TCNQ was reacted with 2-methyl-4-chloroaniline, a similar analogue18 of [2] was obtained, wherein the H atom was found to be shifted from the amino group to the dicyanomethylene end, but the product was nonfluorescent in solution owing to the interruption of π-electron conjugation into the dicyanomethylene group due to the methane hydrogen atom at the δ– carbon. Surprisingly, in our case, in spite of the presence of methane hydrogen at the δ– carbon in [2], still [2] is found to be appreciably fluorescent due to the extended length of conjugation along the δ+ carbon (Figure 4), resulting in the evolution of red fluorescence in [2] while conjugation length is limited in [1]. This is totally a new perspective noted in our work, suggesting that the substituent along the δ+ carbon with considerable conjugation (absent, thus nonfluorescent in a previously reported TCNQ derivative18) could generate optical property in solution of [1]. Thus, the choice of a novel amine (APM) in a single-step reaction could result in [1, yellow] and [2, red] fluorescent compounds. As mentioned before, both [1] and [2] exhibited increased fluorescence intensity in solutions than their solids (Figure 5a), a contrasting feature with reported mono/di-substituted TCNQ derivatives wherein enhancement of fluorescence was observed particularly in solids due to aggregation, H-bonding, supramolecular assemblies, etc. relative to solutions. Rietsch et al. have recently reported that benzene-functionalized DADQ’s, i.e., diaminodicyanoquinones6 (cyclized at δ+), showed substantial fluorescence in solutions. It should be noted that mono/di-substituted TCNQs of analogues like [2] are not known to exhibit fluorescence in solutions; hence, [2] in this article reveals the existence-enhanced fluorescence in solutions also. Increased quantum yields were observed in DMF/DMSO than other solvents. Compound [2] exhibited more fluorescence (Φf ∼ 4%) than [1] (Φf ∼ 0.7%) in DMSO (Table S3). Solids revealed negligible quantum yields. The decreased emission intensity in solution of [2] than [1] could be due to the overlap of S1/T1 vibronic modes with S0.51 Yet, especially in solids, due to the presence of H-bonding and rigorous noncovalent interactions, [1] displayed aggregation-enhanced emission compared to [2]. Lifetime study revealed an exponential decay; solutions of [1, 2] and solid of [2] showed similar and lesser lifetimes (solutions: 0.01 ns [1], 0.05 ns [2], 0.06 ns solid [2]), leading to fast nonradiative decay (Figure 5b); and solid [1] expressed a slightly ∼10 times higher lifetime (0.17 ns) (Figure S6) owing to the strong zwitterionic nature and stability of the excited state, leading to fewer nonradiative decay channels.19 Decay fitting was acquired from the second and third preexponentials in [1, 2], respectively, and the accuracy of fitting was determined by verifying the χ2 value (Table S4). Figure 5c,d shows the emission of solids holding chromaticity plots with obtained color coordinates: x, 0.6118 and y, 0.38843, corresponding to the emission λmax ∼ 576 nm (λexcitation 464 nm) exhibited by [1]; x, 0.6497 and y, 0.3499 associated with λmax emission ∼ 620 nm (λexcitation 424 nm) for [2]. Compounds [1, 2] manifested fluorescence quenching in solids. Usually ascribed owing to various factors like aggregation, excimer complex formation, excited-state energy transfer between the fluorophores, π–π stacking of the planar dye molecules under hydrophobicity, etc.52 Though it is hard to precisely quantify emission from solids (due to scattering, reflection, and possible waveguide effects), Figure 5a demonstrates explicit enhancement of emission in solutions over solids. Control experiments were performed to visualize the effect of aggregation-caused quenching in [1, 2] (commonly observed in dyes such as rhodamine, cyanine, etc.). To a 3.7 × 10–3 M DMSO solution of [1], 0–90% Millipore (18 MΩ) water fraction (fw) was added; the emission intensity strikingly decreased from 10% fw and was almost saturated with a nonlinear feature. Similarly, [2] showed a linear gradual decrease in the emission intensity (Figure S7a–d), perhaps due to divergent intermolecular interactions experienced by [1, 2] as a result of their nonidentical molecular structures, since [1] is strongly zwitterionic and [2] resembles imine.
Figure 3.

Absorption spectra represented as [1] (orange) and [2] (brown-red); normalized emission spectra of [1] (yellow) and [2] (red) in DMF solutions. Substantial Stokes shifts of ∼114 nm in [1] and 134 nm in [2] are noted. The inset shows fluorescence of [1, 2] as (a, b) under visible light and (c, d) under short wavelength UV light respectively.
Figure 4.
Highlighted region shows π-electron conjugation in [1] and extended length of π-conjugation in [2] playing significant roles in the evolution fluorescence in solutions.
Figure 5.
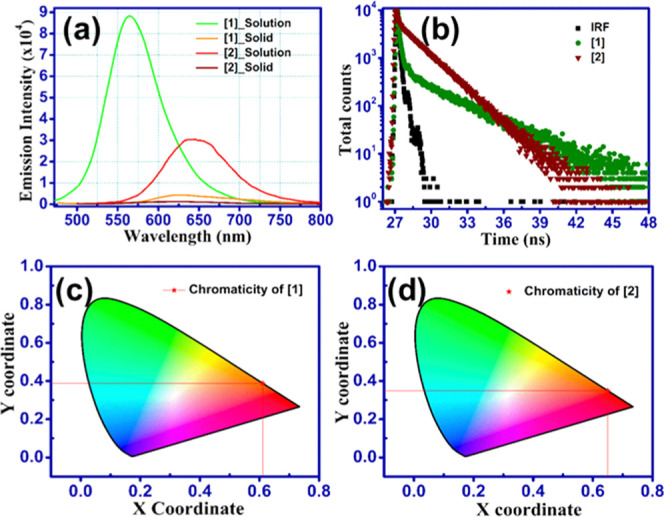
Increased fluorescence intensity noted in all solutions considered in this study compared to solids [1] and [2]. (a) DMF solutions vs solids. Due to the highly zwitterionic nature, solution of [1] exhibited ∼20 times and [2] exhibited ∼4 times enhanced fluorescence than solid state. (b) Life time decay of [1, 2] showing less lifetimes of ∼0.03 ns in solutions. (c, d) CIE chromaticity plots of [1] and [2] corresponding to emission wavelengths as ∼576 and 620 nm in the solid state of both [1] and [2].
