

Abstract
Several X-linked neurodevelopmental disorders including Rett syndrome, induced by mutations in the MECP2 gene, and fragile X syndrome (FXS), caused by mutations in the FMR1 gene, share autism-related features. The mRNA coding for methyl CpG binding protein 2 (MeCP2) has previously been identified as a substrate for the mRNA-binding protein, fragile X mental retardation protein (FMRP), which is silenced in FXS. Here, we report a homeostatic relationship between these two key regulators of gene expression in mouse models of FXS (Fmr1 Knockout (KO)) and Rett syndrome (MeCP2 KO). We found that the level of MeCP2 protein in the cerebral cortex was elevated in Fmr1 KO mice, whereas MeCP2 KO mice displayed reduced levels of FMRP, implicating interplay between the activities of MeCP2 and FMRP. Indeed, knockdown of MeCP2 with short hairpin RNAs led to a reduction of FMRP in mouse Neuro2A and in human HEK-293 cells, suggesting a reciprocal coupling in the expression level of these two regulatory proteins. Intra-cerebroventricular injection of an adeno-associated viral vector coding for FMRP led to a concomitant reduction in MeCP2 expression in vivo and partially corrected locomotor hyperactivity. Additionally, the level of MeCP2 in the posterior cortex correlated with the severity of the hyperactive phenotype in Fmr1 KO mice. These results demonstrate that MeCP2 and FMRP operate within a previously undefined homeostatic relationship. Our findings also suggest that MeCP2 overexpression in Fmr1 KO mouse posterior cerebral cortex may contribute to the fragile X locomotor hyperactivity phenotype.
Graphical Abstract
Graphical Abstract.
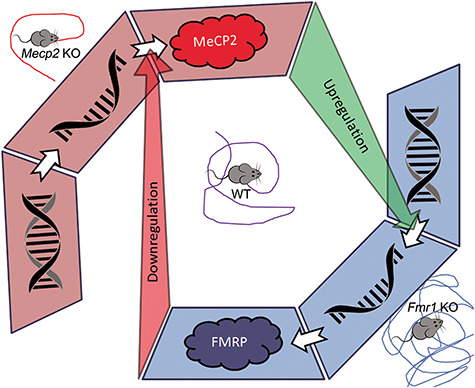
Interregulation between MeCP2 and FMRP. MeCP2, a transcriptional modulator, contributes to the upregulation of FMRP expression. FMRP, a translational modulator, contributes to the downregulation of MeCP2 translation. MeCP2 KO mice are hypoactive. Fmr1 KO mice display hyperactivity.
Introduction
Several X-linked neurodevelopmental disorders, including fragile X syndrome (FXS) and Rett syndrome, display shared symptoms associated with autism spectrum disorders (ASDs) and are characterized by impairments in social interaction and communication and repetitive stereotyped patterns of behavior (1,2). The Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-V) classifies FXS as an intellectual disability disorder on the autism spectrum. The disorder is caused by a CGG expansion of the 5′ region of the FMR1 gene causing impairment of fragile X mental retardation protein (FMRP) protein expression required for normal neurological function. Approximately, 30–40% of FXS patients fit the full diagnostic criteria for ASD, while more than 75% display at least some autistic characteristics (1,3,4). Rett syndrome, caused by de novo mutations that impair methyl CpG binding protein 2 (MeCP2) protein function or expression (5,6), was previously classified as a pervasive developmental disorder but was not included in the ASD classification in the DSM-V (7). Like FXS, this disorder displays overlapping clinical features associated with ASD, including intellectual and locomotor deficits.
FMRP functions as an mRNA-binding protein and acts as a wide-spectrum translational modulator via its ability, among others, to stall ribosomal translation (8–10). FMRP has also been implicated in microRNA (miRNA) processing, maturation and nuclear export, and it participates in RNA-induced silencing complex (RISC)-mediated mRNA processing (11–13). FMRP also plays a role in: (a) mRNA stability (14,15), (b) regulation of intracellular trafficking, (c) as an allosteric modulator of K+ channel activity through protein–protein interactions (16–18) and (d) contributes to intracellular phase separation of neuronal granules (10,19). The absence of FMRP’s pleiotropic activity in FXS results in aberrant up- and downregulation of several proteins required for cellular differentiation, proper synaptic function and neuronal plasticity.
In contrast to FMRP-mediated translational modulation, MeCP2, which binds methylated CpG DNA sequences, both represses and activates gene transcription (20,21). Additional studies suggest that MeCP2 also participates in RNA splicing (22,23) as well as miRNA regulation (24). MeCP2 is posttranscriptionally regulated in a spatiotemporal manner during brain development (25). The necessity for precise control of MeCP2 and FMRP expression levels is highlighted by case studies that reveal the neurodevelopmental consequences of FMR1 (26–29) and MeCP2 gene duplications (30). MeCP2 duplication syndrome is manifested by profound neurodevelopmental abnormalities and shows similarities to FMRP gene duplication where patients display severe intellectual disability, anxiety, impaired language and motor skills, hypotonia and stereotyped behaviors (30–33).
Here, we investigated the interrelationship of these two key proteins in the context of wild-type (WT), Fmr1 KO and MeCP2 KO mice. In the cerebral cortical tissue, MeCP2 protein levels were upregulated in Fmr1 KO mice, while FMRP was downregulated in MeCP2 KO mice. The expression of MeCP2 and FMRP was inversely regulated on a cell autonomous level in WT and in Fmr1 KO mice transduced with an adeno-associated viral (AAV) vector coding for FMRP. Notably, MeCP2 protein overexpression significantly correlated with the severity of locomotor hyperactivity, suggesting a link to FXS symptomology. Our results demonstrate that the brain levels of FMRP and MeCP2 are interlinked with each other, suggesting that elevated MeCP2 may contribute to the FXS phenotype, and conversely, that reduced FMRP may contribute to the Rett syndrome phenotype.
Results
Biochemical and cytological characterization of MeCP2 in WT and Fmr1 KO mice
To evaluate the cellular relationship between FMRP and MeCP2, we used confocal microscopy to determine whether the two proteins are co-expressed within the same cells. In the motor cortex of WT mouse brain, both FMRP and MeCP2 were expressed within the same cells, albeit primarily in different subcellular compartments (FMRP predominantly in the cytosol; MeCP2 in the nucleus). As expected, both proteins were co-expressed primarily within cells that also expressed the neuron-specific marker, NeuN, confirming the neuronal identity of the cells that display elevated MeCP2 or elevated FMRP (Supplementary Material, Fig. S1A and B). Relative NeuN intensity directly correlated with both FMRP and MeCP2 immunoreactivity on the individual cell level (P < 0.01). Neuronal co-expression of FMRP and MeCP2 can also be seen in the cerebral cortex (Fig. 1A), striatum (Fig. 1B) and hippocampus (Fig. 1C) as well the deep cerebellar nuclei (DCN) and the cerebellar cortex (Fig. 1D–F). Thus, previous evidence that MeCP2 mRNA could be a substrate of FMRP (8,34) together with the observation here that both proteins are largely present within the same cells, suggest that they are physically co-expressed to potentially allow interregulation in a cell autonomous fashion.
Figure 1.
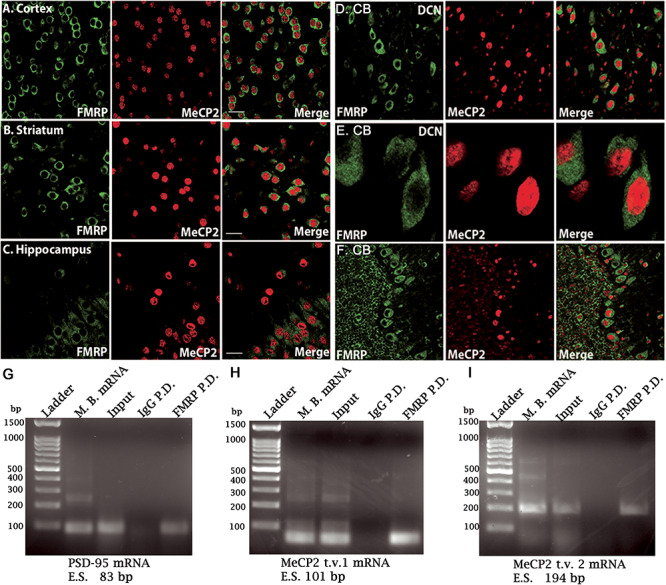
MeCP2 transcript analysis and cellular co-distribution of MeCP2 and FMRP in the WT mouse brain. (A–C) Photomicrographs of the cerebral cortex (A), striatum (B) and hippocampus (C) showing co-localization of MeCP2 and FMRP. (D–F) Photomicrographs of the DCN under low (D) and high magnification (E). (F) Example of cerebellar cortex showing the Purkinje neuron layer situated between the molecular layer (right) and granule cell layer (left). Anti-FMRP is shown in green and anti-MeCP2 is shown in red. White bar = 20 μm; CB, cerebellum; DCN, deep cerebellar nuclei. Reverse transcription of mRNA from WT mouse. (G) PSD-95 mRNA reverse transcribed from total mouse brain mRNA, supernatant (input), non-specific IgG pull-down and anti-FMRP pull-down was used as a positive control. (H) MeCP2 transcript variant 1 reverse transcribed mRNA and (I) MeCP2 transcript variant 2 reverse transcribed mRNA screening. N = 4 mice for all conditions. Input, total isolated mRNA; P.D., pull-down; M.B., mouse brain total mRNA; bp, base pairs; t.v., transcript variant; E.S., expected size of band.
