

Abstract

In the last few decades, many efforts have been made to make poly(3-hydroxybutyrate) (PHB) and its copolymers more suitable for industrial production and large-scale use. Plasticization, especially using biodegradable oligomeric plasticizers, has been one of the strategies for this purpose. However, PHB and its copolymers generally present low miscibility with plasticizers. An understanding of the plasticizer distribution between the mobile and rigid amorphous phases and how this influences thermal, mechanical, and morphological properties remains a challenge. Herein, formulations of poly(hydroxybutyrate-co-valerate) (PHBV) plasticized with an oligomeric polyester based on lactic acid, adipic acid, and 1,2-propanediol (PLAP) were prepared by melt extrusion. The effects of the PLAP content on the processability, miscibility, and microstructure of the semicrystalline PHBV and on the thermal, morphological, and mechanical properties of the formulations were investigated. The compositions of the mobile and rigid amorphous phases of the PHBV/PLAP formulations were easily estimated by combining dynamic mechanical data and the Fox equation, which showed a heterogeneous distribution of PLAP in these two phases. An increase in the PLAP mass fraction in the formulations led to progressive changes in the composition of the amorphous phases, an increase of both crystalline lamellae and interlamellar layer thickness, and a decrease in the melting and glass transition temperatures as well as the PHBV stiffness. The Flory–Huggins interaction parameter varied with the formulation composition in the range of −0.299 to −0.081. The critical PLAP mass fraction of 0.37 obtained from thermodynamic data is close to the value estimated from dynamic mechanical analysis (DMA) data and the Fox equation. The mechanical properties showed a close relationship with the distribution of PLAP in the rigid and mobile amorphous phases as well as with the microstructure of the crystalline phase of PHBV in the formulations.
Introduction
Polyhydroxyalkanoates (PHA) are among the most promising bio-based polymer classes for use as substitutes for conventional petrochemical-based polymers. PHA are biodegradable polyesters produced by a wide range of microorganisms that use plant resources such as carbohydrates and vegetable oils as carbon sources. Poly(3-hydroxybutyrate) (PHB) and its copolymers are isotactic and semicrystalline PHA that have attracted great attention because their mechanical properties are similar to those of conventional fossil-based thermoplastics such as polypropylene.1−3 Despite this, PHB presents some drawbacks that limit its applications. For example, PHB is a rigid and brittle material due to its high stereoregularity, high degree of crystallinity, and formation of very large and overlapped spherulites with a high tendency to crack. These morphological characteristics are due to the high purity of PHB. The absence of impurities that could act as a nucleating agent and the stereoregularity of the PHB lead to a growth rate that is higher than the nucleation rate when PHB is crystallized from the melt.3−6 Another challenge regarding the usability of PHB is related to its thermal stability. PHB has a narrow processing window, and it is susceptible to thermal degradation at temperatures close to the melting point. This leads to degradation during processing, which is detrimental to mechanical properties.2,7,8
The PHB microstructure is reported to be composed of a crystalline phase and two different amorphous phases: the mobile phase, which has the same properties and glass transition temperature (Tg) as the bulk amorphous phase of PHB, and the rigid phase, located adjacent to the crystalline phase, which has reduced chain mobility and, as a consequence, higher Tg.9−11 The Tg of the PHB amorphous phase is close to room temperature. This is responsible for the polymer chains’ mobility and for the embrittlement with time, a phenomenon called aging. The aging could occur through two independent processes: secondary crystallization and physical aging of the amorphous phase. Both phenomena increase the fraction of the crystalline and rigid amorphous phases, resulting in a decrease in ductility and an increase in the stiffness and brittleness of the PHB.2,6,10,12
Despite the promises for PHB applications, these drawbacks must be overcome for industrial and large-scale uses of PHB. Up to now, there have been many efforts by the scientific community to address these drawbacks and both chemical and physical approaches have been used for modulating PHB’s thermal and mechanical properties. Examples of chemical approaches already reported in the literature are copolymerization,13−15 internal plasticization,16,17 and grafting.18,19 Common physical approaches are blending,20−22 external plasticization,23,24 and the use of nucleating agents25 and fillers.26,27 One of the most interesting approaches from an industrial viewpoint is plasticization. Plasticizers comprise one of the major additive industries in the world due to their effectiveness in tuning polymer flexibility and improving processability.28,29 A general rule is that plasticizers decrease polymer–polymer intra- and intermolecular interactions by filling the space between polymer chains, thus increasing the free volume, which leads to a decrease in the glass transition temperature.29,30 Plasticizers can be classified as internal or external plasticizers.29,30 The copolymerization of hydroxybutyrate (HB) and hydroxyvalerate (HV) to form poly(hydroxybutyrate-co-valerate) (PHBV) is a well-known approach to modulate the thermal and mechanical properties of PHB because the HV monomeric units act as internal plasticizers. By increasing the fraction of the HV monomeric units, PHBV becomes more ductile and less brittle, and the melting and glass transition temperature proportionally decrease.16,17 However, the PHBV grades that are commercially available only have up to 20 mol % hydroxyvalerate content, which is not enough to overcome the drawbacks cited above.3 Therefore, external plasticization or a combination of both internal and external plasticization becomes an interesting option. Many different external plasticizers have been used for the plasticization of PHB and PHBV, such as low molar mass phthalates,23,27,31−33 citrates,23,24,27,31,34−38 fatty acids/esters,39−44 esters,31,32,34−36,45−50 vegetable oils and derivatives,23,24,27,46,51−56 terpenes57 and carbohydrates;49,58 and oligomeric plasticizers such as poly(ethylene glycol),24,39,46,48−50,59−61 PHB,62−65 PHA,66 poly(caprolactone), Laprol,48,60 Pluronic,67 aliphatic polyesters,45,48,68,69 poly(adipate),32 polyurethanes,70 and poly(ethylene oxide).71
Most reports in the literature concern the relationship between the plasticizer content and the thermal and mechanical properties of PHB and its copolymers. Generally, the miscibility is analyzed using the criterion of the glass transition temperature depression.23,25,31,32,40,49,50,52,54,57,63,71,72 However, the distribution of the plasticizer in the mobile and rigid amorphous phases and how this affects the PHB mechanical properties have been less studied. Righetti et al.10 reported that PHB and PHBV stiffness increases proportionally with an increase in the sum of crystalline and rigid amorphous phase mass fractions, while the ductility of the materials is proportional to the mass fraction of the mobile amorphous phase. El-Taweel et al.64 determined the mass fraction of the mobile and rigid amorphous phases as well as the fraction of plasticizer in these amorphous phases in PHB plasticized with oligomeric atactic PHB-diol. They concluded that the plasticizer is not homogeneously distributed, and the mobile amorphous phase is richer in the plasticizer. However, they did not correlate the plasticizer distribution with the mechanical properties of the formulations. Crétois et al.47 reported that the addition of a plasticizer to PHB decreases the relaxation temperature of the amorphous phase and prevents the physical aging of the polymer. However, annealing the material induced phase separation and the formation of unplasticized domains that were susceptible to physical aging. Similar results were reported by Kurusu et al.,34 who observed only one glass transition by differential scanning calorimetry (DSC). However, the results of the dynamic mechanical analysis (DMA) led to the conclusion that the amorphous phase of a plasticized PHB was composed of interlamellar amorphous phases with and without the plasticizer, and annealing the sample increased the nonplasticized fraction and polymer brittleness. Recently, Umemura and Felisberti38 studied the effect of aging on the properties of PHB formulations plasticized with triethyl citrate (TEC). They concluded that physical aging and secondary crystallization resulted in the enrichment of the amorphous phase with the plasticizer, and phase separation was reported for the formulation with a TEC mass fraction of 0.3. In general, the aged and plasticized PHB were less brittle and presented a higher capacity to dissipate mechanical energy than the aged pure PHB.
In this work, we have investigated the effect of an oligomeric plasticizer on the PHBV microstructure, morphology, and thermal and mechanical properties of the formulations prepared by extrusion. A biodegradable oligomeric polyester based on lactic acid, adipic acid, and 1,2-propanediol (PLAP) was employed as a plasticizer. The dynamic mechanical analysis (DMA) allowed the identification of the mobile and rigid amorphous phases. The depression of the glass transition temperature of these amorphous phases provided an estimation of the PLAP mass fraction in these two amorphous phases using the Fox equation. Moreover, PLAP phase separation was also observed, and the critical composition was determined and compared with values determined by thermodynamic data from the Flory–Huggins parameter. The influence of PLAP distribution on the morphology of the crystalline phase of PHBV was studied by small-angle X-ray scattering (SAXS), and the spherulite morphology was investigated by polarized optical microscopy (POM). These results were correlated with the thermal properties studied by differential scanning calorimetry (DSC) and with the tensile properties and Izod impact resistance. Using a simple methodology, this work allows the understanding of how the plasticizer is distributed in the mobile and rigid amorphous phases and how this distribution influences the local miscibility and the micro- and macroscopic properties of the PHBV as a function of the plasticizer content.
Results and Discussion
Processability
To investigate the effects of a plasticizer on the processability of PHBV, the force applied by the extruder during the steps of feed (I), melting–compounding (II), and unloading (III) of PHBV and its formulations was monitored, Figure 1a. The feeding stage was performed in two steps to not exceed the maximum torque of the extruder. The maximum force value at the feed stage decreased with the increase in the PLAP mass fraction, indicating easier feeding of the PHBV/PLAP premixture compared with pure PHBV. At the compounding step, the force decreased with the increase in the PLAP mass fraction and mixture time. The force decreased abruptly as the formulations were unloading from the barrel.
Figure 1.
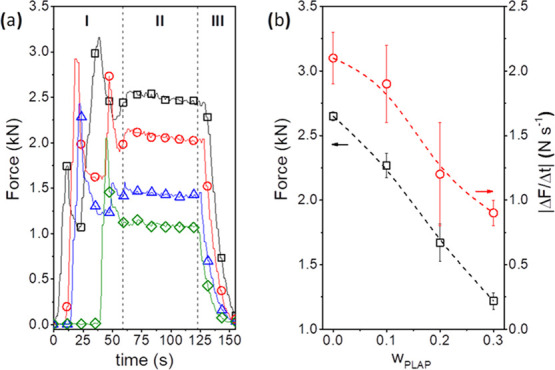
(a) Force applied by the extruder as a function of the processing time for pure PHBV (□) and the formulations with PLAP mass fractions of 0.1 (ring open red), 0.2 (triangle up open blue), and 0.3 (diamond solid green). (b) Force just before the unloading (□) and the modulus of the force decay rate during mixing (ring open red) as a function of the PLAP mass fraction.
