

Preparedness activities against highly transmissible viruses with high mortality rates have been highlighted during the ongoing coronavirus disease 2019 (COVID-19) pandemic. Smallpox, caused by variola virus (VARV) infection, is highly transmissible, with an estimated 30% mortality.
KEYWORDS: animal models, experimental therapeutics, monkeypox, smallpox, virology
ABSTRACT
Smallpox, caused by Variola virus (VARV), was eradicated in 1980; however, VARV bioterrorist threats still exist, necessitating readily available therapeutics. Current preparedness activities recognize the importance of oral antivirals and recommend therapeutics with different mechanisms of action. Monkeypox virus (MPXV) is closely related to VARV, causing a highly similar clinical human disease, and can be used as a surrogate for smallpox antiviral testing. The prairie dog MPXV model has been characterized and used to study the efficacy of antipoxvirus therapeutics, including recently approved TPOXX (tecovirimat). Brincidofovir (BCV; CMX001) has shown antiviral activity against double-stranded DNA viruses, including poxviruses. To determine the exposure of BCV following oral administration to prairie dogs, a pharmacokinetics (PK) study was performed. Analysis of BCV plasma concentrations indicated variability, conceivably due to the outbred nature of the animals. To determine BCV efficacy in the MPXV prairie dog model, groups of animals were intranasally challenged with 9 × 105 plaque-forming units (PFU; 90% lethal dose [LD90]) of MPXV on inoculation day 0 (ID0). Animals were divided into groups based on the first day of BCV treatment relative to inoculation day (ID–1, ID0, or ID1). A trend in efficacy was noted dependent upon treatment initiation (57% on ID–1, 43% on ID0, and 29% on ID1) but was lower than demonstrated in other animal models. Analysis of the PK data indicated that BCV plasma exposure (maximum concentration [Cmax]) and the time of the last quantifiable concentration (AUClast) were lower than in other animal models administered the same doses, indicating that suboptimal BCV exposure may explain the lower protective effect on survival.
IMPORTANCE Preparedness activities against highly transmissible viruses with high mortality rates have been highlighted during the ongoing coronavirus disease 2019 (COVID-19) pandemic. Smallpox, caused by variola virus (VARV) infection, is highly transmissible, with an estimated 30% mortality. Through an intensive vaccination campaign, smallpox was declared eradicated in 1980, and routine smallpox vaccination of individuals ceased. Today's current population has little/no immunity against VARV. If smallpox were to reemerge, the worldwide results would be devastating. Recent FDA approval of one smallpox antiviral (tecovirimat) was a successful step in biothreat preparedness; however, orthopoxviruses can become resistant to treatment, suggesting the need for multiple therapeutics. Our paper details the efficacy of the investigational smallpox drug brincidofovir in a monkeypox virus (MPXV) animal model. Since brincidofovir has not been tested in vivo against smallpox, studies with the related virus MPXV are critical in understanding whether it would be protective in the event of a smallpox outbreak.
INTRODUCTION
Members of the genus Orthopoxvirus (OPXVs) contain many human pathogens, including Variola virus (VARV) (the causative agent of smallpox), Monkeypox virus (MPXV), and Cowpox virus. Smallpox remains a significant concern to the United States and the world as a potential biothreat agent. While smallpox has been eradicated from the human population, recent studies have demonstrated a relatively easy path to virus reconstitution through the use of synthetic biology (1). Additionally, the number of monkeypox cases within Africa is growing, as is the number of African territories reporting the disease, possibly due in part to the waning immunity following smallpox vaccine cessation (2–6). MPXV has also been spread outside Africa, notably during the 2003 U.S. outbreak, as well as from recent imported cases of monkeypox in Israel, the United Kingdom, and Singapore (7–9). Since the eradication of smallpox, MPXV has become the most concerning human pathogen among OPXVs. Although the vaccine Jynneos was recently approved by the U.S. Food and Drug Administration (FDA) for the prevention of both smallpox and monkeypox (10), there is currently no approved therapeutic for the treatment of monkeypox disease. Because genetically, MPXV and VARV are closely related, it would be ideal if multiple antiviral therapeutics were approved to treat both diseases.
The primary objective of smallpox bioterrorism preparedness is to save lives if smallpox somehow reemerges, whether from a natural or a malicious event. The development of antiviral therapeutics is a critical component in outbreak response efforts as well as in disease treatment. As advances in synthetic biology allow for manipulation or recreation of viruses, it is important to remain vigilant in identifying sufficient therapeutic options should an outbreak occur. Current considerations for preparedness response in the event of a smallpox outbreak have suggested the need for two antiviral compounds with separate mechanisms of action to be licensed and ready for use (11). To date, a small-molecule compound, TPOXX (ST-246 or tecovirimat), has recently gained FDA licensure for use against smallpox and remains the only FDA-approved antipoxviral drug (12). TPOXX prevents virus spread by inhibiting the function of the major envelope protein (F13L), thereby preventing the virus from leaving an infected cell. Although TPOXX has shown efficacy against multiple OPXVs in various animal model systems, it has also been noted that a single amino acid alteration to the orthopoxviral F13L protein allows for resistance to TPOXX treatment (13). Additionally, resistance emerged during use of the drug in an extended treatment course of an individual with progressive vaccinia (14). If an outbreak of smallpox were to occur, a multitherapeutic approach would be necessary to successfully control the outbreak.
Brincidofovir (BCV; also referred to as hexadecyloxypropyl-cidofovir [HDP-CDV], or CMX001) is a lipid conjugate of the nucleotide analog cidofovir (CDV). Compared to CDV, BCV has increased cellular uptake and better conversion to the active form by intracellular enzymes (15). Unlike CDV, BCV is orally bioavailable, allowing for tablet and suspension formulations for drug delivery. Additionally, BCV is not actively taken up by the renal organic anion transporter 1 (OAT1) like CDV, which results in substantial improvements in the renal safety profile of BCV (16). The antiviral effect of BCV differs from that of TPOXX in that it causes inhibition of the viral DNA polymerase following incorporation into viral DNA (17). CDV and BCV have previously shown antiviral efficacies against several double-stranded DNA viruses, including adenoviruses and poxviruses (18–20). When tested in vitro against VARV, BCV was active against each of the five strains tested, with 50% effective concentrations (EC50s) ranging from 0.05 μM to 0.21 μM. CDV was also active against each of the five VARV strains but was 97-fold less potent on average (range, 18-fold to 259-fold) (21).
BCV has also shown promising results in different OPXV animal models (15, 22–24), including one MPXV mouse challenge and several Vaccinia virus mouse models. For assessment against MPXV, STAT1 deficient mice were challenged with 5,000 PFU and began BCV treatment on the day of infection (10 mg/kg for the first dose and 2.5 mg/kg every 48 hours for 14 days total) and all mice survived infection (47). However, postexposure treatment was not assessed with this model. BCV was used following a lethal vaccinia virus (IHD-J-Luc strain) challenge in BALB/c mice as well as immunocompromised (BALB/c nu/nu) mice (25). For BALB/c mice, a three-dose regimen of 20, 5, or 2.5 mg of BCV/kg of body weight administered every 48 h (q48h) was assessed starting either on day 1 or day 2 postchallenge. Administration of BCV beginning on day 1 postinfection (p.i.) at doses of 5 and 20 mg/kg protected 100% of mice, while a dose of 2.5 mg/kg BCV provided minimal protection (16% survival). When looking at the effect of delayed treatment (starting on day 2 p.i.), all mice that received 20 mg/kg BCV survived infection; the 5-mg/kg dosing regimen resulted in partial protection (33% survival). The efficacy of BCV within immunocompromised (BALB/c nu/nu) mice was also assessed; a dosing regimen of 20 mg/kg BCV on days 1, 3, and 5 or on days 1, 3, 5, 7, 10, 14, 17, 21, and 24 was used. Although all the immunocompromised mice in these treatment arms perished, both short and extended treatments with BCV significantly increased median survival times compared with results in vehicle-treated mice.
In addition to showing efficacy in vaccinia virus mouse studies, BCV has shown efficacy within the rabbitpox rabbit model and the ectromelia virus (mousepox) mouse model, both of which are considered surrogate smallpox models by the FDA (26–28). In two studies, BCV efficacy in the rabbitpox model demonstrated a statistically significant survival benefit when treatment was initiated at the onset of clinical signs of disease in the rabbits (after the detection of skin lesions) or, in the follow-up study, at up to 48 h after fever onset. When rabbits were treated with BCV immediately upon the development of fever, 100% survival was seen; 93% of those treated 24 h or 48 h after the onset of fever survived. The ectromelia virus model has also demonstrated the promising efficacy of BCV (29). A statistically significant survival benefit of 33% was seen when BCV treatment was started as late as 6 days postinfection with ectromelia virus compared to that of the vehicle treatment control animals.
