

Abstract
Harsh conditions within the tumor microenvironment, such as hypoxia and extracellular acidic pH (pHe), inactivate some chemotherapies, which results in limited or no cytotoxicity. Standard MTT, ATPlite and protease assays that are used to determine the potency of newly developed drugs often give erroneous results when applied under hypoxic or acidic conditions. Therefore, development of a cytotoxicity assay that does not yield false positive or false negative results under circumstances of both hypoxia and acidic pHe is needed. We evaluated currently used cell viability assays as well as neutral red staining to assess viability of ovarian and pancreatic cancer cells grown in an acidic pHe microenvironment after treatment with carboplatin, gemcitabine or chloroquine. We validated cell viability using western blotting of pro-caspase-9 and cleaved-caspase-9, and LC3-I and –II. Standard cell viability assays indicated cell viability accurately at pHe 7.4, but was not correlated with induction of apoptosis or autophagy at acidic pHe. By contrast, our modified neutral red assay detected cell viability accurately over a range of pHe as demonstrated by its correlation with induction of apoptosis and autophagy. Neutral red staining is effective for evaluating the effect of chemotherapeutic agents on cell viability under acidic pHe or hypoxic conditions.
Keywords: cell viability, cytotoxicity, extracellular acidic pH, neutral red, ovarian cancer, pancreatic cancer
By contrast to normal cells, the tumor microenvironment exhibits both an acidic extracellular pH (pHe) and decreased oxygen tension. In response to intracellular lactic acid, the development of an acidic microenvironment around malignant cells may be caused by cellular proton pumps such as lung resistance-related protein (LRP); investigators must account for these features when developing and evaluating efficacy of anticancer drugs (Gatenby et al. 2006; Hockel and Vaupel 2001). For example, carboplatin in combination with paclitaxel currently is a standard first line chemotherapy for treatment of ovarian carcinoma (Marchetti et al. 2019; Thigpen et al. 2011). Efforts to improve antitumor activity and prolong survival by increasing the dose/cycle or by reducing the time interval between dose administrations (dose-dense therapy), however, has failed to meet expectations despite showing significant promise in in vitro systems (LaFargue et al. 2019; van der Burg et al. 2014). Therefore, development of a cell viability protocol that remains effective even under acidic pHe conditions, i.e., those mimicking the tumor microenvironment, is needed to evaluate the effectiveness and therapeutic potential of anti-neoplastic agents.
Currently, several assays can be used to evaluate the antiproliferative or cytotoxic activity of cancer therapeutics. A variety of tetrazolium compounds has been used to detect viable cells. The most commonly used compounds include: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfonyl)-2H-tetrazolium (MTS), and water soluble tetrazolium salt 4-(3-(4-Iodophenyl)-2-(4-nitrophenyl)2H-5-tetrazolio)-1,3-benzene disulfonate (WST-1). These compounds fall into two basic categories: 1) positively charged agents, e.g., MTT, which readily penetrates viable eukaryotic cells and 2) negatively charged agents, e.g., MTS, XTT and WST-1, which do not readily penetrate cells. Category 1 NAD(P)H-dependent cellular oxidoreductase enzymes can reduce tetrazolium dye to the insoluble compound, formazan, which is purple. Category 2 agents, e.g., MTS, XTT, WST-1, typically are used with an intermediate electron acceptor that can transfer electrons from the cytoplasm or plasma membrane to facilitate reduction of the tetrazolium into the colored formazan product. Because these tests only produce a final reading, they are all inherently end point assays (Berridge et al. 2005; Mosmann 1983). Although these colorimetric assays are the most commonly used, they have limitations including inability to account for variation in cellular metabolism throughout the life cycle, significant underestimation of compound potency and efficacy, and non-monotonic dose-response curves due to concentration-dependent phenotypic “switching” (Chan et al. 2013; Quent et al. 2010). Also, the accuracy of MTT, MTS and WST-1 decreases significantly in an acidic cellular environment (Berridge et al. 2005).
Unlike the colorimetric assays described above, the ATPlite assay uses a fluorescence based system to evaluate intracellular levels of ATP, because only viable cells can produce ATP by either anaerobic or aerobic metabolism. Owing to its mechanism of detection of cell viability, the efficacy of the ATPlite assay can be affected by conditions that cause ATP elevations in nonviable cells; autophagy is one such factor in cases of acidic pHe.
Autophagy is a mechanism that enables cells to survive nutrient/growth factor deprivation by recycling cellular components; it also allows cancer cells to manage metabolic stress products, survive in a hypoxic/acidic microenvironment (White and DiPaola 2009), and to produce substrates for oxidation and ATP synthesis in the mitochondria (Lum et al. 2005). The breakdown of cellular components can ensure cell survival during starvation and stress by maintaining cellular energy levels; it also may promote cancer cell survival (Cook et al. 2011). Despite increased cell death associated with acidic pHe, cells undergoing autophagy increase their cellular ATP levels, which results in falsely elevated viability results compared to cells cultured at pHe 7.4.
