

ABSTRACT
Human cytomegalovirus (HCMV) establishes life-long latent infection in hematopoietic progenitor cells and circulating monocytes in infected individuals. Myeloid differentiation coupled with immune dysregulation leads to viral reactivation, which can cause severe disease and mortality. Reactivation of latent virus requires chromatin reorganization and the removal of transcriptional repressors in exchange for transcriptional activators. While some factors involved in these processes are identified, a complete characterization of the viral and cellular factors involved in their upstream regulation remains elusive. Herein, we show the HCMV-encoded G protein-coupled receptor (GPCR), UL33, is expressed during latency. Although this viral GPCR is not required to maintain latent infection, our data reveal UL33-mediated signaling is important for efficient viral reactivation. Additionally, UL33 signaling induces cellular cyclic AMP response element binding protein (CREB1, referred to here as CREB) phosphorylation, a transcription factor that promotes reactivation when recruited to the major immediate early (MIE) enhancer/promoter. Finally, targeted pharmacological inhibition of CREB activity reverses the reactivation phenotype of the UL33 signaling-deficient mutant. In sum, our data reveal UL33-mediated signaling functions to activate CREB, resulting in successful viral reactivation.
KEY WORDS: GPCR, CMV, UL33, Latency reactivation
Summary: The mechanisms dictating CMV reactivation from latency are incompletely understood. Here, we show the viral-encoded GPCR pUL33 is involved in CREB activation, thus aiding in successful reactivation.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus that establishes a latent infection in the majority of adults, with increasing seroprevalence in populations from lower socioeconomic backgrounds (Bate et al., 2010). This latent infection is, for the most part, asymptomatic in healthy individuals, but is a threat to those who are immunocompromised or immunosuppressed. Reactivation of latent virus is triggered by loss of immune control coupled with pro-inflammatory stimulus of latently infected undifferentiated myeloid cells, such as CD34+ hematopoietic progenitor cells (HPCs) and CD14+ monocytes as they differentiate to macrophages and dendritic cells (Collins-McMillen et al., 2018; Elder et al., 2019a). There is currently no vaccine for HCMV, and although antiviral therapies are used in clinical settings, toxicity and antiviral resistance limit treatment options. Furthermore, current therapeutics only target lytic stages of the viral life cycle (Krishna et al., 2019b), when infection is already primed to occur. This highlights the need for novel countermeasures to prevent or reduce viral reactivation, necessitating a better understanding of the molecular mechanisms that control this process.
One hallmark of HCMV latency is repression of the major immediate early (MIE) genes, UL122 and UL123 (Dooley and O'Connor, 2020). These genes are regulated by the ∼1100 bp genomic region upstream, which contains binding sites for a myriad of transcription factors that promote or repress activity of this locus. This MIE enhancer/promoter region contains the canonical MIE promoter (MIEP), as well as several alternative promoters, all of which produce transcripts capable of encoding IE1 and IE2 (Arend et al., 2016), the latter of which is critical for initiating viral early gene transcription and subsequent lytic replication (Malone et al., 1990; Marchini et al., 2001). As such, this locus is tightly regulated during all phases of infection, through chromatin remodeling, chromatin-associated proteins and transcription factor binding (Adamson and Nevels, 2020). During latency, the MIE locus is suppressed by repressive transcription factors, such as YY1 (Liu et al., 1994), and the absence of binding of transcription factors, such as AP-1 (Krishna et al., 2020a) and phosphorylated cyclic AMP (cAMP) response element binding protein (CREB1, referred to here as CREB) (Kew et al., 2014), which would otherwise promote MIE activation. De-repression of this locus likely requires multiple mechanisms acting in concert, a process which is only partly understood (Forte et al., 2020; Hale et al., 2020; Krishna et al., 2020a; Min et al., 2020). Many of these factors are regulated by upstream cellular signaling, thus they are co-opted by the virus to facilitate HCMV reactivation in response to proper cues. These pathways include Src-mitogen-activated protein kinase (MAPK)-CREB (Buehler et al., 2019; Dupont et al., 2019; Kew et al., 2014; Reeves et al., 2005), activator protein-1 (AP-1) (Krishna et al., 2020a), cAMP/protein kinase A (PKA) (Keller et al., 2007), UL135/UL138 control of epidermal growth factor receptor (EGFR)/phosphoinositide 3-kinase (PI3K) (Buehler et al., 2019, 2016), and PKA-CREB-target of rapamycin complex 2 (TORC2) (Yuan et al., 2009). Viral subversion of these cell signaling cascades ultimately transactivate the MIE-derived transcripts, along with other signaling pathways triggered to stimulate differentiation to macrophages (Min et al., 2020), thus contributing to successful HCMV reactivation.
HCMV encodes four viral G protein-coupled receptor (GPCR) homologs: US27, US28, UL33 and UL78 (Chee et al., 1990). Arguably, US28 is the most studied; this viral GPCR signals both in constitutive and ligand-dependent manners during lytic infection (Krishna et al., 2018) and functions to support latent infection (Elder et al., 2019b; Humby and O'Connor, 2015; Krishna et al., 2019a, 2017a,b, 2020b; Wu and Miller, 2016; Zhu et al., 2018). Of the remaining three, the UL33 protein (pUL33) is the only other viral GPCR to directly potentiate cell signaling, despite its classification as an orphan receptor, having no known ligands (Frank et al., 2019). pUL33 therefore potentiates signals constitutively and activates CRE-response elements in luciferase reporter assays in transfected COS-7 cells and AD169-infected U373 cells (Casarosa et al., 2003; Waldhoer et al., 2002). However, pUL33 is dispensable for lytic replication in fibroblasts (Casarosa et al., 2003). Although much of our understanding of UL33 is gleaned from its overexpression (in the absence of viral infection) or infection studies in fibroblasts, work in murine and rat models suggests a physiological role for this viral GPCR. The UL33 ortholog M33, encoded by murine CMV (MCMV), is essential for viral replication in the salivary gland (Bittencourt et al., 2014; Cardin et al., 2009; Farrell et al., 2011), which may result from the inability of M33-deficient virus to disseminate to dendritic cells following inoculation at the mucosa (Farrell et al., 2017). Reactivation assays performed on explanted spleen or lung tissue isolated from mice infected with an M33-deficient virus or an M33 signaling mutant virus failed to reactivate when compared to their wild type-infected counterparts (Cardin et al., 2009; Farrell et al., 2011). However, it is unclear whether this phenotype was due to the failure of the mutant to establish and/or maintain latency or to reactivate, as the authors noted an inability to detect viral genomes in these tissues (Cardin et al., 2009). Nonetheless, this reactivation defect was rescued by HCMV US28 or, to a less significant extent, UL33 expression, in M33-deficient MCMV (Farrell et al., 2011). This suggests these HCMV GPCRs may share some functional properties with M33, although it is also possible factors regulating the transactivation of the murine MIE locus differ from its human counterpart, as these regions and the proteins they encode are distinct (Stinski and Meier, 2007). Similarly, rat CMV (RCMV)-encoded R33 is required for replication in the salivary gland and subsequent disease (Beisser et al., 1998). While these data suggest UL33 orthologs are critical for viral pathogenesis in non-human models, there are some distinct differences between the orthologs and HCMV-encoded UL33. For example, pUL33 couples to Gαq, Gαi/o and Gαs G proteins to constitutively activate CRE-mediated transcription, whereas RCMV pR33 fails to couple to either G protein (Casarosa et al., 2003; Gruijthuijsen et al., 2002). The same is the case for M33, as its signaling features differ from those of pUL33 (Farrell et al., 2011). This highlights the potential species-specific roles for the CMV-encoded GPCRs, and underscores the importance of interrogating their functions in their natural hosts.
Recently, UL33 transcripts were detected in clinical samples of latently infected CD34+ HPCs (Cheng et al., 2017), which led us to interrogate the function(s) pUL33 might have during latency and reactivation. To this end, we confirmed UL33 mRNA and protein expression in latently infected Kasumi-3 cells, a CD34+ cell line that serves as a functional model for latency (O'Connor and Murphy, 2012). We found either deletion of UL33 or mutation of its G protein-coupling domain resulted in a significant impairment of viral reactivation in Kasumi-3 cells, as well as in CD34+ HPCs. We found pUL33 activates CREB, and phosphorylated-CREB (p-CREB) binding to the MIE enhancer/promoter is essential for reactivation in latently-infected Kasumi-3 cells. Importantly, our data show pharmacological inhibition of CREB activity reduces reactivation of wild type-infected Kasumi-3 cells, whereas pharmacological activation of CREB rescues the reactivation-deficient phenotype we observed in cells infected with pUL33 deletion or signaling mutant viruses. Our work highlights the importance of pUL33-mediated signaling in viral reactivation, providing a potential target to prevent HCMV reactivation and subsequent viral pathogenesis.