Further investigation was carried out to access the ability of FMRP to bind MeCP2 mRNA transcript variants. Immuno-precipitations of cortical homogenates from WT mice using an anti-FMRP antibody were performed; PSD-95 mRNA, a well-established substrate of FMRP, was used as a positive control (Fig. 1G). MeCP2 has two major isoforms; MeCP2 transcript isoform 1 is expressed throughout the brain, while isoform 2 is less abundant with restricted expression in the CNS and differs from isoform 1 only in its N-terminus (35,36). Amplification of MeCP2 transcripts 1- (Fig. 1H) and 2-specific mRNAs (Fig. 1I) from the anti-FMRP immunoprecipitate indicated the presence of both variants. Samples containing the non-specific IgG immunoprecipitate did not produce detectable MeCP2 or PSD-95 amplicon, further confirming the specific nature of the FMRP–MeCP2 mRNA interaction. Because of the amplificating nature of the polymerase chain reaction (PCR) reaction, the primers used were sufficient for the qualitative identification of the two MeCP2 isoforms, but the transcripts could not be compared quantitatively by the Q-PCR as they displayed variations in the annealing temperature; thus, only the presence or absence of each mRNA could be confirmed using these probes. Nevertheless, these results support that FMRP is able to bind both MeCP2 transcript variants. Thus, we were also able to detect the presence of MeCP2 mRNA within the specific FMRP immunoprecipitate as was previously observed in a HITS-CLIP assay of whole brain lysate in 2011 (8). However, quantitative reverse transcriptase PCR using primers coding for combined MeCP2 transcript variants 1 and 2 showed no differences in the total MeCP2 mRNA expression in WT compared with Fmr1 KO mice forebrains (Fig. 2A), indicating that FMRP itself, or the homeostatic result of the absence of FMRP, does not alter the transcription rate of MeCP2 DNA into mRNA by a noticeable degree. To examine MeCP2 under conditions of variable FMRP expression, MeCP2 protein quantification was carried out on adult (2-month-old) WT and Fmr1 KO mice that were injected on postnatal day (PND) 0 or 1 with either a control vector carrying no transgene (AAV-empty vector; AAV-EV) or an AAV vector carrying the FMRP transgene (AAV-FMRP). Three test groups of mice were studied: WT mice injected with AAV-EV, Fmr1 KO mice injected with AAV-EV and Fmr1 KO mice injected with AAV-FMRP. Immunoblot analysis demonstrated that, compared with WT controls injected with AAV-EV, MeCP2 protein was overexpressed in the posterior cerebral cortex of the adult Fmr1 KO mice injected with the same control vector (Fig. 2B). Importantly, MeCP2 overexpression in adult Fmr1 KO mice was normalized after treatment with AAV-FMRP (KO + AAV-FMRP group) as would be expected for an FMRP substrate with a lack of translational repression. Densitometric quantification (Fig. 2C) revealed that the elevated MeCP2 protein expression observed in Fmr1 KO mice injected with AAV-EV (132 ± 8%, P < 0.05; N = 12) was significantly reduced in the group of Fmr1 KO mice injected with AAV-FMRP (89 ± 11%, P < 0.01; N = 14). These results demonstrated that MeCP2 overexpression in the Fmr1 KO mice is not caused by an elevation in MeCP2 gene transcription, but rather, is due to a deficiency in posttranscriptional regulation. Together, these findings indicate that the absence of FMRP leads to the upregulation of MeCP2 protein and that this can be corrected in the mouse brain in vivo by the introduction of exogenous AAV-FMRP.
Figure 2.
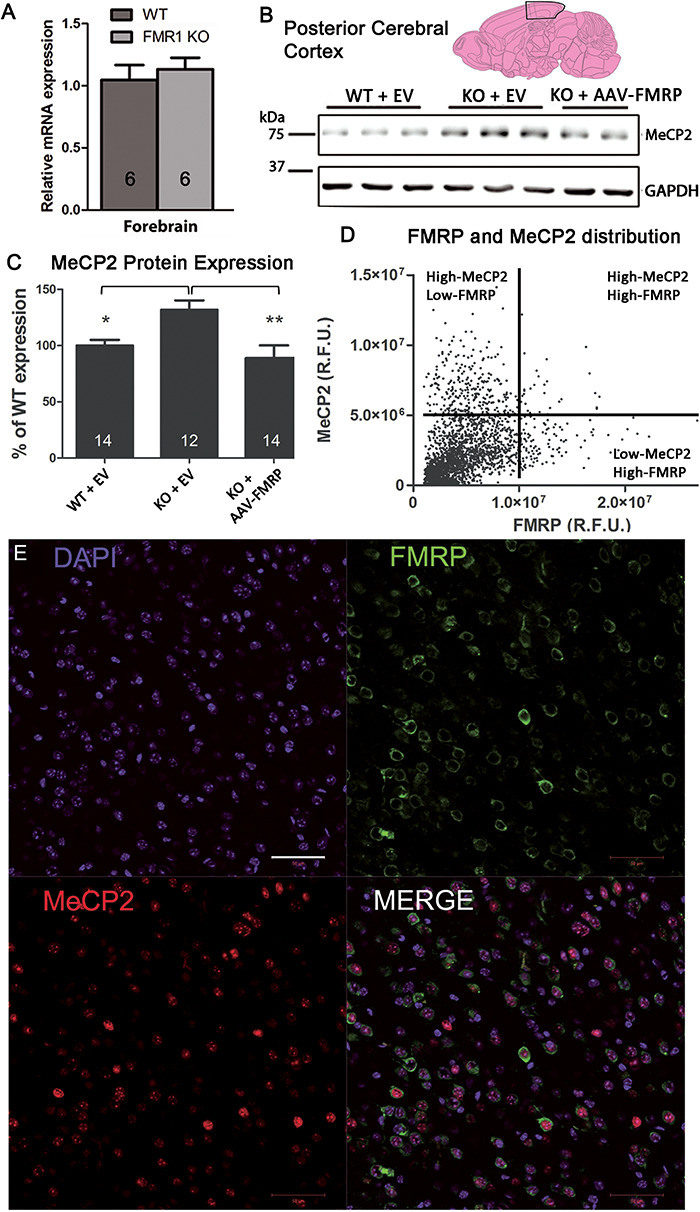
Expression of FMRP and MeCP2 in WT and AAV-FMRP transduced Fmr1 KO mouse brain and individual neurons. (A) Q-RT-PCR of MeCP2 mRNA in WT and Fmr1 KO mouse forebrain. GAPDH mRNA was used to normalize quantification, and the results are presented as relative mRNA expression (arbitrary units; mean ± SEM). (B) Western blot of MeCP2 in the cerebral cortex of WT + EV, KO + EV and KO mice transduced with AAV-FMRP. (C) Densitometric quantification of MeCP2 protein expression from the western blots of the cortices of WT + EV, KO + EV and KO + AAV-FMRP treated mice. GAPDH was used to normalize protein amounts; results are presented as a percentage of WT expression (mean ± SEM). The numbers of replicates (mice) are indicated within the bars (*P < 0.05, **P < 0.01). (D) Scatter plot of quantified FMRP and MeCP2 cellular immunoreactive intensities from three serial coronal slides obtained from four different Fmr1 KO mice injected with AAV-FMRP. MeCP2 and FMRP display an inverse relationship where high expression (arbitrary cutoff) of MeCP2 correlates with low FMRP expression and vice versa. Only 1% of cells exists in the high-FMRP and high-MeCP2 quadrant (top right), indicating that elevated translational modulation (high-FMRP; 7%) generally does not occur in the same cells as those under elevated transcriptional modulation (high-MeCP2; 19%). To assure a proper plane of focus on the nucleus, DAPI staining was used to select cells. R.F.U., relative fluorescence units. (E) Example photomicrograph of the sensorimotor cortex from a 2-month-old Fmr1 KO mouse transduced with AAV-FMRP labeled with anti-FMRP (green) and anti-MeCP2 (red) immunostaining. Nuclei were counterstained with DAPI (blue). Merged image shows the composite overlay. White bar = 50 μm.