Although less commonly used than either of the assays discussed above, the neutral red uptake assay is a well-known and validated quantitative cell viability assay (Repetto et al. 2008; Zhang et al. 1990). The assay relies on the ability of viable cells to take up and bind neutral red dye within lysosomes and, importantly, to maintain pH gradients with respect to their cytoplasm. The dye itself possesses a nearly zero net charge at physiologic pH and becomes charged only within the acidic pH found within the lysosome (pH 4.5–5.0). Therefore, one would not expect its efficacy to be affected significantly even with a 10-fold increase in extracellular acidity (pH 7.4–6.4), which potentially makes it an ideal assay for evaluating the efficacy of anticancer drugs in the characteristic tumor microenvironment.
We have found no report concerning a cell viability assay that retains its accuracy under acidic conditions and thus enables accurate assessment of anticancer drug efficacy. We hypothesized that, unlike commonly used cell viability assays, the neutral red viability assay would retain its accuracy for determining cell viability regardless of pHe.
Material and methods
Cell lines and solutions
We used two ovarian carcinoma cell lines, ES-2 and A2780, and two pancreatic cancer cell lines, S2VP10 and Panc1. ES-2, A2780, and Panc1 cells were obtained from American Type Tissue Culture Collection, ATCC (Manassas, VA). S2VP10 cells were obtained from Michael Hollingsworth, University of Nebraska, as reported earlier (Kimbrough et al. 2015). All cell lines were cultured initially in an atmosphere of 5% CO2 at 37 °C. ES-2, A2780, S2VP10 and Panc1 cells were grown in Dulbecco's modified eagle medium (DMEM) obtained from Life Technologies (Grand Island, NY).
Both pH specific media and phosphate-buffered saline (PBS) were prepared for use in all experiments. Phosphate buffer containing NaH2PO4·H2O and Na2HPO4 (Sigma-Aldrich, St. Louis, MO) was prepared in distilled H2O at pH 7.4, 6.8 and 6.6 and sterilized by autoclaving. DMEM powder, 13.6 g, was dissolved in 1l of the sterilized phosphate buffer (25 mM) at the desired pH. DMEM media solutions then were filtered through sterilized Whatman qualitative filter paper, grade 1 (Sigma-Aldrich) and DMEM solution was mixed with 10% fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and 1% L-glutamine (Life Technologies, Carlsbad, CA). The pH was checked with a pH meter (Hanna Instruments, Woonsocket, RI). The pH was adjusted, if necessary, using sterilized 1M sodium hydroxide or 1 N hydrochloric acid.
ATPlite assay
All four cell lines (ES-2, A2780, S2VP10, and Panc1) were plated in 96-well black plates (poly-L-lysine coated; flat, clear bottom black plates; BD Biosciences, San Jose, CA). In each case, 3 x 103 cells suspended in 100 μl culture medium were plated/well. On day 2, the culture medium was removed and replaced with medium with pH of 7.4, 6.8 or 6.6 and supplemented with 10% FBS and 1% L-glutamine. ES2 and A2780 cell lines were treated with 100 μM carboplatin (Sigma-Aldrich) or 100 μM chloroquine (CQ) (Sigma-Aldrich). Both S2VP10 and Panc1 cell lines were treated with 100 μM CQ or 300 nM gemcitabine (Lilly Oncology, Indianapolis, IN). Immediately following treatment, cells were incubated at 37 °C in the absence of CO2 for the remainder of the study. Cell viability was measured using ATPlite. On day 3, 25 μl lysis buffer from the Luminesence ATP Detection Assay System (PerkinElmer, Waltham, MA) was added to all wells. Plates then were placed on a rocker for 10 min before being sealed with parafilm and incubated at −20 °C for 1 h. Plates were allowed to thaw completely before 25 μl of substrate buffer was added to each well. Luminescence was measured using a BioTek plate reader (BioTek Instruments, Winooski, VT).
MTT assay
ES2, A2780, S2VP10 and Panc1 cells were plated as above in clear 96-well plates. After incubation for 24 h in an atmosphere of 5% CO2 at 37 °C, culture media were removed and replaced with media at pH 7.4, 6.8 and 6.6 supplemented with 10% FBS and 1% L-glutamine. Chemotherapeutic agents were added as described in the ATPlite protocol. Immediately following treatment, cells were incubated at 37 °C in the absence of CO2 for the remainder of the study. Ten microliters 5 mg/ml MTT solution were added to 100 μl of pH 7.4, 6.9 or 6.6 culture media in each well. Plates were incubated for 4 h. We added 50 μl lysis buffer (50 ml 10% SDS, 100 μl 12 N HCl) to each well before plates were read using a BioTek plate reader at 570 nm.
Protease assay
ES2, A2780, and Panc1 cell lines were plated at a cell density of 5 x 105 cells/well, while S2VP10 cells were plated at a cell density of 3 x 103 cells/well in black clear-bottom 96-well plates. After incubation for 24 h in an atmosphere of 5% CO2 at 37 °C, culture media were removed and replaced with media at pH 7.4, 6.8 or 6.6 supplemented with 10% FBS and 1% L-glutamine. Chemotherapeutic agents were added as described above. Immediately following treatment, cells were incubated at 37 °C in the absence of CO2 for the remainder of the study. We then added 40 μl protease diluted 1:100 in pH specific PBS to each well. Plates then were incubated in an incubator at 37 °C with no CO2 for 3 h. The protease containing medium was removed and pH specific PBS was added. The plates then were read using a BioTek plate reader at 497 nm excitation wavelength and 520 nm emission wavelength.