We have previously fully characterized a unique animal model for the study of medical countermeasures (MCMs) against a lethal MPXV challenge (30–33). Through these studies, we have shown that the prairie dog MPXV model displays an incubation period of approximately 10 to 13 days, followed by generalized cutaneous lesions. Systemic lesion formation is a unique characteristic compared to those of other OPXV animal models, and disease progression in the prairie dog MPXV model is remarkably similar to human monkeypox patient disease progression (34). It has been challenging to truly identify a fever stage simply due to broad variations in normal temperature; however, a prodrome characterized by inappetence for ∼2 days before lesion onset is consistently observed with this animal model. The model has allowed for the characterization of monkeypox disease, including virus shedding from the oral cavity (which begins prior to lesion onset), the occurrence of viremia, and finally the serologic detection of anti-OPXV antibodies at or near the onset of lesion formation. Additionally, the prairie dog MPXV model has been utilized to compare observed differences between disease manifestations of the MPXV clades (Congo Basin and West African) as well as comparisons of the levels of transmissibility of the two clades (32, 35, 36). The model has been used to test next-generation vaccines as well as the smallpox therapeutic TPOXX (33, 37). During the TPOXX efficacy study, 75% of infected prairie dogs that received vehicle alone succumbed to MPXV infection. In comparison, 100% of animals that received TPOXX survived challenge, and animals that received treatment before clinical signs remained largely asymptomatic. While this model has not been qualified by the FDA’s voluntary Animal Model Qualification Program, the model is remarkably similar to human disease incubation/presentation and allows for rigorous testing of potential monkeypox and smallpox MCMs. Herein, we describe two studies, first to determine the pharmacokinetics (PK) of BCV within prairie dogs and second to test the efficacy of BCV against a lethal MPXV challenge using the MPXV prairie dog model.
RESULTS
Study 1: exposure to BCV following oral administration to healthy prairie dogs. (i) BCV plasma concentrations following single and repeat oral administration of BCV.
Following single oral administration of BCV at 5 mg/kg or 20 mg/kg, plasma concentrations of BCV were detected in the first sample (1 h) following administration. The median BCV time of maximum concentration (Tmax) ranged between 4 and 8 h. BCV plasma concentrations were generally below the limit of quantification (BLQ) (1 ng/ml) by 24 h following the 5-mg/kg dose and by 36 h following the 20-mg/kg BCV dose (Table 1).
TABLE 1.
Exposure parameters of BCV following oral administration to healthy prairie dogsa
| Dose (mg/kg) | Dose no. | t½ (h) (% of animals) | Tmax (h) (range) | Cmax (ng/ml) (% of animals) | Tlast (h) (range) | AUClast (h · ng/ml) (% of animals) |
|---|---|---|---|---|---|---|
| 5 | 1 | NR | 4.07 (2.92–12.2) | 11.5 (81.0) | 12.2 (8.0–12.2) | 73.4 (62.3) |
| 3 | NR | 3.99 (1.22–7.63) | 9.56 (48.1) | 9.61 (7.63–24.4) | 61.5 (56.2) | |
| 20 | 1 | 3.53 (20.6) | 8.1 (4.07–12.2) | 69.9 (71.5) | 24.2 (12.3–24.2) | 439 (37.1) |
| 3 | 5.94 (36.7) | 5.95 (1.17–24.5) | 53.2 (54.7) | 35.9 (11.8–48.7) | 434 (26.0) |
Animals (n = 16) were dosed with a three-dose (q48h) regimen of 20 (n = 8) or 5 (n = 8) mg/kg BCV via oral gavage so that BCV pharmacokinetics could be determined following the first and last doses. Blood was collected at 8 time points (1, 2, 4, 8, 12, 24, 36, and 48 h) following the first administration and at 9 time points (0, 1, 2, 4, 8, 12, 24, 36, and 48 h) following the last administration. Noncompartmental PK parameter values are means (percent coefficient of variation), except for Tmax and Tlast (discrete time interval parameters), which are medians (ranges). NR, not reported.
Predose plasma concentrations prior to the third administration of BCV at 5 mg/kg or 20 mg/kg (every 48 h [q48h]) were BLQ in the majority of the animals. The BCV Tmax occurred between 4 and 6 h after administration, and BCV concentrations were generally BLQ by 24 h following the third administration of 5 mg/kg/q48h and by 48 h following the third administration of 20 mg/kg/q48h.
(ii) BCV PK parameters following single and repeat oral administration of BCV.
In general, there was no consistent sex effect (as measured by a factor of >2-fold between mean PK parameter values) on BCV PK parameters following the first or last oral gavage administration of a three-dose regimen of 20 or 5 mg/kg BCV (Fig. 1); therefore, data for male and female animals are combined in Table 1. BCV exposure parameters (the maximum concentration of the drug [Cmax] and area under the plasma concentration-time curve [AUC]) increased with increasing dose but in a greater-than-dose-proportional manner; for a 4-fold increase in dose, there was a 6- to 7-fold increase in exposure. As expected, based upon the lack of detectable concentrations of BCV in plasma at 24 to 48 h postdose (Fig. 1), there was no evidence of accumulation of BCV in plasma after the administration of BCV for 3 doses (q48h). Two animals (ID numbers 12054 and 13070) mistakenly received 20 mg/kg instead of 5 mg at the 48-h drug administration point, although the plasma concentrations for those two animals are similar to those of the other animals that received the correct 5-mg/kg dose (Fig. 1b). Half-life (t1/2) was generally not calculated following administration of 5 mg/kg due to inadequate data to characterize the BCV elimination phase; the mean half-life after the administration of 20 mg/kg ranged from 3.5 to 5.9 h. Within a given group, individual animal Cmaxs and areas under the plasma concentration-time curve from time zero to the time of the last quantifiable concentration (AUClasts) were highly varied (the percent coefficient of variation [%CV] ranged from 26% to 81%) (Table 1).
FIG 1.

Animals (n = 16) were dosed with a three-dose (q48h) regimen of 20 (n = 8) or 5 (n = 8) mg/kg BCV via oral gavage, so that BCV pharmacokinetics could be determined following single (a, b) and repeat (c, d) dosing. An increase in concentrations of BCV with increased dose is seen. BCV plasma concentrations were generally below the limit of quantitation (1 ng/ml; BLQ values were not plotted) by 24 h following the 5-mg/kg dose (c) and by 48 h following the 20-mg/kg BCV dose (d). Data for female animals (F) are represented by filled blue symbols, and data for male animals (M) are represented by open red symbols. *, two animals (ID numbers 12054 and 13070) mistakenly received 20 mg/kg instead of 5 mg at the 48-h drug administration point.
The BCV plasma exposure (Cmax and AUClast) observed in prairie dogs following 20 or 5 mg/kg was 2- to 4-fold lower than exposure parameters observed in the mouse (ectromelia) and rabbit (rabbitpox) models administered the same doses (data on file). Although the BCV plasma exposure was not optimal, dosing at higher than 20 mg/kg has been associated with gastrointestinal toxicity, limiting our ability to attempt higher BCV doses within this animal model. Given this limitation, we set out to determine if BCV exposure with a 20/5/5-mg/kg q48h dosing regimen in the prairie dog MPXV model would protect against an MPXV challenge.
Study 2: efficacy of BCV following intranasal challenge with MPXV. (i) Measures of morbidity and mortality varied and were dependent on when BCV treatment began.
Animal 13034 in the vehicle group first began to show clinical signs on day 5 p.i.; numerous other animals within all groups began to show clinical signs on day 8 p.i., with some of these signs necessitating euthanasia beginning day 10 p.i. due to clinical scores. Clinical signs varied for individual animals and between the different groups; however, the animals that began receiving BCV treatment 1 day prior to infection had the lowest average maximum clinical score (n = 7) (Table 2) compared to the animals that received BCV treatment the day of infection (Table 3), animals that received BCV 1 day postinfection (Table 4), or animals in the vehicle group (Table 5) (average maximum clinical scores of 8.1, 8.7, and 9.1, respectively). As seen in Fig. 2, when we compared the survivorship of the four groups, a trend of protection was observed when BCV treatment was administered early. Overall, a 57% survival rate was observed for animals initiating treatment 1 day prior to MPXV challenge, 43% survival was observed for animals that began receiving treatment the day of challenge, and 29% survival was observed for animals that received treatment on day 1 postchallenge. In comparison, a 14% survival rate was seen in the vehicle animals, which was not unexpected as an MPXV challenge was administered at an approximately 90% lethal dose (∼LD90). Additionally, we saw a trend of delay in mortality when BCV treatment was started prior to MPXV infection; for animals treated on day −1, the average day of euthanasia was day 13 p.i. (Table 2), whereas the average euthanasia day for the other groups was approximately day 11 p.i. (Tables 3 to 5) when animals euthanized at the conclusion of the study are excluded.