Single cell analyses of MeCP2 and FMRP expression in the sensorimotor cortex
To assess the effects of intracellular FMRP levels on MeCP2 protein expression, we exploited the cellular variability of transgene uptake and expression previously reported in Fmr1 KO mice transduced with AAV-FMRP (37). Because a synapsin (syn) promoter was used to drive AAV-FMRP neuron-specific transgene expression (34,37), we chose to quantify the MeCP2 expression levels in the nucleus of individual neurons relative to the level of FMRP expressed in the cytosol of those same cells. Eight weeks following a PND 0–1 bilateral intra-cerebroventricular (i.c.v.) injection of AAV-FMRP, immunocytochemistry was performed on coronal sections of fixed brain tissue. Endogenous MeCP2 expression was present in virtually all cells of the sensorimotor cortex and consistently co-localized with DAPI nuclear staining. As seen in WT neurons (e.g. see Fig. 1A), the AAV-FMRP-transduced neurons displayed expected intracellular distribution patterns of the proteins with nuclear localization of MeCP2 and cytosolic localization of FMRP. Three serial coronal sections of the sensorimotor cortex obtained from four KO + AAV-FMRP mice were used in subsequent analyses. MeCP2 immunoreactivity was then analyzed as a function of FMRP immunoreactive intensity within the same cells. An inverse relationship (R = −0.14, P < 0.001; N = 2358 cells; Fig. 2D) between the single cell immunoreactivity of MeCP2 and FMRP (e.g. Fig. 2E) was observed using unbiased MATLAB quantification. Cells labeled with FMRP and NeuN, as well as with MeCP2 and NeuN, displayed a direct correlation where high-FMRP coincided with high-NeuN staining, which confirmed their neuronal identity (Supplementary Material, Fig. S1A and B; left panels). When the cells were co-labeled with the astrocyte marker S100β and FMRP, or with S100β and MeCP2, an inverse relationship was observed (using the MATLAB quantification, Supplementary Material, Fig. S1A and B; right panels). This ‘L-shaped’ scattered distribution indicated that in the motor cortex high-FMRP or high-MeCP2 cells rarely co-localize, with cells expressing a high level of S100β (Supplementary Material, Fig. S1B). More importantly, given that an independent analysis from the scatter plots of the MeCP2 and FMRP immunoreactivity in the posterior cortex also adopts an L-shape (Fig. 2D) and knowing that both high-MeCP2 and high-FMRP cells are predominantly neuronal, we can conclude that the neurons express either high-FMRP or high-MeCP2 but rarely both simultaneously (1%). Flow cytometry of the papain-digested sensorimotor cortex from 2-month-old C57BL/6 WT mice confirmed the reliability of the cell counts (Supplementary Material, Fig. S2), whereby high-FMRP and high-MeCP2 expressing cells also adopted an L-shaped scatter distribution. This relationship implies that as FMRP levels increase, either from endogenous expression or transgenic delivery, MeCP2 protein expression is concomitantly, and proportionally, downregulated.
Characterization of FMRP in MeCP2 KO mice
The observation that FMRP expression in Fmr1 KO mice transduced with AAV-FMRP induced a normalization of MeCP2 levels (Fig. 2B–D) raises the question as to the status of FMRP levels in the absence of MeCP2. Therefore, we performed densitometric quantification of FMRP in the cerebral cortex and cerebellum of adult MeCP2 KO mice using western blotting. As expected, MeCP2 protein was highly expressed in the cerebral cortex and cerebellum of WT mice and was not detected in the strain-matched MeCP2 KO mouse samples (Fig. 3A), thereby confirming the specificity of the MeCP2 antibody used for western blotting. FMRP expression was also quantified and compared in the cerebellum (Fig. 3B and D) and cerebral cortex (Fig. 3C and E) of WT and MeCP2 KO mice. FMRP expression in the MeCP2 KO cerebellum was not significantly different from WT, whereas a significant reduction of FMRP was observed in the cerebral cortex of MeCP2 KO mice (78 ± 5% of WT expression, P < 0.01; N = 9; Fig. 3E). Therefore, in contrast to the upregulation of MeCP2 observed in the Fmr1 KO mouse brain, the absence of MeCP2 in the MeCP2 KO mouse cortex showed significantly reduced FMRP expression.
Figure 3.
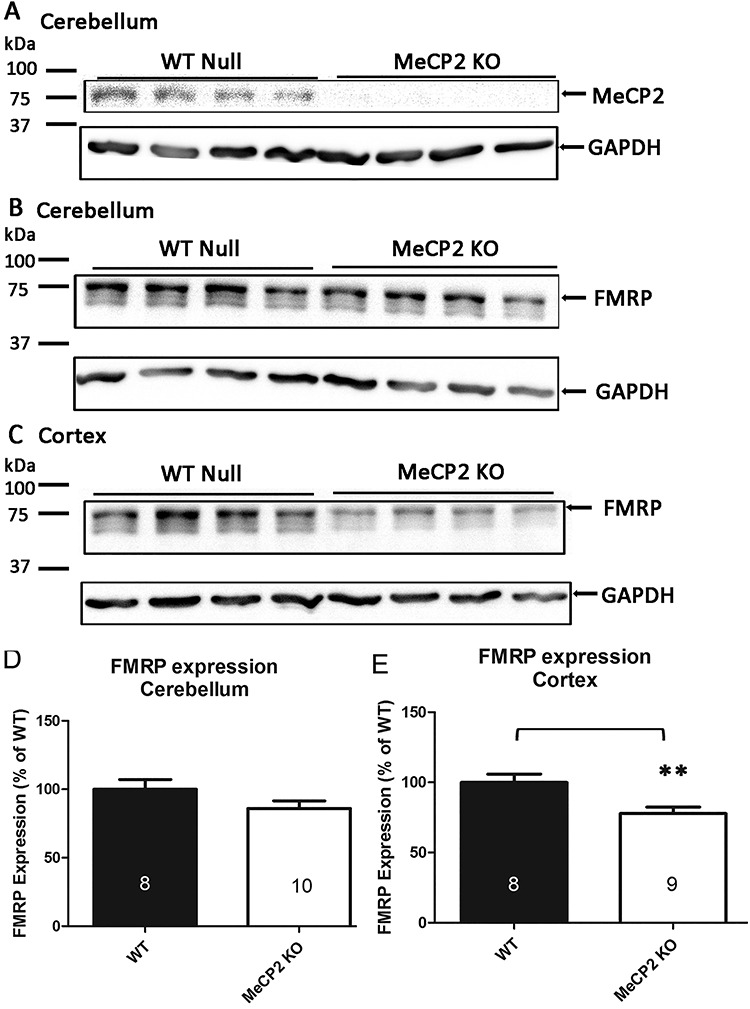
FMRP and MeCP2 protein expression in MeCP2 KO mice. (A) Western blots of MeCP2 in WT and MeCP2 KO mouse cerebellum. (B, C) FMRP expression in WT and MeCP2 KO mouse cerebellum (B) and sensorimotor cortex (C). (D, E) Densitometric quantification of FMRP expression in MeCP2 KO mice from the cerebellum (D; 86 ± 5%, P = 0.1364) and sensorimotor cortex (E; 78 ± 5%; P < 0.01) of WT expression. Expression was normalized to GAPDH. Numbers inside columns indicate the number of mice analyzed in each group. Data are presented as a percentage of WT expression (mean ± SEM). **P < 0.01.
Manipulation of MeCP2 expression in cell lines
To ascertain if anti-sense-mediated downregulation of MeCP2 could directly influence FMRP expression, we assayed five different shRNA plasmids from Sigma’s Mission shRNA library. All five plasmids targeting HUMAN MeCP2 showed significant downregulation of MeCP2 protein expression from plated mouse N2A cells, validating their efficacy in vitro (Fig. 4A and B). Marked downregulation was observed by western immunoblotting and quantification, normalized to GAPDH, showed a significant 70–85% reduction in MeCP2 protein levels in N2A cells with all shRNAs examined (Fig. 4B). Interestingly, FMRP protein levels were also concomitantly downregulated (Fig. 4C). Normalized protein quantification showed a marked 35–45% reduction in FMRP protein expression for each tested MeCP2-specific shRNA, with a significant reduction observed for the 579, 1266 and 1266s21 shRNA plasmids (P < 0.05; Fig. 4C). Taken together with the MeCP2 KO mice quantification data, this observation confirms that reducing MeCP2 protein levels, whether through embryonic mutations or in vitro transfection of immortalized mouse neuroblastomas, leads to a concomitant downregulation of FMRP.