Neutral red assay
Ovarian carcinoma and pancreatic cancer cell lines were plated at a density of 5 x 105 cells/well in 24-well plates. After incubation for 24 h in an atmosphere of 5% CO2 at 37 °C, each well was washed with PBS and culture media were replaced by media at pH 7.4, 6.8 or 6.6 supplemented with 10% FBS and 1% L-glutamine. Chemotherapeutic agents were added at the same concentrations as described above for the ATPlite assay. Plates were incubated at 37 °C in the absence of CO2 for 3 h. We then added 100 μl neutral red (Sigma Aldrich; certified by the Biological Stain Commission) stock solution (3 mg/ml PBS) to 10 ml of medium at each pH to make a 1:100 dilution. The solution was incubated overnight at the same temperature as the cells in the non-CO2 incubator. The medium at each pH was aspirated from the cells and washed with 500 μl PBS with the same pH as the culture medium. The neutral red medium was centrifuged at 583 x g for 5 min and decanted without disturbing the precipitate. Neutral red medium, 500 μl, was added to each well. The plates were incubated for 2 h at 37 °C in an atmosphere without CO2. The neutral red medium was removed and cells were washed with 500 μl PBS at the corresponding pH. Cells were fixed with 1% formaldehyde for 2 min. Fixative was removed and 1% bovine serum albumin was added. Wash solution consisting of deionized water, 48% ethanol and 0.2 % glacial acetic acid then was added to each well. Plates were incubated for 10 min before measurement using a vertical spectrophotometer at 530 nm excitation and 645 nm emission.
Western blot
Western blotting was performed as described earlier (Egger et al. 2013). The primary antibodies were rabbit-anti-human pro-caspase-9, rabbit-anti-human cleaved caspase-9, rabbit anti-LC3 polyclonal antibody (Novus Biologicals, Littleton, CO) and rabbit-anti-human-β-actin (Pierce Scientific, Waltham, MA). Secondary antibodies were rabbit and mouse labeled with either 680 or 800 dyes (LI-COR Biosciences, Lincoln, NE). Blots were developed and analyzed using the Odyssey Infrared Imaging System (LI-COR Biosciences). Dosimetry for each band was measured and compared to B-actin as integrated optical density (I.O.D.).
Statistical analysis
Statistical comparisons were performed using SAS 9.3. The Freidman test was used for statistical analysis of cell viability. Correlation of the cell viability results with apoptotic or autophagic proteins was performed using Spearman rank correlation coefficient. Tukey’s HSD test was used for multivariate adjustments to determine differences. Values for p ≤ 0.05 were considered significant.
Results
Viability of ovarian carcinoma cells following carboplatin or CQ treatment is underestimated or misrepresented by MTT, protease and ATPlite assays under acidic conditions
Ovarian carcinoma cells, ES-2 and A2780, were treated with 100 μM carboplatin or 100 μM CQ under three pH conditions: 7.4, 6.8 and 6.6. Cell viability was evaluated using four methods: MTT, ATPlite, protease and neutral red assays. Cell viability of cells treated with PBS at pH 7.4 was considered the 100% viable standard for each assay. The MTT assay indicated that ES-2 cells were not killed significantly by carboplatin at pH 7.4, which is consistent with the literature; however, cells were killed significantly by CQ (p = 0.0431). The MTT assay for cell viability produced inconsistent results under conditions of acidic pHe. Although viability at pH 6.8 and pH 6.6 was underestimated, the viability was overestimated at pH 6.8 and pH 6.6 when the cells were treated with pH-specific PBS (Figure 1A). The MTT assay indicated a significant reduction of cell viability in A2780 cells at pH 7.4 following carboplatin treatment compared to PBS treatment at 7.4 (p = 0.0460). The combination treatment of acidic pH with carboplatin did not significantly reduce the cell viability with the addition of carboplatin compared to acidic pH alone.
Figure 1.

Assessment of cytotoxicity in ovarian cancer cells cultured at acidic pH. ES-2 and A2780 cells treated with 100 μM carboplatin or 100 μM CQ at pH 7.4, 6.8 or 6.6. Twenty-four hours after treatment, cell viability was evaluated by MTT assay (A), ATPlite assay (B), protease assay (C) and neutral red assay (D). Data are means ± SE for three repeated measurements.
The ATPlite assay produced viability results similar to the MTT assay at pH 7.4; significant cell death of ES-2 cells occurred following CQ treatment (p = 0.0238), but not following carboplatin. At pH 7.4, A2780 cells treated with either carboplatin or CQ exhibited significant cell death (carboplatin, p = 0.0478; CQ, p = 0.0488) (Figure 1B). Unfortunately, the ATPlite assay exhibited a slight, but not significant, decrease in cell viability at pH 6.8 and 6.6 for either cell line or with either carboplatin or CQ (Figure 1B). At pH 6.6, no significant difference in cell viability was observed among all treatment groups using the ATPlite assay (Figure 1B).
The protease assay produced trends similar to the MTT and ATPlite assays at pH 7.4 following carboplatin or CQ treatment for both cell lines (Figure 1C). Also, the protease assay revealed decreased cell viability at pH 6.8 in cells treated with PBS. Treatment with 100 μM carboplatin produced a slight, but not significant, decrease in cell viability (Figure 1C). No change in cell viability was observed between cells treated with PBS at pH 6.8 or pH 6.6.