The force just before unloading and the modulus of the force decay rate (|ΔF/Δt|) during the compounding decreased with the increase in the PLAP mass fraction (Figure 1b). Both parameters may be related to the melt viscosity of the formulation and the thermal stability of PHBV. Thermal degradation of PHB and its copolymers results in a decrease in the molar mass.74 However, the gel permeation chromatography (GPC) data in Table 1 shows similar and small decreases in the molar mass for the processed formulations compared to unprocessed PHBV, with a maximum reduction of 8%. These results indicated that the decrease in the force during processing was mainly due to the PLAP, which decreased the viscosity of the melt. This effect combined with the suitable processing conditions applied resulted in good melt-processing stability with minor chain degradation induced by the shear at high temperatures. A similar tendency (a decrease in the force before unloading and the modulus of the force decay rate as a function of the plasticizer content for formulations of PHB with TEC) was reported in our previous work.75 However, higher molar mass PHB (Mw = 394 kDa) was used, and the GPC analysis showed a higher decrease in the Mw (around 11–16% for plasticized samples) and Đ values, indicating a preferential scission of long polymer chains during processing. A higher extent of PHB degradation (molar mass reduction of 34%) was reported by Garcia-Garcia et al.51 for the extrusion and injection molding of PHB (Mw = 426 kDa) plasticized with epoxidized vegetable oils using similar processing conditions. These results suggest that thermal and shear-induced degradation may be less important for PHB with Mw ≤ 250 kDa.
Table 1. Molar Mass and Molar Mass Dispersity of Unprocessed PHBV, Processed PHBV, and its Formulations with PLAP, as Determined by GPC Analysis.
| wPLAP | Mn (kDa) | Mw (kDa) | Đ |
|---|---|---|---|
| 0.0a | 91.4 | 251.5 | 2.8 |
| 0.0 | 87.1 | 238.4 | 2.7 |
| 0.1 | 84.4 | 263.9 | 3.1 |
| 0.2 | 85.2 | 238.3 | 2.8 |
| 0.3 | 87.9 | 233.6 | 2.7 |
Unprocessed PHBV.
Thermal Stability
The thermal stability of PHBV, PLAP, and their formulations was evaluated by thermogravimetric analysis (TGA). The thermogravimetric curves and their derivatives are presented in Figure S1. The temperatures of the initial mass loss (Tonset) and the maximum mass loss rate (Tmax), the percentage of mass loss for each degradation stage, the residue at 500 °C (R500°C), and the estimated PLAP mass fraction (wplap,real) are summarized in Table 2.
Table 2. Temperature and Mass Loss of Each Degradation Stage of Unprocessed PHBV, Processed PHBV and its Formulations with PLAP, Residual Mass Fraction at 500 °C, and the PLAP Mass Fraction Determined by TGA Analysis.
| 1st
stage |
2nd
stage |
|||||||
|---|---|---|---|---|---|---|---|---|
| wPLAP | Tonset (°C) | Tmax. (°C) | mass loss (%) | Tonset (°C) | Tmax. (°C) | mass loss (%) | R500°C (%) | wPLAP,realc |
| 0.00a | 240 | 300 | 100 | 0 | ||||
| 0.00 | 220 | 295 | 100 | 0 | ||||
| 0.10 | 205 | 260 | 92 | 285 | 345 | 8 | 0 | 0.09 |
| 0.20 | 200 | 255 | 85 | 280 | 350 | 14 | 1 | 0.17 |
| 0.30 | 200 | 255 | 75 | 280 | 350 | 23 | 2 | 0.28 |
| 1.00b | 190 | 280 | 11 | 300 | 385 | 87 | 2 | |
Unprocessed PHBV.
Unprocessed PLAP.
wPLAP,real = {[(mass loss2ndstage/87) × 100] + residue}/100.
In general, PHBV degraded in a single stage due to random chain scission by β-elimination74,76,77 with a Tonset of around 220–240 °C. The PLAP plasticizer presented two overlapped degradation stages with Tonset at around 190 and 300 °C. Cicogna et al.78 reported the Tonset = 184–212 °C for oligomeric poly(lactic acid) and Tonset = 258 °C for oligomeric poly(1,2-propylene adipate), whereas Tonset = 320 °C was reported for polymeric poly(lactic acid). Thus, the first stage of PLAP thermal degradation with a minor mass loss is probably due to the degradation of random segments of poly(lactic acid), and the second stage is related to the overall bulk degradation of the plasticizer.
The plasticized formulations presented two degradation stages with Tonset around 200 and 350 °C. As previously discussed, the first stage is due to the degradation of both PHBV and PLAP, and the second stage is only due to the degradation of PLAP. The initial degradation temperature of the formulations was lower than for pure PHBV, indicating that the introduction of PLAP decreased the thermal stability of the formulations compared with pure PHBV. This is probably due to the presence of carboxylic acid end groups in the PLAP chains. However, this decrease in the thermal stability of the formulations did not affect processing, as observed by the negligible molecular weight decrease for the PHB subjected to extrusion and injection molding. The mass fraction of PLAP in the formulations was estimated using the thermal degradation profile of pure PHBV and PLAP (Table 2). In general, the PLAP mass fraction in the formulations was around 6–10% lower than planned, indicating a loss of PLAP during the preparation and processing of formulations.
Microstructure Analysis
The storage modulus (E′), loss modulus (E″), and loss factor (tan δ = E″/E′) as a function of temperature are presented in Figure 2a–c, respectively. The E′ vs T and E″ vs T curves were vertically shifted to the same modulus at −100 °C to facilitate a comparison of the changes in the relaxation spectrum of the formulations. The glass transition occurred in the temperature range from −50 to 65 °C and was characterized by a drop in E′ and by a peak in E″ and tan δ curves. For PHBV and its plasticized formulations, a shoulder was observed at the main peak in this temperature range. In the E″ curves, the main peak for PHBV presented a maximum at 26 °C. On the other hand, the main peak for the PHBV formulation containing PLAP at a mass fraction of 0.3 presented a maximum at −30 °C and a shoulder at around 6 °C. Similar tendencies were observed for the tan δ curves. There is a clear shift of the maximum of the peak to lower temperatures and an inversion in the intensity of the loss modulus peak and shoulder in this temperature range with the increasing PLAP mass fraction, as indicated by the arrows in Figure 2b,c. Similar behavior was recently reported for PHB plasticized with TEC.38 Secondary relaxations start below −75 °C, but due to the low signal-to-noise ratio, these were unclear. The second drop in E′ and peaks in E″ and tan δ curves were observed at a temperature range from 50 to 125 °C. This is related to the α′ relaxation of the amorphous–crystalline interphase.26
Figure 2.
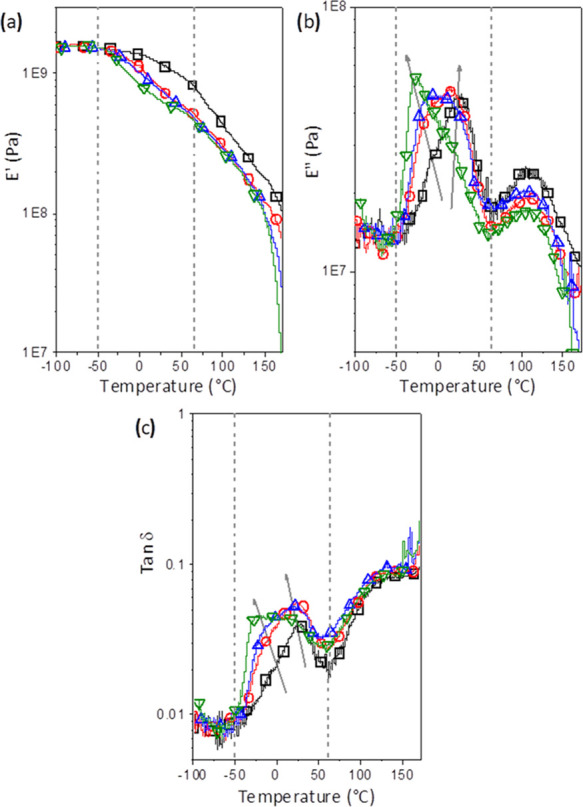
(a) Storage modulus (E′), (b) loss modulus (E″), and (c) loss factor (tan δ = E″/E′) as a function of temperature for processed PHBV (□) and its formulations with PLAP mass fractions of 0.1 (ring open red), 0.2 (triangle up open blue), and 0.3 (triangle down open green). The arrows indicate the presence of multiple glass transitions and their temperature shifts with the increase in the PLAP mass fraction.
To better understand how PLAP affects the glass transition, both tan δ and E″ curves were deconvoluted using Gaussian curves for each formulation, as shown in Figures 3 and S2 (Supporting Information), respectively. In general, for pure PHBV and the formulations with PLAP mass fractions equal to 0.1 and 0.2 (Figures 3a–c and S2a–c), two Gaussian curves described the experimental glass transition region. However, for the formulation with a PLAP mass fraction equal to 0.3 (Figures 3d and S2d), three Gaussian curves should be used for fitting the tan δ vs T and E″ vs T curves.
Figure 3.
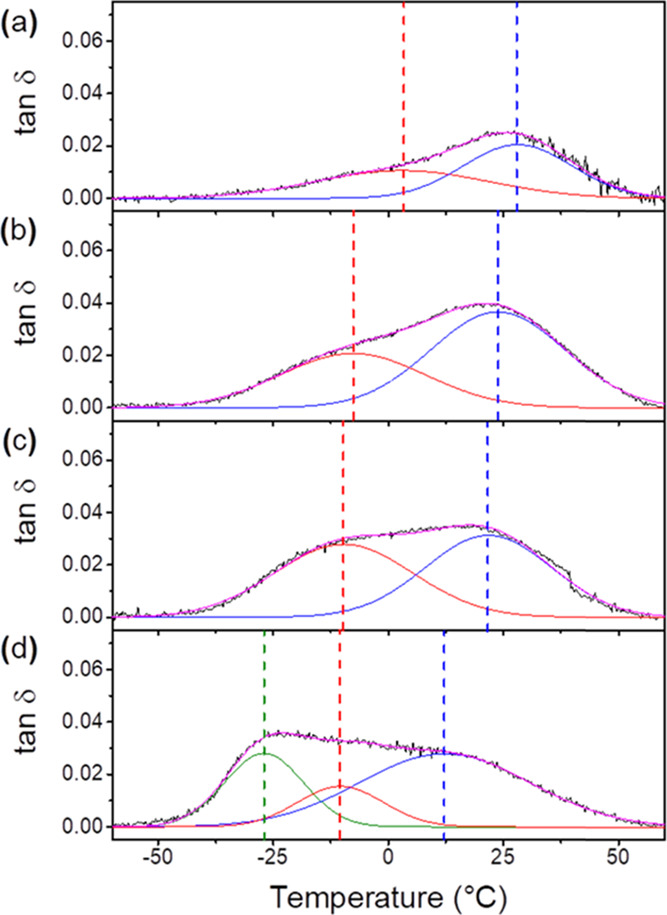
tan δ vs T curves (black line), Gaussians curves (blue and red lines) and the sum of the Gaussians (magenta line) for (a) pure PHBV and its formulations with PLAP mass fractions of (b) 0.1, (c) 0.2, and (d) 0.3.Gaussians curves for (a) pure PHBV and its formulations with PLAP mass fractions of (b) 0.1, (c) 0.2, and (d) 0.3.