Figure 4.
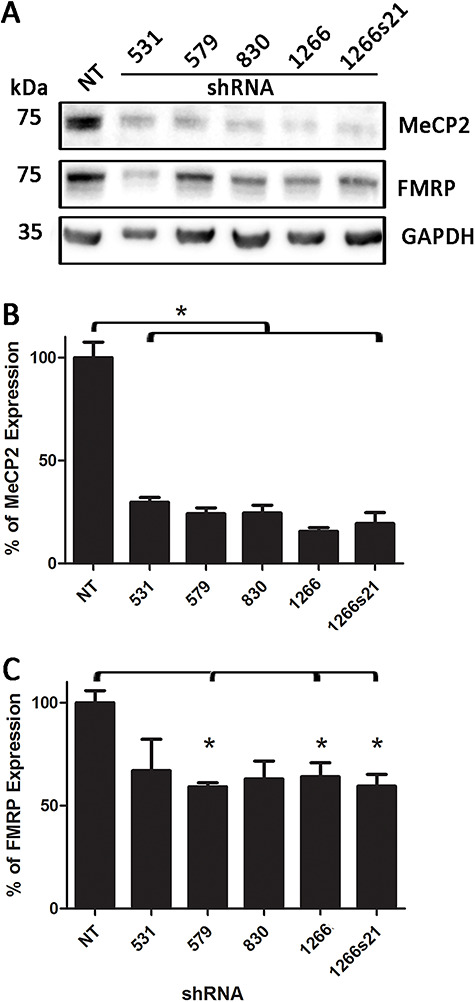
Assay of different shRNA targeting the MeCP2 transcript. (A) Western immunoblot of MeCP2, FMRP and GAPDH from neuroblastoma cells transfected with commercially available shRNAs (full clone IDs are available in Materials and Methods section). (B) Quantification of MeCP2 expression following shRNA transfection. All commercial shRNAs were able to significantly reduce MeCP2 expression compared with no shRNA transfection (NT) control (P < 0.05). (C) Quantification of FMRP following MeCP2 shRNA transfection. shRNA clones 579, 1266 and 1266s21 showed consistent downregulation of FMRP following MeCP2 knockdown.
To further probe this relationship, we also tested a pooled AAV2/9 siRNA virus that specifically targeted human MeCP2 (hMECP2) mRNAs and included a GFP reporter within the same expression cassette. To this end, we used the human embryonic kidney cell line, HEK-293. Using a commercially available anti-hMECP2 AAV2/9, we assayed three different viral doses (2 × 106, 2 × 107 and 6 × 107 GC/ml) by adding it directly to the culture media of HEK-293 cells. In cells collected 42 h post-transfection, western immunoblotting analysis revealed an increase in the positive reporter GFP, downregulation of MeCP2 and a concomitant downregulation of FMRP (Fig. 5A) in a dose-dependent manner (Fig. 5B). MeCP2 protein quantification showed a significant downregulation in the highest viral dose condition (Fig. 5B and C; P < 0.05). As observed in mouse N2A cells, FMRP levels were also decreased relative to GAPDH in this human-derived cell line, but in HEK-293 cells, the FMRP downregulation appeared to be as robust, if not more so, than the specific MeCP2 knockdown itself (Fig. 5B and D). This observation confirms that MeCP2 downregulation also led to FMRP downregulation in human cells, thereby demonstrating that the MeCP2 effects on FMRP levels are not restricted to mouse translational/transcriptional regulatory mechanisms.
Figure 5.
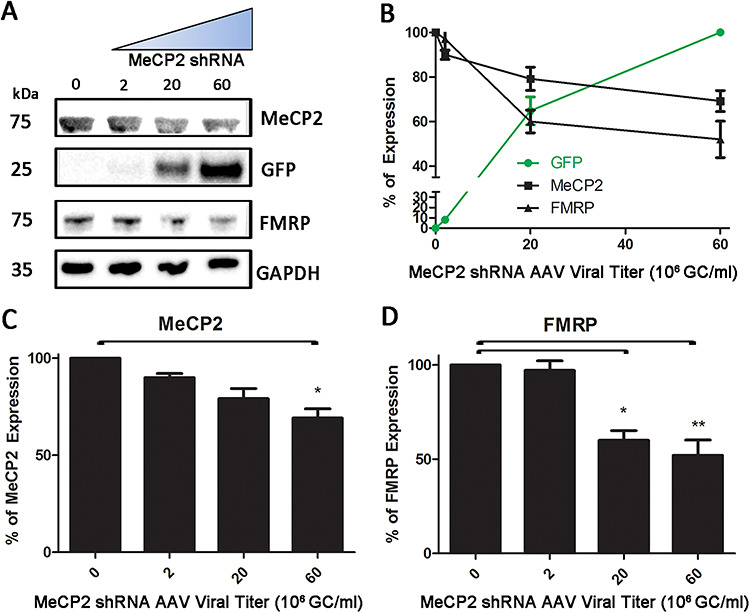
Manipulating MeCP2 protein expression in HEK cells using AAV MeCP2 shRNA. (A) Immunoblots of MeCP2, GFP, FMRP and GAPDH in HEK-293 cells following increasing titers of AAV MeCP2 shRNA. (B) Dose effect curve of AAV MeCP2 shRNA on MeCP2, GFP and FMRP expression. (C) Quantification of MeCP2 knockdown with increasing doses of AAV MeCP2 shRNA. Significant knockdown was observed at the 6 × 107 GC/ml titer (P < 0.05). (D) Quantification of FMRP expression following MeCP2 knockdown. A significant reduction in FMRP expression was observed following a 2 × 107 GC/ml titer administration (P < 0.05) and at the higher titer (P < 0.01). Expression was normalized to GAPDH and control cell (no AAV) expression was set at 100%. Results are presented as mean ± SEM obtained from three independent experiments.
Correlation between FMRP, MeCP2 and motor activity
Locomotor hyperactivity is a key hallmark of human FXS and Fmr1 KO mice (34,37,38). Therefore, we performed open field locomotor activity measurements in conjunction with subsequent quantification of protein expression. Fmr1 KO mice injected with AAV-EV displayed a significant elevation in motor activity compared with WT mice injected with AAV-EV (as indicated by the total distance traveled within 10 min; Fig. 6A and B; P < 0.05; N = 40). Motor hyperactivity was partially normalized in the KO + AAV-FMRP treatment group compared with the KO + AAV-EV group, resulting in the AAV-FMRP treatment group not being significantly different from the WT AAV-EV group (i.e. treatment with AAV-FMRP reduced motor hyperactivity in the KO mice; Fig. 6B). In parallel, i.c.v. injection of AAV-FMRP in the Fmr1 KO mice increased the FMRP expression in the posterior cerebral cortex to 69 ± 13% (n = 14) of WT levels, similar to previously reported (34).
Figure 6.