The neutral red assay revealed a direct correlation between decreased pH and decreased cell viability in both cell lines following treatment with carboplatin or CQ (p < 0.05) (Figure 1D). At pH 7.4, treatment of A2780 cells with carboplatin exhibited significantly decreased cell viability compared to cells treated with PBS (p = 0.0241), but ES-2 cells were resistant to carboplatin (p = 0.363), which was consistent with results of the MTT, ATPlite and protease assays. The neutral red assay was the only assay tested that consistently demonstrated reduced cell viability at pH 6.8 in both ES-2 and A2780 cells following carboplatin or CQ treatment (ES-2: carboplatin, p = 0.0312; CQ, p = 0.022; A2780: carboplatin, p = 0.038; CQ, p = 0.033). At pH 6.6, both ES-2 and A2780 treatment with 100 μM carboplatin caused decreased cell viability (ES-2, p = 0.0181; A2780, p = 0.0235, respectively) (Figure 1D). Treatment of ES-2 ovarian cancer cells with CQ at pH 7.4 also exhibited reduced cell viability as demonstrated by the neutral red assay (p = 0.0282). Our findings suggest that at pH 7.4, all four assays are accurate for determining cell viability, evaluating an apoptotic agent and autophagic agent efficacy. The ATPlite and protease assays, however, overestimated cell viability under conditions of low pH, underestimated the efficacy of chemotherapeutic agents and produced inconsistent results for autophagic agents, which often resulted in overestimating cell viability.
Viability of pancreatic cancer cells following Gemcitabine or CQ treatment is under- or over-estimated by MTT, protease and ATPlite assays under acidic conditions
The MTT assay demonstrated a significant decrease of cell viability after gemcitabine treatment at pH 7.4 (S2VP10, p = 0.012; Panc1, p = 0.014). Cell viability following gemcitabine or CQ, however, was not significantly different at pH 6.8 and pH 6.6 (S2VP10, pH 6.8, p = 0.276; pH 6.6 p = 0.367, respectively; Panc1, pH 6.8, p = 0.387; pH 6.6 p = 0.295, respectively). The MTT assay demonstrated that pH 6.6 exhibited a greater effect on cell viability in S2VP10 (p = 0.367) and Panc1 cells (p = 0.295), but treatment with either gemcitabine or CQ exhibited no significant additional cytotoxic effects (Figure 2A).
Figure 2.

Assessment of low pHe alone and combined with either 300 nM Gemzar or 100 μM on CQ in pancreatic cancer cells. S2VP10 and Panc1 cells were treated with 300 nM Gemzar or 100 μM CQ at pHs of 7.4, 6.8 or 6.6. Twenty-four hours after treatment, cell viability was evaluated by MTT assay (A), ATPlite assay (B), protease assay (C) and neutral red assay (D). Data are means ± SE for three repeated measurements.
The ATPlite assay exhibited significantly decreased cell viability in S2VP10 cells, but not Panc1 cells after treatment with gemcitabine at pH 7.4 (S2VP10, pH 7.4, p = 0.0498; Panc1, pH 7.4, p = 0.364, respectively) (Figure 2B). ATPlite assessment of cell viability exhibited no significant decrease in cells treated with PBS at pH 6.8 and 6.6. Compared to PBS at pH 6.8 and pH 6.6, cells treated with gemcitabine or CQ did not cause decreased cell viability (Figure 2B). This likely was due to increased ATP levels associated with autophagy at acidic pH, which indicates that the ATPlite assay misrepresents cell viability as assessed using western blot (Figure 3).
Figure 3.
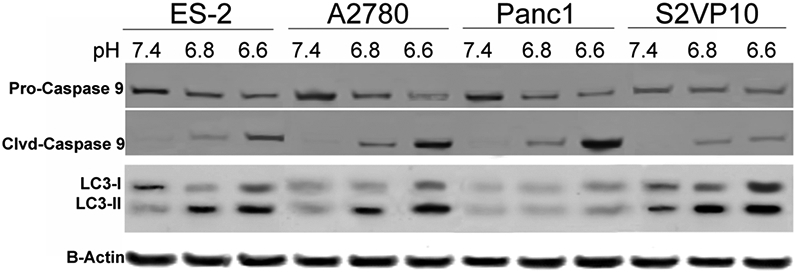
Evaluation of caspase-9 activation and LC3 up-regulation at acidic pH. ES2, A2780, Panc1 and S2VP10 cells were cultured at pH 7.4, 6.8 or 6.6. Twenty-four hours after treatment, expression of pro-caspase-9, caspase-9, and LC3-I and –II were analyzed by western blot. Data are ratios of normalized band intensities of LC3I and LC3II to actin (control). One representative experiment is shown.
The protease assay demonstrated that treatment with gemcitabine at pH 7.4 resulted in significantly decreased S2VP10 cell viability, but not Panc1 (p = 0.0338, p = 0.0842, respectively). We found no significant reduction in cell viability of cells treated with gemcitabine or CQ at pH 6.8 or 6.6 for all other protease samples (Figure 2C).