Peaks at higher temperatures were attributed to the devitrification of the rigid amorphous phase, and peaks centered at lower temperatures were attributed to the glass transition of the mobile amorphous phase. The temperature of both events was denoted as Tg for convenience. Moreover, the tan δ and E″ curves (Figures 3 and S2, respectively) show that these transitions were partially overlapped. According to Esposito et al.,11 this overlap indicates that the boundaries of each phase are not well defined. Otherwise, there is a mobility gradient between the phases and the chain mobility increases progressively from the crystalline phase toward the mobile amorphous phase.
The Tg of the rigid and mobile amorphous phases were taken to be the temperatures corresponding to the maximum of the Gaussian curves in the tan δ and E″ curves (Tables S1 and S2), and they were plotted as a function of the PLAP mass fraction, Figures 4a and S4a (Supporting Information). As observed in Table S1 (Supporting Information), with the increase in the PLAP mass fraction in the formulations, the Tg of the rigid and mobile amorphous phases, determined from tan δ vs T curves, was shifted from 28 to 12 °C and from 3 to −10 °C, respectively. For the formulation with wPLAP = 0.3, the presence of a peak centered at −27 °C (Figure 3) suggests a third phase richer in PLAP, possibly resulting from phase separation. This phase was named the PLAP-rich mobile amorphous phase.
Figure 4.
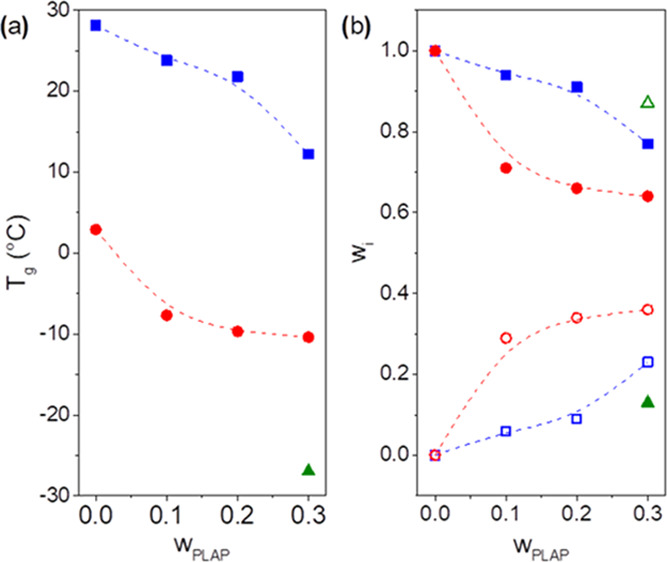
(a) Glass transition temperature and (b) the PHBV (closed symbols) and PLAP (open symbols) mass fractions in the amorphous phases as a function of the PLAP mass fraction in the formulations: the rigid (box solid, box blue), mobile (circle solid, ring open red), and PLAP-rich mobile amorphous phases (triangle up solid, triangle up open green). Data determined from tan δ vs T curves.
The Tg of the mobile and rigid amorphous phases determined from tan δ vs T curves (Table S1) presented a better correlation than Tg determined from E″ vs T curves (Table S2) with the Tg measured from the DSC (Table 3). Because the Fox equation79 was used to estimate the composition of mobile and rigid amorphous phases and for this, the Tg of PLAP should be known, we used the Tg of the amorphous phases for pure PHBV (3 and 28 °C, respectively) determined from the tan δ vs T curve and the Tg of the PLAP (−31 °C) determined by DSC. The glass transition temperature determined by DSC for the PHBV/PLAP formulations follows the Fox equation. Therefore, the composition of the mobile and rigid amorphous phases (Tables S1 and S2) was graphically predicted using the plot in Figure S3 (Supporting Information), which was constructed using the Fox equation, considering that this equation may appropriately describe the dependence of relaxation temperatures of the amorphous phases on the composition too. The Fox equation is the simplest model to predict the Tg of a formulation. It considers the additivity of the free volume of the polymer and of the diluent, which is the plasticizer in this case. This model does not account for polymer–plasticizer interactions, and it is not suitable for systems with strong interactions among the components.29 This may not be the case for the PHBV-PLAP mixture because dipole–dipole interactions predominate for polyester mixtures.68,80−82
Table 3. Glass Transition, Crystallization and Melting Temperatures, Crystallization and Melting Enthalpies, and the Degree of Crystallinity for Unprocessed PHBV and PLAP and for Processed PHBV and its Formulations with PLAP.
| 1st
heating |
cooling |
2nd
heating |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| wPLAP | Tm (°C) | ΔHmc (J g–1) | χc (%) | Tc (°C) | ΔHcd (J g–1) | Tg (°C) | Tcc (°C) | ΔHcce (J g–1) | Tm (°C) | ΔHmc (J g–1) | χc (%) |
| 0a | 174 | 78 | 53 | 60 | 40 | 3 | 44 | 9 | 170 | 82 | 56 |
| 0 | 173 | 80 | 55 | 60 | 19 | 2 | 43 | 31 | 169 | 85 | 58 |
| 0.1 | 170 | 75 | 56 | 58 | 35 | –1 | 42 | 11 | 168 | 78 | 59 |
| 0.2 | 174 | 67 | 56 | 48 | 11 | –4 | 42 | 26 | 167 | 70 | 58 |
| 0.3 | 171 | 63 | 61 | 59 | 20 | –9 | 42 | 16 | 166 | 63 | 61 |
| 1.0b | –31 | ||||||||||
Unprocessed PHBV.
Unprocessed PLAP.
Melting enthalpy.
Crystallization enthalpy.
Cold crystallization enthalpy.
The PHBV and PLAP mass fractions (wi) in the amorphous phase (i) for pure PHBV and its plasticized formulation determined using data from tan δ vs T and E″ vs T curves are presented in Figures 4b and S4b, respectively. Similar profiles were observed for Tg dependence on the composition determined from E″ vs T and tan δ vs T curves.
As observed in Figure 4b and Table S1, increasing the PLAP mass fraction up to 0.3 resulted in an increase of the PLAP mass fraction in the rigid amorphous phase from 0 to 0.23 and in the mobile amorphous phase from 0 to 0.36. The PLAP mass fraction in the mobile amorphous phase was always higher than in the rigid one, suggesting that PLAP is expelled not only from the crystalline phase38 but also from the rigid amorphous phase during sample aging. The phase separation took place for the formulation with a PLAP mass fraction equal to 0.3. This resulted in the rigid and two mobile amorphous phases with PLAP mass fractions of 0.23, 0.36, and 0.87, respectively (Table S1). Therefore, the PHBV/PLAP mixtures are partially miscible, presenting a single and homogeneous mobile amorphous phase for PLAP mass fractions up to 0.2 in the formulation and a critical PLAP concentration of 0.36. The low critical concentration for mixtures of PHB and PHBV and plasticizers may be related to their high degree of crystallinity, the presence of a rigid amorphous phase that accommodates only a small fraction of the plasticizer, and a relatively low fraction of the remaining mobile amorphous phase to accommodate the plasticizer. Physical aging also contributes to the enrichment of the amorphous phase with the plasticizer, which induces phase separation in PHB formulations.38 The low critical concentration for mixtures of PHB and PHBV and plasticizers, even for plasticizers presenting a Hildebrand solubility parameter similar to PHBV, has been reported in the literature.33,34,40,47 In general, it has been reported that PHB is immiscible with poly(lactic acid), PHBV, and other aliphatic polyesters.80,81 On the other hand, PHB is miscible with oligomeric poly(lactic acid) and other aliphatic polyesters in the melt, and it is immiscible or partially miscible upon crystallization.68,82
Regarding the effect of the plasticizer on the microstructure, Crétois et al.47 and Kurusu et al.34 reported only one α-relaxation for PHB plasticized with tri(ethylene glycol) bis(2-ethyl hexanoate) (TEG) and a decrease in the Tg from 20 to 3 °C by the addition of the plasticizer. However, by performing annealing and/or processing of the material, phase separation took place in the amorphous phase with the formation of a pure PHB phase and a rigid and mobile PHB/TEG amorphous phase. El-Taweel et al.64 studied the effect of the addition of oligomeric PHB-diol on both amorphous and semicrystalline PHB samples. They reported that Tg,sample ≈ Tg,Fox for amorphous samples because the PHB-diol is homogeneously distributed over the entire sample. However, Tg-sample < Tg,Fox for the semicrystalline samples because the PHB-diol was expelled from the crystalline phase during the crystallization, and an amorphous phase richer in PHB-diol was formed. They estimated the composition of these phases by determining the Tg of the amorphous phases and combining them with the Fox equation. In general, by increasing the amount of PHB-diol from 10 to 70%, their fraction in the mobile amorphous phase was higher than in the rigid one. This is similar to our results for PHBV plasticized with PLAP.
The influence of the PLAP content on the long period (Lp), crystalline lamellae (lc), and interlamellar amorphous layer (la) thickness of aged specimens and films freshly crystallized at 70 °C for 1 h was investigated by SAXS. The results are presented in Figure 5. The Lorentz-corrected curves, the correlation functions, and the summarized data are presented in Figures S5 and S6 and Table S3 (Supporting Information), respectively.
Figure 5.
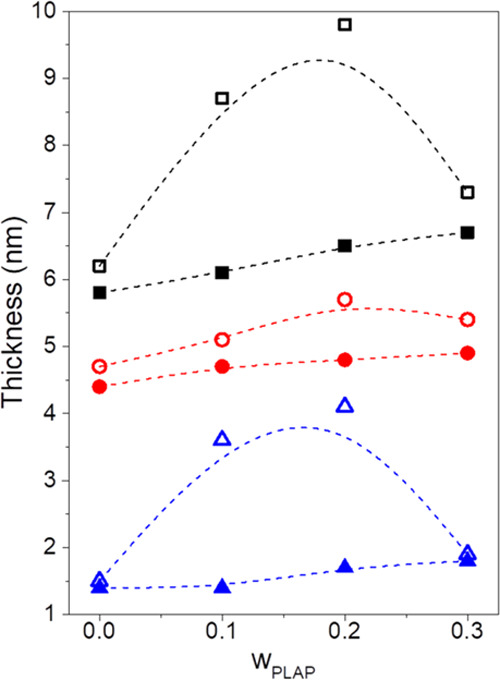
Long period (■), and thickness of crystalline lamellae (circle solid red) and the interlamellar amorphous layer (triangle up solid blue) as a function of the PLAP mass fraction for aged films (closed symbols) and films freshly crystallized at 70 °C for 1 h (open symbols).