RESULTS
UL33 is expressed during latent HCMV infection
A recent report detailed the expression of UL33 transcripts during natural and experimental latency in CD34+ HPCs (Cheng et al., 2017), though a function for this viral GPCR during latent infection and/or reactivation remained unknown. To begin to understand the potential function of pUL33 during these phases of infection, we first confirmed UL33 mRNA and protein expression in latently infected Kasumi-3 cells. To this end, we infected Kasumi-3 cells with TB40/EmCherry (wild type, WT) virus for 7 days and then treated the cultures with either DMSO (vehicle) or the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA) to induce reactivation for an additional 2 days. We then harvested total RNA and quantified UL33 transcription by RT-qPCR (Fig. 1A). As this viral GPCR is expressed during both latent and lytic replication, we quantified its expression relative to UL123, which is robustly expressed during lytic infection and suppressed during latency. Thus, latently expressed genes, such as US28, display a high ratio of their transcripts relative to UL123. However, the relative expression levels are similar following TPA treatment, as both genes are expressed following reactivation (Humby and O'Connor, 2015) (Fig. 1B). Similarly, we confirmed UL33 expression during latency, as evidenced by the increased ratio of UL33 to UL123 mRNA, which like US28, was reduced following reactivation with TPA (Fig. 1A,B). These findings confirm previous results in primary CD34+ HPCs (Cheng et al., 2017), and show UL33 transcripts are expressed in the Kasumi-3 cell model of HCMV latency.
Fig. 1.
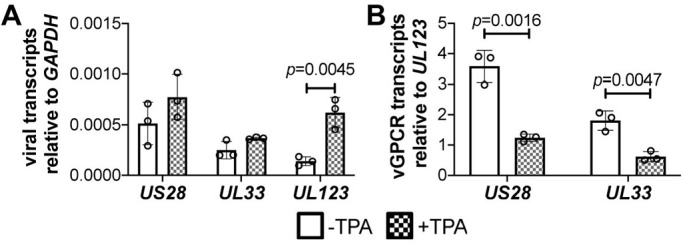
UL33 is expressed during latent HCMV infection. (A,B) Kasumi-3 cells were infected with HCMV TB40/EmCherry (WT; MOI=1.0) in conditions favoring latency. Following 7 days in culture, cells were treated either with vehicle control DMSO (–TPA; open bars) or TPA (+TPA; checked bars) to induce viral reactivation for 2 days. Cells were then collected, total RNA isolated, and (A) US28 or UL33 was quantified relative to GAPDH or (B) the ratio of US28 or UL33 was quantified relative to UL123 and normalized to cellular GAPDH by RT-qPCR. Each data point (circles) is the mean of three technical replicates. Data are mean±s.d. of three biological replicates. Statistical significance was calculated using the two-tailed, unpaired Student's t-test with Benjamini, Krieger and Yekutieli false discovery rate.
To confirm pUL33 expression during latency, we generated an epitope-tagged viral recombinant in the BAC-derived clinical isolate, TB40/EmCherry (O'Connor and Shenk, 2012). Using BAC recombineering, we inserted a triple FLAG epitope tag in-frame with the UL33 open reading frame (ORF) at its C-terminus, yielding TB40/EmCherry-UL33-3xF (UL33-3xF; Fig. 2A,B). Consistent with glycosylation of pUL33 (Margulies et al., 1996), we detected multiple protein bands by immunoblotting with an antibody directed at the triple FLAG epitope (Fig. 2B). However, it is important to note that the isoforms we observed in our total protein lysates are slightly different than those detected previously in infected fibroblast membrane preparations (Margulies et al., 1996). Alternatively, the different banding pattern is possibly due to differences in sensitivity between antibodies, as we used an antibody directed at FLAG, whereas Margulies et al. (1996) raised their own antibody to pUL33 using a synthetic peptide to the 17 C-terminal amino acids of the protein (Margulies et al., 1996). Nonetheless and importantly, our UL33-3xF viral recombinant displays a lytic growth phenotype indistinguishable from WT in highly permissive fibroblasts (Fig. 2C). To evaluate pUL33 expression during latency, we infected Kasumi-3 cells with WT or UL33-3xF for 7 days and then treated each culture with or without TPA for an additional 2 days, as above, and collected protein lysates for immunoblot analyses. Our data show pUL33 is expressed in latently infected Kasumi-3 cells, which is further bolstered following TPA treatment (Fig. 3). Taken together, our data reveal UL33 mRNA and protein are expressed during viral latency in Kasumi-3 cells.
Fig. 2.
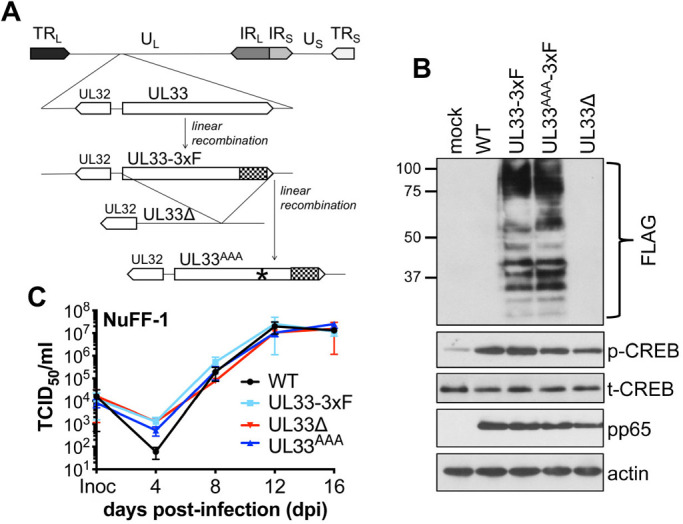
pUL33 mutant viruses grow with wild-type kinetics in lytically infected fibroblasts. (A) Schematic of recombinant viruses. BAC-derived TB40/EmCherry (wild type, WT) was used to generate TB40/EmCherry-UL33-3xF (UL33-3xF) by adding three FLAG epitopes to the C-terminus (denoted by the checkered box), which was then used to delete the entire UL33 ORF [TB40/EmCherry-UL33Δ (UL33Δ)]. Also using UL33-3xF as a backbone, the UL33 G protein-coupling domain (‘DRY’ motif) was replaced with three alanines (denoted by the asterisk), resulting in TB40/EmCherry-UL33AAA-3xF (UL33AAA). TRL, terminal repeat long; UL, unique long; IRL, internal repeat long; IRS, internal repeat short; US, unique short; TRS, terminal repeat short. (B) NuFF-1 fibroblasts were mock infected or infected with the indicated viruses (MOI=0.5). At 96 h post infection, cell lysates (30 µg) were probed for pUL33 expression via the 3xF epitope, pp65, p-CREB, t-CREB and cellular actin. Molecular weight markers (in kDa) are shown to the left of the blot. Representative blots shown. (C) Multistep growth analyses were performed in NuFF-1 fibroblasts infected with the indicated viruses (MOI=0.01) over a 16-day time course. Viral supernatants were collected every 4 days, and viral titers were quantified by TCID50 using naïve NuFF-1 fibroblasts. Representative experiment shown. Samples were analyzed in triplicate. Data are mean±s.d. of the technical replicates.
Fig. 3.
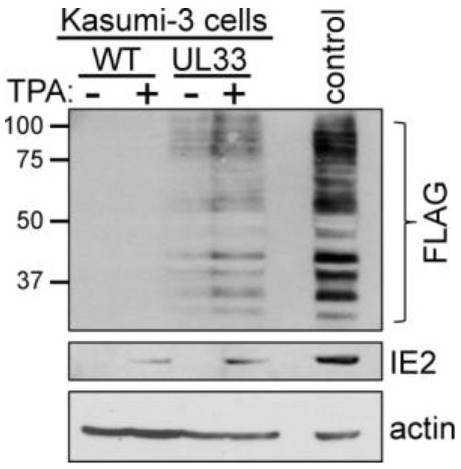
pUL33 is expressed during HCMV latency. Kasumi-3 cells were infected (MOI=1.0) with wild type (WT) or UL33-3xF (UL33) for 7 days, then treated either with DMSO (–TPA) or TPA (+TPA) for an additional 2 days. Cells were then collected, and lysates (60 µg) were assessed for pUL33 expression by immunoblot using an antibody against the C-terminal triple FLAG (3xF) epitope. IE2 served as a control to distinguish latent versus reactivated infections, and cellular actin was probed as a loading control. Lysate from UL33-3xF-lytically infected fibroblasts (MOI=0.5, 96 h, 30 µg) is shown as a positive control for pUL33 expression. Molecular weight markers (in kDa) are shown to the left of the blot. n=3, representative blots shown.
Disruption of the UL33 G protein-coupling domain attenuates HCMV reactivation
To begin to understand the potential function(s) of pUL33 during latency and reactivation, we generated two viral mutants by BAC recombineering (Fig. 2A). Using UL33-3xF as a template, we deleted the complete UL33 ORF, including the C-terminal 3xF epitope tag, yielding TB40/EmCherry-UL33Δ (UL33Δ). As pUL33 potentiates cell signaling constitutively, we also generated a point mutant in the G protein-coupling domain of UL33. This ‘DRY’ motif is a canonical sequence in GPCRs, which when coupled to the appropriate G protein(s) facilitates signaling. Thus, mutating this domain to three alanines ablates the signaling capacity of UL33 (Case et al., 2008; Sherrill and Miller, 2006). To this end, we mutated the UL33 ‘DRY’ motif to AAA in the UL33-3xF background, resulting in TB40/EmCherry-UL33AAA-3xF (UL33AAA; Fig. 2A). We confirmed this signaling mutant retained its ability to express pUL33 in the context of lytically-infected fibroblasts compared to UL33-3xF and also confirmed pUL33 expression is ablated in this context following UL33Δ infection (Fig. 2B). We also evaluated lytic growth of each mutant relative to WT and UL33-3xF in fibroblasts. Our multi-step growth analyses revealed UL33Δ and UL33AAA displayed WT titers over the 16-day time course (Fig. 2C), indicating UL33, and more specifically, UL33-mediated signaling, is not required for TB40/E lytic replication in fibroblasts.