2.3. Cation Recognition Accompanied by a Substantial Enhancement of the Emission Intensity in KX [X = Br, Cl, I] Ground Samples of [1, 2] Overcoming the Quenching Effect in Pure [1, 2]
The unexpected color change detected by the naked eye (within ∼1 min) by simple grinding of [1, 2] with KBr using a mortar and pestle (Figure 6) showed a drastic enhancement in fluorescence, perceived for the first time, distinctly indicating K+-ion sensing/recognition by [1, 2]. This novel and distinctive feature exhibited by [1, 2] warranted extensive investigation. A crystal structure study revealed extensive CH−π and strong to moderate intermolecular interactions in [1] and moderate to numerous weak intermolecular interactions in [2], emerging from the molecular design and assembly (choice of APM), leading to the feasible cooperative effects that could respond to stimuli (mechanical force). One milligram of each [1] and [2] was ground with 100 mg of KX (X = Br, Cl, I) as pellets. FT-IR spectra of pure [1, 2] could not be recorded due to concentration constraint and interaction with KX. Thus, the IR spectrum of pure APM compared with [1, 2] ground KX (X = Cl, Br, I) samples (Figure S8a,b) indicated considerable divergent features supporting the recognition of K+ ion in the presence of donor−π–acceptor framework. The free C≡N stretch (2175, 2135 cm–1) is retained (more intense in [1] than [2] due to the strong zwitterionic nature of [1]) and suggests that the recognition could be likely at the substituent owing to the presence of CO, a highly polarizable group; moreover, CO is not involved in any strong intermolecular interactions/H-bonding and available with the lone pair of electrons. The CO stretch in pure APM (analogue of an amide) was observed at ∼1640 cm–1, whereas in all KX ground samples, the same occurred at 1652 cm–1, also less intense (little redshift of ∼12 cm–1), indicating further elongation of CO bond accompanied by the recognition of K+ ion. Notably, significant new peaks were also noted at approximately 1591, 1560, and 1200 cm–1 in [1] and at 1595, 1564, and 1240 cm–1 in [2] corresponding to the aromatic C=C, C=N, and C–O–C stretch of the ring (in AMP), compared to pure APM, which can be attributed to the alteration in the dipole moment of CO due to recognition. Control experiments were carried out to verify whether a similar phenomenon is occurring in (i) solutions and (ii) with pure APM itself. First, DMSO solutions of [1, 2] were mixed with aqueous solution of KX (based on solubility), no color change was observed either immediately or up to 1 month (Figure S9a–d), and the emission spectra of the same looked identical to pure [1, 2] (Figure S9e). Second, KX (X = Br, Cl, I) ground pristine APM pellets did not show any color change (Figure S9f), which implies that either pure APM or solely mechanical forces do not suffice the recognition phenomenon; furthermore, a donor−π–acceptor framework is essential. Consequently, recognition of K+ ion is ruled out in solution state and with pure APM, further materialized exclusively in the solid state. Also, along with the CO, aromatic C=C stretch indicates its role as well, leading to the evolution of cation−π interaction followed by recognition with CO. To our knowledge we believe that in pure APM, the lone pair of electrons on −NH2 is in resonance with the π-electron cloud of the phenyl ring, whereas in [1, 2], such an effect might be very much minimized, due to the δ+ charge on the carbon, as the K+ ion approaches the phenyl ring of the APM moiety in the obtained ground products. It is known that K+ ion binds to the benzene ring with ΔH° = −19 kcal mol–1; besides, at present, cation−π interaction is now confessed by the scientific community as a major strength in molecular recognition and important in a range of biological processes.53 Till to date, there is only one report, where an organometallic zwitterion has undergone profound solid-state rearrangement with KBr, while IR spectroscopy sample preparation.42 Nevertheless, no reports are found amidst any TCNQ derivatives yet. At present, mechanically/pressure-induced solid-state reactions are attracting scientists engaged in the pursuit for solvent-free chemical procedures, which are important receiving both the environmental and topochemical perspectives.42 Mechanofluorochromic materials have attracted reasonable interest in smart materials.54−56 Although fluorescence response of molecular materials with regard to mechanochromic effects are widely reported,57−59 cation sensing/recognition in TCNQ derivatives is so far unrevealed. The λmax,abs value of pristine [1] was ∼464 nm, and the λmax,abs values of all KX ground samples (∼450 nm) of [1] (2K+BAPMDQ) showed little blueshift (∼8–12 nm) with respect to pure [1] due to the environment of the zwitterionic molecule,22 leading to the formation of H-aggregates (Figure 7). Pristine [2] showed λmax,abs at ∼424 nm, while its KX ground samples (K+APMTQ) showed the corresponding value at ∼450 nm, with a redshift (∼26 nm) forming J-aggregates.51 λmax emission for pristine [1] was ∼576 and ∼555 nm for the corresponding 2K+BAPMDQ samples. Similarly, λmax emission for pristine [2] was observed at ∼620 nm, whereas it was ∼595 nm (Figure 7b) for K+APMTQ samples. Interestingly, in the emission spectra of KX ground samples, blueshifts of ∼21 nm in [1] (2K+BAPMDQ) and ∼25 nm in [2] (K+APMTQ), both leading to the formation of H-aggregates,60 were observed, ascribed to the increasing rigidification in the crystal lattice related to the decrease of vibrational relaxation19,61,62 and restricted nonradiative decay21,22,35 (Figure 7a and Table S5). Identical λmax emission among all of the KX (X = Br, Cl, I) ground samples of [1, 2] suggests that emission is from an indistinguishable locally excited state,51 ∼5-fold enhanced emission intensity was noted in KBr ground [1], and ∼2-fold in KCl, KI ground [1]. On a similar note, ∼6 times enhancement in the emission intensity in KI ground [2] and ∼3.5 times in KCl and KBr ground [2] than pristine [2] were found. Thus, exclusive solid-state recognition has eventually led to a drastic enhancement of fluorescence in all 2K+BAPMDQ, K+APMTQ conquering the quenching of fluorescence in pristine [1, 2], thereby resulting in aggregation-enhanced fluorescence due to the divergent solid-state packing and intermolecular interactions.63 Large Stokes shifts of ∼100 nm (4435 cm–1) and ∼143 nm (5195 cm–1) suggest specific and strong intermolecular interactions42 in all 2K+BAPMDQ, K+APMTQ solid samples. Notably, 140 nm is the largest Stokes shift in solids to date, reported by Hiromu et al.64 In 2,6,10-trisubstituted 4,8,12-triazatriangulene (complex molecules), we could achieve ∼143 nm (5195 cm–1) Stokes shift in a facile and feasible manner, in our small single molecules. Narrow feature65 was identified in the emission spectra of KX ground samples, indicating color purity.51 Also, we exploited the cation recognition phenomenon with several other salts (NaN3, K2C2O4, K2SO4, K2CO3, KSCN, NH4Br, NH4Cl, etc.) as well. Surprisingly, a similar observation was noted (Figure S10 and Table S6) in these also. Compound [1] exhibited significant binding capability with a wide range of salts, while [2] was selective; due to contrasting intermolecular interactions envisaged in pure solids, since [1] possessed extensive CH···π and strong hydrogen bonding, [2] exhibited moderate to weak interactions. Moreover, [1] is strongly zwitterionic and [2] is similar to imine.
Figure 6.

KX (X = Br, Cl, I) ground samples of [1] and [2] manifesting color change within ∼1 min of grinding, detected by the naked eye.
Figure 7.

Absorption and emission spectra of pristine [1, 2] (black dotted line) and their KX ground samples. Shifts in λmax absorption and substantially enhanced fluorescence in ground samples compared to pristine indicate strong electrostatic interaction with KX. (a) [1] ground with KBr (red), KCl (green), and KI (blue), blueshift of ∼12 nm (in absorption) and ∼22 nm (in emission) with ∼554 nm being λmax, emission. (b) [2] ground with KBr (red), KCl (green), and KI (blue). A redshift of ∼25 nm (in absorption), a blue shift of ∼24 nm (in emission), and a maximum emission wavelength of ∼596 nm are observed. Similar wavelengths of emission in each [1, 2] designate fixed excited states in each solid.
X-ray fluorescence analysis disclosed a considerable percentage composition of K+ in all samples (Figure S11 and Table S7) and simultaneous existence of halides too, trapped in the solid matrix. A remarkable variation in the emission intensity of [1, 2] ground KX and other salts shows the influence of anions as well. In the current article, we focus particularly on K+-ion recognition envisaged for the first time. Powder X-ray diffraction (PXRD) of pure [1, 2] match with the simulated pattern generated from SCXRD (Figure S12a,b), which indicates that crystalline nature is retained in powders also. The PXRD pattern of KBr ground samples (2K+BAPMDQ, K+APMTQ) was similar to KBr alone (Figure S12c,d) without any discrete feature, probably due to very small amounts of [1, 2] considered for recognition purpose (1 mg of pure [1, 2] ground with 100 mg of KX, considered throughout the study). Optical band gaps of [1, 2] ground KX and other salts hold a similar value, ∼3.4 eV (Figure S13), resembling the optical band gap of pure [1, 2] (∼3.56 eV), supporting the existence of the core donor−π–acceptor framework. A computational study also supported and revealed that K+ ion undergoes strong electrostatic (K+–O–) and cation−π interactions (K+-phenyl ring of APM), discussed below in detail.