The neutral red assay revealed decreased cell viability with decreased pH after treatment of both cell lines compared to the pH 7.4 control (S2VP10, pH 6.8, p = 0.0356; pH 6.6, p = 0.0372, respectively; Panc1, pH 6.8, p = 0.0420; pH 6.6, p = 0.0463, respectively) (Figure 1D). Also, the neutral red assay demonstrated that gemcitabine treatment at all three experimental pHs decreased cell viability significantly compared to pH-specific PBS controls in both pancreatic cancer cell lines (S2VP10, pH 7.4, p = 0.0365; pH 6.8, p = 0.0267; pH 6.6, p = 0.0145; Panc1, pH 7.4p = 0.0237, pH 6.8, p = 0.0218 and pH 6.6p = 0.0186 (Figure 2D). Furthermore, treatment with CQ at each experimental pH also decreased cell viability compared to the pH-specific PBS treatment (p < 0.05).
Neutral red assay demonstrates potential of CQ as a chemotherapeutic agent for pancreatic cancer
The interior cells of pancreatic tumors are believed to undergo autophagy continually to maintain homeostasis under the hypoxic conditions resulting from insufficient tumor vascularity. We reported earlier that under hypoxic conditions CQ inhibits autophagy in pancreatic cancer cells (Frieboes et al. 2014). These investigators reported that they evaluated cytotoxic activity of CQ under acidic conditions as described above. Our MTT assay revealed that CQ did not decrease cell viability significantly at any experimental pH compared to PBS treatment (Figure 2A) Similar results were reported using the ATPlite assay in which high levels of ATP produced by autophagy artificially augmented ATP levels and therefore increased cell viability (Figure 2B). Although the protease assay demonstrated significant reduction in cell viability at pH 7.4 in both S2VP10 and Panc1 cells following gemcitabine and CQ treatments (p < 0.05), no significant changes in cell viability was observed at acidic pH using the protease assay (Figure 2C).
The neutral red assay demonstrated reduced cell viability following CQ treatment compared to PBS treated controls in both S2VP10 and Panc1 cells at all experimental pHs (S2VP10, pH 7.4, p = 0.0376; pH 6.8, p = 0.0275; pH 6.6, p = 0.0312, respectively; Panc1, pH 7.4 p = 0.0317; pH 6.8 p = 0.0366; pH 6.6 p = 0.0297, respectively (Figure 2D).
Western blot assessment of apoptotic and autophagic proteins
To compare the results of the cell viability assays that we evaluated, we correlated the cell viability based on relative levels of pro-caspase 9 compared to cleaved-caspase 9, and autophagy based upon expression of LC3-I and LC3–II. We evaluated whether ovarian and pancreatic cancer cell viability is altered at low pH. The immunoblot assay revealed decreased pro-caspase-9 and increased caspase-9 activation in both cell lines when tested at pH 6.8. Although caspase-9 activation was increased further at pH 6.6 in the Panc1 cell line, a decrease in pH did not result in further caspase-9 activation in S2VP10 cells (Figure 3). These findings suggest that the cytotoxicity associated with pH observed in Figures 1 and 2 was at least in part due to acidic pHe induced initiation of apoptosis. Our analysis of LC3-I to LC3-II conversion, a marker of autophagy, showed that LC3-II accumulation increased as extracellular pH decreased in the S2VP10 cell line, but remained unchanged in Panc1 cells (Figure 3).
The MTT, ATPlite, and protease assay results with cells treated at pH 7.4 and with an apoptosis inducer were correlated with apoptotic protein levels identified using western blot. The results of the three standard assays, however, did not correlate with decreases in cell viability under conditions of acidic pH or autophagy. For example, the MTT assay did not accurately reflect cell viability and, by extension, treatment efficacy of gemcitabine compared to western blot, especially under conditions of acidic pH (Figure 3). The ATPlite and protease assays underestimated CQ treatment related cell death based on western blot levels (Figure 3). The neutral red assay demonstrated greater accuracy for assessing cell viability (Figure 2D) and for evaluating the cytotoxic activity of chloroquine and gemcitabine under acidic conditions.
The reduction of cell viability with gemcitabine treatment corresponded to increases in cleaved caspase-9, which indicated apoptosis (Figure 3). Our findings suggest that the MTT assay is less reliable for assessing the cytotoxic effect of gemcitabine at low pH. Also, ATPlite and protease assays underestimate gemcitabine cytotoxic activity under low pH conditions. By contrast, the neutral red assay accurately assessed cell viability at all pHs tested with corresponding levels of apoptosis observed in the western blot (Fig 3). The neutral red assay indicated that gemcitabine retains its cytotoxic activity at low pH.
Acidic conditions cause morphological changes consistent with both apoptosis and autophagy
To evaluate the morphological changes caused by acidic culture conditions, S2VP10 cells were photographed immediately prior to cell harvest for western blot. Morphologic analysis revealed that cells cultured at pH 7.4 exhibited normal morphology (Figure 4A), while those cultured at pH 6.6 exhibited large autophagic vesicles and the rounded morphology characteristic of cells undergoing apoptosis (Figure 4B). Our findings establish a correlation between acidic environment, caspase-9 activation, LC3-II up-regulation and morphological changes consistent with apoptosis and autophagy.