We next investigated whether pUL33 expression and signaling contribute to HCMV latency and reactivation. To this end, we infected Kasumi-3 cells with WT, UL33-3xF, UL33Δ or UL33AAA for 7 days, and then quantified the frequency of infectious centers by extreme limiting dilution analysis (ELDA) in the presence or absence of TPA. Both UL33 mutants established and maintained a latent infection similar to WT and UL33-3xF. Although WT and UL33-3xF-infected Kasumi-3 cells treated with TPA displayed a significant increase in the frequency of infectious centers, UL33Δ- and UL33AAA-infected counterpart cultures failed to show a similar phenotype (Fig. 4A), suggesting these mutant viruses do not efficiently reactivate from latency. This is not due to a loss of the virus during latency, as both UL33Δ- and UL33AAA-infected Kasumi-3 cells maintain the viral genome over 10 days in latent culture (Fig. 4B). Since there was no discernible difference in latency or reactivation in UL33-3xF-infected cells relative to WT, the remainder of the experiments were performed using WT virus. Finally, we confirmed our findings in primary CD34+ HPCs isolated from cord blood. Similar to our findings in Kasumi-3 cells, UL33Δ- and UL33AAA-infected CD34+ HPCs did not reactivate to WT levels following stimulus with reactivation medium when quantified by ELDA (Fig. 4C). In sum, our findings suggest that while UL33 is not required to establish or maintain viral latency, its signaling abilities aid in efficient viral reactivation.
Fig. 4.

Disruption of the UL33 G protein-coupling domain attenuates HCMV reactivation. (A) Kasumi-3 cells were infected (MOI=1.0) with the indicated viruses. At 7 dpi, half of each infected population was cultured for an additional 2 days with vehicle (DMSO; –TPA, open bars) or TPA (+TPA, checked bars). Cells were then co-cultured with naïve NuFF-1 fibroblasts to quantify the fold change in the frequency of infectious centers [relative to wild type (WT) –TPA; open gray bar] by ELDA. (B) Kasumi-3 cells were infected (MOI=1.0) as indicated over a 7-day time course in conditions favoring latency. At the indicated times, cells were harvested and cell-associated viral genomes were quantified by qPCR using primers directed at the MIE locus. Viral genome copies are shown relative to those in WT-infected cells (gray bars) at each time point. (C) CD34+ HPCs were infected as indicated (MOI=2.0; 7 days). Cells were then co-cultured with fibroblasts in medium favoring latency (pre-reactivation; open bars) or in culture conditions favoring reactivation (reactivation; checked bars). Fold change in the frequency of infectious centers was quantified by ELDA and is shown relative to WT, pre-reactivation (open gray bar). (A-C) Each data point (circles) is the mean of three technical replicates. Data are mean±s.d. of three biological replicates. Statistical significance was calculated using two-way ANOVA followed by Tukey's post-hoc analysis (*P<0.05; **P<0.01; ****P<0.005; ns, not significant).
pUL33 expression and signaling induces UL123 transcription in THP-1 cells
To further interrogate the potential function(s) of pUL33 during reactivation, we generated THP-1 cells stably expressing UL33-3xF or UL33AAA-3xF under the control of a doxycycline (DOX) promoter, termed THP-1-pSLIK-UL33-3xF and THP-1-pSLIK-UL33AAA-3xF, respectively. To confirm expression of pUL33 in each line, we treated each cell line, as well as the empty vector control (THP-1-pSLIK), ±DOX for 24 h. DOX treatment induced the expression of pUL33 in each THP-1 cell line, similar to pUL33 expression following UL33-3xF lytic infection of fibroblasts (Fig. 5A). To determine whether pUL33 provided in trans complements the reactivation phenotype we observed with the UL33 mutant viruses, we infected each of these THP-1 cell lines with wild type, UL33Δ, or UL33AAA. At 7 days post infection (dpi), the cultures were treated ±DOX and/or TPA for an additional 2 days. We then isolated RNA to evaluate UL123 expression as a measure of MIE locus activity. Consistent with our data in Kasumi-3 cells, TPA treatment of UL33Δ- and UL33AAA-infected THP-1-pSLIK (empty vector) cultures did not result in robust UL123 expression when compared to counterpart WT cultures (Fig. 5B). However, providing full-length, wild-type pUL33 in trans complemented the phenotype of UL33Δ- and UL33AAA-infected THP-1-pSLIK-UL33-3xF cells (Fig. 5C). Importantly, THP-1-pSLIK-UL33AAA-3xF cells, which express the signaling-deficient mutant form of this protein, did not provide complementation, as UL33Δ- and UL33AAA-infected cells treated with TPA and DOX failed to express UL123 to WT levels (Fig. 5D). Consistent with our above findings in the context of infection, these data demonstrate pUL33 signaling induces MIE-driven gene expression upon TPA treatment of THP-1 cells.
Fig. 5.

pUL33-mediated signaling induces UL123 transcription in THP-1 cells. (A) THP-1 cells transduced with the indicated lentiviral vectors were treated with DMSO (–DOX) or +DOX, and harvested 24 h post treatment. Lysates (30 μg) were probed with a FLAG antibody to detect pUL33 and pUL33AAA, each with a C-terminal 3xFLAG (3xF) tag. Cell lysate (10 μg) from NuFF-1 fibroblasts lytically infected with TB40/EmCherry-UL33-3xF (UL33-3xF; MOI=0.5, 96 h post infection) is shown as a control for pUL33 expression (control). Cellular actin is shown as a loading control. Molecular weight markers (in kDa) are shown to the left of the blot. Representative blots are shown (n=3). (B) THP-1-pSLIK, (C) THP-1-pSLIK-UL33-3xF, or (D) THP-1-pSLIK-UL33AAA-3xF cells were infected with the indicated viruses (MOI=1.0; 7 days) and maintained under conditions favoring latency. (B-D) At 7 dpi, half of each infected population was cultured for an additional 2 days with vehicle (DMSO) to maintain latency (–TPA, black bars), or with TPA to induce reactivation (+TPA, blue bars) in the absence or presence of DOX. Total RNA was harvested and UL123 was measured by RT-qPCR relative to cellular GAPDH. Each data point (circles) is the mean of three technical replicates. Data are mean±s.d. of three biological replicates, and statistical significance was calculated using two-way ANOVA followed by Tukey's post-hoc analysis (*P<0.05; **P<0.01; ***P<0.001).
pUL33 activates CREB and enhances its recruitment to the MIE enhancer/promoter
Our data thus far suggest pUL33-mediated signaling supports reactivation from latency through de-repression of MIE transcription. pUL33 activates CREB response elements in transfected COS-7 cells (Waldhoer et al., 2002) and AD169-infected U373 glioblastoma cells (Casarosa et al., 2003). Additionally, pUL33 induces phosphorylation of CREB in lytically-infected fibroblasts, which is attenuated in cells infected with UL33AAA (Fig. 2B). These observations, together with the requirement for p-CREB recruitment to the MIE locus for reactivation in primary CD14+ monocytes (Kew et al., 2014), led us to hypothesize pUL33 may regulate this transcription factor to facilitate reactivation from latency. Using our THP-1 cell lines (Fig. 5A), we investigated whether pUL33 stimulates CREB phosphorylation. We induced pUL33 expression in each cell line with DOX for 24 h, and then measured CREB phosphorylation by immunoblot. Although the expression of wild-type pUL33 activated CREB, neither the control nor UL33AAA-expressing cells treated with DOX induced CREB phosphorylation (Fig. 6A). Next, to test whether pUL33 stimulates recruitment of p-CREB to the MIE locus, we infected Kasumi-3 cells with WT, UL33Δ, or UL33AAA for 7 days, and then treated each culture with or without TPA for an additional 2 days. We then quantified p-CREB recruitment to the MIE locus by chromatin immunoprecipitation (ChIP) followed by quantitative PCR (qPCR). In WT-infected cells, TPA treatment induced p-CREB binding to the MIE enhancer/promoter (Fig. 6B). However, UL33Δ- and UL33AAA-infected cells treated with TPA failed to display a significant increase in p-CREB recruitment to this promoter (Fig. 6B). These findings suggest pUL33-induced signaling results in CREB activation, in turn facilitating its binding to the MIE enhancer/promoter.
Fig. 6.