TABLE 2.
Day −1 treatment group resultsa
| Animal | Onset of systemic rash (day p.i.) | No. of lesions | Maximum clinical score (day[s] recorded)b | Mortality (day p.i.) | MPXV DNA in blood (range of days p.i.) | Viable MPXV in blood (range of days p.i.) | Specimens in which VV was found at necropsy (positive samples) | Peak viral load at necropsy (source) |
|---|---|---|---|---|---|---|---|---|
| PD9245 | NA | 1 (tongue) | 4 (14/17) | NA | 5–21§ | Neg | Neg | NA |
| PD13117 | 11 | 5 | 4 (14/17) | NA | 5–21§ | Neg | Neg | NA |
| PD13074 | NA | 0 | ≥10 (11) | 11 | 5–11* | 5* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Sm. Int., Mes LN | 1 × 109 (nose) |
| PD13086 | 11 | 25 | 6 (14/16) | NA | 5–21 | 5 | Nasal | 1.4 × 103 (nose) |
| PD13083 | 11 | 20 | ≥10 (14) | 14 | 5–14* | 8* | Nasal, SM LN, tongue, lung, skin, Mes LN | 2.4 × 109 (nose) |
| PD13055 | 11 | >100 | ≥10 (14) | 14 | 3–14* | Neg* | Liver, nasal, SM LN, tongue, lung, skin, Sm. Int., Mes LN | 4.3 × 108 (skin) |
| PD13149 | 14 | 14 | 5 (14) | NA | 5–21§ | Neg | Neg | NA |
Prairie dogs were dosed with 20 mg/kg brincidofovir via oral gavage on the day before monkeypox virus challenge (day −1) (n = 7). Following the initial dose, animals were dosed (q48h) two additional times with 5 mg/kg and 5 mg/kg. Animals were observed for clinical signs, including lesion presentation, and the clinical score was assessed. If animals survived challenge, they were humanely euthanized on day 25 p.i. Postmortem samples were evaluated for the presence of virus. Clinical observations and viral DNA/tissue culture results are summarized within the table. Blood was collected on p.i. days 1, 3, 5, 8, 11, 14, 17, and 21 (and the day of euthanasia if different than the blood collection day). * indicates that an animal on at least one of these points was not available for testing. § indicates that the specimen at one of these time points tested negative. VV, viable virus; NA, not applicable; Neg, negative; SM LN, submandibular lymph node; Mes LN, mesenteric lymph node; Sm. Int., small intestine.
The average clinical score was 7.
TABLE 3.
Day 0 treatment groupa
| Animal | Onset of systemic rash (day p.i.) | No. of lesions | Maximum clinical score (day recorded)b | Mortality (day p.i.) | MPXV DNA in blood (range of days p.i.) | Viable MPXV in blood (range of days p.i.) | Specimens in which VV was found at necropsy (positive samples) | Peak viral load at necropsy (source) |
|---|---|---|---|---|---|---|---|---|
| PD9254 | NA | 8 | 6 (11) | NA | 5–21 | Neg | Neg | NA |
| PD13046 | NA | 0 | 3 (11) | NA | 8–17*§ | Neg* | Neg | NA |
| PD13008 | 10 | 3 | ≥10 (11) | 11 | 5–11* | Neg* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Sm. Int., Mes LN | 4.9 × 108 (nose) |
| PD13064 | 11 | 15 | 8 (17) | NA | 5–21 | Neg | Neg | NA |
| PD13092 | 11 | 7 | ≥10 (12) | 12 | 3–12 | Neg* | Liver, nasal, SM LN, tongue, lung, skin, Mes LN | 1.6 × 109 (skin) |
| OD13076 | 11 | 10 | ≥10 (14) | 14 | 3–14* | 5* | Nasal, SM LN, tongue, skin | 3.3 × 108 (skin) |
| PD13066 | 10 | 15 | ≥10 (10) | 10 | 3–11* | 5* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Sm. Int., Mes LN | 6.9 × 108 (spleen) |
Prairie dogs were dosed with 20 mg/kg brincidofovir via oral gavage on the day of monkeypox virus challenge (day 0) (n = 7). Following the initial dose, animals were dosed (q48h) two additional times with 5 mg/kg and 5 mg/kg. Animals were observed for clinical signs, including lesion presentation, and the clinical score was assessed. If animals survived challenge, they were humanely euthanized on day 25 p.i. Postmortem samples were evaluated for the presence of virus. Clinical observations and viral DNA/tissue culture results are summarized within the table. Blood was collected on p.i. days 1, 3, 5, 8, 11, 14, 17, and 21 (and the day of euthanasia if it was different than the blood collection day). * indicates at that an animal on at least one of these time points was not available for testing. § indicates that a specimen at one of these time points tested negative. VV, viable virus; NA, not applicable; SM LN, submandibular lymph node; Mes LN, mesenteric lymph node; Sm. Int., small intestine.
The average clinical score was 8.1.
TABLE 4.
Day 1 treatment groupa
| Animal | Onset of systemic rash (day p.i.) | No. of lesions | Maximum clinical score (day recorded)b | Mortality (day p.i.) | MPXV DNA in blood (range of days p.i.) | Viable MPXV in blood (day[s] p.i.) | Specimens in which VV was found at necropsy (positive samples) | Peak viral load at necropsy |
|---|---|---|---|---|---|---|---|---|
| PD9260 | NA | 0 | ≥10 (11) | 11 | 3–11* | 5, 11* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Sm. Int., Mes LN | 1.9 × 109 (spleen) |
| PD13038 | 10 | 4 | ≥10 (10) | 10 | 3–11* | Neg* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Sm. Int. | 6.1 × 107 (tongue) |
| PD13027 | 14 | 5 | 3 (11/14) | NA | 5–21 | Neg | Neg | NA |
| PD13116 | 8 | 6 | ≥10 (10) | 10 | 3–11* | 5* | Liver, nasal, SM LN, tongue, lung, skin | 1.1 × 108 (skin) |
| PD13108 | 13 | 25 | 8 (14/16) | NA | 5–21*§ | Neg* | Neg | NA |
| PD13148 | 11 | ≥50 | ≥10 (14) | 14 | 5–14* | 14* | Nasal, SM LN, tongue, lung, skin, Sm. Int. | 3.7 × 108 (tongue) |
| PD13063 | 11 | 12 | ≥10 (12) | 12 | 5–12* | Neg* | Nasal, SM LN, tongue, lung, skin, Mes LN | 4.4 × 107 (skin) |
Prairie dogs were dosed with 20 mg/kg brincidofovir via oral gavage on the day following monkeypox virus challenge (day 1) (n = 7). Following the initial dose, animals were dosed (q48h) two additional times with 5 mg/kg and 5 mg/kg. Animals were observed for clinical signs, including lesion presentation, and the clinical score was assessed. If animals survived challenge, they were humanely euthanized on day 25 p.i. Postmortem samples were evaluated for presence of virus. Clinical observations and viral DNA/tissue culture results are summarized within the table. Blood was collected on p.i. days 1, 3, 5, 8, 11, 14, 17, and 21 (and the day of euthanasia if it was different than a blood collection day). * indicates that an animal on at least one of these points was not available for testing. § indicates that a specimen at one of these time points tested negative. VV, viable virus; NA, not applicable; SM LN, submandibular lymph node; Mes LN, mesenteric lymph node; Sm. Int., small intestine.
The average clinical score was 8.7.
TABLE 5.