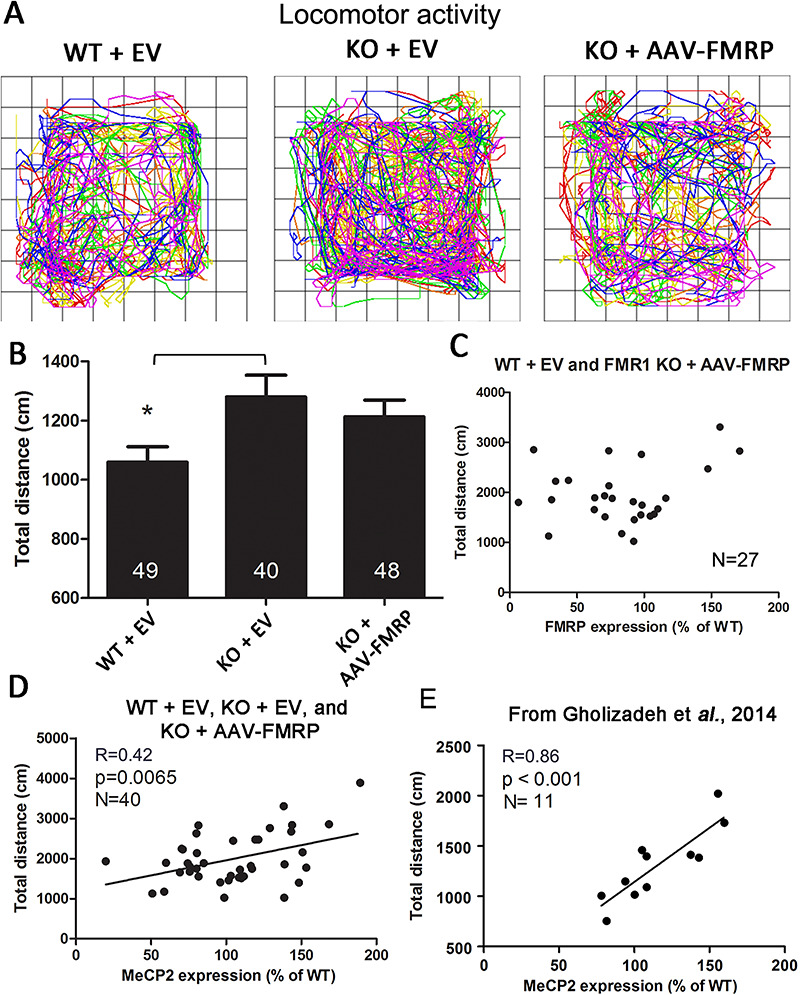
Locomotor activity and brain MeCP2 expression in AAV-transduced WT and Fmr1 KO mice. (A) Locomotor path plotter for typical WT + EV-, KO + EV- and KO + AAV-FMRP-treated mice. Rainbow gradient shows the start (blue) to end of trial (red). (B) Total distance traveled within the first 10 min shows significant locomotor hyperactivity in the Fmr1 KO mice injected with the AAV control null vector (*P < 0.05). (C) Scatter plot distribution of the total distance traveled and FMRP expression in the cortex of individual mice. (D) Correlation between the total distance traveled and the MeCP2 expression in individual mice; MeCP2 protein expression significantly correlated (P < 0.01) with the severity of the locomotor hyperactivity phenotype. (E) Correlation between the expression of MeCP2 in individual mice from the PBS-injected WT and Fmr1 KO + AAV-FMRP groups and total distance traveled (previously unpublished analysis) from our previous study (37).
Given that reintroduction of FMRP via AAV-FMRP may lead to the expression of variable levels of cortical FMRP transgene expression from mouse to mouse, we endeavored to explore potential correlations between the expression level and motor activity. No significant linear correlation was observed between the levels of FMRP and total distance traveled (Fig. 6C). However, given the observed inverse relationship between FMRP and MeCP2 expression, we also analyzed the relationship between locomotion and MeCP2 levels in individual mice (Fig. 6D). Remarkably, when the total distance traveled was analyzed as a function of MeCP2 expression in the posterior cerebral cortex of individual mice (WT + EV, Fmr1 KO + EV, Fmr1 KO + AAV-FMRP; Fig. 6D), we found a significant correlation between these two parameters (Fig. 6D, R = 0.42, P < 0.01; N = 40). To further substantiate this finding, we examined previously unpublished motor activity and MeCP2 protein expression results obtained from a separate group of injected mice from our previous study (37). WT controls and Fmr1 KO mice both injected with AAV-FMRP were analyzed for total distance travel as a function of MeCP2 expression levels; this analysis also showed a direct correlation between motor activity and MeCP2 expression (Fig. 6E; R = 0.86, P < 0.001; N = 11). Finally, we note that the converse of the results described here was reported by Wither et al. (39), who studied MeCP2 haplo-insufficient mice (hypoactive) and reported that MeCP2 protein expression correlated directly with the locomotor activity.
To determine whether the MeCP2 overexpression might be induced from the innate increased locomotor activity in Fmr1 KO mice, naive WT and Fmr1 KO mice were maintained under acute sedation with ketamine and xylazine for 10 h, after which the expression of MeCP2 was quantified in the cortex. Acute suppression of locomotor activity did not significantly alter the MeCP2 expression compared with non-sedated littermates (Supplementary Material, Fig. S3). This observation suggests that the MeCP2 overexpression in Fmr1 KO mice was not driven by the elevated locomotor activity per se, but rather MeCP2 overexpression is directly related to the absence of FMRP in neurons.
Discussion
In this study, we sought to investigate the possibility of interregulation or cross talk between FMRP and MeCP2. Our findings reveal a previously undocumented homeostatic relationship between MeCP2 and FMRP expression. The observation that both proteins are widely co-expressed within the same cells indicated that an interrelationship between the two could occur on a cell autonomous level. We determined that MeCP2 protein expression was upregulated in the Fmr1 KO mouse, whereas FMRP expression was reduced in MeCP2 KO mouse cortex. Since pathologies emerge when the expression of MeCP2 and FMRP are either hypo- or hyper-expressed, the discovery that Fmr1 KO mice display overexpression of MeCP2 while MeCP2 KO mice display reduced FMRP, implies potential cross-contributions to the phenotypes of these two neurodevelopmental disorders.
Motor hyperactivity is a salient characteristic of FXS. This study is the first to link cortical MeCP2 levels to locomotor activity in Fmr1 KO mice. This finding was revealed by plotting the relationship between motor activity and cortical MeCP2 levels in individual mice injected with AAV-FMRP; MeCP2 expression levels in these mice in the posterior cerebral cortex correlated directly with the locomotor activity. We also investigated whether MeCP2 expression in the posterior cortex correlated with other behaviors in the same cohort of mice, but we did not observe additional significant correlations outside of locomotor activity. A link between MeCP2 in the cortex and motor activity was also indicated in a previous study conducted in MeCP2 haplo-insufficient female mice, where locomotor hypoactivity and low levels of MeCP2 were correlated (39), and in another study, where hypoactivity was rescued following the reintroduction of transgenic MeCP2 (40). In contrast to MeCP2, we did not observe a correlation between FMRP expression in the posterior cortex and motor activity. However, in our earlier work, we demonstrated that FMRP expression levels in the striatum of AAV-FMRP-treated Fmr1 KO mice correlated with the time and number of entries into the open arm of the elevated plus maze test of non-social anxiety (34).
Although the underlying biochemical pathways mediating interregulation of FMRP and MeCP2 were not the focus of this study, several mechanistic processes can be postulated. For instance, a number of MeCP2 binding elements are present upstream of the FMR1 gene (41–43). Moreover, the loss of FMRP-associated non-coding RNA regulation in FXS and Fmr1 KO mice, including long non-coding RNAs and micro-RNAs (12,44,45), could potentially alter MeCP2 expression.
Previous work from our laboratory demonstrated that mild-to-moderate AAV-mediated overexpression of FMRP (less than 1.5-fold over WT) did not lead to severe adverse effects in WT or Fmr1 KO mice (34). However, very high level FMRP overexpression in Fmr1 KO mice induced by AAV-FMRP administration (>2.5-fold over WT) caused aberrant behaviors (e.g. abnormal startle responses), and transgenic mice overexpressing FMRP severalfold over WT displayed increased anxiety and a reversal of the motor activity rescue (46). In comparison with FMRP where abnormal behaviors were only seen after very high overexpression, mild-to-modest upregulation of MeCP2 in transgenic mice has been shown to induce seizures and motor hypoactivity (40,47,48). The severe consequences of MeCP2 overexpression are further illustrated by case studies where patients with chromosomal duplications overlapping the MECP2 gene were found to induce profound intellectual disability, speech deficits and impaired immune function (31,33). Our findings raise an intriguing question: could the suppression of brain MeCP2 expression be therapeutically beneficial in FXS? The higher demographic prevalence of autism in males and the finding that MeCP2 and FMRP display interregulation suggests that the conserved placement of Fmr1 and MeCP2 on the X chromosome in mammals might not be a stochastic happenstance of evolution, but rather, a conserved homeostatic mechanism. A balance between the transcriptional output of MeCP2 and the translational modulation by FMRP, as well as other X-linked transcriptional and translational modulators, may represent an important regulatory mechanism implicated not only in maintaining normal neurological function but possibly also in the sexual dimorphism of some behaviors (49,50).
Materials and Methods
Animals
WT and Fmr1 KO mice, both on the C57BL/6 background, were used in this study as described elsewhere (34). MeCP2 KO mice were obtained from Jackson Laboratories (B6.129P2(C)-Mecp2tm1.1Bird (51). All protocols were approved by the University of Toronto Animal Care Committee and are in accordance with the Canadian Council on Animal Care guidelines.