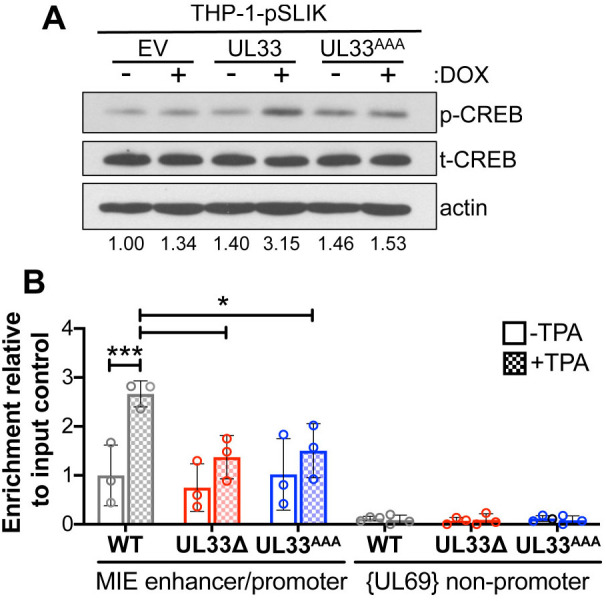
pUL33 activates CREB and enhances its recruitment to the MIE enhancer/promoter. (A) THP-1-pSLIK (empty vector, EV), THP-1-pSLIK-UL33-3xF (UL33), or THP-1-pSLIK-UL33AAA-3xF (UL33AAA) cells were treated with vehicle (DMSO; –DOX) or DOX (+DOX) for 24 h. Lysates (30 µg) were analyzed by immunoblot using antibodies directed at phosphorylated CREB (p-CREB), total CREB (t-CREB), and cellular actin as a loading control. Representative blots are shown (n=3). p-CREB expression relative to t-CREB was quantified by densitometry from two independent experiments and indicated below the blots. (B) Kasumi-3 cells (MOI=1.0; 7 days) were infected with the indicated viruses. At 7 dpi, half of each infected population was cultured for an additional 2 days with vehicle (DMSO; –TPA, open bars) or TPA (+TPA, checked bars). Binding of p-CREB to the MIE enhancer/promoter was quantified by ChIP using an anti-p-CREB antibody. Co-precipitated MIE enhancer/promoter was quantified by qPCR, and data are shown as fold change relative to input for WT –TPA cultures (open gray bar). Each data point (circles) is the mean of three technical replicates. Data are mean±s.d. of three biological replicates, and the statistical significance was calculated using two-way ANOVA followed by Tukey's post-hoc analysis (*P<0.05; ***P<0.001).
Pharmacological-induced CREB phosphorylation complements reactivation in pUL33 signaling-deficient infected Kasumi-3 cells treated with TPA
Our results thus far suggest pUL33 signaling stimulates CREB phosphorylation and its recruitment to the MIE locus following reactivation stimuli, which aids in activation of transcription from this region and reactivation of latent cultures. Thus, we reasoned that inhibiting CREB would reduce HCMV reactivation from latency, whereas its activation would rescue the reactivation deficiency we observed with the UL33 mutant viruses. To this end, we used two pharmacological compounds: (1) forskolin, which phosphorylates CREB; and (2) 666-15, which inhibits CREB phosphorylation. We first confirmed the effects of both pharmacological agents on p-CREB in Kasumi-3 cells (Fig. 7A). Importantly, neither compound impacted cell viability at the concentrations used in our study (Fig. S1). Next, to evaluate the functionality of p-CREB after each drug treatment, we evaluated the expression of two cellular genes, B cell lymphoma-2 (Bcl-2) and cyclooxygenase-2 (COX-2; also known as Ptgs2), which are both driven by CREB-responsive promoters (e.g. Klein et al., 2007; Wilson et al., 1996). We assessed the expression of these cellular transcripts using our DOX-inducible THP-1 cells to (1) demonstrate that the pharmacological compounds functionally affect known target genes and (2) determine whether pUL33 activation of p-CREB influences cellular CREB-responsive promoters. Treatment of each of our THP-1 cell lines with forskolin, which activates CREB, induced a significant upregulation of both Bcl-2 and COX-2 gene expression, irrespective of DOX treatment or cell line (Fig. 7B). Following DOX addition, expression of the wild-type pUL33, but not the signaling mutant, upregulated expression of each cellular transcript, which was reversed when also treated with the p-CREB inhibitor, 666-15 (Fig. 7B). These data confirm the functionality of each pharmacological compound and reveal CREB-responsive cellular genes, like MIE-driven gene expression (Fig. 5B-D), are upregulated with pUL33 expression. Finally, to determine the impact these compounds have on viral reactivation, we infected Kasumi-3 cells with WT, UL33Δ or UL33AAA for 7 days, and quantified the frequency of infectious centers by ELDA in the presence or absence of TPA and/or 666-15 or forskolin (Fig. 7C). WT-infected Kasumi-3 cells treated with both 666-15 and TPA showed a significant reduction in the frequency of infectious centers, similar to levels we observed in UL33Δ- and UL33AAA-infected cells in the absence of these compounds (Fig. 7C). Importantly, treatment of the UL33Δ- and UL33AAA-infected cultures with both forskolin and TPA resulted in a significant increase in the frequency of infectious centers (Fig. 7C), suggesting the activation of CREB rescues the inability of these infected cells to reactivate in the presence of a reactivation stimulus. It is important to note that although forskolin treatment resulted in reactivation of the pUL33 viral mutants in the presence of TPA, forskolin treatment alone (–TPA) was not sufficient to induce reactivation of the WT-infected cells (Fig. 7C), suggesting that although p-CREB is an important factor in reactivation, it is not sufficient for this process. Together, these data suggest pUL33-mediated signaling activates CREB in myeloid cells to facilitate reactivation given the proper cues.
Fig. 7.
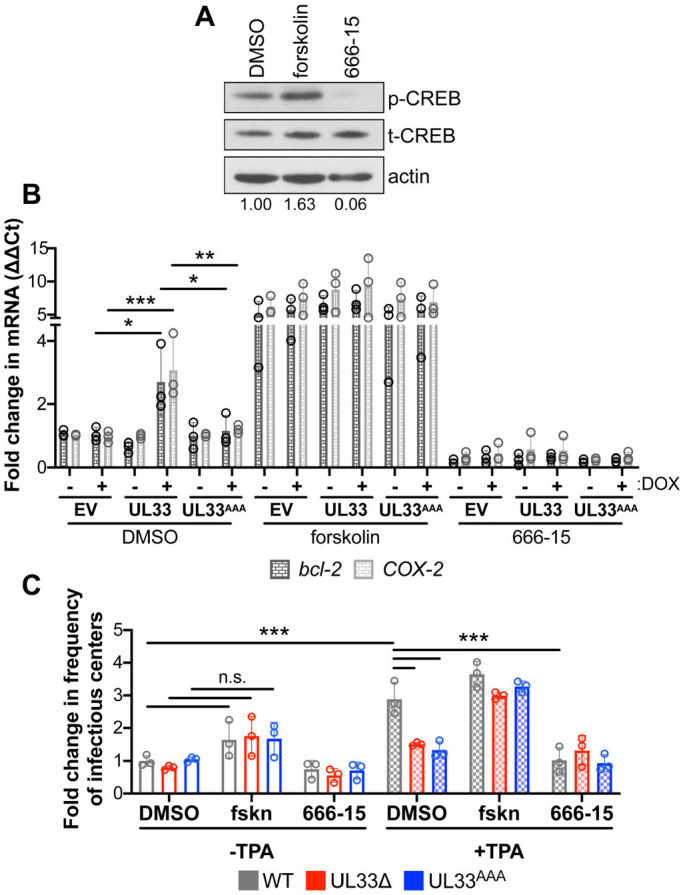
Pharmacological stimulation of p-CREB rescues the reactivation deficiency of UL33Δ- and UL33AAA-infected Kasumi-3 cells treated with TPA. (A) Kasumi-3 cells were treated for 24 h with vehicle control (DMSO), forskolin (20 µM) or 666-15 (1.0 µM). Lysates were harvested for immunoblot and assessed for p-CREB and t-CREB expression. Cellular actin is shown as a loading control. p-CREB expression was quantified by densitometry relative to t-CREB, indicated below the blots. Representative blots are shown (n=3). (B) THP-1-pSLIK (empty vector; EV), THP-1-pSLIK-UL33-3xF (UL33) and THP-1-pSLIK-UL33AAA-3xF (UL33AAA) cells were treated with vehicle (DMSO; –DOX) or DOX (+DOX) in the absence (DMSO) or presence of forskolin (20 µM) or 666-15 (1.0 µM) for 24 h, as indicated. Total RNA was harvested, and Bcl-2 (black) and COX-2 (light gray) were measured by RT-qPCR and plotted as ΔΔCt relative to EV +DMSO/–DOX. (C) Kasumi-3 cells (MOI=1.0) were infected with wild type (WT, gray), UL33Δ (red) and UL33AAA (blue). At 7 dpi, half of each infected population was cultured for an additional 2 days with vehicle (DMSO, –TPA; open bars) or TPA (+TPA; checked bars) in the presence of vehicle control (DMSO), forskolin (20 µM) or 666-15 (1.0 µM). Cells were then co-cultured with naïve NuFF-1 cells to quantify the fold change in the frequency of infectious centers (relative to WT –TPA/DMSO; open gray bar) by ELDA. (B,C) Each data point (circles) is the mean of three technical replicates. Data are mean±s.d. *P<0.05; **P<0.01; ***P<0.001; n.s., not significant (two-way ANOVA and Tukey's post-hoc analysis).