Vehicle groupa
| Animal | Onset of systemic rash (day p.i.) | No. of lesions | Maximum clinical score (day recorded)b | Mortality (day p.i.) | MPXV DNA in blood (range of days p.i.) | Viable MPXV in blood (day p.i.) | Specimens in which VV was found at necropsy (positive samples) | Peak viral load at necropsy (source) |
|---|---|---|---|---|---|---|---|---|
| PD9203 | 8 | 5 | ≥10 (14) | 14 | 3–14* | Neg* | Nasal, SM LN, tongue, skin | 4.1 × 107 (skin) |
| PD13053 | 11 | 3 | ≥10 (11) | 11 | 3–11* | Neg* | Nasal, SM LN, tongue, lung, skin | 6.8 × 107 (nose) |
| PD13114 | 8 | 9 | 4 (14) | NA | 5–14 | Neg | Neg | NA |
| PD13028 | NA | 0 | ≥10 (10) | 10 | 3–11* | Neg* | Nasal, SM LN, tongue, lung, skin, Sm. Int. | 3.7 × 108 (tongue) |
| PD13131 | 11 | 11 | ≥10 (11) | 11 | 3–11* | Neg* | Liver, nasal, SM LN, spleen, tongue, lung, skin, Mes LN | 1.4 × 109 (nose) |
| PD13034 | 11 | 6 | ≥10 (11) | 11 | 3–14* | 8* | Nasal, SM LN, tongue, lung, skin, Sm. Int. | 8.2 × 107 (tongue) |
| PD13115 | 11 | 5 | ≥10 (12) | 12 | 3–12* | 5* | Nasal, SM LN, tongue, lung, skin, Mes LN | 3.8 × 108 (nose) |
Prairie dogs were dosed with the vehicle via oral gavage and challenged with monkeypox virus (n = 7). Following the initial dose, animals were dosed (q48h) two additional times with the vehicle. Animals were observed for clinical signs, including lesion presentation, and the clinical score was assessed. If animals survived challenge, they were humanely euthanized on day 25 p.i. Postmortem samples were evaluated for the presence of virus. Clinical observations and viral DNA/tissue culture results are summarized within the table. Blood was collected on p.i. days 1, 3, 5, 8, 11, 14, 17, and 21 (and the day of euthanasia if it was different than a blood collection day). * indicates that an animal on at least one of these time points was not available for testing. § indicates that a specimen at one of these time points tested negative. VV, viable virus; NA, not applicable; SM LN, submandibular lymph node; Mes LN, mesenteric lymph node; Sm. Int., small intestine.
The average clinical score was 9.1.
FIG 2.

Survival curve for groups of MPXV-challenged prairie dogs. Animals were treated with brincidofovir starting 1 day preinfection (–1), on the day of infection (day 0), or 1 day postinfection (day 1) with MPXV. Placebo (vehicle-only) animals were challenged with MPXV and treated with the vehicle to serve as positive controls. Differences in mortality rates and day of euthanasia among all groups (day −1, day 0, day +1, vehicle only) were not statistically significant (P = 0.56 and P = 0.61, respectively). There was no significant difference in survivorship between the treatment timing groups and vehicle only group (P = 0.42). A survival trend was observed, with more animals surviving the sooner brincidofovir treatment began.
(ii) Molecular results (ELISA, real-time PCR, and tissue culture).
To analyze immune response, an enzyme-linked immunosorbent assay (ELISA) was performed. This assay typically captures IgM but does not capture IgG as well, which is therefore a limitation of our assay. An immune response (IgM) is typically seen when animals start getting cutaneous lesions, which is between days 9 and 13 postinfection (32). Due to these reasons and a lack of animal numbers at later time points, day 11 and day 14 were chosen for comparisons of immune responses between the groups. We found that treatment with BCV did not impact the overall immune response. Regardless of when treatment began, there was no statistical difference from the vehicle group on these 2 days (day 11 P value range, 0.14 to 0.76; day 14 P value range, 0.19 to 0.8) (Fig. 3). We did, however, see variations in immune responses, which was expected due to the nature of the outbred animal model. In order to ensure that the immune response was not impacted due to BCV treatment, several additional comparisons were performed. The anti-OPXV immune responses as measured by the optical density (OD) levels for all treatment groups were combined and compared to those of the vehicle-only group (data not shown). No significant differences were noted. Additionally, the fold rise in the OD minus the cutoff value (OD–COV) between day 11 and day 14 was calculated for each prairie dog for which both values were available. The fold rises in titer were compared between the groups (day −1, day 0, day 1, and in the vehicle-only group) using Wilcoxon rank sum tests. No significant differences were detected (P value range, 0.06 to 0.86 [data not shown]). Finally, fold differences from the vehicle-only group were determined for all treatment groups combined, and again, no significant differences were noted (data not shown).
FIG 3.
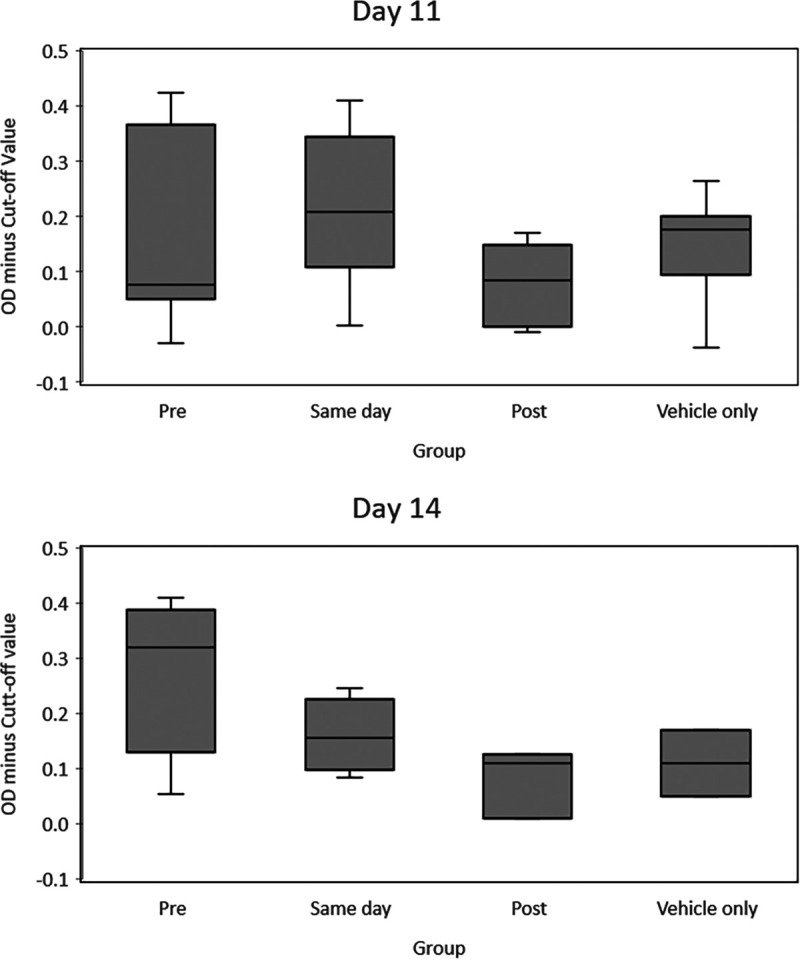
Comparison of immune responses (days 11 and 14) between groups of MPXV-challenged prairie dogs, using ELISA. Optical density minus the cutoff value (OD–COV) levels were compared between treatment groups (preinfection [day −1], day of infection [day 0], and 1 day postinfection [day 1] groups and the placebo group [vehicle]) using the Wilcoxon rank sum test, as data were not normally distributed.
Necropsy samples were first tested for the presence of viral DNA, and if positive, they were tested for viable virus by a standard plaque assay. Animals that survived infection were negative for viable virus in most necropsy samples tested, regardless of treatment group (Tables 2 to 5). For those animals that succumbed to infection, regardless of treatment group, viable virus levels were high in most tissues tested (Fig. 4; Tables 2 to 5). When determining which tissues showed the highest viral loads for animals that had succumbed to disease (n = 18), we found the nasal tissue to most often contain the highest levels of virus (n = 6), followed by skin (n = 6), spleen (n = 3), and tongue (n = 3) (Tables 2 to 5). To compare viral loads in animals that succumbed to infection, results from animals that died on day 14 p.i. (which is typically around the peak of infection in this animal model) were compared. Similar levels of virus were found in tissues from each of the four groups on day 14, and the variability seen was comparable to those of previous studies with this outbred model (Fig. 5).
FIG 4.

Viable virus was detected in necropsy samples from MPXV-challenged prairie dogs. Animals were treated with BCV starting 1 day preinfection (day −1) (a), on the day of infection (day 0) (b), or 1 day postinfection (day 1) (c). (d) Placebo (vehicle-only) animals were challenged with MPXV and treated with the vehicle to serve as positive controls. * indicates that one or more of those samples had the cell culture monolayer destroyed or that plaques were present but below the limit of detection (LOD) for this assay. † indicates that the animal died before the study end (day 25 p.i.). SM Ln, SM LN, or Sm Ln, submandibular lymph node; Sm. Int., small intestine; Mes LN, mesenteric lymph node.