2.4. Computational Analysis—Dipole Moment and Binding Mode of K+
Molecule [1] and its K+-ion-bound forms (2K+BAPMDQ) showed significant dipole moments. Molecule [2] in its optimized geometry (Figure S14) shows a very small dipole moment (3.93 D), which is significantly enhanced (15.56 D) when it is bound to K+ ion (K+APMTQ). The magnitude and the direction of the dipole moments are shown in Figure S15. In molecule [1], both the carbonyl groups are found to bind to one K+ ion each. In this case, the O–K distance is found to be smaller (2.47 Å) compared to molecule [2]. The partial charges on the O atom of carbonyl and potassium ions were estimated as −0.77 and +0.99, respectively, an indication of somewhat stronger electrostatic forces between the two atoms, compared to molecule [2]. Unlike molecule [2], the cation−π interaction in molecule [1] is found to be somewhat weaker, with the K+ ion at a distance of 4.70 Å from the phenyl group. The binding free energy of the K+ ion in molecule [1] is calculated as −16.3 kcal mol–1. The optimized geometry of K+-bound molecule [2] shows a stabilizing interaction between the carbonyl group of molecule [2] and the K+ ion, with an O–K distance of 2.53 Å. The partial charges on the carbonyl O atom and potassium ions were calculated as −0.75 and +0.97, respectively, suggesting a strong electrostatic interaction between the two. The K+ ion is found to adopt a conformation that provides additional stabilization energy via the cation−π interaction between the K+ ion and the phenyl ring. The optimized geometry shows that K+ ion is about 3.05 Å away from the phenyl ring.
The binding free energy of K+ ion is estimated as −25.2 kcal mol–1. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of [1, 2] with and without K+ are shown in Figures 8 and 9, respectively. For the first time, we discovered cation recognition and cation−π interaction in TCNQ derivatives. Compound [2] showed a stronger cation−π interaction than [1]; therefore, we further investigated if the cation is dynamically stable in the molecular assembly, and the atom-centered density matrix propagation (ADMP)66 was carried out by introducing one potassium ion into the asymmetric unit of the molecule [2] as obtained from the X-ray crystal structure. Geometry optimization of this complex was carried out with the B3LYP functional and 6-31G basis set. Starting from this optimized geometry, the ADMP trajectory was calculated at 298.15 K temperature and 1 atm pressure, using the B3LYP/6-31 G method. The trajectory was calculated for 160 fs with a time step of 0.1 fs. The time evolution of the distance of the potassium ion from molecule [2] shows that the cation is dynamically stable inside the molecular assembly of [2] (Figure 10). In fact, over time, it is observed that the cation moves closer to the aromatic ring (decrease in the K+-ring distance) with an accompanying increase in the distance between O– and K+ separation. Thus, it is understood that carbonyl lone pair plays a significant role in the solid-state recognition.8,10−15 Eventually, K+-ion recognition (Figure 11) has overcome fluorescence quenching in pure [1, 2] and furthermore resulted in substantial enhancement of fluorescence by the occupancy of K+ ions amidst [1, 2] layers. The K+APMTQ samples showed an ∼8-fold increase of emission intensity than 2K+BAPMDQ samples probably due to the stronger cation−π interaction noted in [2] than in [1]. Thus, the potassium ion is stabilized by the ligand by an electrostatic interaction between the K+ ion and the carbonyl group as well as between the K+ ion and the π-cloud of the phenyl ring of APM.
Figure 8.

HOMO and LUMO of molecule [1] in the absence of potassium ions (a, b) and in the presence of potassium ions (c, d).
Figure 9.

HOMO and LUMO of molecule [2] in the absence of potassium ions (a, b) and in the presence of potassium ions (c, d).
Figure 10.

Dynamic stability of potassium ion in [2] assembly. The time evolution of the distance between potassium ion and O atom and the perpendicular distance of the potassium ion from the aromatic ring obtained from atom-centered density matrix propagation. The inset shows the last snapshot of the trajectory. The hydrogen atoms are not shown for clarity.
Figure 11.
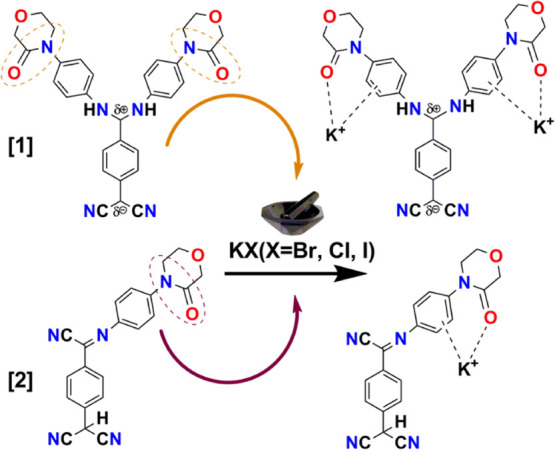
Plausible molecular structures emanating from solid-state grinding of KX (X = Br, Cl, I) with [1] and [2] as a recognition phenomenon due to key role played by the carbonyl functionality on the ring. The anions are supposed to be trapped in the solid matrix.
To gain an insight into the fabrication of micro/nanostructures, 1 mmol of acetonitrile, methanol, and DMF solutions was considered. A drop-cast acetonitrile solution of [1] demonstrated beautiful “micro-roses” and methanol “square blocks”, whereas [2] resembled dense uniform aggregates in acetonitrile and isolated spherical/oval aggregates in methanol. The DMF solution of [1] showed a closely packed uniformly dispersed feature, and [2] did not give a considerable aspect (Figure 12). Variation in morphology is attributed to different evaporation rates experienced by the solvents contemplated with different polarities, and consequently molecular assembly resulting from the interaction of the molecules with solvent emanating in different growth kinetics.11 Nanosized particles have been observed in films from the acetonitrile and methanol solutions. The SEM images of pure [1, 2] and their ground solids (Figure 13) were studied under the same (5 μm) magnification. Compounds [1, 2] disclose dispersed crystallites, and their ground samples indicate microaggregates, suggesting the binding of [1, 2] with KX in the solid state. Moreover, Figure 13c,g,h displays certain voids.
Figure 12.

Scanning electron microscope images of [1, 2] drop-cast on silicon wafer showing discrete morphological features. Drop-cast solutions of (a–c) [1] and (d–f) [2] in acetonitrile, methanol, and DMF, respectively.
Figure 13.

Scanning electron microscope images of [1, 2] in pristine and KX (X = Br, Cl, I) ground samples obtained by grinding 1 mg of pristine sample with 100 mg of KX considered in our study. (a, e) Pure [1, 2]; (b–d) and (f–h) KX ground sample of [1] and [2], respectively, with X = Br, Cl, I. Pure sample reveals crystalline morphology, and ground samples in either indicate the binding of [1, 2] with KX in the solid state manifesting closely packed aggregates. The highlighted region shows certain voids in (c), (g), and (h).
2.5. Electrochemical Study
The electrochemical properties of [1] and [2] were explored by cyclic voltammetry (CV), and the results obtained therein are shown in Figure 14a. Measurements were accomplished at room temperature in dry CH3CN. Multiple reversible reduction waves with half-wave potential E1/2 at −0.11 and −0.88 V (vs Ag/AgCl) along with multiple irreversible oxidation peaks, Eox = 2.16 and 2.85 V were showed by [1, 2]. The former is attributed to the successive reductions of the dicyano part into analogous radical anions within the sample,67 and the latter is attributed to the presence of donor APM functionality, specifically selected as a design element in this study. In our previously reported BHEPDQ,68 the oxidation–reduction phenomenon occurred at low potentials, but in [1, 2], due to the C=O functionality, more potentials were needed. The HOMO and LUMO energy levels of [1, 2] from CV (Figure 14a) gave greater insight concerning energy levels. Picking the equivalent extreme oxidation and reduction onsets and using empirical equations, the HOMO and LUMO energy levels and electrochemical band gaps (ΔEgel) of [1] and [2] were calculated as follows.69,70
Figure 14.

(a) Cyclic voltammogram (CV) of [1, 2] in dry acetonitrile recorded at 25 mV s–1 scan rate with the supporting electrolyte being 0.1 M tetrabutylammonium perchlorate solution. (b, c) Kubelka–Munk plots of [1, 2] achieved from the diffuse reflectance study (DRS) spectra for ΔEgopt calculation.