DISCUSSION
Reactivation of HCMV from latency is a significant clinical risk for immunocompromised and immunosuppressed individuals. As such, therapeutics that suppress viral reactivation could prove beneficial and undoubtedly improve patient quality of life and outcomes, underscoring the need to better understand this phase of infection. Herein, we show the HCMV-encoded GPCR, UL33, contributes to reactivation through stimulation of the cellular transcription factor, CREB, facilitating its binding to the MIE locus. Deletion of the UL33 ORF or mutation of the ‘DRY’ motif to prevent signal transduction leads to deficient viral reactivation in latently infected Kasumi-3 cells (Fig. 4A) and primary CD34+ HPCs (Fig. 4C). Using THP-1 complementing cell lines, expression of wild type, but not the signaling deficient pUL33, rescues the reactivation phenotype, as measured by de-repression of the MIE-driven gene, UL123 (Fig. 5B-D). As observed in other cell types (Casarosa et al., 2003; Waldhoer et al., 2002), ectopic expression of pUL33 in THP-1 cells phosphorylates cellular CREB (Fig. 6A), and although p-CREB is recruited to the MIE locus following TPA treatment of WT-infected Kasumi-3 cells, we observed significantly less p-CREB binding in UL33Δ- and UL33AAA-infected cultures treated with TPA (Fig. 6B). Finally, inducing p-CREB with the addition of forskolin allowed UL33Δ- and UL33AAA-infected Kasumi-3 cells to reactivate in the presence of TPA (Fig. 7C). Together, these results highlight a pivotal role for pUL33-mediated activation of CREB during HCMV reactivation from latency.
Consistent with previous observations in naturally and experimentally infected CD34+ HPCs (Cheng et al., 2017), we detected UL33 mRNA expression in latently infected Kasumi-3 cells (Fig. 1). Although we detect pUL33 expression during latency, its levels are increased upon TPA stimulation in Kasumi-3 cells (Fig. 3). As our data argue pUL33-mediated signaling is required to phosphorylate CREB during reactivation, it is perhaps not surprising the virus devised a mechanism to express pUL33 during latency for immediate action given the proper cues during reactivation. This may also explain the lesser levels to which pUL33 is expressed during latent infection versus reactivation (Fig. 3), as excessive expression of pUL33 could otherwise compromise repression of the MIE locus during latency.
Importantly, pUL33-mediated CREB activation is consistent with previous studies. This viral GPCR activates CREB following transient expression of pUL33 in COS-7 cells and infection of U373 cells with AD169 (Casarosa et al., 2003; Waldhoer et al., 2002) or fibroblasts (Fig. 2B). Our finding that pUL33 phosphorylates CREB in myeloid cells (Fig. 6A) suggests the ability of this viral GPCR to activate this signaling pathway is irrespective of cell type. This is in stark contrast to US28, which displays cell type-specific signaling (Krishna et al., 2018). So how exactly does pUL33 alter cell signaling to regulate CREB? While the precise mechanism by which pUL33 activates CREB in myeloid cells is unknown, inhibition of p38 MAPK in UL33-expressing COS-7 cells significantly repressed CREB activation by luciferase reporter assay (Waldhoer et al., 2002). p38 MAPK is activated by a variety of upstream pathways, initiated by several cell membrane proteins stimulated by growth factors, inflammatory cytokines, as well as environmental stress (e.g. DNA damage) (Martinez-Limon et al., 2020). Cellular GPCRs that couple to Gαo activate p38 MAPK, and ultimately CREB, via thousand and one amino acid (TAO) kinase (Chen et al., 2003). It is therefore attractive to speculate pUL33 may similarly activate CREB in this fashion, although blocking Gαi/o receptor coupling with pertussis toxin failed to attenuate pUL33-mediated CREB activity in transiently transfected COS-7 cells (Waldhoer et al., 2002). Furthermore, although recruitment of p-CREB to the MIE locus is critical for reactivation in monocytes (Kew et al., 2014), which we confirmed in Kasumi-3 cells herein (Fig. 6B), inhibiting p38 MAPK had a minimal effect on attenuating p-CREB binding (Kew et al., 2014). Future work aimed at understanding the pathway upstream of CREB, through which pUL33 potentiates this signal in the context of latency and reactivation in myeloid cells, will further reveal the additional cellular, and perhaps viral, proteins important for viral reactivation.
Our findings also confirm work by Kew et al. (2014) detailing the importance of p-CREB binding to the MIE locus for reactivation in latently infected monocytes. In line with our data and this previous study, treatment of infected fibroblast and NTera2-derived neuronal cells with forskolin, which phosphorylates CREB, results in transcription from the MIE locus. Furthermore, this was dependent on the CRE-binding sites located between positions –300 and –579 of the MIE distal enhancer (Keller et al., 2003). There are additional binding sites for p-CREB in the MIE proximal enhancer, and while they may not contribute to transactivation of MIE-driven transcription in fibroblasts or NTera 2 cells, it is possible these other sites, either alone or in combination with those in the distal enhancer, function in other cell types, such as myeloid cells. This is not without precedence, as we recently showed AP-1 transcription factor binding to the MIE enhancer is important for reactivation from latency in myeloid cells, and recruitment to the locus during lytic infection of fibroblast or epithelial cells was not required for replication (Krishna et al., 2020a). These findings, as well as work from other groups, highlight the complexity of the MIE enhancer/promoter region. It is likely successful reactivation from latency requires many factors working in concert. For example, p-CREB may influence the binding of other transcription factors and/or chromatin-associated proteins required for transactivation of the MIE locus. Answers to such questions remain outstanding, but further interrogation into the regulation of the MIE region during latency and reactivation will not only reveal newly identified transcription factors, such as FOXO family members (Hale et al., 2020), but heighten our understanding of the factors that dictate these phases of infection.
Our data argue that pUL33 phosphorylates CREB, a transcription factor whose recruitment to the MIE locus is critical for reactivation in myeloid cells following reactivation stimuli. However, it is possible pUL33 signaling deficient viral recombinants fail to replicate in more differentiated myeloid cells, such as monocyte-derived macrophages, which support lytic replication following differentiation of latently infected myeloid cells given proper reactivation cues. This possibility is plausible if pUL33-mediated phosphorylation of CREB was perhaps required for binding and activating the MIE locus in these cells. Although pUL33 seems to activate CREB in a variety of cell types (Casarosa et al., 2003; Waldhoer et al., 2002; Fig. 6A), this viral GPCR is dispensable for lytic replication in fibroblasts (Casarosa et al., 2003; Fig. 2C). There is certainly precedent for the viral GPCRs to display cell-specific growth properties (O'Connor and Shenk, 2011, 2012), although whether this is true for pUL33 remains elusive. It is feasible to test lytic replication of our mutants in pre-differentiated monocyte-derived macrophages, although de novo infection of these cells likely will not faithfully mimic reactivation of virus from latently infected less-differentiated myeloid cells (e.g. CD34+ HPCs and monocytes).
In sum, our study reveals that the HCMV-encoded GPCR, UL33, plays a significant role in reactivation from latency through its regulation of p-CREB. Future studies aimed at understanding exactly how pUL33 modulates cellular signaling in this process may reveal cellular and/or viral factors to leverage as novel therapeutic targets to prevent HCMV reactivation and subsequent disease.
MATERIALS AND METHODS
Cell culture
Primary newborn human foreskin fibroblasts (NuFF-1, passage 16-28; GlobalStem) were cultured in Dulbecco's modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 10 mM HEPES, 0.1 mM non-essential amino acids and 100 U/ml each of penicillin and streptomycin. Human Embryonic Kidney (HEK) 293T cells [American Type Culture Collection (ATCC)] were cultured in DMEM, supplemented with 10% newborn calf serum (NCS) and 100 U/ml each of penicillin and streptomycin. THP-1 cells (ATCC) were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% FBS and 100 U/ml each of penicillin and streptomycin and maintained at densities between 3×105 and 8×105 cells/ml. Kasumi-3 cells (ATCC) were maintained in RPMI-1640 medium (ATCC), supplemented with 20% FBS, 100 µg/ml gentamicin and 100 U/ml each of penicillin and streptomycin and maintained at densities between 5×105 and 1×106 cells/ml. SI/SI and M2-10B4 (MG3) stromal cells were kind gifts from the Terry Fox Laboratories, BC Cancer Agency (Vancouver, BC, Canada). SI/SI cells were expanded in Iscove's modified Dulbecco's medium (IMDM), supplemented with 10% FBS, 1 mM sodium pyruvate and 100 U/ml each of penicillin and streptomycin. MG3 cells were cultured in RPMI-1640, supplemented with 10% FBS, 100 µg/ml gentamicin and 100 U/ml each of penicillin and streptomycin. Primary CD34+ HPCs were isolated from cord blood mononuclear cells by Ficoll Paque (GE Healthcare) separation and magnetic activated cell sorting (MACS, Miltenyi Biotech), as described previously (Umashankar and Goodrum, 2014). Cord blood was obtained from the Abraham J. and Phyllis Katz Cord Blood Foundation dba Cleveland Cord Blood Center and Volunteer Donating Communities in Cleveland and Atlanta. Cells used in this study were not independently authenticated. All cells were maintained at 37°C and 5% CO2.