FIG 5.

Viral titration comparison of treatment groups on day 14 p.i. One animal from each group (vehicle, 1 day postinfection [TX], day of infection [day 0], or preinfection [day −1]) succumbed on day 14. Viral titer results are shown as PFU per gram of tissue for comparison between the animals. TX, treatment; SM LN, submandibular lymph node; Sm. Int., small intestine; Mes LN, mesenteric lymph node.
Levels of viremia were also similar between animals and groups. As the viremia phase is generally short and variable during OPXV infections (38, 39), we missed the viremic period for some animals and observed viable virus only within the blood on 1 day for the majority of other animals (most often on day 5 p.i., although days 8, 11, and 14 were also observed) (Tables 2 to 5). Viral DNA in blood was detected for longer periods than viable virus, beginning on day 3 or 5 p.i. for all animals except animal 13046 (this animal was negative for viral DNA in its blood on day 3, and no sample was available for testing on day 5 p.i.). Viral DNA was detectable within the blood until the study end (day 25 p.i.) for all animals that survived infection except for two animals (13046 on day 17 and 13114 on day 14). As this is an outbred model, the variability observed between animals and groups was similar to that of previous studies (30, 32).
DISCUSSION
BCV is an antiviral therapeutic that has a mechanism of action which differs from that of TPOXX; it inhibits viral genome replication by the viral DNA polymerase following incorporation of cidofovir into viral DNA (17). This therapeutic has previously shown efficacy within OPXV animal models, including the rabbitpox rabbit model and the ectromelia virus mouse model. Based on these results and the characteristics of the prairie dog MPXV model showing pathogenesis similar to that of human monkeypox cases, we believe that this novel animal model is valuable to ascertain the effectiveness of treatments against OPXV infection. As MPXV is a human health threat of increasing importance, testing BCV against this virus in vivo would be informative.
In this study, we first determined the PK of BCV within the prairie dog. Due to the outbred nature of the animals, we saw animal-to-animal variation in PK results, which was expected and can be considered a limitation of the model. Unexpectedly, the BCV plasma exposure (Cmax and AUClast) observed in prairie dogs following 20 or 5 mg/kg was 2- to 4-fold lower than exposure parameters observed in mice (ectromelia) and rabbits (rabbitpox) administered the same doses. These findings suggested a potentially suboptimal BCV exposure in the prairie dog following the 20- or 5-mg/kg dosing regimen. Due to toxicity issues identified in other animal species and the concern that higher doses may have caused significant toxicity in the prairie dog, we selected safe doses for administration that achieved lower exposures than had previously been demonstrated as efficacious in other animal models. These selected doses were evaluated to determine if lower BCV exposures would be effective against MPXV in this model. Animals were challenged with an LD90 of MPXV, and groups were treated with BCV as a three-dose (q48h) regimen of 20 mg/kg, followed by 5 mg/kg and 5 mg/kg. BCV treatment was started on day −1 (n = 7) prior to challenge, on day 0 (n = 7) of challenge, and on day 1 (n = 7) postchallenge. During the efficacy study, trends of protection against a lethal MPXV challenge were seen when BCV was administered early. A 57% survival rate was observed for animals initiating treatment 1 day prior to MPXV challenge, a 43% survival rate was observed for animals that began receiving treatment the day of challenge, and a 29% survival rate was observed on day 1 postchallenge. In comparison, a 14% survival rate was seen in the vehicle (control) animals.
Although only a trend of protection was observed with BCV in the prairie dog MPXV model, primarily when the drug was given preinfection, the minimal protection seen suggests a potentially suboptimal BCV exposure in the prairie dog. If higher BCV exposure levels had been achieved, potentially higher levels of protection may have been attained. It is possible that the lower BCV plasma exposure observed in prairie dogs than in the mouse and rabbit models administered the same doses was due to differences in the ways the prairie dogs metabolized the drug. No determination of the PK for the active metabolite cidofovir diphosphate (CDV-PP) was performed in this study due to difficulties of getting blood samples out of the biosafety level 3 (BSL-3) laboratory. As species differences in the conversion of BCV to CDV-PP have been reported (18), it is also possible that inefficient conversion of BCV to CDV-PP in prairie dogs may play a role in survivorship during this MPXV efficacy study. Unfortunately, doses of BCV higher than 20 mg when given during the preclinical program resulted in gastrointestinal toxicity within different animal models (unpublished data); therefore, doing follow-up studies with higher BCV doses in the prairie dog MPXV model is likely not feasible. However, the conversion of BCV to CDV-PP within the prairie dog may be evaluated with naive animals in future studies to compare the results with human drug data. As the outbred prairie dog MPXV model mimics human disease so closely, these additional studies might be informative in helping to predict the efficacy of BCV to treat human monkeypox and smallpox cases. Another worthwhile study would be BCV used in combination with a second drug, such as TPOXX. Although we observed low efficacy of BCV alone within the prairie dog MPXV model, synergistic efficacy may be achieved if the two drugs are given in combination. BCV inhibits OPXV replication at a different stage in the viral life cycle compared to TPOXX, and in vivo studies with mice have shown a synergistic effect when both drugs were administered (40). It would therefore be worthwhile to determine if a similar benefit was achieved with combination treatment within other animal models, such as the prairie dog MPXV model.
Within the current study, lower/suboptimal BCV exposures resulted in lower efficacy in the outbred prairie dog MPXV model. Such findings may be meaningful when considering the likely efficacy within the human population for both MPXV and smallpox. However, previous studies found that, at higher exposures with the same administered doses, BCV was efficacious within other animal models approved for use as surrogates via the FDA Animal Rule. As BCV is an oral antiviral possessing a different mechanism of action from that of the approved smallpox drug TPOXX, it warrants future evaluations to fully characterize its anti-OPXV properties for protective benefits.
MATERIALS AND METHODS
Study 1: exposure of BCV following oral administration to healthy prairie dogs. (i) Animals.
For the first study (study 1), the PK of BCV orally administered was determined in healthy, noninfected prairie dogs. Sixteen wild-caught, juvenile, black-tailed prairie dogs (Cynomys ludovicianus) obtained from Texas were used for this portion of the study, with 8 males and 8 females evenly distributed between the two dosing regimens. At the time of the study, animals were approximately 2 to 4 years old, had been prescreened by a veterinarian, and were determined to be in good health. A sterile passive integrated transponder (PIT) tag was injected subcutaneously at the base of the neck for animal identification. The average starting weight for animals was 1,064 g (range of 913 to 1,230 g). During the study, animals were housed individually in a large HEPA-filtered ventilated cage rack system within an animal biological safety level 2 (ABSL-2) facility. The study was approved by the CDC Institutional Animal Care and Use Committee (IACUC) guidelines under an approved protocol (2627HUTPRAC). In addition to being given prairie dog chow, animals were provided with monkey biscuits and apple slices.
(ii) Drug dosing and serial blood collection.
For all procedures (drug dosing and blood collection), animals were anesthetized via inhalation of isoflurane gas (1 to 5% within their cages) and then maintained on a nose cone during the procedure (1 to 5% isoflurane). Animals (n = 16) were dosed with a three-dose (q48h) regimen of 20 (n = 8) or 5 (n = 8) mg/kg BCV via oral gavage, so that BCV pharmacokinetics could be determined following single and repeated doses. Blood (the maximum allowed per the IACUC protocol was ∼0.5 ml) was collected at 8 time points (1, 2, 4, 8, 12, 24, 36, and 48 h) following the first BCV administration and at 9 time points (0, 1, 2, 4, 8, 12, 24, 36, and 48 h) following the last BCV administration via venipuncture (either the saphenous or jugular vein). To prevent dehydration and pain from the repeated blood collection, animals were given DietGel Criticare (ClearH2O; Portland, ME, USA) gel cup supplements, subcutaneous fluids (lactated ringers fluid), and meloxicam (4 mg/ml) every 24 h.
(iii) Blood sample preparation.
Blood was collected in EDTA tubes, kept on ice, and processed within 1 h of each blood draw. Blood was centrifuged at 2,000 rpm at 4°C for 20 min. After centrifugation, plasma was removed, placed into a clean tube, and frozen at –80°C.
(iv) PK analysis.