Using the above equations, the HOMO and LUMO energy levels of [1] were estimated to be −6.86 and −2.58 eV, respectively, generating an ΔEgel of 4.28 eV, and for [2], the corresponding values were −6.88 and −2.61 eV, resulting in a ΔEgel of 4.27 eV. Hence, [1] and [2] fall in the category between those of conventional semiconductors with a wide band gap. A diffuse reflectance study (DRS) was carried out on a JASCO V-670 UV–visible spectrophotometer for the evaluation of optical band gap (ΔEg [eV]) obtained using the Kubelka–Munk method presented in Figure 14b,c. The Kubelka–Munk factor (K) was calculated using K = (1 – R)2/2R (R is % reflectance), and E represents the energy of the incident radiation. ΔEgopt was determined by taking the intersection point of extrapolated lines keeping in view wide-band-gap molecular materials. Thus, the above method resulted ΔEg = 3.56 and 3.57 eV for [1] and [2], respectively. The electrochemical band gaps slightly differ from the optical band gaps, as the bulk effects are neglected at the electrodes6,71 (Table S8). To have a mechanistic perception on the electrochemical operation of [1, 2], a scan-rate-dependent CV study was also performed (Figure S16a,b). The anodic peak currents were expanded with a step-by-step increment of scan rate, and plots of anodic current vs scan rate as well as square root of scan rate were linearly fitted (Figure 15) for both compounds. We observed that the anodic peak current at 2.85 V was best fitted with square root of scan rate because the minimum mean-square error (R2) was closest to 1 (0.998 in Figure 15b,d) vs ∼0.9825 in Figure 15a,c, and generally for a stereotypical oxidation–reduction process, if the anodic peak current is in proportion to scan rate, the rate-determining step is considered to be an electron transfer onto the electrode, whereas if proportional to square root of scan rate, the rate-determining step is surmised on the diffusion speed of the electroactive species.70,72 Therefore, we presume that our sample is understandably a diffusion-controlled system in nature.
Figure 15.

Scan-rate-dependent cyclic voltammograms of the samples (a) [1] and (c) [2] in a scan rate range of 50–500 mV s–1 for [1] and 50–450 mV s–1 for [2]; variation of anodic peak current at 2.85 V with a square root of scan rate (b) for [1] and (d) for [2].
3. Conclusions
Reactions of TCNQ, mostly with primary/secondary amines and a few aromatic amines, are known to produce mono/di-substituted TCNQ derivatives and adducts. The choice of 4-(4-aminophenyl)morpholin-3-one (APM) possessing considerable π-conjugation and carbonyl functionality in the ring has totally changed the scenario, resulting in two well-defined novel optical molecular materials [1] and [2]. Enhanced fluorescence, large Stokes shift in solutions (ca. 110–140 nm), and exclusive cation recognition in solids (ca. 110–143 nm) via cation−π interaction (K+ with phenyl of APM) and strong electrostatic attraction (K+ with CO) have not been described yet among any TCNQ derivatives. Cation recognition was accompanied by a substantial increase in the fluorescence intensity overcoming aggregation quenching in pure [1] and [2]. Furthermore, density functional theory (DFT) calculations supported the cation (K+) binding/interaction capability, wherein the K+ ion is stabilized with the ligand by an electrostatic interaction with the CO group and the π-electron cloud of the phenyl ring (APM). Therefore, experimental and computation studies together suggest the role of electrostatic and cation−π interactions in the recognition event. The electrochemical band gap (4.275 eV) was in close proximity to the optical band gap (∼3.565 eV) of pristine [1] and [2]. However, the optical band gaps of the ground samples of [1, 2] with KX/other salts (∼3.42 eV) remained the same as pristine samples, indicating that these molecular materials fall under the category of wide-band-gap materials, which can hopefully be utilized as acceptors in solar cell application. The SEM images of ground samples showed certain voids. Overall, we conclude that, through this work, we could successfully design and develop novel TCNQ derivatives in a single-step reaction, possessing contrasting optical properties in solutions (yellow, red) and suggesting their possible application in fluorescence imaging; moreover, this work opens up a gateway to explore TCNQ derivatives as solid-state recognition agents.
4. Experimental Section
4.1. Materials
Tetracyanoquinodimethane (TCNQ) and 4-(4-aminophenyl)morpholin-3-one (APM) were purchased from TCI Chemicals. TCNQ was recrystallized from acetonitrile, and APM was used without further purification. Pyridine (Hychem Laboratories) and all solvents used were commercially available of high purity grade.
4.2. Synthesis
7,7-Bis(4-(4-aminophenyl)morpholin-3-ono)dicyanoquinodimethane (BAPMDQ) and 7,7,8-(4-(4-aminophenyl)morpholin-3-ono)tricyanoquinodimethane (APMTQ) were synthesized by minor modification of the reported procedure.17,19 The reaction of TCNQ with APM (Scheme 1) resulted in a mixture of products, isolated and well-defined as [1, 2]. APM (0.206 g, 2.05 mmol) was added to the hot acetonitrile solution of TCNQ (0.200 g, 0.979 mmol) under N2 atmosphere. (Caution: HCN is liberated, removed by connecting a round-bottom flask to the scrubber filled with ferrous sulfate solution.) After 10 min, pyridine (∼1 mL) was added to the reaction mixture at 75 °C and the temperature was increased to 100–105 °C for 2 h, subsequently again increased to 120 °C for 1 h. After completion, the reaction mixture was cooled to 30 °C and allowed to stand for 2 days under N2 atmosphere. Orange-red microcrystals settled at the bottom of solution and separated by filtration. The filtrate was concentrated under vacuum at 70–75 °C and became a solid was washed with DMF to produce green BAPMDQ [1], which on recrystallization from DMF resulted in pure [1] as orange-red crystals. Surprisingly, adding a 1:1 mixture of ether/ethylacetate and hexane to the filtrate obtained above yielded brown-red pure APMTQ [2]. Henceforth, in a single step, a 7:3 ratio of [1] and [2] was achieved, with [1] as the major product (Scheme 1). Remarkably, the reaction of TCNQ with only 1.0 equiv of amine (APM) in a similar route followed a dark brown-red solution with little undissolved [1], which was separated by filtration. At this stage, an immediate workup by evaporating the solvent under vacuum without allowing it to stand for 2 days under nitrogen resulted in a dark brown-red solid [2] in a 3:7 ratio, with [2] being the major product. Finally, [1] and [2] were recrystallized as orange-red blocks (from DMF) and dark brown-red flakes (from acetonitrile), respectively, by slow evaporation at room temperature. Figure 16 shows the protocol we followed to separate [1] and [2] from the reaction mixture. The mechanisms of formation of [1] and [2] are shown in Figure S17a,b, respectively.
Figure 16.

Protocol followed for the separation and isolation of the products [1] and [2].
4.3. Characterization
[1]: Yield 75%; (0.392 g); m.p. 305.2–310.4 °C (decomp.); 1H NMR (400 MHz, DMSO-d6, 25 °C, tetramethylsilane (TMS)): δ = 11.27 (s, 2H, −NH), 7.47 (d, J = 6.7 Hz, 2H, Aromatic-CH), 7.29 (d, J = 7.8 Hz, 4H, phenyl-CH), 7.18 (broad d, 2H, phenyl-CH), 6.83 (d, J = 8.4 Hz, 2H, aromatic-CH), 4.18 (s, 4H, −CH2 of adjacent to carbonyl), 3.97–3.90 (broad t, 4H, aliphatic −CH2), 3.68–3.63 (broad t, 4H, aliphatic −CH2). 13C NMR (101 MHz, DMSO-d6): δ = 166.40, 159.73, 150.79, 139.87, 135.49, 132.16, 126.16, 124.22, 123.18, 117.89, 79.63, 68.16, 63.88, 49.26 (Figure S18a,b); FT-IR (KBr): υ̅ = 2177 cm–1, 2139 (substituted CN stretch) cm–1, 3182 (−NH– stretch) cm–1, 1661 (amide carbonyl stretch) cm–1, 1581 (aromatic C=C stretch) cm–1, 1322 (C–N bending) cm–1 and 1123 (C–O–C stretch) cm–1. UV–vis (CH3CN): λmax = 442 nm, mass (electrospray ionization (ESI)): m/z = 536 [M + H]+, HRMS (ESI): m/z [M + H]+, calculated for molecular formula C30H27N6O4 is 535.2094, found: 535.2091 (Figure S18c,d).