Regarding aged samples, the progressive increase in the PLAP content up to wPLAP = 0.3 resulted in a slight increase of the Lp from 5.8 to 6.7 nm, in the lc from 4.4 to 4.9 nm, and in the la from 1.4 to 1.8 nm. For films freshly crystallized at 70 °C for 1 h, Lp and la increased from 6.2 to 9.8 nm and from 1.5 to 4.1 nm, respectively, with the increase in the PLAP mass fraction up to 0.2. However, a smaller increase of the lc from 4.7 to 5.7 nm was observed. A further increase in the PLAP mass fraction to wPLAP = 0.3 led to a decrease in Lp, lc, and la to 7.3, 5.4, and 1.9 nm, respectively. This is probably due to phase separation, which led to the formation of amorphous domains with sizes out of the limit of detection range of the SAXS analysis, as will be discussed further in the POM analysis. These results for the recrystallized samples are in agreement with those reported by Ambrosi et al.67 for PHB samples plasticized with oligomeric Pluronic (F68 and F127). They reported that a complete incorporation of the plasticizer in the lamellar stack would be followed by a monotonic increase of Lp and la, as observed for the recrystallized samples with PLAP mass fractions up to 0.2. However, when compared to the pure PHB, slight changes in the morphological parameters upon the addition of the plasticizer, as observed for the recrystallized sample with wPLAP = 0.3, indicate the presence of amorphous domains richer in the plasticizer outside the lamellar stacks that are not detected by SAXS. Comparing the aged films and those freshly crystallized at 70 °C, the smaller Lp, lc, and la for aged samples can be attributed to PHBV secondary crystallization that creates thinner crystalline lamellae within the interlamellar amorphous phase.83
Morphology
Solvent-cast films of unprocessed PHBV, processed PHBV, and its formulations with PLAP were isothermally crystallized from the melt at 55, 65, and 75 °C for 60 min and analyzed by POM. The POM images are presented in Figure 6.
Figure 6.

(a) POM images of solvent-cast samples of unprocessed PHBV, processed PHBV, and its formulations with PLAP isothermally crystallized at 55, 65, and 75 °C. (b) Zoomed-in view of the region (highlighted in yellow) of the spherulites in the formulation with wPLAP = 0.30 crystalized at 55 °C (left) and 65 °C (right) with arrows indicating the presence of circular dark spots.
In general, the spherulites are large with the characteristic Maltese cross and with or without the presence of bands. Generally, the spherulite size tends to increase with a decrease in the nucleation rate and/or an increase in the growth rate, which occurs by decreasing the supercooling degree (ΔT = Tm – Tic, where Tm is the melting temperature and Tic is the isothermal crystallization temperature).33,75 For a given crystallization temperature (horizontal sequence of POM images in Figure 6a), the size of the spherulites is little affected by processing or PLAP content because Tm is slightly affected by the presence of the plasticizer (as will be discussed further in the Thermal Properties section) and the degree of supercooling is maintained to be almost constant. This result is quite different from that observed for PHB plasticized with TEC, for which the spherulite diameter presented a remarkable increase with the increase in the TEC mass fraction due to the decrease in the supercooling degree,75 On the other hand, for a given composition (vertical sequence), an increase in the crystallization temperature led to an increase in the spherulite size. This was due to a decrease in the degree of supercooling. Other works also reported that by varying the crystallization temperature between the Tg and Tm range, the highest crystallization growth rate is around 75–85 °C for PHB, PHBV, and plasticized formulations.17,25,33,75 For the formulation with wPLAP = 0.3, some circular dark spots in the microscopic scale are observed throughout the images (Figure 6b). These are probably due to the presence of an amorphous phase resulting from phase separation, as discussed above. The PHBV/PLAP formulations formed spherulites large enough to be observed by the naked eye. Digital photographs of the spherulites are shown in Figure S7 (Supporting Information).
Thermal Properties
The influence of the PLAP mass fraction on PHBV thermal properties was investigated by DSC. The DSC curves for the first heating, cooling, and second heating scans are presented in Figure 7a–c, respectively. The phase transition temperatures, the enthalpies, and the crystallization degree (χc) are summarized in Table 3.
Figure 7.

(a) First heating, (b) cooling, and (c) second heating DSC scans of unprocessed PHBV (□) and processed PHBV (○) and its formulations with PLAP mass fractions of 0.1 (triangle up open red), 0.2 (triangle down open blue), and 0.3 (tilted square open green).
The first heating scan (Figure 7a) reflected not only the intrinsic properties of the formulations but also their thermal history, such as the processing and aging of the samples. By increasing the PLAP mass fraction, the Tm for the first scan varied randomly within a narrow range of 4 °C, and χc tended to slightly increase in the range from 55 to 61%. However, the melting peak in the first scan was broad with a shoulder at lower temperatures, which suggests recrystallization during heating. The χc was slightly smaller than in the second scan.
In the second heating scan (Figure 7c), the glass transition and the melting temperatures of the processed samples systematically decreased from 2 to −9 °C and from 169 to 166 °C, respectively, with the increase in the PLAP mass fraction up to 0.3, evidencing the capacity of PLAP to act as a plasticizer for PHBV. For the formulation with wPLAP = 0.3, despite the phase separation, only one glass transition could be observed by DSC, probably because of the overlap of the glass transition of two mobile amorphous phases. For the plasticized PHBV, ΔHm decreased due to the increase in the PLAP mass fraction. However, the degree of crystallinity (χc) was constant at around 58–61%. Generally, the plasticizer caused either a decrease in χc due to the dilution effect or an increase in χc due to the decrease in melting viscosity, which results in higher chain diffusion and a faster crystallization rate. In the cooling step (Figure 7b), PHBV crystallized at the same peak temperature (Tc = 60 °C) for the pure polymer and for polymer plasticized with PLAP at a mass fraction of 0.1. A further increase in the plasticizer to wPLAP = 0.2 caused the Tc to decrease to 48 °C. Concerning the degree of crystallinity, the addition of PLAP at 0.1 and 0.2 mass fractions caused an increase and a decrease, respectively (Table 3). Therefore, the concentration of PLAP had a complex influence on the crystallization kinetics of PHBV. For the formulation with wPLAP = 0.3, due to the phase separation, the crystallization occurred at Tc = 59 °C and ΔHc = 20 J g–1, similar to the crystallization of the PHBV processed in the absence of the plasticizer. The cold crystallization in the second heating scan was around 42 °C for all formulations, and the values of ΔHcc followed the inverse tendency of ΔHc, i.e., the fraction of PHBV that did not crystallize during cooling crystallized further in the second heating scan. This behavior can be attributed to the presence of a rigid amorphous phase. In semicrystalline polymers crystallized from the melt, the presence of a rigid amorphous phase makes the crystallization under cooling difficult. However, the rigid amorphous phase starts to gain some mobility as the sample is heated, and at the end of the glass transition (around 40–50 °C, Figure 3), cold crystallization will occur. This crystallization generates a new rigid amorphous phase layer that will further gain mobility and crystallize at higher temperatures, between the cold crystallization and the melting temperatures.9,12,84 Therefore, in the temperature range between cold crystallization and the melting temperature, the low intense exothermic peak observed in the second heating scan (highlighted by the dashed lines in Figure 7c) was attributed to crystallization of the rigid amorphous phase.12 This thermal event overlapped with the beginning of melting, which imparts an uncertainty to the calculated degree of crystallization.
The Flory–Huggins interaction parameter (χ1,2) was estimated from the melting point depression using the Nishi–Wang equations, eq S5 (Supporting Information).85 Conventionally, the use of the Nishi–Wang equation requires the determination of the equilibrium melting temperature provided by Hoffman–Weeks plots.86 However, PHB and PHBV are highly susceptible to thermal degradation at temperatures close to Tm, making the use of this procedure impracticable.87 Therefore, an estimate of the value of χ1,2 was performed as suggested by Pizzoli et al.33 for PHB and for PHB plasticized with di-n-butyl phthalate (DBP) using the nonequilibrium melting peak temperatures determined by DSC. The χ1,2 values were dependent on the composition of the plasticized formulations. However, they were negative for all cases, indicating miscibility between PHBV and PLAP, Figure S8 (Supporting Information). The Flory–Huggins interaction parameter for the formulation with PLAP mass fractions equal to 0.1, 0.2, and 0.3 were χ1.2 = −0.299, −0.147, and −0.081, respectively. This tendency can be due to the fact that the Nishi–Wang equation does not consider the enthalpic effects on the entropy of the mixture, and the occurrence of phenomena, such as recrystallization and phase separation, are not considered.22,88 Flory–Huggins interaction parameters were already reported in the literature for other PHB/plasticizer systems. For example, Pizzoli et al.33 reported χ1.2 = −0.1 for the PHB plasticized with DBP, and Saad89 reported χ1.2 = −0.48 for mixtures of PHB and oligomeric PHB-diol.
By plotting χ1.2 as a function of volume fraction of PLAP (øPLAP), Figure 8, the critical PLAP content of øPLAP = 0.38 (wPLAP = 0.37) was obtained by extrapolation to χ1.2 = 0.00. This value is close to the value of wPLAP = 0.36 estimated from DMA data and the Fox equation.
Figure 8.
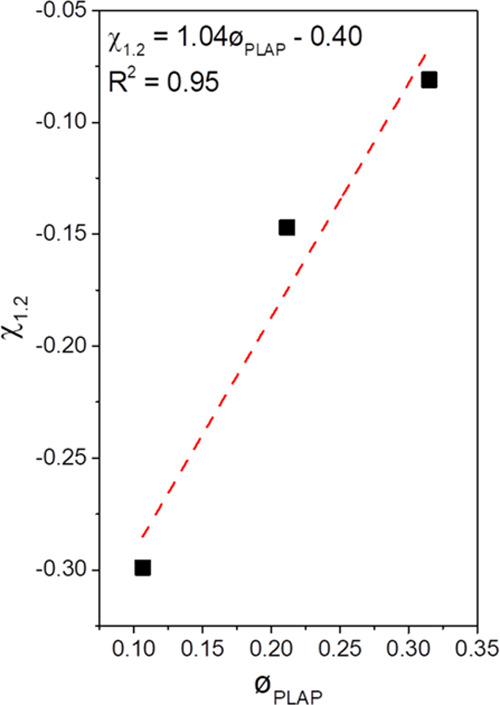
Composition-dependence of the Flory–Huggins interaction parameter (■) and linear fit (--).
Mechanical Properties
The influence of the PLAP content and its distribution in the mobile and rigid amorphous phases on the mechanical properties of the formulations was investigated by analyzing the impact resistance capability, elastic modulus, tensile strength, and elongation at break. The results are presented in Figure 9a–c and summarized in Table S4 (Supporting Information).
Figure 9.

(a) Impact resistance, (b) elastic modulus, (c) tensile strength, and elongation at break as a function of the PLAP mass fraction in the formulations.