All collected plasma samples were analyzed in six batches for BCV (CMX001) using a qualified liquid chromatography-tandem mass spectrometry (LC-MS/MS) method (41) with a calibration range of 1 to 1,500 ng/ml. The interassay accuracy of the analytical quality controls (percent bias) for the six batches ranged from −16.3% to 3.0%, with an interassay precision (percent coefficient of variation [CV]) ranging from 3.0% to 17%. A noncompartmental pharmacokinetic analysis was performed on BCV plasma drug concentration data using Phoenix WinNonlin (version 6.3 or later; Certara USA, Inc., Princeton, NJ). All concentrations below the limit of quantitation (BLQ) were treated as zero if they occurred prior to the Tmax and were treated as missing if they occurred after the Tmax.
Study 2: efficacy of BCV following intranasal challenge with MPXV. (i) Animal groups and drug dosing.
In study 2, prairie dogs similar to those utilized in study 1 were wild caught, obtained from Texas, 2 to 4 years old, and determined to be negative for the presence of anti-OPXV antibodies. They had a PIT tag implanted subcutaneously. Study 2 was approved by the CDC IACUC under an approved protocol (2679HUTPRAC). Procedures throughout the study were performed according to IACUC standards in a BSL-3 enhanced (BSL-3E) laboratory and utilizing BSL-3 personal protective equipment (PPE). As no consistent BCV plasma concentration differences were observed between sexes in study 1, only female animals were used for study 2. Twenty-eight female animals were divided into 4 experimental groups: positive-control/vehicle-only animals (n = 7) and animals that began receiving BCV treatment on day −1 (n = 7), on day 0 (n = 7), and on day 1 (n = 7) relative to MPXV challenge (day 0). Four drug-only controls animals were also included; however, since no side effects from the drug were seen during the first study reported herein (exposure of BCV following oral administration to prairie dogs), data are not shown for these animals. For all groups that received BCV, animals were dosed via oral gavage with a three-dose (q48h) regimen of 20 mg/kg for the first dose, 5 mg/kg for the second dose, and 5 mg/kg for the third dose based on efficacy studies within the rabbitpox model (27). Prior to all animal procedures, animals were anesthetized by inhalation of isoflurane gas (1 to 5% within their cages) and then maintained on a nose cone with 1 to 5% isoflurane during procedures. Surviving animals were euthanized on day 25 p.i.
(ii) Virus.
The West African MPXV strain used to challenge animals, MPXV-USA-2003-044 (GenBank accession no. DQ011153), was isolated during the 2003 U.S. outbreak (42, 43). The virus has been fully sequenced and underwent two passages in African green monkey kidney cells (BSC-40) prior to seed pool production; sucrose cushion semipurified preparations of virus were used for animal challenges.
(iii) Animal inoculation.
Inoculum dosages (8.8 × 105 PFU) were calculated based on the morbidity and mortality rates observed in the authors’ previous studies with this animal model. Briefly, a challenge dose of 6 × 105 PFU WA MPXV resulted in disease morbidity, including skin lesions, in 100% of animals, with 75% mortality (31). As our goal was to achieve an LD90, we extrapolated from the previous study to determine the challenge dose. The viral strain stock was diluted in phosphate-buffered saline (PBS). Inoculum titers were immediately reconfirmed following challenge by a standard plaque assay (as described below). Animals were infected via an intranasal (i.n.) route of inoculation with a total volume of 10 μl (5 μl in each nostril) while under anesthesia with 1 to 5% isoflurane.
(iv) Observations and sampling.
For all 25 days of the study, animals were observed daily for signs of morbidity (inappetence, decreased activity, recumbence with a reluctance to move, etc.) and clinical lesions, including skin rash. Prebleed specimens and swabs were collected prior to the start of the study. On sample days (1, 3, 5, 8, 11, 14, 17, and 22 postinoculation [p.i.]), oral swabs, blood, weights, and lesion counts were collected from all animals while under general anesthesia (as described above). Blood (target, ∼1 ml) was collected from the saphenous vein using a 25- or 26-gauge needle for ELISA and PCR/tissue culture analysis.
(v) Pain and euthanasia criteria.
Strict euthanasia criteria specific to the prairie dog MPXV model were applied throughout the study, with pain scores assigned to four categories (body weight, appearance, behavior, and clinical signs). A score of 4 to 6 resulted in performing a complete physical assessment at least every 48 h, offering additional approved palatable treats to encourage eating, and administering subcutaneous (s.c.) or oral fluids if dehydration was observed. A score of 7 to 9 necessitated continuing the precautions listed above for a score of 4 to 6, with the following adjustments/additions: increasing observation to at least twice daily, administering an analgesic (buprenorphine) if clinical signs warranted it, and/or considering euthanasia. A previously conducted study had determined that buprenorphine does not alter the disease course within the prairie dog following MPXV challenge (44). A score of ≥10 resulted in immediate euthanasia. For euthanasia, 100 mg/kg of sodium pentobarbital was administered via cardiocentesis while the animals were maintained under deep anesthesia (5% isoflurane).
(vi) Necropsy and tissue specimen collection.
Complete necropsies were performed following euthanasia. Instruments were cleaned and decontaminated by rinsing them in 5% Micro-Chem Plus–70% ethanol and then with sterile water between collections of each tissue. Tissues were frozen at –70°C prior to further processing for virology assessments. Swabs (oral and skin lesion swabs, if applicable) were collected with sterile individual Dacron swabs and stored frozen without diluent. Serum was separated from whole blood and then kept refrigerated at 4°C prior to testing by an appropriate serologic assay. Tissues and swabs were subsequently processed and further prepared for DNA analysis and virus isolation.
(vii) Sample preparation for real-time PCR and viral growth.
Sample processing was performed under BSL-2 conditions with BSL-3 work practices. For whole-blood samples, 100 μl was used for DNA extraction, and the remaining blood was used for tissue culture propagation. For each swab collected, 400 μl of PBS was added. The swab extraction tube system (SETS) (Roche) protocol was used to recover sample from the swab. DNA was extracted from 100 μl of the swab lysate. The remaining swab eluate was used for virus isolation. For tissue preparation, tissue samples were weighed and 1-ml aliquots of PBS and SPEX beads (SPEX SamplePrep, Metuchen, NJ, USA) were prepared. The PBS/bead aliquot was then poured into a tube containing the tissue sample. The GenoGrinder 2000 (SPEX SamplePrep) was used according to the manufacturer’s instructions to create a tissue homogenate. One hundred microliters of the homogenate was used for extraction of DNA. The remaining homogenate was used for virus isolation. Sample aliquots were incubated at 56°C for 1 h to inactivate viable virus particles prior to DNA extraction. The BioRobot EZ-1 workstation (Qiagen) was used for DNA extraction of all blood, swab, and tissue samples.
(viii) Real-time PCR analysis.
Samples were tested by real-time PCR using forward and reverse primers and probes complementary to the conserved OPXV E9L (DNA polymerase) gene (45). Purified MPXV DNA (10 fg to 1 ng) was used as standard controls to allow quantification of viral DNA within samples. A sample was considered positive if real-time PCR produced a threshold cycle (CT) value (in duplicate) of 37 or earlier. A weakly positive sample displayed CT values of 38 to 39 (in duplicate).
(ix) Virus tissue infectivity.
All samples were stored at –70°C until virus isolation was attempted. Previous analyses demonstrated that real-time PCR detects trace amounts of MPXV DNA and is more sensitive than tissue culture detection of viable virus (7). Therefore, specimens were first tested for the presence of OPXV DNA by real-time PCR, and if samples were positive, they were subsequently evaluated for viable virus by a standard plaque assay Each blood, swab, or tissue sample was titrated in duplicate using 10-fold dilutions of swab eluate or tissue slurry on BSC-40 cell monolayers, incubated at 35.5°C and in 6% CO2 for 72 h, and subsequently stained with crystal violet stain containing formalin to visualize plaques. Titers were expressed as PFU per milliliter of blood or swab eluate or as PFU per gram of tissue.
(x) Serologic analysis.
A modified ELISA was used for analysis of anti-OPXV immunoglobulins in separated sera as previously described in detail (30, 46).
Statistical analyses.