Figure 4.
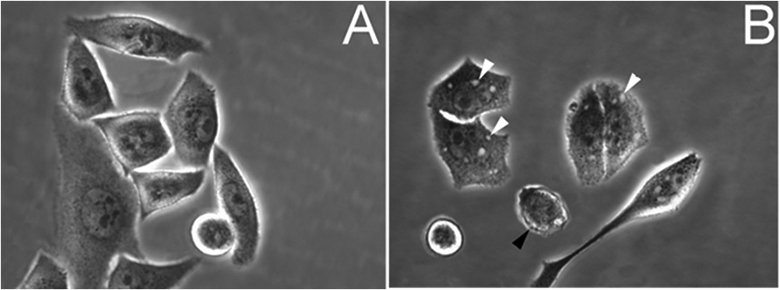
Analysis of morphological changes in pancreatic cancer cells cultured at acidic pH. S2VP10 cells were cultured at 7.4 pH medium (A) or 6.6 pH medium (B). Black arrows, apoptotic cells; white arrows, large autophagic vesicles.
Discussion
The acidity of the tumor microenvironment significantly impacts the efficacy of antineoplastic agents in vivo as a result of protonation and subsequent inactivation (Marino et al. 2012). Acidic conditions have been reported to cause metabolic stress and to activate the caspase pathway, which leads to induction of apoptosis (Park et al. 1999). To deal with the stress caused by these conditions, cancer cells undergo autophagy to manage waste products and optimize nutrient utilization (Pellegrini et al. 2014). Therefore, the cytotoxicity of anticancer therapies should be evaluated in vitro under conditions of low pH to reflect better their potential efficacy in vivo to account for the acidic tumor microenvironment. Also, mathematical models have indicated that pancreatic tumors are sensitive to gemcitabine in vitro, but resistant to gemcitabine treatment in vivo (Lee et al. 2013). Because the acidic pH of the tumor microenvironment could contribute to chemotherapy resistance, we evaluated gemcitabine cytotoxicity in pancreatic cancer cells at low pH using the MTT, ATPlite, protease and neutral red assays.
The MTT assay has been the standard method for evaluating cell viability following treatment with chemotherapeutic agents (Quent et al. 2010). Although this assay generally works well under physiologic conditions, it suffers from several critical limitations. The MTT assay relies on the ability of mitochondrial enzymes in living cells to reduce MTT to formazan, thereby using cellular metabolism as an indicator for cell proliferation and viability (Berridge et al. 2005). Therefore, the MTT assay tends to overestimate the efficacy of drugs that alter cell metabolism; it also fails to differentiate cell death from growth inhibition (Smith et al. 2011). Particularly germane to our study, acidic culture conditions decrease the accuracy of the MTT assay, because acidic pH negatively impacts the production of formazan (Johno et al. 2010).
Therapeutically inducing or blocking autophagy has been reported as an alternative strategy for cancer treatment (Boone et al. 2015). Autophagy is a mechanism by which cells degrade unnecessary or dysfunctional cellular components. This breakdown of cellular components can enhance cell survival during starvation and stress by maintaining cellular energy levels (Gozuacik and Kimchi 2007). Under such conditions, autophagy provides the fuel required to maintain cell metabolism and production of ATP (Scarlatti et al. 2009). ATP based assays of cell viability, e.g., ATPlite, use the presence of intracellular ATP as an indicator for cell viability. Therefore, although cancer cells experience autophagy, high levels of ATP in autophagic cells may produce false positive results. A comparative study of MTT and ATPlite viability assays revealed that ATPlite assay significantly underestimated antineoplastic agent (gemcitabine and etoposide) potency and efficacy for treatment of cancer cells (Chan et al. 2013). Our data support these findings for various antineoplastic agents. Our findings appear to be correlated with the increased autophagy observed in acidic environments as evidenced by the accumulation of LC3-II or up-regulation of both LC3-I and LC3–II at low pHe.
Like the MTT assay, the protease cell viability assay relies on intracellular enzymes. The fluorogenic protease substrate, glycylphenylalanyl-aminofluorocoumarin (GF-AFC), used in the assay detects protease activity in viable cells selectively (Niles et al. 2007). Upon cellular penetration, cytoplasmic aminopeptidases remove glycine and phenylalanine to release aminofluorocoumarin (AFC) and generate a fluorescent signal proportional to the number of viable cells. The GF-AFC enzyme can be affected by physicochemical parameters, however, e.g., changes in culture medium pH and alteration by pharmacologic agents. Enzyme activity also may decay over time after exposure to the extracellular milieu, particularly in the setting of events that result in the production of cellular metabolites and alterations of ion gradients, such as large volume cell death and rapid metabolism, both of which are prominent in tumor microenvironments (Galluzzi et al. 2009).
Unlike the typical assays, our findings suggest that the neutral red assay detects cell viability more accurately over a broad range of pHe; it detects decreased cell viability accurately even with the increased extracellular acidity that accompanies apoptosis and autophagy. Also, unlike viability tests that use tetrazolium salts, e.g., MTT assay, the neutral red assay does not use unstable reagents. Despite its substantial advantages over other cell viability assays, the neutral red assay has limitations. Most important, it must be performed on freshly isolated cells. Freezing cells or use of any form of fixative renders the assay ineffective.