The addition of PLAP to PHBV at a mass fraction up to 0.2 resulted in an impact resistance increase of 120% and elastic modulus and tensile strength decrease of 27%, while the elongation at break remained constant around 8.0–8.2%. This means that PLAP improves the capacity to dissipate mechanical energy, decreases the PHBV stiffness, and slightly improves the ductility. These results can be explained based on the microstructure of the samples. As reported by Righetti et al.,10 both the elastic modulus and tensile strength are proportional to the sum of the fraction of crystalline and rigid amorphous phases, whereas the elongation at break is proportional to the mass fraction of the mobile amorphous phase. The composition of the mobile and rigid amorphous phase progressively changed as the PLAP mass fraction increased. As shown in Figure 4a, this led to a progressive shift of the Tg of the rigid amorphous phase from 28 to 12 °C with the increase in the PLAP mass fraction explaining the observed decrease in the stiffness of the formulations at 25 °C (the temperature at which mechanical tests were performed). Simultaneously, the PLAP mass fraction in the mobile amorphous phase in the rubbery state at 25 °C increased with the PLAP mass fraction in the formulations. Consequently, an increase in the impact resistance was observed. However, the ductility of the material was little affected because the major fraction of the material is still in the crystalline phase, which remains in a rigid state.
For the formulations with wPLAP = 0.3, the impact resistance did not show a further increase and the elongation at break decreased to 6.6%. This was due to the presence of an amorphous phase richer in PLAP resulting from the phase separation. This phase should be mechanically fragile and responsible for the decrease in the cohesive energy between the amorphous phases, facilitating crack propagation.90 Ambrosi et al.67 also reported a decrease in the tensile strength and in the elongation at break for formulations of PHB/Pluronic where phase separation and a microstructure composed of poorly defined spherulites were observed. A loss in the mechanical properties of plasticized PHB (or PHBV) formulations upon an increase in the plasticizer content is frequently reported in the literature.24,32,39,51,52 This tendency of the mechanical properties to deteriorate with an increase in the plasticizer content is usually attributed to the low solubility/miscibility of the plasticizer and/or to plasticizer phase separation. However, in these studies, just one Tg was observed in the DSC curves, and no conclusive investigation about the miscibility or the plasticizer distribution in the microstructure was performed.
The efficiency of PLAP to decrease Tg and Tm and to tune the mechanical properties of the formulations was compared with other oligomeric and low molar mass plasticizers reported in the literature at the same mass fractions. A short review about the efficiency of plasticizers for PHB and PHBV can be found in Tables S5 and S6. Compared to other oligomeric plasticizers, PLAP acts to decrease Tg, Tm, and the elastic modulus and to increase the elongation at break similarly to poly[di(ethylene glycol) adipate],46 poly(caprolactone)-triol,69 Pluronic F68 and F127,67 Laprol 503 and 5003,48 TolonateXFLO100,70 and poly(ethylene glycol) (PEG) with the molar mass in the range from 0.2 to 6.0 kDa.24,39,46,48,50 For these plasticizers, a maximum decrease in Tg of around 15 °C, Tm in the range of 3–8 °C, the elastic modulus in the range from 35 to 60%, and an increase in the elongation at break lower than 10% have been reported. On the other hand, low molar mass plasticizers such as phthalates,32 citrates,31,35,75 glycerol esters,32,43,45 vegetable oils,52,54 terpenes,57 and other esters31,32,35,48 are more effective in decreasing Tg and Tm and in tuning the mechanical properties of formulations when compared to the oligomeric plasticizers. Using low molar mass plasticizers, a decrease in the range of 20–40 °C in Tg, around 10–20 °C in Tm, and from 60 to 80% in the elastic modulus, and increase of the elongation at break of more than 10% are frequently reported. However, low molar mass plasticizers have some disadvantages such as higher volatility, lower resistance to migration, and a higher tendency to be exuded when compared to the oligomeric plasticizers. This makes oligomeric plasticizers more suitable for high-performance applications.91
Conclusions
PLAP, an oligomeric polyester based on lactic acid, adipic acid, and 1,2-propanediol with a number average molar mass of 6.5 kDa, acted as a plasticizer for PHBV. It improved the processability in the melt and preserved the polymer against thermomechanical degradation. Moreover, the addition of PLAP resulted in a decrease in the glass transition and melting temperatures and the elastic modulus and an increase in the impact resistance. Dynamic dynamical analysis revealed the complexity of the amorphous phase of the formulations and, combined with the Fox equation, allowed an estimation of the composition of the mobile and rigid amorphous phases. PLAP was heterogeneously distributed in these phases. Despite this, the Flory–Huggins interaction parameter was negative and varied with the formulation composition in the range of −0.299 to −0.081. The critical PLAP mass fraction of 0.37 was obtained from thermodynamic data, and it was close to the value estimated from the DMA data and the Fox equation, indicating that this estimation was reasonable. PLAP contents also influenced the microstructure of the semicrystalline PHBV, progressively increasing the thickness of both the crystalline lamellae and the interlamellar layer. The mechanical properties showed a close relationship with the distribution of PLAP in the rigid and mobile amorphous phases as well as with the microstructure of the crystalline phase of PHBV in the formulations. The progressive shift of the Tg of the rigid amorphous phase from 28 to 12 °C with the increase in the PLAP mass fraction was responsible for the decrease in the stiffness and for the increase in the impact resistance of the plasticized PHBV. Ductility was not changed, because of the high degree of crystallization, even for plasticized PHBV. Overall, PLAP acted as an effective plasticizer. It decreased Tg, Tm, and the elastic modulus and increased the elongation at break. Also, its efficiency was comparable with other oligomeric plasticizers reported in the literature. This work demonstrated that the knowledge of plasticizer distribution in the mobile and rigid amorphous phases and how this affects the thermal properties of the amorphous phases is of great importance for understanding and finely tuning the mechanical properties of PHB and PHBV formulations.
Experimental Section
Materials
Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV) with a valerate content of around 3.0 mol % (determined by 1H and 13C RMN, Figure S9—Supporting Information) was kindly supplied by PHB Industrial S/A. PHBV was dried at 70 °C for 24 h before use. The random polyester based on lactic acid, adipic acid, and 1,2-propanediol (PLAP) at a molar ratio of 20:40:40 (determined by 1H and 13C RMN, Figure S10—Supporting Information) is an amorphous oligomer (Tg = −31 °C) with an average molar mass of 6.5 kDa and molar mass dispersity of 2.6. Chloroform (99.9%, LabSynth) and CDCl3 (99.8% D atom, 1% TMS (v/v) Sigma-Aldrich) were used without any further treatment.
Preparation of PHBV/PLAP Formulations
PHBV with PLAP in mass fractions of 0.1, 0.2, and 0.3 (total mass of 12.0 g for each batch) were manually premixed (24 h before processing). This was followed by processing in a laboratory twin screw extruder, DSM Xplore Microcompounder (length to diameter ratio equal to 18, volume capacity of 15 cm3). The formulations were fed at 170 °C and 150 rpm and the molten mixtures were compounded for 1 min at 170 °C at a screw speed of 250 rpm. The specimens were injection molded from the molten formulations into tensile (ASTM D638-14, specimen type V) and impact resistance (ASTM D286-10) test bars in a laboratory-scale injection machine, DSM Micro Injection Molder (volume capacity = 12 cm3), with the following parameters: barrel temperature of 170 °C, mold temperature of 60 °C, cooling time of 10 s, and initial and hold pressures of 4 and 4.5 bars, respectively. The specimens were stored in a desiccator at room temperature for at least 1 month to minimize the effects of aging on the results, as reported in our previous work.38
Characterization
The number average molar mass (Mn), mass average molar mass (Mw), and the molar mass dispersity (Đ) of PHBV and its plasticized formulations were determined by gel permeation chromatography (GPC) performed on a Viscotek GPCmax VE2001 instrument equipped with three columns (Shodex K-802, K-803, and K804) operating at 40 °C and using chloroform as the eluent at a flow rate of 0.5 mL min–1. The detection was performed using a Viscotek VE3580 refractive index detector. Solutions in chloroform were prepared at a concentration of 5.0 mg mL–1, and they were filtered in poly(tetrafluoroethylene) (PTFE) filters (0.45 μm) before analysis. Polystyrene standards (Viscotek) with molar masses ranging from 935 to 1 790 000 g mol–1 were used to determine the relative molar mass of the samples. OmniSEC. 4.6.2 software (Viscotec, Malvern) was used for data collection and processing.
Proton nuclear magnetic resonance (1H NMR) analyses were performed on a Bruker Avance 500 MHz spectrometer operating at 25 °C with acquisition parameters of 1.6 s acquisition time, 1.0 s recycle delay, spectra width of 10 302 Hz, 16 scans, 32 000 points, and a free induction decay (FID) resolution of 0.63 Hz. Polymer solutions of ca. 10 mg mL–1 in chloroform-d1 were used. Chemical shifts (δ) in ppm were assigned to the TMS signal at δ = 0.00 ppm. Carbon-13 nuclear magnetic resonance (13C NMR) analyses were performed on a Bruker Avance 500 MHz spectrometer operating at 25 °C with acquisition parameters of 1.0 s acquisition time, 60 s recycle delay, the spectral width of 32 894 Hz, 960 scans, 64 k points, and an FID resolution of 1.0 Hz without nuclear Overhauser enhancement. Polymer solutions of ca. 50 mg mL–1 in chloroform-d1 were used. Chemical shifts in ppm were assigned to the residual solvent proton of the chloroform at δ = 77.16 ppm.
The thermal stabilities of PHBV and its formulations were evaluated by thermogravimetric analysis (TGA) using a TGA 2950 TA Instruments thermobalance under an argon atmosphere (flow of 100 mL min–1). The samples of 5–10 mg were heated from 30 to 600 °C at a heating rate of 10 °C min–1.
Dynamic mechanical analysis (DMA) of rectangular specimens with dimensions of 25 mm × 10 mm × 3 mm was performed on DMTA V equipment (Rheometric Scientific) operating in a single cantilever configuration with the following conditions: the temperature range from −140 to 175 °C, 2 °C min–1 heating rate, 0.05 % strain, a frequency of 1 Hz, and 8 mm gap between clamps. Fityk73 software was used to deconvolute the loss modulus and loss factor curves by adjusting the baseline and fitting the signals with Gaussian functions and the Lev-Mar method.
Thin films were prepared by solvent casting from a 40 mg mL–1 solution in chloroform followed by melting at 185 °C, compression between glass slides, and quenching to 55, 65, or 75 °C for isothermal crystallization over 60 min. These films were analyzed by polarized optical microscopy (POM) using a Nikon 80i optical microscope. These films were also put between two polarizers, and images of the spherulites were captured using a digital camera.
Small-angle X-ray scattering (SAXS) experiments were performed on the D01ASAXS2 beamline of the Brazilian Synchrotron Light Laboratory (LNLS; Campinas, Brazil) using films freshly crystallized at 70 °C for 1 h according to the procedure described for films prepared for POM analyses and films from the injection-molded specimens aged for at least 1 month. The specimens were placed between two mica sheets and subjected to synchrotron light radiation of 0.1488 nm wavelength for 30 s. The scattering vector (q = (4π/λ)senθ) evaluated in SAXS measurement ranged from 0.01 to 5 nm–1. Using the Fit2D program, the scattering patterns were radially averaged and subtracted from the background. The long period (Lp), the crystalline lamella (lc), and the amorphous layer (la) were determined using SAXDAT software.