As data were not normally distributed; nonparametric statistical analyses were used. The Wilcoxon rank sum test was utilized to compare ELISA results. A P value of ≤0.05 was considered statistically significant. Data analysis was performed using SAS v9.3. Descriptive statistics, including mean, standard deviation, and coefficient of variation, were used to summarize the BCV PK parameters. Differences in mortality rates among the treatment timing groups and compared to the vehicle only group were analyzed using Fisher's exact test, due to small sample size. Treatment timing groups' day of euthanasia (postinfection) for prairie dogs was made using the Wilcoxon rank-sum test, as data were not normally distributed. Kaplan-Meier survivorship curves were calculated and compared using the log-rank test. A P value less than 0.06 was considered statistically significant. Data analysis was performed using SAS version 9.4 (SAS Institute, Cary, NC).
ACKNOWLEDGMENTS
We acknowledge Nicole L. Lukovsky-Akhsanov, Cynthia D. Cary, Nishi Patel, Shannon Keckler, Michael Townsend, Laura Hughes-Baker, Theodora Khan, Lawrence Trost, Katherine V. Sickle, and Timothy Tippin.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
REFERENCES
- 1.Noyce RS, Lederman S, Evans DH. 2018. Construction of an infectious horsepox virus vaccine from chemically synthesized DNA fragments. PLoS One 13:e0188453. doi: 10.1371/journal.pone.0188453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogoina D, Izibewule JH, Ogunleye A, Ederiane E, Anebonam U, Neni A, Oyeyemi A, Etebu EN, Ihekweazu C. 2019. The 2017 human monkeypox outbreak in Nigeria—report of outbreak experience and response in the Niger Delta University Teaching Hospital, Bayelsa State, Nigeria. PLoS One 14:e0214229. doi: 10.1371/journal.pone.0214229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye F, Song J, Zhao L, Zhang Y, Xia L, Zhu L, Kamara IL, Ren J, Wang W, Tian H, Wu G, Tan W. 2019. Molecular evidence of human monkeypox virus infection, Sierra Leone. Emerg Infect Dis 25:1220–1222. doi: 10.3201/eid2506.180296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reynolds MG, Wauquier N, Li Y, Satheshkumar PS, Kanneh LD, Monroe B, Maikere J, Saffa G, Gonzalez JP, Fair J, Carroll DS, Jambai A, Dafae F, Khan SH, Moses LM. 2019. Human monkeypox in Sierra Leone after 44-year absence of reported cases. Emerg Infect Dis 25:1023–1025. doi: 10.3201/eid2505.180832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doshi RH, Guagliardo SAJ, Doty JB, Babeaux AD, Matheny A, Burgado J, Townsend MB, Morgan CN, Satheshkumar PS, Ndakala N, Kanjingankolo T, Kitembo L, Malekani J, Kalemba L, Pukuta E, N'Kaya T, Kangoula F, Moses C, McCollum AM, Reynolds MG, Mombouli JV, Nakazawa Y, Petersen BW. 2019. Epidemiologic and ecologic investigations of monkeypox, Likouala Department, Republic of the Congo, 2017. Emerg Infect Dis 25:281–289. doi: 10.3201/eid2502.181222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sklenovska N, Van Ranst M. 2018. Emergence of monkeypox as the most important orthopoxvirus infection in humans. Front Public Health 6:241. doi: 10.3389/fpubh.2018.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutson CL, Lee KN, Abel J, Carroll DS, Montgomery JM, Olson VA, Li Y, Davidson W, Hughes C, Dillon M, Spurlock P, Kazmierczak JJ, Austin C, Miser L, Sorhage FE, Howell J, Davis JP, Reynolds MG, Braden ZH, Karem KL, Damon IK, Regnery RL. 2007. Monkeypox zoonotic associations: insights from laboratory evaluation of animals associated with the multi-state US outbreak. Am J Trop Med Hyg 76:757–767. doi: 10.4269/ajtmh.2007.76.757. [DOI] [PubMed] [Google Scholar]
- 8.Vaughan A, Aarons E, Astbury J, Balasegaram S, Beadsworth M, Beck CR, Chand M, O'Connor C, Dunning J, Ghebrehewet S, Harper N, Howlett-Shipley R, Ihekweazu C, Jacobs M, Kaindama L, Katwa P, Khoo S, Lamb L, Mawdsley S, Morgan D, Palmer R, Phin N, Russell K, Said B, Simpson A, Vivancos R, Wade M, Walsh A, Wilburn J. 2018. Two cases of monkeypox imported to the United Kingdom, September 2018. Euro Surveill 23:1800509. doi: 10.2807/1560-7917.ES.2018.23.38.1800509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Health Organization. 2019. Monkeypox—Singapore. World Health Organization, Geneva, Switzerland: https://www.who.int/csr/don/16-may-2019-monkeypox-singapore/en/. [Google Scholar]
- 10.Food and Drug Administration. 2019. FDA approves first live, non-replicating vaccine to prevent smallpox and monkeypox. https://www.fda.gov/news-events/press-announcements/fda-approves-first-live-non-replicating-vaccine-prevent-smallpox-and-monkeypox. FDA, Bethesda, MD. [Google Scholar]
- 11.Delaune D, Iseni F. 2020. Drug development against smallpox: present and future. Antimicrob Agents Chemother 64:e01683-19. doi: 10.1128/AAC.01683-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merchlinsky M, Albright A, Olson V, Schiltz H, Merkeley T, Hughes C, Petersen B, Challberg M. 2019. The development and approval of tecoviromat (TPOXX®), the first antiviral against smallpox. Antiviral Res 168:168–174. doi: 10.1016/j.antiviral.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duraffour S, Lorenzo MM, Zoller G, Topalis D, Grosenbach D, Hruby DE, Andrei G, Blasco R, Meyer H, Snoeck R. 2015. ST-246 is a key antiviral to inhibit the viral F13L phospholipase, one of the essential proteins for orthopoxvirus wrapping. J Antimicrob Chemother 70:1367–1380. doi: 10.1093/jac/dku545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lederman ER, Davidson W, Groff HL, Smith SK, Warkentien T, Li Y, Wilkins KA, Karem KL, Akondy RS, Ahmed R, Frace M, Shieh WJ, Zaki S, Hruby DE, Painter WP, Bergman KL, Cohen JI, Damon IK. 2012. Progressive vaccinia: case description and laboratory-guided therapy with vaccinia immune globulin, ST-246, and CMX001. J Infect Dis 206:1372–1385. doi: 10.1093/infdis/jis510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hostetler KY, Beadle JR, Trahan J, Aldern KA, Owens G, Schriewer J, Melman L, Buller RM. 2007. Oral 1-O-octadecyl-2-O-benzyl-sn-glycero-3-cidofovir targets the lung and is effective against a lethal respiratory challenge with ectromelia virus in mice. Antiviral Res 73:212–218. doi: 10.1016/j.antiviral.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tippin TK, Morrison ME, Brundage TM, Mommeja-Marin H. 2016. Brincidofovir is not a substrate for the human organic anion transporter 1: a mechanistic explanation for the lack of nephrotoxicity observed in clinical studies. Ther Drug Monit 38:1–786. doi: 10.1097/FTD.0000000000000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrei G, Snoeck R. 2010. Cidofovir activity against poxvirus infections. Viruses 2:2803–2830. doi: 10.3390/v2122803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lanier R, Trost L, Tippin T, Lampert B, Robertson A, Foster S, Rose M, Painter W, O'Mahony R, Almond M, Painter G. 2010. Development of CMX001 for the treatment of poxvirus infections. Viruses 2:2740–2762. doi: 10.3390/v2122740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wold WS, Toth K. 2015. New drug on the horizon for treating adenovirus. Expert Opin Pharmacother 16:2095–2099. doi: 10.1517/14656566.2015.1083975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster SA, Parker S, Lanier R. 2017. The role of brincidofovir in preparation for a potential smallpox outbreak. Viruses 9:320. doi: 10.3390/v9110320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olson VA, Smith SK, Foster S, Li Y, Lanier ER, Gates I, Trost LC, Damon IK. 2014. In vitro efficacy of brincidofovir against variola virus. Antimicrob Agents Chemother 58:5570–5571. doi: 10.1128/AAC.02814-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Israely T, Paran N, Lustig S, Erez N, Politi B, Shafferman A, Melamed S. 2012. A single cidofovir treatment rescues animals at progressive stages of lethal orthopoxvirus disease. Virol J 9:119. doi: 10.1186/1743-422X-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rice AD, Adams MM, Wallace G, Burrage AM, Lindsey SF, Smith AJ, Swetnam D, Manning