[2]: Yield 70% (0.25 g); m.p. 145.2–155.2 °C (decomp.); 1H NMR (400 MHz, DMSO) δ = 7.74 (d, J = 8.6 Hz, 2H, phenyl-CH), 7.48 (d, J = 8.8 Hz, 2H, aromatic-CH), 7.20 (d, J = 8.7 Hz, 2H, phenyl-CH), 6.91 (d, J = 8.3 Hz, 2H, aromatic-CH), 5.61 (s, 1H, 3CH), 4.23 (s, 2H, −CH2 of adjacent to carbonyl), 4.06–3.91 (t, 2H, aliphatic −CH2), 3.83–3.68 (t, 2H, aliphatic −CH2). 13C NMR (101 MHz, DMSO) δ = 166.50, 147.31, 139.92, 138.55, 129.53, 127.16, 126.36, 124.26, 121.70, 119.53, 112.05, 68.23, 63.97, 49.34 ppm (Figure S19a,b). FT-IR (KBr): υ̅ = 2176, 2133 (substituted nitrile stretch) cm–1, 1647 (amide carbonyl stretch) cm–1, 1565 (aromatic C=C stretch) cm–1, 1328 (C–N bending) cm–1 and 1181 (C–O–C stretch) cm–1. UV–vis (CH3CN): λmax = 470 nm, mass (ESI): m/z = 368 [M – H]+, HRMS (ESI): m/z [M – H]+, calculated for molecular formula C21H15N5O2 is 368.1147, found: 368.1217 (Figure S19c,d).
4.4. Crystallographic Studies
Suitable crystals of [1] and [2] were subjected to single-crystal X-ray diffraction using a Rigaku Oxford XtaLAB Pro: Kappa dual offset/far diffractometer. Data were collected at 293 (K) with the radiation source as Mo Kα (0.71073 Å) for [1] and Cu Kα (1.54184 Å) for [2]. Using spherical harmonics, empirical absorption correction was implemented in SCALE3 ABSPACK scaling algorithm. Using Olex2,73 the structure was solved with the ShelXS74 structure solution program utilizing Intrinsic Phasing and refined with the ShelXL75 refinement package using least-squares minimization. All nonhydrogen atoms were refined applying anisotropic displacement parameters. Compounds [1, 2] belonged to the P21/c space group. Both molecules had one full structure in the asymmetric unit. Oak Ridge thermal ellipsoid plot (ORTEP) molecular structure diagrams with 50% probability thermal ellipsoids are provided in Figure S20, and basic crystallographic data are given in Table S9. The details of data collection, solution, and refinement along with the CIF files are provided in the Supporting Information (Tables S10–S19).
4.5. Spectroscopy
Electronic absorption spectra were recorded on a UV-3600 Plus UV–vis spectrophotometer (Shimadzu). IR stretching frequencies were recorded on a JASCO FT/IR-4200 Fourier transform infrared spectrometer (400–4000 cm–1) on solid samples prepared as KBr pellets. A Fluorolog-HORIBA was used to determine absolute quantum yields, and emission studies and a time-correlated single-photon count instrument, Deltaflex Horiba, were used for lifetime measurements. The fluorescence quantum yield of solutions was calculated in comparison to quinine sulfate in 1 N H2SO4 (ϕ = 0.546),76 and in solid samples, the absolute value of quantum yield was estimated using an integrating sphere and the photoluminescence quantum yield (PLQY) calculator v.3 software. X-ray fluorescence spectroscopy was performed on an ED-XRF EPSILON-1 (PANalytical) to estimate the chemical composition of the KBr ground samples of [1, 2]. Detection of mass (m/z) was conducted using a Shimadzu device following ESI mode and high-resolution mass spectrometry (HRMS) for both [1, 2] and determined by electrospray ionization using a quadrupole time-of-flight (Q-TOF) mass spectrometer in a negative mode (M – H). 1H and 13C NMR spectra were recorded on a Bruker AVANCE Neo-400.
4.6. Microscopy
Scanning electron microscopy (SEM) images were recorded on an Oxford X-maxN LEICA EM ACE200 scanning electron microscope with 10 kV operating voltage. Silicon wafer used as substrate was cleaned thoroughly with water, soap solution, and acetone and dried in an oven at 100 °C for 1 h prior to coating. Solutions [1, 2] of various solvents were drop-cast on a silicon wafer and gold-sputtered for about 5 min.
4.7. Electrochemical Study
Electrochemical measurement was performed using an Autolab potentiostat PGSTAT128N by a conventional three-electrode system. A glassy carbon electrode (GCE) with a 3 mm electrode diameter (electrode surface area, 0.07 cm2) was used as the working electrode. GCE was polished carefully with 1, 0.3, and 0.05 mm alumina powder step by step, until the mirror finish was acquired. GCE was washed with water and ethanol mixture, and finally with acetone several times, and dried in air at room temperature. A saturated Ag/AgCl electrode and a Pt wire were used as the reference and counter electrodes, respectively. Electrolyte solution (0.1 M tetrabutylammonium perchlorate solution in acetonitrile) was mixed with the sample solution to achieve 6 μM final concentration of the sample. Cyclic voltammetry (CV) was performed in a potential window of −3.5 to 3.5 V in 25 mV s–1 scan rate. The scan-rate-dependent study was performed at different scan rates starting from 50 to 500 mV s–1.
4.8. Computational Method
The molecules [1] and [2] were optimized in their ground electronic state with density functional theory (DFT) by employing wb97xd functional77 and 6-31G(d,p) basis set.78 Hessian calculation was carried out at the optimized geometry to verify that optimized state is not associated with any imaginary frequency. Potassium ions were introduced to the optimized geometries of these molecules, and the resulting structures were subjected to geometry optimization at the above-mentioned level of theory. Hessian calculations at these optimized geometries showed no imaginary frequency, suggesting a stable complex of the cation with the molecules. All calculations were carried out using Gaussian 16.79
Acknowledgments
S.J. thanks Dr. Chanchal Chakraborty for the fruitful discussion regarding electrochemical study. S.J. is also thankful to Sivakumar K.V.S. for discussions regarding cation sensing. The DST-FIST grant facility to the Department of Chemistry, BITS Pilani, Hyderabad campus, is highly acknowledged. The Central Analytical Laboratory (CAL) facility provided by the BITS Pilani, Hyderabad campus, is also gratefully acknowledged by the authors.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05486.
Detailed mechanism of formation of products, ORTEP diagrams, 1H and 13C NMR spectra of [1, 2], emission spectra of solutions, lifetime decay measurements, emission spectra of ground solid samples, images of solid-state color change by the naked eye, dipole moment study, crystallographic details (PDF)
Author Contributions
The manuscript was overall handled and written by S.J., except the computational part, which was written by S.M.
S.J. thanks SERB-DST, Government of India, for funding (DST-EMR/2016/002209). S.M. acknowledges support from SERB-DST, Government of India (EMR/2015/001890 and SR/FST/CSII-026/2013), and CSIR, Government of India (01(2897)/19/EMRII).
The authors declare no competing financial interest.