AAV vectors and AAV vector injections
Single-stranded AAV2/9 vector containing the human syn-1 promoter and the mouse coding region of FMRP (isoform 1) was supplied by the University of Pennsylvania Vector Core Facility (Philadelphia, PA). This AAV2/9 vector (AAV-FMRP), which has been described in our previous work (34,37), was used at a titer of 1 × 1013 genomes/ml in sterile phosphate-buffered saline (PBS; 140 mM NaCl, 2.7 mM KCl, 10 mM HNa2PO4, 2 mM KH2PO4, pH 7.4) and stored at −80°C. Another single stranded AAV vector of the same serotype containing the cytomegalovirus promoter but without the FMRP transgene (AAV-EV) was used as a control as previously described (34) and was also prepared at the University of Pennsylvania Vector Core Facility.
A single bilateral i.c.v. injection of AAV-EV or AAV-FMRP was administered to WT and Fmr1 KO mice on PND 0 or 1, as previously described (37,52). WT and Fmr1 KO mice injected with the AAV-null vector served as control groups or as otherwise indicated.
Qualitative and quantitative reverse transcriptase polymerase chain reaction of forebrain and cortical mRNA
Immunoprecipitation followed by quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed as described previously (34) using a modification of the method described by Pacey et al. (53). The primers, specifically recognizing MeCP2 transcript variants 1 and 2 as well as the primers specific for GAPDH, used for quantitative PCR were described previously (34). The housekeeping gene GAPDH mRNA was used as a normalizing control. The cDNA was diluted 1:25 and quantitative RT-PCR was performed using an ABI Prism 7900 HT (Applied Biosystems) using the Sybr Green detection system (53). Relative mRNA levels were calculated using the relative threshold cycle method (53). Results are shown as mean relative mRNA level (arbitrary units) ± SEM. Data were analyzed using the unpaired student t-test.
Cell culture and MeCP2 shRNA assay
N2A and HEK-293 cells were grown and processed as previously reported for N2A cells (54). Briefly, cells were grown in a 37°C incubator with 5% CO2. Cells were grown in high glucose DMEM (Gibco) supplemented with 5% fetal calf serum (Gibco) and 1% penicillin/streptomycin (Thermo Fisher) and passaged when confluence reached over 90%. For downregulation assays, cells were seeded at 50 000 cells/well in uncoated 12 well plates (1 mL). For transfection, 1.2ug of shRNA plasmids (Sigma; Mission shRNA) were added per well 24 h after plating and transfected according to manufacturer’s protocol. The shRNAs used in this study were: (clone ID): NM_010788.3-1266s21c1 (abbr. 1266 s21); NM_010788.3-830s21c1 (830); NM_010788.3-579s21c1 (579); NM_010788.1-531s1c1 (531) and NM_010788.1-1266s1c1 (1266). For viral transduction, a 109 GC/ml concentration of the AAV2/9-siRNA(MeCP2)-GFP, purchased from ABM Good (iAAV04936009), was added directly to the plated cells. Forty-two hours after transfection or viral transduction, the cells were lysed with homogenization buffer (50 mM Tris, 1% SDS, pH 7.4) supplemented with protease cocktail inhibitor (Roche). Approximately, 2 μg of protein per condition was resolved on SDS-polyacrylamide gel electrophoresis (SDS-PAGE) as described in the following text. All cell measurements were averaged from three replicates.
Immunocytochemistry and confocal microscopy
At 65 ± 2 days postinjection, the AAV-FMRP- and AAV-EV-injected mice were anesthetized using a ketamine–xylazine solution, then transcardially perfused with PBS, followed by 4% paraformaldehyde in PBS (pH 7.4). Serial coronal sections were cut at a thickness of 25 μm using a cryostat (Leica Microsystems, Wetzlar, Germany) as previously described (37,55). Free-floating sections were washed with PBS, and antigen retrieval was performed as described elsewhere (56). Monoclonal mouse anti-FMRP 5c2 (57) was used along with monoclonal rabbit anti-MeCP2 D4F3 (Cell Signaling, Beverly, USA) at a 1:1000 dilution. To confirm the neuronal lineage of the cells, anti-NeuN (MAB377 from Millipore and ab177487 from Abcam) and anti-S100β (ab868 from Abcam and S2532 from Sigma) were used at 1:1000 dilutions to label neurons and astrocytes, respectively. The sections were labeled with goat anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 594 (1:4000; Jackson Immunoresearch Laboratories, West Grove, USA). DAPI (Sigma-Aldrich) was used to label nuclei. Cell populations quantification was implemented in MATLAB version R2017a (9.2.0) and the code was included in the supplementary information. Only cells fully within this focal plane were quantified. Linear regression was performed using Graphpad Prism 5 to calculate the MeCP2/FMRP correlation parameters.
Quantitative western blotting
Samples of the cerebellum and cortex were collected and stored at −80°C. The caudal segment of the cerebral cortex which encompasses the retrosplenial area, the anterior cingulate area, the visual area and the posterior somatomotor area, herein referred to as the posterior cerebral cortex for brevity, was also harvested and stored at −80°C as previously described (34). Mouse brain anatomical regions and nomenclature were obtained from the Allen Mouse Brain Reference Atlas (58). Protein concentration was determined using the BCA assay. To properly quantify bands, 10 μg of protein were loaded per lane. After SDS-PAGE, the proteins were transferred onto PVDF membranes, blocked with a 5% milk (BioShop, Burlington, Canada) solution in PBS and incubated overnight with the primary antibodies listed above at 1:1000 dilutions, with the addition of mouse monoclonal anti-GAPDH (GAPDH-71.1; Sigma-Aldrich) at 1:40 000 dilution which was used as a loading control. Optimum antibody concentration was determined to avoid oversaturation of signal (34,37,53,59,60). The AlphaEase SA software was used to quantify the density of the protein bands. Only unmodified tiff images were used to quantify band intensity. Sample values were averaged from two quantifications. N value represents the number of animals used. Quantification of protein expression was normalized using GAPDH immunoreactivity, and data are presented as a percentage of WT expression. Results are presented as mean ± SEM. Statistical significance was determined with Graphpad Prism 5 by using one-way ANOVA and Tukey’s multiple comparison tests.
Open field testing
Testing was conducted on adult age-matched (2–4 months) animals. All mice were naive to the test. The experimenters were blind to the vector treatment at the time of testing. Locomotor activity testing was performed in an open field locomotor monitor system (Accuscan Images, Salt Lake City, USA). Testing was performed between 13:00 and 17:00 h on PND 55 ± 2. Mice were acclimated to the testing room for 5 min, then placed in the open field and monitored for 20 min in low light conditions (~40 lux). The total distance traveled over 20 min was recorded using the Fusion software (Fusion software, Johannesburg, South Africa) and the first 10 min was compared between groups using one-way ANOVA and Tukey’s multiple comparison tests. Two-way ANOVA excluded sex as a variable, thus male and female homozygous-null mice data from the same groups were combined. The locomotor path plotter function was used to visualize the total exploratory trajectory of individual mice. Results are presented as mean ± SEM.
Statistical analyses
For all parametric data, one-way ANOVA was performed followed by Tukey’s post hoc test unless otherwise indicated. Correlation and normality distribution was determined using protein expression as a function of the behavioral data or the cognate protein expression obtained from the same mouse. GraphPad Prism 5 was used to perform the statistical analyses. A P-value of <0.05 was deemed statistically significant.
Supplementary Material
Acknowledgements
The authors thank Dr Stephane Anger for the use of the confocal microscope. J.A. and A.H. were the recipients of the Fragile X Research Foundation of Canada’s postdoctoral fellowship.
Conflict of Interest statement. None.
Contributor Information
Jason Arsenault, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada; Department of Physiology, University of Toronto, Toronto, ON M5S 1A8, Canada; Program in Neurosciences and Mental Health, Hospital for Sick Children Research Institute, Toronto, ON M5G 0A4, Canada.
Alexander W M Hooper, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada.
Shervin Gholizadeh, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada.
Tian Kong, Department of Physiology, University of Toronto, Toronto, ON M5S 1A8, Canada; Program in Neurosciences and Mental Health, Hospital for Sick Children Research Institute, Toronto, ON M5G 0A4, Canada.
Laura K Pacey, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada.
Enea Koxhioni, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada.
Yosuke Niibori, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada.
James H Eubanks, Division of Genetics and Development, Krembil Research Institute, University Health Network, Toronto, ON M5T 0S8, Canada; Department of Physiology, University of Toronto, Toronto, ON M5S 1A8, Canada; Department of Surgery, Division of Neurosurgery, University of Toronto, Toronto, ON M5T 1P5, Canada.