The thermal properties of PHBV and its formulations were determined by differential scanning calorimetry (DSC) on a DSC Q2000, TA Instruments (New Castle, DE). The samples, 5–10 mg, were hermetically sealed in aluminum pans and analyzed according to the following program: (i) equilibrium at 25 °C; (ii) heating to 200 °C at 20 °C min–1; (iii) isotherm at 200 °C for 2 min; (iv) cooling to −150 °C at 20 °C min–1; (v) isotherm at −150 °C for 2 min; and (v) heating to 200 °C at 20 °C min–1.
The PHBV degree of crystallinity (χc) was calculated by the ratio of the experimental melting enthalpy (ΔHm) and the melting enthalpy of completely crystalline PHB (ΔHmo = 146 J g–1)4 multiplied by the PHB mass fraction, eq 1
| 1 |
Tensile tests were conducted on an Instron series EMIC 23–20 universal testing machine with a load cell of 500 N and a rate of 5 mm min–1. Specimens of Type V were conditioned for 72 h at 25 °C and 50% moisture before testing according to ASTM D638-14. The Izod impact resistance test of notched injection-molded specimens was conducted using EMIC AIC-1 equipment with a 2.7 J hammer. Tests were performed according to ASTM D256-10 method E, in which the hammer impacts the side of the specimen as opposed to the notched one.
Acknowledgments
The authors would like to thank FAPESP (Grants #2018/19091-0, #2018/01562-6, and #2015/25406–5) and Capes (Finance Code “001”) for the financial funding, and M.Sc. Airton Siqueira for supplying the plasticizer PLAP.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c05765.
Thermogravimetric curves and their derivative curves; deconvolution of DMA curves; estimation of the PLAP composition in amorphous phases; SAXS analysis; digital images of the spherulites; determination of the Flory–Huggins interaction parameter; mechanical properties; efficiency of plasticizers—a short review; 1H and 13C NMR spectra of PHBV; and 1H and 13C NMR spectra of PLAP (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Sudesh K.; Abe H.; Doi Y. Synthesis, Structure and Properties of Polyhydroxyalkanoates: Biological Polyesters. Prog. Polym. Sci. 2000, 25, 1503–1555. 10.1016/S0079-6700(00)00035-6. [DOI] [Google Scholar]
- Chodak I.Polyhydroxyalkanoates: Origin, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Belgacem M. N.; Gandini A., Eds.; Pergamon Press, Elsevier Ltd.: Oxford, 2008; chapter 2, pp 451–477. [Google Scholar]
- Liu Q.; Zhang H.; Deng B.; Zhao X. Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate): Structure, Property, and Fiber. Int. J. Polym. Sci. 2014, 374368 10.1155/2014/374368. [DOI] [Google Scholar]
- Barham P. J.; Keller A.; Otun E. L.; Holmes P. A. Crystallization and Morphology of a Bacterial Thermoplastic: Poly-3-Hydroxybutyrate. J. Mater. Sci. 1984, 19, 2781–2794. 10.1007/BF01026954. [DOI] [Google Scholar]
- Barham P. J.; Barker P.; Organ S. J. Physical Properties of Poly(Hydroxybutyrate) and Copolymers of Hydroxybutyrate and Hydroxyvalerate. FEMS Microbiol. Lett. 1992, 103, 289–298. 10.1016/0378-1097(92)90322-F. [DOI] [Google Scholar]
- Di Lorenzo M. L.; Androsch R.. Crystallization of Poly[(R)-3-Hydroxybutyrate]. In Thermal Properties of Bio-based Polymers, Advances in Polymer Science; Di Lorenzo M. L.; Androsch R., Eds.; Springer Nature: Switzerland AG: Cham, 2019; Vol. 283, pp 119–142. [Google Scholar]
- Pachekoski W. M.; Dalmolin C.; Agnelli J. A. M. The Influence of the Industrial Processing on the Degradation of Poly(Hydroxybutyrate) - PHB. Mater. Res. 2013, 16, 327–332. 10.1590/S1516-14392012005000180. [DOI] [Google Scholar]
- Renstad R.; Karlsson S.; Albertsson A. C.; Werner P. E.; Westdahl M. Influence of Processing Parameters on the Mass Crystallinity of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate). Polym. Int. 1999, 43, 201–209. 10.1002/(sici)1097-0126(199707)43:33.0.co;2-7. [DOI] [Google Scholar]
- Righetti M. C.; Tombari E.; Di Lorenzo M. L. The Role of the Crystallization Temperature on the Nanophase Structure Evolution of Poly[(R)-3-Hydroxybutyrate]. J. Phys. Chem. B 2013, 117, 12303–12311. 10.1021/jp4063127. [DOI] [PubMed] [Google Scholar]
- Righetti M. C.; Aliotta L.; Mallegni N.; Gazzano M.; Passaglia E.; Cinelli P.; Lazzeri A. Constrained Amorphous Interphase and Mechanical Properties of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate). Front. Chem. 2019, 7, 1–16. 10.3389/fchem.2019.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito A.; Delpouve N.; Causin V.; Dhotel A.; Delbreilh L.; Dargent E. From a Three-Phase Model to a Continuous Description of Molecular Mobility in Semicrystalline Poly(Hydroxybutyrate-Co-Hydroxyvalerate). Macromolecules 2016, 49, 4850–4861. 10.1021/acs.macromol.6b00384. [DOI] [Google Scholar]
- Di Lorenzo M. L.; Righetti M. C. Evolution of Crystal and Amorphous Fractions of Poly[(R)-3-Hydroxybutyrate] upon Storage. J. Therm. Anal. Calorim. 2013, 112, 1439–1446. 10.1007/s10973-012-2734-3. [DOI] [Google Scholar]
- Adamus G.; Sikorska W.; Janeczek H.; Kwiecień M.; Sobota M.; Kowalczuk M. Novel Block Copolymers of Atactic PHB with Natural PHA for Cardiovascular Engineering: Synthesis and Characterization. Eur. Polym. J 2012, 48, 621–631. 10.1016/j.eurpolymj.2011.12.017. [DOI] [Google Scholar]
- Don T. M.; Liao K. H. Studies on the Alcoholysis of Poly(3-Hydroxybutyrate) and the Synthesis of PHB-b-PLA Block Copolymer for the Preparation of PLA/PHB-b-PLA Blends. J. Polym. Res. 2018, 25, 1–10. 10.1007/s10965-017-1432-z. [DOI] [Google Scholar]
- Bergamaschi J. M.; Pilau E. J.; Gozzo F. C.; Felisberti M. I. Synthesis of Polyurethane from Poly(3-Hydroxybutyrate) and Poly(p-Dioxanone): Molar Mass Reduction via Sodium Borohydrate. Macromol. Symp. 2011, 299–300, 10–19. 10.1002/masy.200900103. [DOI] [Google Scholar]
- Savenkova L.; Gercberga Z.; Bibers I.; Kalnin M. Effect of 3-Hydroxy Valerate Content on Some Physical and Mechanical Properties of Polyhydroxyalkanoates Produced by Azotobacter chroococcum. Process Biochem. 2000, 36, 445–450. 10.1016/S0032-9592(00)00235-1. [DOI] [Google Scholar]
- You J.-W.; Chiu H.-J.; Shu W.-J.; Don T.-M. Influence of Hydroxyvalerate Content on the Crystallization Kinetics of Poly(Hydroxybutyrate-Co-Hydroxyvalerate). J. Polym. Res. 2003, 10, 47–54. 10.1023/A:1023958014221. [DOI] [Google Scholar]
- Zhijiang Z.; Guoxiang C.; Mingming G.; Zhiyuan T. Surface Modification of Poly(3-Hydroxybutyrate) (PHB) by Photografting and Its Properties Evaluation. J. Polym. Res. 2004, 11, 99–104. 10.1023/B:JPOL.0000031065.88526.92. [DOI] [Google Scholar]
- Hong S.-G.; Lin Y.-C.; Lin C.-H. Improvement of the Thermal Stability of Polyhydroxybutyrates by Grafting with Maleic Anhydride by Different Methods: Differential Scanning Calorimetry, Thermogravimetric Analysis, and Gel Permeation Chromatography. J. Appl. Polym. Sci. 2008, 110, 2718–2726. 10.1002/app.28782. [DOI] [Google Scholar]
- Antunes M. C. M.; Felisberti M. I. Blends of Poly(Hydroxybutyrate) and Poly (ε-Caprolactone) Obtained from Melting Mixture. Polímeros 2005, 15, 134–138. 10.1590/S0104-14282005000200014. [DOI] [Google Scholar]
- Ha C. S.; Cho W. J. Miscibility, Properties, and Biodegradability of Microbial Polyester Containing Blends. Prog. Polym. Sci. 2002, 27, 759–809. 10.1016/S0079-6700(01)00050-8. [DOI] [Google Scholar]
- Quental A. C.; De Carvalho F. P.; Dos Santos Tada E.; Felisberti M. I. Blendas de PHB e Seus Copolímeros: Miscibilidade e Compatibilidade. Quim. Nova 2010, 33, 438–446. 10.1590/S0100-40422010000200035. [DOI] [Google Scholar]
- Choi J. S.; Park W. H. Effect of Biodegradable Plasticizers on Thermal and Mechanical Properties of Poly(3-Hydroxybutyrate). Polym. Test. 2004, 23, 455–460. 10.1016/j.polymertesting.2003.09.005. [DOI] [Google Scholar]
- Jost V.; Langowski H. C. Effect of Different Plasticisers on the Mechanical and Barrier Properties of Extruded Cast PHBV Films. Eur. Polym. J. 2015, 68, 302–312. 10.1016/j.eurpolymj.2015.04.012. [DOI] [Google Scholar]
- Brunel D. G.; Pachekoski W. M.; Dalmolin C.; Agnelli J. A. M. Natural Additives for Poly (Hydroxybutyrate – CO - Hydroxyvalerate) – PHBV: Effect on Mechanical Properties and Biodegradation. Mater. Res. 2014, 17, 1145–1156. 10.1590/1516-1439.235613. [DOI] [Google Scholar]