Construction of viral recombinants and reconstitution of infectious virus
HCMV BAC-derived strain TB40/E, clone 4 (Sinzger et al., 2008), was previously engineered to express mCherry to monitor infection (TB40/EmCherry; O'Connor and Shenk, 2011). TB40/EmCherry was used to insert a triple FLAG epitope tag (3xF) in-frame with the UL33 ORF at the C-terminus, yielding TB40/EmCherry-UL33-3xF. Briefly, the 3xF epitope tag and kanamycin (Kan)-flipase recognition target (frt) cassette were PCR amplified from pGTE-3xFLAG-Kan-frt (O'Connor and Shenk, 2011) (Table S1). Methods for Kan-frt selection and counterselection were described previously (Lee et al., 2001; Miller et al., 2012; O'Connor and Shenk, 2011, 2012). We then made two additional recombinants by galactokinase (galK) recombineering using TB40/EmCherry-UL33-3xF as the backbone. These methods were described in detail previously (O'Connor and Miller, 2014; Warming et al., 2005). First, we generated a complete UL33 ORF deletion inclusive of the epitope tag (TB40/EmCherry-UL33Δ). Finally, we mutated the ‘DRY’ motif, to which G proteins couple, from DRY to AAA, resulting in TB40/EmCherry-UL33AAA-3xF. All primers used for recombineering and sequencing are shown in Table S1. All viral stocks were propagated in NuFF-1 fibroblasts, as described previously (O'Connor and Shenk, 2012), and titered by 50% tissue culture infectious dose (TCID50).
Latent infection and reactivation analyses by ELDA
Kasumi-3 cells were cultured in serum-low medium (X-VIVO 15; Lonza) for 48 h before infection. Cells were then infected at a multiplicity of infection (MOI) of 1.0 TCID50/cell at a density of 5.0×105 cells/ml by centrifugal enhancement at 1000 g for 30 min at room temperature, as described previously (O'Connor and Murphy, 2012). Infected cells were cultured for 7 d in X-VIVO 15 and then treated with 20 nM TPA or vehicle (DMSO) for an additional 2 days. Where indicated, cells were treated with forskolin (20 μM) or 666-15 (1.0 μM; both from Selleckchem).
Primary CD34+ HPCs were infected (MOI=2.0) by centrifugal enhancement at 450 g for 30 min, followed by overnight incubation at 37°C and 5% CO2. In parallel, SI/SI and MG3 cells were plated in a 1:1 ratio (1.5×105 cells each) onto collagen-coated (1 mg/ml) six-well plates in human CD34+ long-term culture medium (hLTCM), comprised of MyeloCult H5100 (Stem Cell Technologies) supplemented with 1 µM hydrocortisone and 100 U/ml each of penicillin and streptomycin. Before co-culture with CD34+ HPCs, stromal cell growth was arrested by irradiation using a fixed source 137Cesium Shepherd Mark I Irradiator at 20 Gy, after which the cells were washed three times with 1× PBS. Fresh hLTCM was added and cells were returned to culture overnight at 37°C and 5% CO2. The next day, viral inoculum was removed from the CD34+ HPCs and the cells were washed. Infected CD34+ HPCs were then cultured in transwells above the irradiated SI/SI:MG3 murine stromal cells, and fresh hLTCM was provided every 5 days.
The frequency of infectious centers was quantified by ELDA by co-culture with naïve NuFF-1 fibroblasts, essentially as described previously (Umashankar and Goodrum, 2014) using online software (http://bioinf.wehi.edu.au/software/elda/). At 7 dpi, a portion of each infected myeloid population was treated with reactivation stimulus. For CD34+ HPCs, cells were cultured in reactivation medium (RPMI-1640 containing 20% FBS, 10 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM non-essential amino acids, and 100 U/ml each penicillin and streptomycin), with 15 ng/ml each (all from R&D Systems) of IL-6, G-CSF (also known as CSF3), GM-CSF (also known as CSF2), or IL-3, or cultured in hLTCM to maintain latency. For Kasumi-3 cells, reactivation was stimulated with TPA (20 nM) or treated with DMSO (equivalent v/v) to maintain latency.
Generation of THP-1 cell lines
The lentiviral vectors pSLIK-UL33-3xF and pSLIK-UL33AAA-3xF were generated by gateway cloning. In brief, UL33-3xF and UL33AAA-3xF were PCR amplified from the respective TB40/E recombinant viruses described above, using primers with 5′ Spe1 and 3′ Xba1 restriction enzyme sites. DNA fragments were purified using the GFX PCR purification kit (GE Healthcare), according to the manufacturer's instructions. The resulting fragments and the pEN-TmiRC3 entry vector were digested with SpeI and XbaI (New England Biolabs), after which the entry vector was dephosphorylated using calf intestinal phosphatase (New England Biolabs). Following gel purification of the DNA, the insert fragments were ligated with the entry vector using T4 ligase (New England Biolabs). After confirming ligation, the insert-containing entry vector was used for gateway recombination with the destination vector pSLIK-Venus, using the Gateway LR Clonase Enzyme Mix (Thermo Fisher Scientific), according to the manufacturer's protocol. The resulting pSLIK-UL33-3xF and pSLIK-UL33AAA-3xF sequences were confirmed by Sanger sequencing, using the primers listed in Table S1. pEN-TmiRC3 and pSLIK-Venus were deposited in Addgene by Iain Fraser (Addgene plasmid 25748 and 25734, respectively; Shin et al., 2006).
To generate the THP-1 expressing cell lines, HEK293T cells were transfected using Fugene 6 Transfection Reagent (Promega), following the manufacturer's protocol with pCMV-ΔR8.91 packaging (Addgene plasmid 2221) and pCMV-VSV-G envelope plasmids, and either pSLIK-Venus, pSLIK-UL33-3xF or pSLIK-UL33AAA-3xF at a ratio of 3:5:10. pCMV-VSV-G was deposited in Addgene by Bob Weinberg (Addgene plasmid 8454; Stewart et al., 2003). Medium was collected 48 h and 72 h post transfection, clarified through a 0.45 μm filter, and concentrated by ultracentrifugation (82,200 g at 4°C for 90 min), after which the viral pellet was resuspended in X-VIVO15 (Lonza). The concentrated lentiviruses were then used to transduce 106 THP-1 cells by centrifugal enhancement (1000 g) in the presence of polybrene (4 μg/ml) for 1 h at room temperature, and were then returned to culture overnight at 37°C and 5% CO2. The following day, cells were clarified by centrifugation onto Ficoll Paque (GE Healthcare), washed three times in 1× PBS, and cultured at 37°C and 5% CO2 in RPMI-1640 THP-1 cell medium, as described above. Transduced cells were selected 3 d later by fluorescence-activated cell sorting (FACS) by isolating Venus+ cells at 511-543 nm into RPMI-1640 THP-1 cell growth medium. Cells were washed three times in 1× PBS and resuspended in RPMI-1640 THP-1 cell medium at 37°C and 5% CO2. Expression of pUL33-3xF and pUL33AAA-3xF was assessed by immunoblot (described below), following treatment with or without DOX (1.0 µg/ml) for 24 h.
Viral growth and genomic maintenance
Multi-step growth assays were performed by infecting NuFF-1 cells at a MOI of 0.01 TCID50/cell. Supernatants from each infection were collected over a time course and stored at –80°C. Infectious virus was then titrated on naïve NuFF-1 cells and quantified by TCID50.
To quantify viral genomes, Kasumi-3 cells were infected (MOI=1.0), as detailed above. At each indicated time point, cells were washed three times in 1× PBS and treated with trypsin for 5 min at 37°C to remove cell-associated virus that had not entered. Cells were again washed three times in 1× PBS and lysed in lysis buffer (400 mM NaCl, 10 mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.5% SDS and 100 µg/ml proteinase K) at 37°C overnight. Samples were then treated with RNaseA (100 µg/ml) for 1 h at 37°C to remove contaminating RNA, and DNA was purified by phenol-chloroform-isoamyl alcohol extraction, followed by ethanol precipitation. DNA was resuspended in 10 mM Tris-HCl, pH 8.0 with 0.1 mM EDTA, and equal volumes were used in the qPCR reaction. Absolute HCMV genome quantities and MDM2 were calculated against a standard curve using a BAC standard that contains an MDM2 gene fragment (Krishna et al., 2020a), using primers specific for the MIE enhancer region (to prevent detection of transcribed mRNA) and MDM2 as a cellular control (Table S1).
RNA and protein analyses
Total RNA was collected from infected cells and extracted using the High Pure RNA Isolation kit (Roche), according to the manufacturer's instructions. cDNA was generated from 1 µg of RNA using TaqMan Reverse Transcription (RT) Reagents and random hexamer primers (Roche), and 5 ng of cDNA was used for qPCR using gene-specific primers (Table S1), as indicated in the text. Absolute quantities for each transcript were calculated using a standard curve with a tenfold serial dilution of a BAC-standard that contains a fragment of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) sequence (Krishna et al., 2020a). Viral gene expression was normalized to cellular GAPDH for each sample in all experiments. Each primer set had a similar linear range of detection for the BAC standard (linear between 12 and 35 copies; r2>0.95 for all experiments). Samples were analyzed in triplicate using a 96-well-format CFX Connect (Bio-Rad).
For protein analyses, cells were lysed in radioimmunoprecipitation assay (RIPA) buffer [1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.01 M NaPO4 (pH 7.2) and 2 mM EDTA] on ice for 1 h, with vortexing every 15 min. Samples were denatured in 2% SDS at 95°C for 10 min for all proteins except pUL33, which was denatured at 42°C for 10 min. Equal concentrations of lysates were separated by 10% SDS-PAGE and proteins detected using the following antibodies: anti-FLAG M2 (Sigma-Aldrich, F3165; 1:7500), anti-p-CREB (Cell Signaling Technology, 9198; 1:1000), anti-t-CREB (Cell Signaling Technology, 9197; 1:1000), anti-actin-horseradish peroxidase (HRP; Sigma-Aldrich; 1:20,000), anti-IE2 (clone 3A9, Nitzsche et al., 2008), anti-pp65 (clone 8A8, Bechtel and Shenk, 2002) and goat-anti-mouse-HRP or goat-anti-rabbit-HRP (both Jackson ImmunoResearch Labs; 1:10,000).