Lu-Yang Wang, Department of Physiology, University of Toronto, Toronto, ON M5S 1A8, Canada; Program in Neurosciences and Mental Health, Hospital for Sick Children Research Institute, Toronto, ON M5G 0A4, Canada.
David R Hampson, Department of Pharmaceutical Sciences, Leslie Dan Faculty of Pharmacy, University of Toronto, Toronto, ON M5S 3M2, Canada; Department of Pharmacology, Faculty of Medicine, University of Toronto, Toronto, ON M5S 1A8, Canada.
Funding
Canadian Institutes of Health Research (to J.H.E., L.Y.W. and D.R.H.); Canada Research Chair Program (to L.Y.W.).
References
- 1. Budimirovic, D.B. and Kaufmann, W.E. (2011) What can we learn about autism from studying fragile X syndrome? Dev. Neurosci., 33, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liyanage, V.R. and Rastegar, M. (2014) Rett syndrome and MeCP2. Neuromolecular Med., 16, 231–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lozano, R., Rosero, C.A. and Hagerman, R.J. (2014) Fragile X spectrum disorders. Intractable Rare Dis. Res., 3, 134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Loesch, D.Z., Huggins, R.M. and Hagerman, R.J. (2004) Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev., 10, 31–41. [DOI] [PubMed] [Google Scholar]
- 5. Guy, J., Gan, J., Selfridge, J., Cobb, S. and Bird, A. (2007) Reversal of neurological defects in a mouse model of Rett syndrome. Science, 315, 1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Amir, R.E., Van den Veyver, I.B., Wan, M., Tran, C.Q., Francke, U. and Zoghbi, H.Y. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet., 23, 185–188. [DOI] [PubMed] [Google Scholar]
- 7. Abbeduto, L.O., Susan and Thurman, A.J. (2014). Chapter 8: Neurodevelopmental Disorders. In Hales, R.E., Yudofsky, S.C., Roberts, L.W. and American Psychiatric Publishing (eds), The American Psychiatric Publishing Textbook of Psychiatry. American Psychiatric Pub, Washington, DC. [Google Scholar]
- 8. Darnell, J.C., Van Driesche, S.J., Zhang, C., Hung, K.Y., Mele, A., Fraser, C.E., Stone, E.F., Chen, C., Fak, J.J., Chi, S.W. et al. (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell, 146, 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ascano, M., Jr., Mukherjee, N., Bandaru, P., Miller, J.B., Nusbaum, J.D., Corcoran, D.L., Langlois, C., Munschauer, M., Dewell, S., Hafner, M. et al. (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature, 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsang, B., Arsenault, J., Vernon, R.M., Lin, H., Sonenberg, N., Wang, L.Y., Bah, A. and Forman-Kay, J.D. (2019) Phosphoregulated FMRP phase separation models activity-dependent translation through bidirectional control of mRNA granule formation. Proc. Natl. Acad. Sci. U. S. A., 116, 4218–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jin, P., Alisch, R.S. and Warren, S.T. (2004) RNA and microRNAs in fragile X mental retardation. Nat. Cell Biol., 6, 1048–1053. [DOI] [PubMed] [Google Scholar]
- 12. Liu, T., Wan, R.P., Tang, L.J., Liu, S.J., Li, H.J., Zhao, Q.H., Liao, W.P., Sun, X.F., Yi, Y.H. and Long, Y.S. (2015) A MicroRNA profile in Fmr1 knockout mice reveals microRNA expression alterations with possible roles in fragile X syndrome. Mol. Neurobiol., 51, 1053–1063. [DOI] [PubMed] [Google Scholar]
- 13. Smalheiser, N.R. and Lugli, G. (2009) microRNA regulation of synaptic plasticity. Neuromolecular Med., 11, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Westmark, C.J. (2013) FMRP: a triple threat to PSD-95. Front. Cell. Neurosci., 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zalfa, F., Eleuteri, B., Dickson, K.S., Mercaldo, V., De Rubeis, S., di Penta, A., Tabolacci, E., Chiurazzi, P., Neri, G., Grant, S.G. et al. (2007) A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci., 10, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adinolfi, S., Ramos, A., Martin, S.R., Dal Piaz, F., Pucci, P., Bardoni, B., Mandel, J.L. and Pastore, A. (2003) The N-terminus of the fragile X mental retardation protein contains a novel domain involved in dimerization and RNA binding. Biochemistry, 42, 10437–10444. [DOI] [PubMed] [Google Scholar]
- 17. Zhang, Y., Brown, M.R., Hyland, C., Chen, Y., Kronengold, J., Fleming, M.R., Kohn, A.B., Moroz, L.L. and Kaczmarek, L.K. (2012) Regulation of neuronal excitability by interaction of fragile X mental retardation protein with slack potassium channels. J. Neurosci., 32, 15318–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang, Y.M., Arsenault, J., Bah, A., Krzeminski, M., Fekete, A., Chao, O.Y., Pacey, L.K., Wang, A., Forman-Kay, J., Hampson, D.R. et al. (2020) Identification of a molecular locus for normalizing dysregulated GABA release from interneurons in the fragile X brain. Mol. Psychiatry, 25, 2017-2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khayachi, A., Gwizdek, C., Poupon, G., Alcor, D., Chafai, M., Casse, F., Maurin, T., Prieto, M., Folci, A., De Graeve, F. et al. (2018) Sumoylation regulates FMRP-mediated dendritic spine elimination and maturation. Nat. Commun., 9, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chahrour, M., Jung, S.Y., Shaw, C., Zhou, X., Wong, S.T., Qin, J. and Zoghbi, H.Y. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science, 320, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sugino, K., Hempel, C.M., Okaty, B.W., Arnson, H.A., Kato, S., Dani, V.S. and Nelson, S.B. (2014) Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci., 34, 12877–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li, R., Dong, Q., Yuan, X., Zeng, X., Gao, Y., Chiao, C., Li, H., Zhao, X., Keles, S., Wang, Z. et al. (2016) Misregulation of alternative splicing in a mouse model of Rett syndrome. PLoS Genet., 12, e1006129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Young, J.I., Hong, E.P., Castle, J.C., Crespo-Barreto, J., Bowman, A.B., Rose, M.F., Kang, D., Richman, R., Johnson, J.M., Berget, S. et al. (2005) Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U. S. A., 102, 17551–17558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu, H., Tao, J., Chen, P.J., Shahab, A., Ge, W., Hart, R.P., Ruan, X., Ruan, Y. and Sun, Y.E. (2010) Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A., 107, 18161–18166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodrigues, D.C., Kim, D.S., Yang, G., Zaslavsky, K., Ha, K.C., Mok, R.S., Ross, P.J., Zhao, M., Piekna, A., Wei, W. et al. (2016) MECP2 is post-transcriptionally regulated during human neurodevelopment by combinatorial action of RNA-binding proteins and miRNAs. Cell Rep., 17, 720–734. [DOI] [PubMed] [Google Scholar]
- 26. Hickey, S.E., Walters-Sen, L., Mosher, T.M., Pfau, R.B., Pyatt, R., Snyder, P.J., Sotos, J.F. and Prior, T.W. (2013) Duplication of the Xq27.3-q28 region, including the FMR1 gene, in an X-linked hypogonadism, gynecomastia, intellectual disability, short stature, and obesity syndrome. Am. J. Med. Genet. A, 161A, 2294–2299. [DOI] [PubMed] [Google Scholar]
- 27. Nagamani, S.C., Erez, A., Probst, F.J., Bader, P., Evans, P., Baker, L.A., Fang, P., Bertin, T., Hixson, P., Stankiewicz, P. et al. (2012) Small genomic rearrangements involving FMR1 support the importance of its gene dosage for normal neurocognitive function. Neurogenetics, 13, 333–339. [DOI] [PubMed] [Google Scholar]
- 28. Rio, M., Malan, V., Boissel, S., Toutain, A., Royer, G., Gobin, S., Morichon-Delvallez, N., Turleau, C., Bonnefont, J.P., Munnich, A. et al. (2010) Familial interstitial Xq27.3q28 duplication encompassing the FMR1 gene but not the MECP2 gene causes a new syndromic mental retardation condition. Eur. J. Hum. Genet., 18, 285–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vengoechea, J., Parikh, A.S., Zhang, S. and Tassone, F. (2012) De novo microduplication of the FMR1 gene in a patient with developmental delay, epilepsy and hyperactivity. Eur. J. Hum. Genet., 20, 1197–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Esch, H. (2012) MECP2 duplication syndrome. Mol. Syndromol., 2, 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smyk, M., Obersztyn, E., Nowakowska, B., Nawara, M., Cheung, S.W., Mazurczak, T., Stankiewicz, P. and Bocian, E. (2008) Different-sized duplications of Xq28, including MECP2, in three males with mental retardation, absent or delayed speech, and recurrent infections. Am. J. Med. Genet. B. Neuropsychiatr. Genet., 147B, 799–806. [DOI] [PubMed] [Google Scholar]