- Scalioni L. V.; Gutiérrez M. C.; Felisberti M. I. Green Composites of Poly(3-Hydroxybutyrate) and Curaua Fibers: Morphology and Physical, Thermal, and Mechanical Properties. J. Appl. Polym. Sci. 2016, 134, 44676 10.1002/app.44676. [DOI] [Google Scholar]; 1–13
- Slongo M. D.; Brandolt S. D. F.; Daitx T. S.; Mauler R. S.; Giovanela M.; Crespo J. S.; Carli L. N. Comparison of the Effect of Plasticizers on PHBV—and Organoclay—Based Biodegradable Polymer Nanocomposites. J. Polym. Environ. 2018, 26, 2290–2299. 10.1007/s10924-017-1128-9. [DOI] [Google Scholar]
- Rahman M.; Brazel C. S. The Plasticizer Market: An Assessment of Traditional Plasticizers and Research Trends to Meet New Challenges. Prog. Polym. Sci. 2004, 29, 1223–1248. 10.1016/j.progpolymsci.2004.10.001. [DOI] [Google Scholar]
- Wypych G.Handbook of Plasticizers, 3rd ed.; Wypych G., Ed.; ChemTec Publishing: Toronto, 2017; Vol. 1. [Google Scholar]
- Wilkes C. A.; Summers J. W.; Daniels C. E.. Plasticizers. In PVC Handbook; Wilkes C. A.; Summers J. W.; Daniels C. E., Eds.; Hanser Gardner Publications: Munich, 2005. [Google Scholar]
- Wang L.; Zhu W.; Wang X.; Chen X.; Chen G.-Q.; Xu K. Processability Modifications of Poly(3-Hydroxybutyrate) by Plasticizing, Blending, and Stabilizing. J. Appl. Polym. Sci. 2008, 107, 166–173. 10.1002/app.27004. [DOI] [Google Scholar]
- Baltieri R. C.; Mei L. H. I.; Bartoli J. Study of the Influence of Plasticizers on the Thermal and Mechanical Properties of Poly(3-Hydroxybutyrate) Compounds. Macromol. Symp. 2003, 197, 33–44. 10.1002/masy.200350704. [DOI] [Google Scholar]
- Pizzoli M.; Scandola M.; Ceccorulli G. Crystallization and Melting of Isotactic Poly(3-Hydroxy Butyrate) in the Presence of a Low Molecular Weight Diluent. Macromolecules 2002, 35, 3937–3941. 10.1021/ma012122x. [DOI] [Google Scholar]
- Kurusu R. S.; Siliki C. A.; David É.; Demarquette N. R.; Gauthier C.; Chenal J. M. Incorporation of Plasticizers in Sugarcane-Based Poly(3-Hydroxybutyrate)(PHB): Changes in Microstructure and Properties through Ageing and Annealing. Ind. Crops Prod. 2015, 72, 166–174. 10.1016/j.indcrop.2014.12.040. [DOI] [Google Scholar]
- Kunze C.; Freier T.; Kramer S.; Schmitz K. P. Anti-Inflammatory Prodrugs as Plasticizers for Biodegradable Implant Materials Based on Poly(3-Hydroxybutyrate). J. Mater. Sci.: Mater. Med. 2002, 13, 1051–1055. 10.1023/A:1020392606225. [DOI] [PubMed] [Google Scholar]
- Râpă M.; Darie-ni R. N. Ţ. Ă.; Grosu E.; Tănase E. E.; Trifoi A. R.; Pap T.; Vasile C. Effect of Plasticizers on Melt Processability and Properties of PHB. J. Optoelectron. Adv. Mater. 2015, 17, 1778–1784. [Google Scholar]
- Erceg M.; Kovačić T.; Klarić I. Thermal Degradation of Poly(3-Hydroxybutyrate) Plasticized with Acetyl Tributyl Citrate. Polym. Degrad. Stab. 2005, 90, 313–318. 10.1016/j.polymdegradstab.2005.04.048. [DOI] [Google Scholar]
- Umemura R. T.; Felisberti M. I. Modeling of the Properties of Plasticized Poly(3-Hydroxybutyrate) as a Function of Aging Time and Plasticizer Content. Mater. Today Commun. 2020, 25, 101439 10.1016/j.mtcomm.2020.101439. [DOI] [Google Scholar]
- Requena R.; Jiménez A.; Vargas M.; Chiralt A. Effect of Plasticizers on Thermal and Physical Properties of Compression-Moulded Poly[(3-Hydroxybutyrate)-Co-(3-Hydroxyvalerate)] Films. Polym. Test. 2016, 56, 45–53. 10.1016/j.polymertesting.2016.09.022. [DOI] [Google Scholar]
- Yoshie N.; Nakasato K.; Fujiwara M.; Kasuya K.; Abe H.; Doi Y.; Inoue Y. Effect of Low Molecular Weight Additives on Enzymatic Degradation of Poly(3-Hydroxybutyrate). Polymer 2000, 41, 3227–3234. 10.1016/S0032-3861(99)00547-9. [DOI] [Google Scholar]
- Ashby R. D.; Solaiman D. K. Y.; Liu C. K.; Strahan G.; Latona N. Sophorolipid-Derived Unsaturated and Epoxy Fatty Acid Estolides as Plasticizers for Poly(3-Hydroxybutyrate). J. Am. Oil Chem. Soc. 2016, 93, 347–358. 10.1007/s11746-015-2772-7. [DOI] [Google Scholar]
- Abe H.; Doi Y.; Satkowski M. M.; Noda I. Morphology and Enzymatic Degradation of Poly[(R)-3-Hydroxybutyrate] Plasticized with Acylglycerols. Stud. Polym. Sci. 1994, 12, 591–595. 10.1016/b978-0-444-81708-2.50070-1. [DOI] [Google Scholar]
- Ishikawa K.; Kawaguchi Y.; Doi Y. Pasticization of Bacterial Polyester by the Addition of Acylglycerols and Its Enzymatic Degradability. Kobunshi Ronbunshu 1991, 48, 221–226. 10.1295/koron.48.221. [DOI] [Google Scholar]
- Nosal H.; Moser K.; Warzała M.; Holzer A.; Stańczyk D.; Sabura E. Selected Fatty Acids Esters as Potential PHB-V Bioplasticizers: Effect on Mechanical Properties of the Polymer. J. Polym. Environ. 2021, 29, 38–53. 10.1007/s10924-020-01841-5. [DOI] [Google Scholar]
- Seoane I. T.; Manfredi L. B.; Cyras V. P. Effect of Two Different Plasticizers on the Properties of Poly(3- Hydroxybutyrate) Binary and Ternary Blends. J. Appl. Polym. Sci. 2017, 135, 46016 10.1002/app.46016. [DOI] [Google Scholar]; 1–12
- Panaitescu D. M.; Nicolae C. A.; Frone A. N.; Chiulan I.; Stanescu P. O.; Draghici C.; Iorga M.; Mihailescu M. Plasticized Poly(3-Hydroxybutyrate) with Improved Melt Processing and Balanced Properties. J. Appl. Polym. Sci. 2017, 134, 1–14. 10.1002/app.44810. [DOI] [Google Scholar]
- Crétois R.; Chenal J. M.; Sheibat-Othman N.; Monnier A.; Martin C.; Astruz O.; Kurusu R.; Demarquette N. R. Physical Explanations about the Improvement of Polyhydroxybutyrate Ductility: Hidden Effect of Plasticizer on Physical Ageing. Polymer 2016, 102, 176–182. 10.1016/j.polymer.2016.09.017. [DOI] [Google Scholar]
- Bibers I.; Tupureina V.; Dzene A.; Kalnins M. Improvement of the Deformative Characteristics of Poly-β-Hydroxybutyrate by Plasticization. Mech. Compos. Mater. 1999, 35, 357–364. 10.1007/BF02259726. [DOI] [Google Scholar]
- Fernandes E. G.; Pietrini M.; Chiellini E. Thermo-Mechanical and Morphological Characterization of Plasticized Poly[(R)-3-Hydroxybutyric Acid]. Macromol. Symp. 2004, 218, 157–164. 10.1002/masy.200451416. [DOI] [Google Scholar]
- Martino L.; Berthet M. A.; Angellier-Coussy H.; Gontard N. Understanding External Plasticization of Melt Extruded PHBV-Wheat Straw Fibers Biodegradable Composites for Food Packaging. J. Appl. Polym. Sci. 2014, 132, 41611 10.1002/app.41611. [DOI] [Google Scholar]; 1–11
- Garcia-Garcia D.; Ferri J. M.; Montanes N.; Lopez-Martinez J.; Balart R. Plasticization Effects of Epoxidized Vegetable Oils on Mechanical Properties of Poly(3-Hydroxybutyrate). Polym. Int. 2016, 65, 1157–1164. 10.1002/pi.5164. [DOI] [Google Scholar]
- Garcia-Garcia D.; Fenollar O.; Fombuena V.; Lopez-Martinez J.; Balart R. Improvement of Mechanical Ductile Properties of Poly(3-Hydroxybutyrate) by Using Vegetable Oil Derivatives. Macromol. Mater. Eng. 2016, 302, 1600330 10.1002/mame.201600330. [DOI] [Google Scholar]; 1-12
- Özgür Seydibeyoğlu M.; Misra M.; Mohanty A. Synergistic Improvements in the Impact Strength and % Elongation of Polyhydroxybutyrate-Co-Valerate Copolymers with Functionalized Soybean Oils and Poss. Int. J. Plast. Technol. 2010, 14, 1–16. 10.1007/s12588-010-0005-3. [DOI] [Google Scholar]
- Choi J. S.; Park W. H. Thermal and Mechanical Properties of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate) Plasticized by Biodegradable Soybean Oils. Macromol. Symp. 2003, 197, 65–76. 10.1002/masy.200350707. [DOI] [Google Scholar]
- Audic J. L.; Lemiègre L.; Corre Y. M. Thermal and Mechanical Properties of a Polyhydroxyalkanoate Plasticized with Biobased Epoxidized Broccoli Oil. J. Appl. Polym. Sci. 2013, 131, 39983 10.1002/app.39983. [DOI] [Google Scholar]; 1–7
- Giaquinto C. D. M.; De Souza G. K. M.; Caetano V. F.; Vinhas G. M. Evaluation of the Mechanical and Thermal Properties of PHB/Canola Oil Films. Polímeros 2017, 27, 201–207. 10.1590/0104-1428.10716. [DOI] [Google Scholar]
- Mangeon C.; Michely L.; Rios De Anda A.; Thevenieau F.; Renard E.; Langlois V. Natural Terpenes Used as Plasticizers for Poly(3-Hydroxybutyrate). ACS Sustainable Chem. Eng. 2018, 6, 16160–16168. 10.1021/acssuschemeng.8b02896. [DOI] [Google Scholar]
- Jenkins M. J.; Fitzgerald A. V. L.; Kelly C. A. Reduction of Poly(Hydroxybutyrate-Co-Hydroxyvalerate) Secondary Crystallisation through Blending with Saccharides. Polym. Degrad. Stab. 2019, 159, 116–124. 10.1016/j.polymdegradstab.2018.11.020. [DOI] [Google Scholar]