ChIP
Cells were infected and harvested as indicated above, fixed in 1% formaldehyde for 15 min at room temperature, and quenched with 125 mM glycine. Cells were lysed in immunoprecipitation (IP) buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.5% Igepal-CA630 and 1% Triton X-100), and the supernatant was clarified of debris by centrifugation at 20,000 g for 30 min. DNA was sheared to 0.3- to 1-kb fragments with a MiSonix Sonicator 3000 (20% output, 0.5 s on/off, 1 min) and aliquots stored as input controls. Anti-p-CREB (Cell Signaling Technology, 9198; 1:100) was used to precipitate p-CREB-associated DNA using protein A agarose (MilliporeSigma). Normal rabbit serum (Cell Signaling Technology) was used as a control for non-specific antibody binding. DNA was eluted by boiling and proteinase K treatment (0.2 ng/μl), and was then ethanol precipitated and assessed by PCR using primers directed at the MIE enhancer/promoter or the UL69 non-promoter region (Table S1).
Statistical analyses
All statistical analyses were performed using Prism 8 software (GraphPad). Analyses specific for each experiment are indicated in the figure legends. Each experiment was performed three independent times, unless indicated as representative data shown. Normal distribution of data was assumed, but not formally tested.
Supplementary Material
Acknowledgements
The authors would like to thank Amy Graham and Eric Schultz in the Cleveland Clinic LRI Flow Cytometry Core for assistance with FACS sorting and analyses.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: B.A.K., C.M.O.; Methodology: B.A.K., A.B.W., C.M.O.; Validation: B.A.K., A.B.W.; Formal analysis: B.A.K.; Investigation: B.A.K., A.B.W.; Resources: B.A.K., A.B.W., A.L.D.; Writing - original draft: B.A.K.; Writing - review & editing: B.A.K., A.B.W., A.L.D., C.M.O.; Visualization: B.A.K., C.M.O.; Supervision: C.M.O.; Project administration: C.M.O.; Funding acquisition: C.M.O., A.L.D.
Funding
This work was supported by the Cleveland Clinic (to C.O.), National Institutes of Health (AI153348 to C.O.), and the American Heart Association and the Hablitzel Family (20PRE35080060 to A.D.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.254268.supplemental
References
- Adamson, C. S. and Nevels, M. M. (2020). Bright and early: inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses 12, 110 10.3390/v12010110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend, K. C., Ziehr, B., Vincent, H. A. and Moorman, N. J. (2016). Multiple transcripts encode full-length human cytomegalovirus IE1 and IE2 proteins during lytic infection. J. Virol. 90, 8855-8865. 10.1128/JVI.00741-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bate, S. L., Dollard, S. C. and Cannon, M. J. (2010). Cytomegalovirus seroprevalence in the United States: the national health and nutrition examination surveys, 1988-2004. Clin. Infect. Dis. 50, 1439-1447. 10.1086/652438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtel, J. T. and Shenk, T. (2002). Human cytomegalovirus UL47 tegument protein functions after entry and before immediate-early gene expression. J. Virol. 76, 1043-1050. 10.1128/JVI.76.3.1043-1050.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisser, P. S., Vink, C., Van Dam, J. G., Grauls, G., Vanherle, S. J. V. and Bruggeman, C. A. (1998). The R33 G protein-coupled receptor gene of rat cytomegalovirus plays an essential role in the pathogenesis of viral infection. J. Virol. 72, 2352-2363. 10.1128/JVI.72.3.2352-2363.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt, F. M., Wu, S.-E., Bridges, J. P. and Miller, W. E. (2014). The M33 G protein-coupled receptor encoded by murine cytomegalovirus is dispensable for hematogenous dissemination but is required for growth within the salivary gland. J. Virol. 88, 11811-11824. 10.1128/JVI.01006-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler, J., Zeltzer, S., Reitsma, J., Petrucelli, A., Umashankar, M., Rak, M., Zagallo, P., Schroeder, J., Terhune, S., Goodrum, F.et al. (2016). Opposing regulation of the EGF receptor: a molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog. 12, e1005655 10.1371/journal.ppat.1005655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buehler, J., Carpenter, E., Zeltzer, S., Igarashi, S., Rak, M., Mikell, I., Nelson, J. A. and Goodrum, F. (2019). Host signaling and EGR1 transcriptional control of human cytomegalovirus replication and latency. PLoS Pathog. 15, e1008037 10.1371/journal.ppat.1008037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin, R. D., Schaefer, G. C., Allen, J. R., Davis-Poynter, N. J. and Farrell, H. E. (2009). The M33 chemokine receptor homolog of murine cytomegalovirus exhibits a differential tissue-specific role during in vivo replication and latency. J. Virol. 83, 7590-7601. 10.1128/JVI.00386-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarosa, P., Gruijthuijsen, Y. K., Michel, D., Beisser, P. S., Holl, J., Fitzsimons, C. P., Verzijl, D., Bruggeman, C. A., Mertens, T., Leurs, R.et al. (2003). Constitutive signaling of the human cytomegalovirus-encoded receptor UL33 differs from that of its rat cytomegalovirus homolog R33 by promiscuous activation of G proteins of the Gq, Gi, and Gs classes. J. Biol. Chem. 278, 50010-50023. 10.1074/jbc.M306530200 [DOI] [PubMed] [Google Scholar]
- Case, R., Sharp, E., Benned-Jensen, T., Rosenkilde, M. M., Davis-Poynter, N. and Farrell, H. E. (2008). Functional analysis of the murine cytomegalovirus chemokine receptor homologue M33: ablation of constitutive signaling is associated with an attenuated phenotype in vivo. J. Virol. 82, 1884-1898. 10.1128/JVI.02550-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee, M. S., Satchwell, S. C., Preddie, E., Weston, K. M. and Barrell, B. G. (1990). Human cytomegalovirus encodes three G protein-coupled receptor homologues. Nature 344, 774-777. 10.1038/344774a0 [DOI] [PubMed] [Google Scholar]
- Chen, Z., Raman, M., Chen, L., Lee, S. F., Gilman, A. G. and Cobb, M. H. (2003). TAO (thousand-and-one amino acid) protein kinases mediate signaling from carbachol to p38 mitogen-activated protein kinase and ternary complex factors. J. Biol. Chem. 278, 22278-22283. 10.1074/jbc.M301173200 [DOI] [PubMed] [Google Scholar]
- Cheng, S., Caviness, K., Buehler, J., Smithey, M., Nikolich-Žugich, J. and Goodrum, F. (2017). Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 114, E10586-E10595. 10.1073/pnas.1710522114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-McMillen, D., Buehler, J., Peppenelli, M. and Goodrum, F. (2018). Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses 10, E444 10.3390/v10080444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley, A. L. and O'Connor, C. M. (2020). Regulation of the MIE locus during HCMV latency and reactivation. Pathogens 9, E869 10.3390/pathogens9110869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, L., Du, L., Poulter, M., Choi, S., McIntosh, M. and Reeves, M. B. (2019). Src family kinase activity drives cytomegalovirus reactivation by recruiting MOZ histone acetyltransferase activity to the viral promoter. J. Biol. Chem. 294, 12901-12910. 10.1074/jbc.RA119.009667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder, E., Krishna, B., Williamson, J., Aslam, Y., Farahi, N., Wood, A., Romashova, V., Roche, K., Murphy, E., Chilvers, E.et al. (2019a). Monocytes latently infected with human cytomegalovirus evade neutrophil killing. iScience 12, 13-26. 10.1016/j.isci.2019.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder, E. G., Krishna, B. A., Williamson, J., Lim, E. Y., Poole, E., Sedikides, G. X., Wills, M., O'Connor, C. M., Lehner, P. J., Sinclair, J.et al. (2019b). Interferon-responsive genes are targeted during the establishment of human cytomegalovirus latency. mBio 10, e02574-e02519 10.1128/mBio.02574-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell, H. E., Abraham, A. M., Cardin, R. D., Sparre-Ulrich, A. H., Rosenkilde, M. M., Spiess, K., Jensen, T. H., Kledal, T. N. and Davis-Poynter, N. (2011). Partial functional complementation between human and mouse cytomegalovirus chemokine receptor homologues. J. Virol. 85, 6091-6095. 10.1128/JVI.02113-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell, H. E., Bruce, K., Lawler, C., Oliveira, M., Cardin, R., Davis-Poynter, N. and Stevenson, P. G. (2017). Murine cytomegalovirus spreads by dendritic cell recirculation. mBio 8, e01264-e01217 10.1128/mBio.01264-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forte, E., Zhang, Z., Thorp, E. B. and Hummel, M. (2020). Cytomegalovirus latency and reactivation: an intricate interplay with the host immune response. Front. Cell Infect. Microbiol. 10, 130 10.3389/fcimb.2020.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, T., Niemann, I., Reichel, A. and Stamminger, T. (2019). Emerging roles of cytomegalovirus-encoded G protein-coupled receptors during lytic and latent infection. Med. Microbiol. Immunol. 208, 447-456. 10.1007/s00430-019-00595-9 [DOI] [PubMed] [Google Scholar]