Supplementary Material
References
- Liu J.; Zhang H.; Dong H.; Meng L.; Jiang L.; Jiang L.; Wang Y.; Yu J.; Sun Y.; Hu W.; Heeger A. J. High Mobility Emissive Organic Semiconductor. Nat. Commun. 2015, 6, 10032 10.1038/ncomms10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-T.; Liao C.-H.; Tsai C.-H.; Chen C. H. Highly Efficient, Deep-Blue Doped Organic Light-Emitting Devices. Adv. Mater. 2005, 17, 2493–2497. 10.1002/adma.200501169. [DOI] [Google Scholar]
- Yang Y.; Zhao Q.; Feng W.; Li F. Luminescent Chemodosimeters for Bioimaging. Chem. Rev. 2013, 113, 192–270. 10.1021/cr2004103. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Lavis L. D.; Betzig E. Imaging Live-Cell Dynamics and Structure at the Single-Molecule Level. Mol. Cell 2015, 58, 644–659. 10.1016/j.molcel.2015.02.033. [DOI] [PubMed] [Google Scholar]
- Szablewski M.; Thomas P. R.; Thornton A.; Bloor D.; Cross G. H.; Cole J. M.; Howard J. A. K.; Malagoli M.; Meyers F.; Brédas J.-L.; Wenseleers W.; Goovaerts E. Highly Dipolar, Optically Nonlinear Adducts of Tetracyano-p-Quinodimethane: Synthesis, Physical Characterization, and Theoretical Aspects. J. Am. Chem. Soc. 1997, 119, 3144–3154. 10.1021/ja963923w. [DOI] [Google Scholar]
- Rietsch P.; Witte F.; Sobottka S.; Germer G.; Becker A.; Güttler A.; Sarkar B.; Paulus B.; Resch-Genger U.; Eigler S. Diaminodicyanoquinone: Fluorescent Dyes with High Dipole Moments and Electron-Acceptor Properties. Angew. Chem., Int. Ed. 2019, 58, 8235–8239. 10.1002/anie.201903204. [DOI] [PubMed] [Google Scholar]
- Ravi M.; Cohen S.; Agranat I.; Radhakrishnan T. P. Molecular and Crystal Structures of a Class of Push-Pull Quinonoid Compounds with Potential Nonlinear Optical Applications. Struct. Chem. 1996, 7, 225–232. 10.1007/BF02281233. [DOI] [Google Scholar]
- Ravi M.; Radhakrishnan T. P. Analysis of the Large Hyperpolarizabilities of Push-Pull Quinonoid Molecules. J. Phys. Chem. A. 1995, 17624–17627. 10.1021/j100049a023. [DOI] [Google Scholar]
- Ravi M. A Simple Method for the Estimation of Hyperpolarisibilities: Application to Diamino Substituted Dicyanoquinodimethane Molecules. Proc. Indian Acad. Sci., Chem. Sci. 1998, 110, 133–141. [Google Scholar]
- Srujana P.; Radhakrishnan T. P. Establishing the Critical Role of Oriented Aggregation in Molecular Solid State Fluorescence Enhancement. Chem. – Eur. J. 2018, 24, 1784–1788. 10.1002/chem.201705041. [DOI] [PubMed] [Google Scholar]
- Boyineni A.; Jayanty S. Supramolecular Helical Self-Assemblies and Large Stokes Shift in 1-(2-Cyanophenyl)Piperazine and 4-Piperidinopiperidine Bis-Substituted Tetracyanoquinodimethane Fluorophores. Dyes Pigm. 2014, 101, 303–311. 10.1016/j.dyepig.2013.10.023. [DOI] [Google Scholar]
- Bloor D.; Kagawa Y.; Szablewski M.; Ravi M.; Clark S. J.; Cross G. H.; Pålsson L. O.; Beeby A.; Parmer C.; Rumbles G. Matrix Dependence of Light Emission from TCNQ Adducts. J. Mater. Chem. 2001, 11, 3053–3062. 10.1039/b104992p. [DOI] [Google Scholar]
- Szablewski M.; Bloor D.; Kagawa Y.; Mosurkal R.; Cole J. M.; Clark S. J.; Cross G. H.; Pålsson L. O. Matrix Dependence of Blue Light Emission from a Novel NH2-Functionalized Dicyanoquinodimethane Derivative. J. Phys. Org. Chem. 2006, 19, 206–213. 10.1002/poc.1020. [DOI] [Google Scholar]
- Srujana P.; Gera T.; Radhakrishnan T. P. Fluorescence Enhancement in Crystals Tuned by a Molecular Torsion Angle: A Model to Analyze Structural Impact. J. Mater. Chem. C 2016, 4, 6510–6515. 10.1039/C6TC01610C. [DOI] [Google Scholar]
- Raghavaiah P.; Kuladeep R.; Narayana Rao D.; Jyothi Lakshmi A.; Srujana P.; Subbalakshmi J. Bis-(1-(2-Aminoethyl)Piperidino), (2-(2-Aminoethyl)Pyridino) and (1-(2-Aminoethyl)Pyrrolidino)-Substituted Dicyanoquinodimethanes: Consequences of Flexible Ethylene Spacers with Heterocyclic Moieties and Amine Functionalities. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 709–715. 10.1107/S2052520616009537. [DOI] [PubMed] [Google Scholar]
- Gangopadhyay P.; Sharma S.; Rao A. J.; Rao D. N.; Cohen S.; Agranat I.; Radhakrishnan T. P. Optical Second Harmonic Generation in Achiral Bis (n-Alkylamino)Dicyanoquinodimethanes: Alkyl Chain Length as the Design Element. Chem. Mater. 1999, 11, 466–472. 10.1021/cm980676h. [DOI] [Google Scholar]
- Hertler W. R.; Hartzler H. D.; Acker D. S.; Benson R. E. Substituted Quinodimethanes. III. Displacement Reactions of 7, 7,8,8-Tetracyanoquinodimethane. J. Am. Chem. Soc. 1962, 84, 3387–3393. 10.1021/ja00876a030. [DOI] [Google Scholar]
- Patra A.; Radhakrishnan T. P. Molecular Materials with Contrasting Optical Responses from a Single-Pot Reaction and Fluorescence Switching in a Carbon Acid. Chem. – Eur. J. 2009, 15, 2792–2800. 10.1002/chem.200801878. [DOI] [PubMed] [Google Scholar]
- Patra A.; Hebalkar N.; Sreedhar B.; Sarkar M.; Samanta A.; Radhakrishnan T. P. Tuning the Size and Optical Properties in Molecular Nano/Microcrystals: Manifestation of Hierarchical Interactions. Small 2006, 2, 650–659. 10.1002/smll.200500385. [DOI] [PubMed] [Google Scholar]
- Patra A.; Hebalkar N.; Sreedhar B.; Radhakrishnan T. P. Formation and Growth of Molecular Nanocrystals Probed by Their Optical Properties. J. Phys. Chem. C 2007, 111, 16184–16191. 10.1021/jp075103j. [DOI] [Google Scholar]
- Chandaluri C. G.; Radhakrishnan T. P. Zwitterionic Diaminodicyanoquinodimethanes with Enhanced Blue–Green Emission in the Solid State. Opt. Mater. 2011, 34, 119–125. 10.1016/j.optmat.2011.07.033. [DOI] [Google Scholar]
- Srujana P.; Sudhakar P.; Radhakrishnan T. P. Enhancement of Fluorescence Efficiency from Molecules to Materials and the Critical Role of Molecular Assembly. J. Mater. Chem. C 2018, 6, 9314–9329. 10.1039/C8TC03305F. [DOI] [Google Scholar]
- Chandaluri C. G.; Radhakrishnan T. P. Amorphous-to-Crystalline Transformation with Fluorescence Enhancement and Switching of Molecular Nanoparticles Fixed in a Polymer Thin Film. Angew. Chem., Int. Ed. 2012, 51, 11849–11852. 10.1002/anie.201205081. [DOI] [PubMed] [Google Scholar]
- Chandaluri C. G.; Patra A.; Radhakrishnan T. P. Polyelectrolyte-Assisted Formation of Molecular Nanoparticles Exhibiting Strongly Enhanced Fluorescence. Chem. – Eur. J. 2010, 16, 8699–8706. 10.1002/chem.201000502. [DOI] [PubMed] [Google Scholar]
- Patra A.; Chandaluri C. G.; Radhakrishnan T. P. Optical Materials Based on Molecular Nanoparticles. Nanoscale 2012, 4, 343–359. 10.1039/C1NR11313E. [DOI] [PubMed] [Google Scholar]