Our assessment of the cytotoxic potential of CQ for treatment of pancreatic cancer reflects the utility of the neutral red assay for evaluating treatment efficacy. We demonstrated inhibition of pancreatic cancer cells by CQ in an acidic environment. Previous reports have demonstrated the antitumor effects of CQ on pancreatic cancer cells in vitro (Zeilhofer et al. 1989). Subsequently, CQ has been shown to inhibit autophagy in breast cancer and glioblastoma cells (Cufi et al. 2011; Firat et al. 2012). In addition to the effects of CQ on autophagy, CQ treatment also can cause cell death by its inhibition of CXCR4 and hedgehog signaling (Balic et al. 2014). Together with earlier reports, our findings suggest that chloroquine could be effective for treating pancreatic cancer in vivo; our findings also support further exploration of use of CQ in the clinical setting.
Although the neutral red assay can be used to assess cell viability and antineoplastic drug activity accurately under low pHe, acidity represents only one aspect by which the tumor microenvironment differs from the normal extracellular milieu. Assessment of the accuracy of the neutral red assay under conditions of hypoxia, elevated extracellular lactate concentration, glucose deprivation and the combination of all of these would enable evaluation of its accuracy for assessing cell viability regardless of the actual tumor microenvironment. Like any cell viability assay for assessing cytotoxicity of antineoplastic agents, results must be correlated with the clinical efficacy of these drugs.
The neutral red assay is a sensitive and easily quantifiable cell viability assay that is accurate over a range of pH and in the context of either apoptotic or autophagic cell death. Its use enables more accurate evaluation of the cytotoxicity of chemotherapeutic agents under acidic conditions such as those within the tumor microenvironment. Although we repeated the neutral red assay at least five times and obtained accurate evaluation with both apoptotic or autophagic modulating therapies and acidic pHe, we did not evaluate the accuracy of the neutral red assay following treatment with necrotic inducing agents at pH > pH 6.6 or in nonmalignant cells. Future uses of the neutral red assay are likely to be especially beneficial for investigations of either the effects of acidic pHe or autophagy related to cancer.
We report here that a modified neutral red staining protocol can be used to evaluate cell viability under acidic conditions more accurately than the most common cell viability assays, i.e., MTT, ATPlite and protease assays, after treatment with carboplatin, gemcitabine or CQ.
Acknowledgments
Jorge G. Gomez-Gutierrez and Neal Bhutiani contributed equally to this work.
Funding
This work was supported in part by NIH grants R01CA205941, R01CA212350, R01EB020125, and R25CA134283.
References
- Balic A, Sorensen MD, Trabulo SM, Sainz B Jr., Cioffi M, Vieira CR, Miranda-Lorenzo I, Hidalgo M, Kleeff J, Erkan M, Heeschen C, 2014. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther 13:1758–1771. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Herst PM, Tan AS, 2005. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Ann Rev. 11, 127–152. [DOI] [PubMed] [Google Scholar]
- Boone BA, Bahary N, Zureikat AH, Moser AJ, Normolle DP, Wu WC, Singhi AD, Bao P, Bartlett DL, Liotta LA, Espina V, Loughran P, Lotze MT, Zeh HJ 3rd, 2015. Safety and biologic response of pre-operative autophagy inhibition in combination with gemcitabine in patients with pancreatic adenocarcinoma. Ann Surg Oncol. 22:4402–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan GK, Kleinheinz TL, Peterson D, Moffat JG, 2013. A simple high-content cell cycle assay reveals frequent discrepancies between cell number and ATP and MTS proliferation assays. PloS One 8, e63583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KL, Shajahan AN, Clarke R, 2011. Autophagy and endocrine resistance in breast cancer. Exp Rev Anticancer Ther. 11:1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA, 2011. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. 10:3871–3885. [DOI] [PubMed] [Google Scholar]
- Egger ME, Huang JS, Yin W, McMasters KM, McNally LR, 2013. Inhibition of autophagy with chloroquine is effective in melanoma. J Surg Res. 184:274–281. [DOI] [PubMed] [Google Scholar]
- Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G, 2012. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PloS One 7, e47357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieboes HB, Huang JS, Yin WC, McNally LR, 2014. Chloroquine-mediated cell death in metastatic pancreatic adenocarcinoma through inhibition of autophagy. J Panc. 15:189–197. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Castedo M, Cidlowski JA, Ciechanover A, Cohen GM, De Laurenzi V, De Maria R, Deshmukh M, Dynlacht BD, El-Deiry WS, Flavell RA, Fulda S, Garrido C, Golstein P, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Jaattela M, Kepp O, Kimchi A, Klionsky DJ, Knight RA, Kornbluth S, Kumar S, Levine B, Lipton SA, Lugli E, Madeo F, Malomi W, Marine JC, Martin SJ, Medema JP, Mehlen P, Melino G, Moll UM, Morselli E, Nagata S, Nicholson DW, Nicotera P, Nunez G, Oren M, Penninger J, Pervaiz S, Peter ME, Piacentini M, Prehn JH, Puthalakath H, Rabinovich GA, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Scorrano L, Simon HU, Steller H, Tschopp J, Tsujimoto Y, Vandenabeele P, Vitale I, Vousden KH, Youle RJ, Yuan J, Zhivotovsky B, Kroemer G, 2009. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 16:1093–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ, 2006. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 66:5216–5223. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A, 2007. Autophagy and cell death. Curr Top Dev Biol. 78:217–245. [DOI] [PubMed] [Google Scholar]