- Kelly C. A.; Fitzgerald A. V. L.; Jenkins M. J. Control of the Secondary Crystallisation Process in Poly(Hydroxybutyrate-Co-Hydroxyvalerate) through the Incorporation of Poly(Ethylene Glycol). Polym. Degrad. Stab. 2018, 148, 67–74. 10.1016/j.polymdegradstab.2018.01.003. [DOI] [Google Scholar]
- Bibers I.; Tupureina V.; Dzene A.; Savenkova L.; Kalnins M. Biodegradable Materials from Plasticized PHB Biomass. Macromol. Symp. 2001, 170, 61–71. 10.1002/1521-3900(200106)170:13.0.CO;2-7. [DOI] [Google Scholar]
- Parra D. F.; Fusaro J.; Gaboardi F.; Rosa D. S. Influence of Poly(Ethylene Glycol) on the Thermal, Mechanical, Morphological, Physical-Chemical and Biodegradation Properties of Poly (3-Hydroxybutyrate). Polym. Degrad. Stab. 2006, 91, 1954–1959. 10.1016/j.polymdegradstab.2006.02.008. [DOI] [Google Scholar]
- Cal A. J.; Grubbs B.; Torres L. F.; Riiff T. J.; Kibblewhite R. E.; Orts W. J.; Lee C. C. Nucleation and Plasticization with Recycled Low-Molecular-Weight Poly-3-Hydroxybutyrate Toughens Virgin Poly-3-Hydroxybutyrate. J. Appl. Polym. Sci. 2018, 136, 47432 10.1002/app.47432. [DOI] [Google Scholar]; 1–7
- Hong S. G.; Hsu H. W.; Ye M. T. Thermal Properties and Applications of Low Molecular Weight Polyhydroxybutyrate. J. Therm. Anal. Calorim. 2013, 111, 1243–1250. 10.1007/s10973-012-2503-3. [DOI] [Google Scholar]
- El-Taweel S. H.; Höhne G. W. H.; Mansour A. A.; Stoll B.; Seliger H. Glass Transition and the Rigid Amorphous Phase in Semicrystalline Blends of Bacterial Polyhydroxybutyrate PHB with Low Molecular Mass Atactic R, S-PHB-Diol. Polymer 2004, 45, 983–992. 10.1016/j.polymer.2003.12.007. [DOI] [Google Scholar]
- El-Taweel S. H.; Stoll B.; Höhne G. W. H.; Mansour A. A.; Seliger H. Stress-Strain Behavior of Blends of Bacterial Polyhydroxybutyrate. J. Appl. Polym. Sci. 2004, 94, 2528–2537. 10.1002/app.21215. [DOI] [Google Scholar]
- Lukasiewicz B.; Basnett P.; Nigmatullin R.; Matharu R.; Knowles J. C.; Roy I. Binary Polyhydroxyalkanoate Systems for Soft Tissue Engineering. Acta Biomater. 2018, 71, 225–234. 10.1016/j.actbio.2018.02.027. [DOI] [PubMed] [Google Scholar]
- Ambrosi M.; Raudino M.; Diañez I.; Martínez I. Non-Isothermal Crystallization Kinetics and Morphology of Poly(3-Hydroxybutyrate)/Pluronic Blends. Eur. Polym. J. 2019, 120, 109189 10.1016/j.eurpolymj.2019.08.016. [DOI] [Google Scholar]
- Hsieh Y. T.; Woo E. M. Phase Diagrams in Blends of Poly(3-Hydroxybutyric Acid) with Various Aliphatic Polyesters. Express Polym. Lett. 2011, 5, 570–580. 10.3144/expresspolymlett.2011.56. [DOI] [Google Scholar]
- Wessler K.; Nishida M. H.; Da Silva J.; Pezzin A. P. T.; Pezzin S. H. Thermal Properties and Morphology of Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate) with Poly(Caprolactone Triol) Mixtures. Macromol. Symp. 2007, 245–246, 161–165. 10.1002/masy.20065132. [DOI] [Google Scholar]
- Reddy C. S.; O’Connor K.; Babu P. R. The Influence of Biobased Oligomeric Diisocyanate on Thermal and Mechanical Properties of Poly(3-Hydroxybutyrate). Macromol. Symp. 2016, 365, 223–229. 10.1002/masy.201650008. [DOI] [Google Scholar]
- You J. W.; Chiu H. J.; Don T. M. Spherulitic Morphology and Crystallization Kinetics of Melt-Miscible Blends of Poly(3-Hydroxybutyrate) with Low Molecular Weight Poly(Ethylene Oxide). Polymer 2003, 44, 4355–4362. 10.1016/S0032-3861(03)00348-3. [DOI] [Google Scholar]
- Silvino A. C.; Cruz J. Preparation of Blends of Oligo ([ R, S] -3-Hydroxybutyrate) and Poly ([ R]-3-Hydroxybutyrate): Thermal Properties and Molecular Dynamic Studies. Polym. Test. 2015, 42, 144–150. 10.1016/j.polymertesting.2015.01.013. [DOI] [Google Scholar]
- Wojdyr M. Fityk: A General-Purpose Peak Fitting Program. J. Appl. Crystallogr. 2010, 43, 1126–1128. 10.1107/S0021889810030499. [DOI] [Google Scholar]
- Grassie N.; Murray E. J.; Holmes P. A. The Thermal Degradation of Poly(-(d)-β-Hydroxybutyric Acid): Part 2-Changes in Molecular Weight. Polym. Degrad. Stab. 1984, 6, 95–103. 10.1016/0141-3910(84)90075-2. [DOI] [Google Scholar]
- Umemura R. T.; Felisberti M. I. Plasticization of Poly(3-Hydroxybutyrate) with Triethyl Citrate: Thermal and Mechanical Properties, Morphology, and Kinetics of Crystallization. J. Appl. Polym. Sci. 2021, 138, e49990 10.1002/app.49990. [DOI] [Google Scholar]
- Grassie N.; Murray E. J.; Holmes P. A. The Thermal Degradation of Poly(-(d)-β-Hydroxybutyric Acid): Part 1-Identification and Quantitative Analysis of Products. Polym. Degrad. Stab. 1984, 6, 47–61. 10.1016/0141-3910(84)90016-8. [DOI] [Google Scholar]
- Grassie N.; Murray E. J.; Holmes P. A. The Thermal Degradation of Poly(-(d)-β-Hydroxybutyric Acid): Part 3-The Reaction Mechanism. Polym. Degrad. Stab. 1984, 6, 127–134. 10.1016/0141-3910(84)90032-6. [DOI] [Google Scholar]
- Cicogna F.; Coiai S.; De Monte C.; Spiniello R.; Fiori S.; Franceschi M.; Braca F.; Cinelli P.; Fehri S. M. K.; Lazzeri A.; Oberhauser W.; Passaglia E. Poly(Lactic Acid) Plasticized with Low-Molecular-Weight Polyesters: Structural, Thermal and Biodegradability Features. Polym. Int. 2017, 66, 761–769. 10.1002/pi.5356. [DOI] [Google Scholar]
- a Fox T. G. Influence of Diluent and of Copolymer Composition on the Glass Temperature of a Polymer System. Bull. Am. Phys. Soc. 1956, 1, 123–135. [Google Scholar]; b Wypych G.Handbook of Plasticizers, 3rd ed.; Wypych G., Ed.; ChemTec Publishing: Toronto, 2017; Vol. 1. [Google Scholar]
- Zembouai I.; Kaci M.; Bruzaud S.; Benhamida A.; Corre Y.; Grohens Y. A Study of Morphological, Thermal, Rheological and Barrier Properties of Poly(3-Hydroxybutyrate-Co-3- Hydroxyvalerate)/Polylactide Blends Prepared by Melt Mixing. Polym. Test. 2013, 32, 842–851. 10.1016/j.polymertesting.2013.04.004. [DOI] [Google Scholar]
- Kumagai Y.; Doi Y. Enzymatic Degradation of Binary Blends of Microbial Poly(3-Hydroxybutyrate) with Enzymatically Active Polymers. Polym. Degrad. Stab. 1992, 37, 253–256. 10.1016/0141-3910(92)90167-4. [DOI] [Google Scholar]
- Blümm E.; Owen A. J. Miscibility, Crystallization of Poly(3-Hydroxybutyrate)/Poly(L-Lactide) Blends. Polymer 1995, 36, 4077–4081. 10.1016/0032-3861(95)90987-D. [DOI] [Google Scholar]
- Heo K.; Yoon J.; Jin K. S.; Jin S.; Sato H.; Ozaki Y.; Satkowski M. M.; Noda I.; Ree M. Structural Evolution in Microbial Polyesters. J. Phys. Chem. B 2008, 112, 4571–4582. 10.1021/jp711136x. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo M. L.; Gazzano M.; Righetti M. C. The Role of the Rigid Amorphous Fraction on Cold Crystallization of Poly(3-Hydroxybutyrate). Macromolecules 2012, 45, 5684–5691. 10.1021/ma3010907. [DOI] [Google Scholar]
- Nishi T.; Wang T. T. Melting Point Depression and Kinetic Effects of Cooling on Crystallization in Poly(Vinylidene Fluoride)-Poly(Methyl Methacrylate) Mixtures. Macromolecules 1975, 8, 909–915. 10.1021/ma60048a040. [DOI] [Google Scholar]
- Hoffman J. D.; Weeks J. J. Rate of Spherulitic Crystallization with Chain Folds in Polychlorotrifluoroethylene. J. Chem. Phys. 1962, 37, 1723–1741. 10.1063/1.1733363. [DOI] [Google Scholar]
- Kunioka M.; Doi Y. Thermal Degradation of Microbial Copolyesters: Poly(3-Hydroxybutyrate-Co-3-Hydroxyvalerate) and Poly(3-Hydroxybutyrate-Co-4-Hydroxybutyrate). Macromolecules 1990, 23, 1933–1936. 10.1021/ma00209a009. [DOI] [Google Scholar]
- Silva M. A.; De Paoli M. A.; Felisberti M. I. Flory-Huggins Interaction Parameter of Poly(Ethylene Oxide)/Poly(Epichlorohydrin) and Poly(Ethylene Oxide)/Poly(Epichlorohydrin-Co-Ethylene Oxide) Blends. Polymer 1998, 39, 2551–2556. 10.1016/S0032-3861(97)00574-0. [DOI] [Google Scholar]
- Saad G. R. Blends of Bacterial Poly[(R)-3-Hydroxybutyrate] with Oligo[(R,S)-3-Hydroxybutyrate]-Diol. Polym. Int. 2002, 51, 338–348. 10.1002/pi.844. [DOI] [Google Scholar]
- Thomas S.; Grohens Y.; Jyotishkumar P. In Characterization Of Polymer Blends: Miscibility, Morphology and Interfaces; Thomas S.; Grohens Y.; Jyotishkumar P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2015. [Google Scholar]
- Godwin A. D.Plasticizers. In Applied Plastics Engineering Handbook: Processing, Materials, and Applications, 2nd ed.; Kutz M., Ed.; Elsevier Inc.: United Kingdom, 2017; chapter 24, pp 533–553. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.