- Srujana P.; Radhakrishnan T. P. Extensively Reversible Thermal Transformations of a Bistable, Fluorescence-Switchable Molecular Solid: Entry into Functional Molecular Phase–Change Materials. Angew. Chem. 2015, 127, 7378–7382. 10.1002/ange.201501032. [DOI] [PubMed] [Google Scholar]
- Sudhakar P.; Radhakrishnan T. P. Stimuli Responsive and Reversible Crystalline–Amorphous Transformation in a Molecular Solid: Fluorescence Switching and Enhanced Phosphorescence in the Amorphous State. J. Mater. Chem. C 2019, 7, 7083–7089. 10.1039/C9TC01600G. [DOI] [Google Scholar]
- Chandaluri C. G.; Radhakrishnan T. P. Hierarchical Assembly of a Molecular Material through the Amorphous Phase and the Evolution of Its Fluorescence Emission. J. Mater. Chem. C 2013, 1, 4464. 10.1039/c3tc30615a. [DOI] [Google Scholar]
- Radhakrishnan T. P. Molecular Structure, Symmetry, and Shape as Design Elements in the Fabrication of Molecular Crystals for Second Harmonic Generation and the Role of Molecules-in-Materials. Acc. Chem. Res. 2008, 41, 367–376. 10.1021/ar7002072. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Tang H.; Zhang J.; Wang Z. Y.; Campo J.; Wenseleers W.; Goovaerts E. Functionalized Picolinium Quinodimethane Chromophores for Electro-Optics: Synthesis, Aggregation Behavior, and Nonlinear Optical Properties. Chem. Mater. 2008, 20, 7465–7473. 10.1021/cm802341j. [DOI] [Google Scholar]
- Ravi M.; Rao D. N.; Cohen S.; Agranat I.; Radhakrishnan T. P. Push–Pull Quinonoid Compounds: Enhanced Powder SHG Utilizing the Effect of Chiral Centers on the Dipole Alignment. Chem. Mater. 1997, 9, 830–837. 10.1021/cm960530n. [DOI] [Google Scholar]
- Ravi M.; Gangopadhyay P.; Rao D. N.; Cohen S.; Agranat I.; Radhakrishnan T. P. Dual Influence of H-Bonding on the Solid-State Second-Harmonic Generation of a Chiral Quinonoid Compound. Chem. Mater. 1998, 10, 2371–2377. 10.1021/cm9800128. [DOI] [Google Scholar]
- Sudhakar P.; Radhakrishnan T. P. A Strongly Fluorescent Molecular Material Responsive to Physical/Chemical Stimuli and Their Coupled Impact. Chem. – Asian J. 2019, 14, 4754–4759. 10.1002/asia.201901086. [DOI] [PubMed] [Google Scholar]
- Jayanty S.; Radhakrishnan T. P. Enhanced Fluorescence of Diaminodicyano quinodimethanes in the solid state and Fluorescence Switching in a Doped Polymer by Solvent Vapors. Chem. – Eur. J. 2004, 10, 791–797. 10.1002/chem.200305123. [DOI] [PubMed] [Google Scholar]
- Mizobe Y.; Ito H.; Hisaki I.; Miyata M.; Hasegawa Y.; Tohnai N. A Novel Strategy for Fluorescence Enhancement in the Solid-State: Affording Rigidity to Fluorophores Packing. Chem. Commun. 2006, 2126. 10.1039/b517687e. [DOI] [PubMed] [Google Scholar]
- Scott J. L.; Yamada T.; Tanaka K. Guest Specific Solid-State Fluorescence Rationalised by Reference to Solid-State Structures and Specific Intermolecular Interactions. New J. Chem. 2004, 28, 447. 10.1039/b312931d. [DOI] [Google Scholar]
- Yoshida K.; Ooyama Y.; Tanikawa S.; Watanabe S. Heterocyclic Quinol-Type Fluorophores. Part 2. Solid-State Fluorescence Enhancement Behaviour of Benzofurano[3,2-b]Naphthoquinol-TypeThe IUPAC Name for the Parent Benzofurano[3,2-b]Naphthoquinone Is Naphtho[2,3-b]Benzofuran-6,11-Dione. Clathrate Hosts Upo. J. Chem. Soc., Perkin Trans. 2 2002, 708–714. 10.1039/b109202m. [DOI] [Google Scholar]
- Mederski W. W. K. R.; Wendel P. L.; Woissyk M. Practical and efficiant processes for the preparation of 4-(4-aminophenylmorpholine-3-ones on a larger scale: precursor for factor Xa inhibitors. Heterocycles 2007, 74, 437–445. 10.3987/COM-07-S(W)22. [DOI] [Google Scholar]
- Roehrig S.; Straub A.; Pohlmann J.; Lampe T.; Pernerstorfer J.; Schlemmer K.; Reinemer P.; Perzborn E. Discovery of the Novel Antithrombotic Agent 5-Chloro- N -({(5S)-2-Oxo-3-[4-(3-Oxomorpholin-4-Yl)Phenyl]-1,3-Oxazolidin-5-Y1}methyl)Thiophene-2-Carboxamide (BAY 59-7939): An Oral, Direct Factor Xa Inhibitor. J. Med. Chem. 2005, 48, 5900–5908. 10.1021/jm050101d. [DOI] [PubMed] [Google Scholar]
- Mizukami S.; Nagano T.; Urano Y.; Odani A.; Kikuchi K. A Fluorescent Anion Sensor That Works in Neutral Aqueous Solution for Bioanalytical Application. J. Am. Chem. Soc. 2002, 124, 3920–3925. 10.1021/ja0175643. [DOI] [PubMed] [Google Scholar]
- Nadella S.; Selvakumar P. M.; Suresh E.; Subramanian P. S.; Albrecht M.; Giese M.; Fröhlich R. Lanthanide(III) Complexes of Bis-Semicarbazone and Bis-Imine-Substituted Phenanthroline Ligands: Solid-State Structures, Photophysical Properties, and Anion Sensing. Chem. – Eur. J. 2012, 18, 16784–16792. 10.1002/chem.201201705. [DOI] [PubMed] [Google Scholar]
- Braga D.; Maini L.; Grepioni F.; Selmi V. Vienna, V. Unexpected Solid–Solid Reaction upon Preparation of KBr Pellets and Its Exploitation in Supramolecular Cation Complexation. Chem. Commun. 2002, 2302–2303. 10.1039/B207493A. [DOI] [PubMed] [Google Scholar]
- Balema V. P.; Wiench J. W.; Pruski M.; Pecharsky V. K. Solvent-Free Mechanochemical Synthesis of Two Pt Complexes: Cis-(Ph3P)2PtCl2 and Cis-(Ph3P)2PtCO3. Chem. Commun. 2002, 1606–1607. 10.1039/b203694k. [DOI] [PubMed] [Google Scholar]
- Balema V. P.; Wiench J. W.; Pruski M.; Pecharsky V. K. Solvent-Free Mechanochemical Synthesis of Phosphonium Salts. Chem. Commun. 2002, 1, 724–725. 10.1039/b111515d. [DOI] [PubMed] [Google Scholar]
- Umezawa Y.; Nishio M. CH/π Interactions in the Crystal Structure of TATA-Box Binding Protein/DNA complexes. Bioorg. Med. Chem. 2000, 8, 2643–2650. 10.1016/S0968-0896(00)00197-8. [DOI] [PubMed] [Google Scholar]
- Tewari A. K.; Dubey R.. Conformational Tuning of Molecular Network Stabilized via C–H·π and π–π interaction in 2-[2-(3-cyano-4,6-dimethyl-2-oxo-2H-pyridin1-yl-methyl)-benzyloxyl]-4,6-demethylnicotinonitrile; Arkivoc: Gainesville, FL, 2009; pp 283–291. [Google Scholar]
- Kim E.-i.; Paliwal S.; Wilcox C. S. Measurements of Molecular Electrostatic Field Effects in Edge-to-Face Aromatic Interactions and CH-π Interactions with Implications for Protein Folding and Molecular Recognition. J. Am. Chem. Soc. 1998, 120, 11192–11193. 10.1021/ja982620u. [DOI] [Google Scholar]
- Ramirez-Gualito K.; Alonso-Rios R.; Quiroz-Garcia B.; Rojas-Aguilar A.; Diaz D.; Jimenez-Barbero J.; Cuevas G. Enthalpic Nature of the CH/π Interaction Involved in the Recognition of Carbohydrates by Aromatic Compounds, Confirmed by a Novel Interplay of NMR, Calorimetry, Theoretical calculations. J. Am. Chem. Soc. 2009, 131, 18129–18138. 10.1021/ja903950t. [DOI] [PubMed] [Google Scholar]