- Gruijthuijsen, Y. K., Casarosa, P., Kaptein, S. J. F., Broers, J. L. V., Leurs, R., Bruggeman, C. A., Smit, M. J. and Vink, C. (2002). The rat cytomegalovirus R33-encoded G protein-coupled receptor signals in a constitutive fashion. J. Virol. 76, 1328-1338. 10.1128/JVI.76.3.1328-1338.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale, A. E., Collins-McMillen, D., Lenarcic, E. M., Igarashi, S., Kamil, J. P., Goodrum, F. and Moorman, N. J. (2020). FOXO transcription factors activate alternative major immediate early promoters to induce human cytomegalovirus reactivation. Proc. Natl. Acad. Sci. USA 117, 18764-18770. 10.1073/pnas.2002651117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humby, M. S. and O'Connor, C. M. (2015). Human cytomegalovirus US28 is important for latent infection of hematopoietic progenitor cells. J. Virol. 90, 2959-2970. 10.1128/JVI.02507-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, M. J., Wheeler, D. G., Cooper, E. and Meier, J. L. (2003). Role of the human cytomegalovirus major immediate-early promoter's 19-base-pair-repeat cyclic AMP-response element in acutely infected cells. J. Virol. 77, 6666-6675. 10.1128/JVI.77.12.6666-6675.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, M. J., Wu, A. W., Andrews, J. I., McGonagill, P. W., Tibesar, E. E. and Meier, J. L. (2007). Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 81, 6669-6681. 10.1128/JVI.01524-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew, V. G., Yuan, J., Meier, J. and Reeves, M. B. (2014). Mitogen and stress activated kinases act co-operatively with CREB during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog. 10, e1004195 10.1371/journal.ppat.1004195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, T., Shephard, P., Kleinert, H. and Kömhoff, M. (2007). Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim. Biophys. Acta 1773, 1605-1618. 10.1016/j.bbamcr.2007.09.001 [DOI] [PubMed] [Google Scholar]
- Krishna, B. A., Poole, E. L., Jackson, S. E., Smit, M. J., Wills, M. R. and Sinclair, J. H. (2017a). Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 8, e01754-e01717 10.1128/mBio.01754-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Spiess, K., Poole, E. L., Lau, B., Voigt, S., Kledal, T. N., Rosenkilde, M. M. and Sinclair, J. H. (2017b). Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat. Commun. 8, 14321 10.1038/ncomms14321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Miller, W. E. and O'Connor, C. M. (2018). US28: HCMV's swiss army knife. Viruses 10, 445 10.3390/v10080445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Humby, M. S., Miller, W. E. and O'Connor, C. M. (2019a). Human cytomegalovirus G protein-coupled receptor US28 promotes latency by attenuating c-fos. Proc. Natl. Acad. Sci. USA 116, 1755-1764. 10.1073/pnas.1816933116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Wills, M. R. and Sinclair, J. H. (2019b). Advances in the treatment of cytomegalovirus. Br. Med. Bull. 131, 5-17. 10.1093/bmb/ldz031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Wass, A. B. and O'Connor, C. M. (2020a). Activator protein-1 transactivation of the major immediate early locus is a determinant of cytomegalovirus reactivation from latency. Proc. Natl. Acad. Sci. USA 117, 20860-20867. 10.1073/pnas.2009420117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna, B. A., Wass, A. B., Sridharan, R. and O'Connor, C. M. (2020b). The requirement for US28 during cytomegalovirus latency is independent of US27 and US29 gene expression. Front. Cell Infect. Microbiol. 10, 186 10.3389/fcimb.2020.00186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E.-C., Yu, D., Martinez de Velasco, J., Tessarollo, L., Swing, D. A., Court, D. L., Jenkins, N. A. and Copeland, N. G. (2001). A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56-65. 10.1006/geno.2000.6451 [DOI] [PubMed] [Google Scholar]
- Liu, R., Baillie, J., Sissons, J. G. P. and Sinclair, J. H. (1994). The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in nonpermissive cells. Nucleic Acids Res. 22, 2453-2459. 10.1093/nar/22.13.2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone, C. L., Vesole, D. H. and Stinski, M. F. (1990). Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: mutational analysis of the viral proteins. J. Virol. 64, 1498-1506. 10.1128/JVI.64.4.1498-1506.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini, A., Liu, H. and Zhu, H. (2001). Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J. Virol. 75, 1870-1878. 10.1128/JVI.75.4.1870-1878.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies, B. J., Browne, H. and Gibson, W. (1996). Identification of the human cytomegalovirus G protein-coupled receptor homologue encoded by UL33 in infected cells and enveloped virus particles. Virology 225, 111-125. 10.1006/viro.1996.0579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Limon, A., Joaquin, M., Caballero, M., Posas, F. and de Nadal, E. (2020). The p38 pathway: from biology to cancer therapy. Int. J. Mol. Sci. 21, 1913 10.3390/ijms21061913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, W. E., Zagorski, W. A., Brenneman, J. D., Avery, D., Miller, J. L. C. and O'Connor, C. M. (2012). US28 is a potent activator of phospholipase C during HCMV infection of clinically relevant target cells. PLoS ONE 7, e50524 10.1371/journal.pone.0050524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min, C.-K., Shakya, A. K., Lee, B.-J., Streblow, D. N., Caposio, P. and Yurochko, A. D. (2020). The differentiation of human cytomegalovirus infected-monocytes is required for viral replication. Front. Cell Infect. Microbiol. 10, 368 10.3389/fcimb.2020.00368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitzsche, A., Paulus, C. and Nevels, M. (2008). Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 82, 11167-11180. 10.1128/JVI.01218-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, C. M. and Miller, W. E. (2014). Methods for studying the function of cytomegalovirus GPCRs. Methods Mol. Biol. 1119, 133-164. 10.1007/978-1-62703-788-4_10 [DOI] [PubMed] [Google Scholar]
- O'Connor, C. M. and Murphy, E. A. (2012). A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 86, 9854-9865. 10.1128/JVI.01278-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, C. M. and Shenk, T. (2011). Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J. Virol. 85, 3700-3707. 10.1128/JVI.02442-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor, C. M. and Shenk, T. (2012). Human cytomegalovirus pUL78 G protein-coupled receptor homologue is required for timely cell entry in epithelial cells but not fibroblasts. J. Virol. 86, 11425-11433. 10.1128/JVI.05900-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, M. B., MacAry, P. A., Lehner, P. J., Sissons, J. G. P. and Sinclair, J. H. (2005). Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 102, 4140-4145. 10.1073/pnas.0408994102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrill, J. D. and Miller, W. E. (2006). G protein-coupled receptor (GPCR) kinase 2 regulates agonist-independent Gq/11 signaling from the mouse cytomegalovirus GPCR M33. J. Biol. Chem. 281, 39796-39805. 10.1074/jbc.M610026200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, K.-J., Wall, E. A., Zavzavadjian, J. R., Santat, L. A., Liu, J., Hwang, J.-I., Rebres, R., Roach, T., Seaman, W., Simon, M. I.et al. (2006). A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. USA 103, 13759-13764. 10.1073/pnas.0606179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzger, C., Hahn, G., Digel, M., Katona, R., Sampaio, K. L., Messerle, M., Hengel, H., Koszinowski, U., Brune, W. and Adler, B. (2008). Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89, 359-368. 10.1099/vir.0.83286-0 [DOI] [PubMed] [Google Scholar]
- Stewart, S. A., Dykxhoorn, D. M., Palliser, D., Mizuno, H., Yu, E. Y., An, D. S., Sabatini, D. M., Chen, I. S. Y., Hahn, W. C., Sharp, P. A.et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493-501. 10.1261/rna.2192803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinski, M. F. and Meier, J. L. (2007). Immediate-early viral gene regulation and function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis (ed. Arvin A., Campadelli-Fiume G., Mocarski E., Moore P. S., Roizman B., Whitley R. and Yamanishi K.), pp. 241-263. Cambridge University Press, Cambridge. [PubMed] [Google Scholar]
- Umashankar, M. and Goodrum, F. (2014). Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol. Biol. 1119, 99-112. 10.1007/978-1-62703-788-4_7 [DOI] [PubMed] [Google Scholar]
- Waldhoer, M., Kledal, T. N., Farrell, H. and Schwartz, T. W. (2002). Murine Cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J. Virol. 76, 8161-8168. 10.1128/JVI.76.16.8161-8168.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming, S., Costantino, N., Court, D. L., Jenkins, N. A. and Copeland, N. G. (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33, e36 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, B. E., Mochon, E. and Boxer, L. M. (1996). Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol. Cell Biol. 16, 5546-5556. 10.1128/MCB.16.10.5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, S.-E. and Miller, W. E. (2016). The HCMV US28 vGPCR induces potent Gαq/PLC-β signaling in monocytes leading to increased adhesion to endothelial cells. Virology 497, 233-243. 10.1016/j.virol.2016.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan, J., Liu, X., Wu, A. W., McGonagill, P. W., Keller, M. J., Galle, C. S. and Meier, J. L. (2009). Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 83, 6391-6403. 10.1128/JVI.00061-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, D., Pan, C., Sheng, J., Liang, H., Bian, Z., Liu, Y., Trang, P., Wu, J., Liu, F., Zhang, C.-Y.et al. (2018). Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat. Microbiol. 3, 503-513. 10.1038/s41564-018-0131-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.