BR, Gray SA, Lampert B, Foster S, Lanier R, Robertson A, Painter G, Moyer RW. 2011. Efficacy of CMX001 as a post exposure antiviral in New Zealand White rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 3:47–62. doi: 10.3390/v3010047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rice AD, Adams MM, Lampert B, Foster S, Robertson A, Painter G, Moyer RW. 2011. Efficacy of CMX001 as a prophylactic and presymptomatic antiviral agent in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses 3:63–82. doi: 10.3390/v3020063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaitseva M, McCullough KT, Cruz S, Thomas A, Diaz CG, Keilholz L, Grossi IM, Trost LC, Golding H. 2015. Postchallenge administration of brincidofovir protects healthy and immune-deficient mice reconstituted with limited numbers of T cells from lethal challenge with IHD-J-Luc vaccinia virus. J Virol 89:3295–3307. doi: 10.1128/JVI.03340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trost LC, Rose ML, Khouri J, Keilholz L, Long J, Godin SJ, Foster SA. 2015. The efficacy and pharmacokinetics of brincidofovir for the treatment of lethal rabbitpox virus infection: a model of smallpox disease. Antiviral Res 117:115–121. doi: 10.1016/j.antiviral.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Grossi IM, Foster SA, Gainey MR, Krile RT, Dunn JA, Brundage T, Khouri JM. 2017. Efficacy of delayed brincidofovir treatment against a lethal rabbitpox virus challenge in New Zealand White rabbits. Antiviral Res 143:278–286. doi: 10.1016/j.antiviral.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 28.Parker S, Touchette E, Oberle C, Almond M, Robertson A, Trost LC, Lampert B, Painter G, Buller RM. 2008. Efficacy of therapeutic intervention with an oral ether–lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res 77:39–49. doi: 10.1016/j.antiviral.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker S, Chen NG, Foster S, Hartzler H, Hembrador E, Hruby D, Jordan R, Lanier R, Painter G, Painter W, Sagartz JE, Schriewer J, Mark Buller R. 2012. Evaluation of disease and viral biomarkers as triggers for therapeutic intervention in respiratory mousepox—an animal model of smallpox. Antiviral Res 94:44–53. doi: 10.1016/j.antiviral.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutson CL, Olson VA, Carroll DS, Abel JA, Hughes CM, Braden ZH, Weiss S, Self J, Osorio JE, Hudson PN, Dillon M, Karem KL, Damon IK, Regnery RL. 2009. A prairie dog animal model of systemic orthopoxvirus disease using West African and Congo Basin strains of monkeypox virus. J Gen Virol 90:323–333. doi: 10.1099/vir.0.005108-0. [DOI] [PubMed] [Google Scholar]
- 31.Hutson CL, Carroll DS, Self J, Weiss S, Hughes CM, Braden ZH, Olson VA, Smith SK, Karem KL, Regnery RL, Damon IK. 2010. Dosage comparison of Congo Basin and West African strains of monkeypox virus using a prairie dog model of systemic orthopoxvirus disease. Virology 402:72–82. doi: 10.1016/j.virol.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutson CL, Carroll DS, Gallardo-Romero N, Drew C, Zaki SR, Nagy T, Hughes C, Olson VA, Sanders J, Patel N, Smith SK, Keckler MS, Karem K, Damon IK. 2015. Comparison of monkeypox virus clade kinetics and pathology within the prairie dog animal model using a serial sacrifice study design. Biomed Res Int 2015:965710. doi: 10.1155/2015/965710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keckler MS, Carroll DS, Gallardo-Romero NF, Lash RR, Salzer JS, Weiss SL, Patel N, Clemmons CJ, Smith SK, Hutson CL, Karem KL, Damon IK. 2011. Establishment of the black-tailed prairie dog (Cynomys ludovicianus) as a novel animal model for comparing smallpox vaccines administered preexposure in both high- and low-dose monkeypox virus challenges. J Virol 85:7683–7698. doi: 10.1128/JVI.02174-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huhn GD, Bauer AM, Yorita K, Graham MB, Sejvar JJ, Likos A, Damon IK, Reynolds MG, Kuehnert MJ. 2005. Clinical characteristics of human monkeypox and risk factors for severe disease. Clin Infect Dis 41:1742–1751. doi: 10.1086/498115. [DOI] [PubMed] [Google Scholar]
- 35.Hutson CL, Gallardo-Romero N, Carroll DS, Clemmons C, Salzer JS, Nagy T, Hughes CM, Olson VA, Karem KL, Damon IK. 2013. Transmissibility of the monkeypox virus clades via respiratory transmission: investigation using the prairie dog-monkeypox virus challenge system. PLoS One 8:e55488. doi: 10.1371/journal.pone.0055488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutson CL, Carroll DS, Gallardo-Romero N, Weiss S, Clemmons C, Hughes CM, Salzer JS, Olson VA, Abel J, Karem KL, Damon IK. 2011. Monkeypox disease transmission in an experimental setting: prairie dog animal model. PLoS One 6:e28295. doi: 10.1371/journal.pone.0028295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith SK, Self J, Weiss S, Carroll D, Braden Z, Regnery RL, Davidson W, Jordan R, Hruby DE, Damon IK. 2011. Effective antiviral treatment of systemic orthopoxvirus disease: ST-246 treatment of prairie dogs infected with monkeypox virus. J Virol 85:9176–9187. doi: 10.1128/JVI.02173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho CT, Wenner HA. 1973. Monkeypox virus. Bacteriol Rev 37:1–18. doi: 10.1128/BR.37.1.1-18.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mucker EM, Wollen-Roberts SE, Kimmel A, Shamblin J, Sampey D, Hooper JW. 2018. Intranasal monkeypox marmoset model: prophylactic antibody treatment provides benefit against severe monkeypox virus disease. PLoS Negl Trop Dis 12:e0006581. doi: 10.1371/journal.pntd.0006581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quenelle DC, Prichard MN, Keith KA, Hruby DE, Jordan R, Painter GR, Robertson A, Kern ER. 2007. Synergistic efficacy of the combination of ST-246 with CMX001 against orthopoxviruses. Antimicrob Agents Chemother 51:4118–4124. doi: 10.1128/AAC.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Painter W, Robertson A, Trost LC, Godkin S, Lampert B, Painter G. 2012. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob Agents Chemother 56:2726–2734. doi: 10.1128/AAC.05983-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Likos AM, Sammons SA, Olson VA, Frace AM, Li Y, Olsen-Rasmussen M, Davidson W, Galloway R, Khristova ML, Reynolds MG, Zhao H, Carroll DS, Curns A, Formenty P, Esposito JJ, Regnery RL, Damon IK. 2005. A tale of two clades: monkeypox viruses. J Gen Virol 86:2661–2672. doi: 10.1099/vir.0.81215-0. [DOI] [PubMed] [Google Scholar]
- 43.Reed KD, Melski JW, Graham MB, Regnery RL, Sotir MJ, Wegner MV, Kazmierczak JJ, Stratman EJ, Li Y, Fairley JA, Swain GR, Olson VA, Sargent EK, Kehl SC, Frace AM, Kline R, Foldy SL, Davis JP, Damon IK. 2004. The detection of monkeypox in humans in the Western Hemisphere. N Engl J Med 350:342–350. doi: 10.1056/NEJMoa032299. [DOI] [PubMed] [Google Scholar]
- 44.Hutson CL, Gallardo-Romero N, Carroll DS, Salzer JS, Ayers JD, Doty JB, Hughes CM, Nakazawa Y, Hudson P, Patel N, Keckler MS, Olson VA, Nagy T. 2019. Analgesia during monkeypox virus experimental challenge studies in prairie dogs (Cynomys ludovicianus). J Am Assoc Lab Anim Sci 58:485–500. doi: 10.30802/AALAS-JAALAS-18-000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Olson VA, Laue T, Laker MT, Damon IK. 2006. Detection of monkeypox virus with real-time PCR assays. J Clin Virol 36:194–203. doi: 10.1016/j.jcv.2006.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karem KL, Reynolds MG, Braden ZH, Lou G, Bernard N, Patton J, Damon IK. 2005. Characterization of acute-phase humoral immunity to monkeypox: use of immunoglobulin M enzyme-linked immunosorbent assay for detection of monkeypox infection during the 2003 North American outbreak. Clin Diagn Lab Immunol 12:867–872. doi: 10.1128/CDLI.12.7.867-872.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stabenow J, Buller RM, Schriewer J, West C, Sagartz JE, Parker S. 2010. A mouse model of lethal infection for evaluating prophylactics and therapeutics against Monkeypox virus. J Virol 84:3909–3920. doi: 10.1128/JVI.02012-09. [DOI] [PMC free article] [PubMed] [Google Scholar]