- 32. Lombardi, L.M., Baker, S.A. and Zoghbi, H.Y. (2015) MECP2 disorders: from the clinic to mice and back. J. Clin. Invest., 125, 2914–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fieremans, N., Bauters, M., Belet, S., Verbeeck, J., Jansen, A.C., Seneca, S., Roelens, F., De Baere, E., Marynen, P. and Froyen, G. (2014) De novo MECP2 duplications in two females with intellectual disability and unfavorable complete skewed X-inactivation. Hum. Genet., 133, 1359–1367. [DOI] [PubMed] [Google Scholar]
- 34. Arsenault, J., Gholizadeh, S., Niibori, Y., Pacey, L.K., Halder, S.K., Koxhioni, E., Konno, A., Hirai, H. and Hampson, D.R. (2016) FMRP expression levels in mouse central nervous system neurons determine behavioral phenotype. Hum. Gene Ther., 27, 982–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Olson, C.O., Zachariah, R.M., Ezeonwuka, C.D., Liyanage, V.R. and Rastegar, M. (2014) Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS One, 9, e90645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Orlic-Milacic, M., Kaufman, L., Mikhailov, A., Cheung, A.Y., Mahmood, H., Ellis, J., Gianakopoulos, P.J., Minassian, B.A. and Vincent, J.B. (2014) Over-expression of either MECP2_e1 or MECP2_e2 in neuronally differentiated cells results in different patterns of gene expression. PLoS One, 9, e91742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gholizadeh, S., Arsenault, J., Xuan, I.C., Pacey, L.K. and Hampson, D.R. (2014) Reduced phenotypic severity following adeno-associated virus-mediated FMR1 gene delivery in fragile x mice. Neuropsychopharmacology, 39, 3100–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding, Q., Sethna, F. and Wang, H. (2014) Behavioral analysis of male and female Fmr1 knockout mice on C57BL/6 background. Behav. Brain Res., 271, 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wither, R.G., Lang, M., Zhang, L. and Eubanks, J.H. (2013) Regional MeCP2 expression levels in the female MeCP2-deficient mouse brain correlate with specific behavioral impairments. Exp. Neurol., 239, 49–59. [DOI] [PubMed] [Google Scholar]
- 40. Jugloff, D.G., Vandamme, K., Logan, R., Visanji, N.P., Brotchie, J.M. and Eubanks, J.H. (2008) Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum. Mol. Genet., 17, 1386–1396. [DOI] [PubMed] [Google Scholar]
- 41. de Paz, A.M., Khajavi, L., Martin, H., Claveria-Gimeno, R., Dieck, S.T., Cheema, M.S., Sanchez-Mut, J.V., Moksa, M.M., Carles, A., Brodie, N.I. et al. (2019) MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenetics Chromatin, 12, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Couvert, P., Bienvenu, T., Aquaviva, C., Poirier, K., Moraine, C., Gendrot, C., Verloes, A., Andres, C., Le Fevre, A.C., Souville, I. et al. (2001) MECP2 is highly mutated in X-linked mental retardation. Hum. Mol. Genet., 10, 941–946. [DOI] [PubMed] [Google Scholar]
- 43. Tzeng, C.C., Liou, C.P., Li, C.F., Lai, M.C., Tsai, L.P., Cho, W.C. and Chang, H.T. (2009) Methyl-CpG-binding PCR of bloodspots for confirmation of fragile X syndrome in males. J. Biomed. Biotechnol., 2009, 643692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Edbauer, D., Neilson, J.R., Foster, K.A., Wang, C.F., Seeburg, D.P., Batterton, M.N., Tada, T., Dolan, B.M., Sharp, P.A. and Sheng, M. (2010) Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron, 65, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou, Y., Hu, Y., Sun, Q. and Xie, N. (2019) Non-coding RNA in fragile X syndrome and converging mechanisms shared by related disorders. Front. Genet., 10, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Peier, A.M., McIlwain, K.L., Kenneson, A., Warren, S.T., Paylor, R. and Nelson, D.L. (2000) (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum. Mol. Genet., 9, 1145–1159. [DOI] [PubMed] [Google Scholar]
- 47. Bodda, C., Tantra, M., Mollajew, R., Arunachalam, J.P., Laccone, F.A., Can, K., Rosenberger, A., Mironov, S.L., Ehrenreich, H. and Mannan, A.U. (2013) Mild overexpression of Mecp2 in mice causes a higher susceptibility toward seizures. Am. J. Pathol., 183, 195–210. [DOI] [PubMed] [Google Scholar]
- 48. Collins, A.L., Levenson, J.M., Vilaythong, A.P., Richman, R., Armstrong, D.L., Noebels, J.L., David Sweatt, J. and Zoghbi, H.Y. (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet., 13, 2679–2689. [DOI] [PubMed] [Google Scholar]
- 49. Retico, A., Giuliano, A., Tancredi, R., Cosenza, A., Apicella, F., Narzisi, A., Biagi, L., Tosetti, M., Muratori, F. and Calderoni, S. (2016) The effect of gender on the neuroanatomy of children with autism spectrum disorders: a support vector machine case-control study. Mol. Autism., 7, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mottron, L., Duret, P., Mueller, S., Moore, R.D., Forgeot d'Arc, B., Jacquemont, S. and Xiong, L. (2015) Sex differences in brain plasticity: a new hypothesis for sex ratio bias in autism. Mol. Autism., 6, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guy, J., Hendrich, B., Holmes, M., Martin, J.E. and Bird, A. (2001) A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet., 27, 322–326. [DOI] [PubMed] [Google Scholar]
- 52. Gholizadeh, S., Tharmalingam, S., Macaldaz, M.E. and Hampson, D.R. (2013) Transduction of the central nervous system after intracerebroventricular injection of adeno-associated viral vectors in neonatal and juvenile mice. Hum. Gene Ther. Methods, 24, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pacey, L.K., Doss, L., Cifelli, C., van der Kooy, D., Heximer, S.P. and Hampson, D.R. (2011) Genetic deletion of regulator of G-protein signaling 4 (RGS4) rescues a subset of fragile X related phenotypes in the FMR1 knockout mouse. Mol. Cell. Neurosci., 46, 563–572. [DOI] [PubMed] [Google Scholar]
- 54. Arsenault, J., Ferrari, E., Niranjan, D., Cuijpers, S.A., Gu, C., Vallis, Y., O'Brien, J. and Davletov, B. (2013) Stapling of the botulinum type a protease to growth factors and neuropeptides allows selective targeting of neuroendocrine cells. J. Neurochem., 126, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gholizadeh, S., Halder, S.K. and Hampson, D.R. (2015) Expression of fragile X mental retardation protein in neurons and glia of the developing and adult mouse brain. Brain Res., 1596, 22–30. [DOI] [PubMed] [Google Scholar]
- 56. Gabel, L.A., Won, S., Kawai, H., McKinney, M., Tartakoff, A.M. and Fallon, J.R. (2004) Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J. Neurosci., 24, 10579–10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. LaFauci, G., Adayev, T., Kascsak, R., Kascsak, R., Nolin, S., Mehta, P., Brown, W.T. and Dobkin, C. (2013) Fragile X screening by quantification of FMRP in dried blood spots by a Luminex immunoassay. J. Mol. Diagn., 15, 508–517. [DOI] [PubMed] [Google Scholar]
- 58. Lein, E.S., Hawrylycz, M.J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A.F., Boguski, M.S., Brockway, K.S., Byrnes, E.J. et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature, 445, 168–176. [DOI] [PubMed] [Google Scholar]
- 59. Pacey, L.K., Xuan, I.C., Guan, S., Sussman, D., Henkelman, R.M., Chen, Y., Thomsen, C. and Hampson, D.R. (2013) Delayed myelination in a mouse model of fragile X syndrome. Hum. Mol. Genet., 22, 3920–3930. [DOI] [PubMed] [Google Scholar]
- 60. Pacey, L.K., Guan, S., Tharmalingam, S., Thomsen, C. and Hampson, D.R. (2015) Persistent astrocyte activation in the fragile X mouse cerebellum. Brain Behav., 5, e00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.