- Hockel M, Vaupel P, 2001. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Nat Cancer Inst. 93:266–276. [DOI] [PubMed] [Google Scholar]
- Johno H, Takahashi S, Kitamura M, 2010. Influences of acidic conditions on formazan assay: a cautionary note. Appl Biochem Biotechnol. 162:1529–1535. [DOI] [PubMed] [Google Scholar]
- Kimbrough CW, Khanal A, Zeiderman M, Khanal BR, Burton NC, McMasters KM, Vickers SM, Grizzle WE, McNally LR, 2015. Targeting acidity in pancreatic adenocarcinoma: multispectral optoacoustic tomography detects ph-low insertion peptide probes in vivo. Clin Cancer Res. 21:4576–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFargue CJ, Dal Molin GZ, Sood AK, Coleman RL, 2019. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 20:e15–e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Huang J, England CG, McNally LR, Frieboes HB, 2013. Predictive modeling of in vivo response to gemcitabine in pancreatic cancer. PLoS Comput Biol. 9, e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB, 2005. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev. 6:439–448. [DOI] [PubMed] [Google Scholar]
- Marchetti C, De Felice F, Perniola G, Palaia I, Musella A, Di Donato V, Cascialli G, Muzii L, Tombolini V, Benedetti Panici P 2019. Role of intraperitoneal chemotherapy in ovarian cancer in the platinum-taxane-based era: a meta-analysis. Crit Rev Oncol Hematol. 136:64–69. [DOI] [PubMed] [Google Scholar]
- Marino ML, Pellegrini P, Di Lernia G, Djavaheri-Mergny M, Brnjic S, Zhang X, Hagg M, Linder S, Fais S, Codogno P, De Milito A, 2012. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J Biol Chem. 287:30664–30676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T, 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. [DOI] [PubMed] [Google Scholar]
- Niles AL, Moravec RA, Eric Hesselberth P, Scurria MA, Daily WJ, Riss TL, 2007. A homogeneous assay to measure live and dead cells in the same sample by detecting different protease markers. Anal Biochem. 366:197–206. [DOI] [PubMed] [Google Scholar]
- Park HJ, Lyons JC, Ohtsubo T, Song CW, 1999. Acidic environment causes apoptosis by increasing caspase activity. Br J Cancer. 80:1892–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini P, Strambi A, Zipoli C, Hagg-Olofsson M, Buoncervello M, Linder S, De Milito A, 2014. Acidic extracellular pH neutralizes the autophagy-inhibiting activity of chloroquine: implications for cancer therapies. Autophagy. 10:562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quent VM, Loessner D, Friis T, Reichert JC, Hutmacher DW, 2010. Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. J Cell Mol Med 14:1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repetto G, del Peso A, Zurita JL, 2008. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 3:1125–1131. [DOI] [PubMed] [Google Scholar]
- Scarlatti F, Granata R, Meijer AJ, Codogno P, 2009. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 16:12–20. [DOI] [PubMed] [Google Scholar]
- Smith SM, Wunder MB, Norris DA, Shellman YG, 2011. A simple protocol for using a LDH-based cytotoxicity assay to assess the effects of death and growth inhibition at the same time. PloS One 6, e26908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thigpen T, duBois A, McAlpine J, DiSaia P, Fujiwara K, Hoskins W, Kristensen G, Mannel R, Markman M, Pfisterer J, Quinn M, Reed N, Swart AM, Berek J, Colombo N, Freyer G, Gallardo D, Plante M, Poveda A, Rubinstein L, Bacon M, Kitchener H, Stuart GC, 2011. First-line therapy in ovarian cancer trials. Int J Gynecol Cancer. 21:756–762. [DOI] [PubMed] [Google Scholar]
- van der Burg ME, Onstenk W, Boere IA, Look M, Ottevanger PB, de Gooyer D, Kerkhofs LG, Valster FA, Ruit JB, van Reisen AG, Goey SH, van der Torren AM, ten Bokkel Huinink D, Kok TC, Verweij J, van Doorn HC, 2014. Long-term results of a randomised phase III trial of weekly versus three-weekly paclitaxel/platinum induction therapy followed by standard or extended three-weekly paclitaxel/platinum in European patients with advanced epithelial ovarian cancer. Eur J Cancer. 50:2592–2601. [DOI] [PubMed] [Google Scholar]
- White E, DiPaola RS, 2009. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 15:5308–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Mollenhauer J, Brune K, 1989. Selective growth inhibition of ductal pancreatic adenocarcinoma cells by the lysosomotropic agent chloroquine. Cancer Lett. 44:61–66. [DOI] [PubMed] [Google Scholar]
- Zhang SZ, Lipsky MM, Trump BF, Hsu IC, 1990. Neutral red (NR) assay for cell viability and xenobiotic-induced cytotoxicity in primary cultures of human and rat hepatocytes. Cell Biol Toxicol. 6:219–234. [DOI] [PubMed] [Google Scholar]