- Le Barbu-Debus K.; Broquier M.; Mahjoub A.; Zehnacker-Rentien A. Chiral Recognition in Jet-Cooled Complexes of (1R,2S)-(+)-cis-1-amino-2-indanol and methyl lactate: on the Importance of the CH·π Interaction. Phys. Chem. Chem. Phys. 2009, 11, 7589–7598. 10.1039/b906834a. [DOI] [PubMed] [Google Scholar]
- Reichardt C. Solvatochromic Dyes as Solvent Polarity Indicators. Chem. Rev. 1994, 94, 2319–2358. 10.1021/cr00032a005. [DOI] [Google Scholar]
- Lackowicz Joseph R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Perepichka D. F.; Bryce M. R.; Pearson C.; Petty M. C.; McInnes E. J. L.; Zhao J. P. A Covalent Tetrathiafulvalene–Tetracyanoquinodimethane Diad: Extremely Low HOMO–LUMO Gap, Thermoexcited Electron Transfer, and High-Quality Langmuir–Blodgett Films. Angew. Chem., Int. Ed. 2003, 42, 4636–4639. 10.1002/anie.200351876. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. The Cation−π interaction. Acc. Chem. Res. 2013, 46, 885–893. 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp G.Organic Solid-State Reactions. Encyclopedia of Physical Organic Chemistry, 1st ed.; John-Wiley & Sons, 2017; pp 1–79. [Google Scholar]
- Chi Z.; Zhang X.; Xu B.; Zhou X.; Ma C.; Zhang Y.; Liu S.; Xu J. Recent Advances in Organic Mechanofluorochromic Materials. Chem. Soc. Rev. 2012, 41, 3878–3896. 10.1039/c2cs35016e. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Chi Z.; Zhang Y.; Liu S.; Xu J. Recent Advances in Mechanochromic Luminescent Metal complexes. J. Mater. Chem. C 2013, 1, 3376–3390. 10.1039/c3tc30316k. [DOI] [Google Scholar]
- Sudhakar P.; Neena K. K.; Thilagar P. H-bonded assisted mechanoluminescence of borylated arylamines:tunable emission and polymorphism. J. Mater. Chem. C 2017, 5, 6537–6546. 10.1039/C7TC01676J. [DOI] [Google Scholar]
- Zhou L.; Xu D.; Gao H.; Han A.; Liu X.; Zhang C.; Li Z.; Yang Y. Triphenylamine functionalized β-ketoiminate boron complex exhibiting aggregation-induced emission and mechanofluorochromism. Dyes Pigm. 2017, 137, 200–207. 10.1016/j.dyepig.2016.10.019. [DOI] [Google Scholar]
- Tanioka M.; Kamino S.; Muranaka A.; Ooyama Y.; Ota H.; Shirasaki Y.; Horigome J.; Ueda M.; Uchiyama M.; Sawada D.; Enomoto S. Reversible near-Infrared/blue mechanofluorochromism of aminobenzopyranoxanthene. J. Am. Chem. Soc. 2015, 137, 6436–6439. 10.1021/jacs.5b00877. [DOI] [PubMed] [Google Scholar]
- Varughese S. Non-covalent routes to tune the optical properties of molecular materials. J. Mater. Chem. C 2014, 2, 3499–3516. 10.1039/c3tc32414a. [DOI] [Google Scholar]
- Kasai H.; Kamatani H.; Yoshikawa Y.; Okada S.; Oikawa H.; Watanabe A.; Itoh O.; Nakanishi H. Crystal Size Dependence of Emission from Perylene Microcrystals. Chem. Lett. 1997, 26, 1181–1182. 10.1246/cl.1997.1181. [DOI] [Google Scholar]
- Xiao D.; Xi L.; Yang W.; Fu H.; Shuai Z.; Fang Y.; Yao J. Size-Tunable Emission from 1, 3-diphenyl-5-(2-anthryl)-2-pyrazoline Nanoparticles. J. Am. Chem. Soc. 2003, 125, 6740–6745. 10.1021/ja028674s. [DOI] [PubMed] [Google Scholar]
- Neena K. K.; Sudhakar P.; Dipak K.; Thilagar P. H-bonded assisted mechanoluminescence of borylated arylamines:tunable emission and polymorphism. Diarylboryl-phenothiazine based multifunctional molecular siblings. Chem. Commun. 2017, 53, 3641–3644. 10.1039/C6CC09717K. [DOI] [PubMed] [Google Scholar]
- Hiromu N.; Takashi H.; Soichi Y.; Kenji M. Fluorescence Behavior of 2, 6, 10-trisubstituted 4,8,12-triazatriangulene Cations in Solutions and in the Solid state. Cryst. Eng. Comm. 2016, 18, 7377–7383. 10.1039/C6CE00703A. [DOI] [Google Scholar]
- Li B.; Zhou J.; Bai F.; Xing Y. Lanthanide–organic Framework based on a 4, 4-(9, 9-dimethyl-9H-fluorene-2, 7-diyl) dibenzoic acid: Synthesis, Structure and Fluorescent Sensing for a Variety of Cations and Anions Simultaneously. Dyes Pigm. 2020, 172, 107862–107873. 10.1016/j.dyepig.2019.107862. [DOI] [Google Scholar]
- Iyengar S. S.; Schlegel H. B.; Millam J. M.; Voth G. A.; Scuseria G. E.; Frisch M. J. Ab Initio Molecular Dynamics: Propagating the Density Matrix with Gaussian Orbitals. II. Generalizations based on Mass-Weighting, Ide potency, Energy Conservation and Choice of Initial Conditions. J. Chem. Phys. 2001, 115, 10291–10302. 10.1063/1.1416876. [DOI] [Google Scholar]
- Keshtov M. L.; Kuklin S. A.; Radychev N. A.; Nikolaev A. Y.; Ostapov I. E.; Krayushkin M. M.; Konstantinov I. O.; Koukaras E. N.; Sharma A.; Sharma G. D. New Low Bandgap Near-IR Conjugated D–A Copolymers for BHJ Polymer Solar Cell Applications. Phys. Chem. Chem. Phys. 2016, 18, 8389–8400. 10.1039/C5CP07705B. [DOI] [PubMed] [Google Scholar]
- Anwarhussaini S.; Battula H.; Boppidi P. K. R.; Kundu S.; Chakraborty C.; Jayanty S. Photophysical, Electrochemical and Flexible Organic Resistive Switching Memory Device Application of a Small Molecule: 7,7-Bis(Hydroxyethylpiperazino) Dicyanoquinodimethane. Org. Electron. 2020, 76, 105457–105468. 10.1016/j.orgel.2019.105457. [DOI] [Google Scholar]
- Chakraborty C.; Layek A.; Ray P. P.; Malik S. Star-Shaped Polyfluorene: Design, Synthesis, Characterization and Application towards Solar Cells. Eur. Polym. J. 2014, 52, 181–192. 10.1016/j.eurpolymj.2014.01.008. [DOI] [Google Scholar]
- Roy S.; Chakraborty C. Nanostructured Metallo-Supramolecular Polymer-Based Gel-Type Electrochromic Devices with Ultrafast Switching Time and High Colouration Efficiency. J. Mater. Chem. C 2019, 7, 2871–2879. 10.1039/C8TC06138F. [DOI] [Google Scholar]
- Levitskiy O. A.; Dulov D. A.; Nikitin O. M.; Bogdanov A. V.; Eremin D. B.; Paseshnichenko K. A.; Magdesieva T. V. Competitive Routes for Electrochemical Oxidation of Substituted Diarylamines:the Guidelines. ChemElectroChem 2018, 5, 3391–3410. 10.1002/celc.201801177. [DOI] [Google Scholar]
- Roy S.; Chakraborty C. Metallo-Macrocycle Camouflages: Multicolored Electrochromism in a Fe(II) Based Metallo-Supramolecular Macrocycle Utilizing the Redox of Metal Centers and Carbazole Containing Ligand. ACS Appl. Electron. Mater. 2019, 1, 2531–2540. 10.1021/acsaelm.9b00556. [DOI] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas J. N.; Crosby G. A. The measurement of photoluminescence quantum yields. J. Phys. Chem. B. 1971, 75, 991–1024. 10.1021/j100678a001. [DOI] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Petersson G. A.; Bennett A.; Tensfeldt T. G.; Al-Laham M. A.; Shirley W. A.; Mantzaris J. A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row atoms. J. Chem. Phys. 1988, 89, 2193–2218. 10.1063/1.455064. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, revision C.01; Gaussian Inc.: Wallingford, CT, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.