

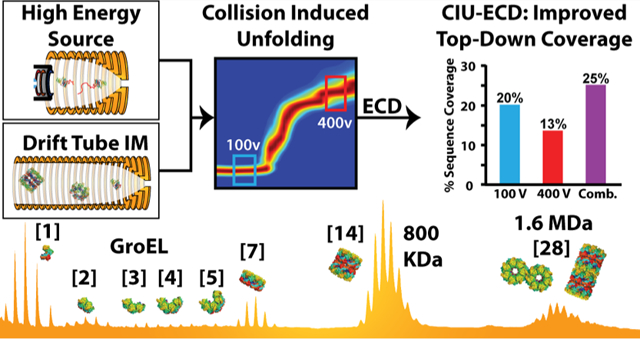
Abstract
Native ion mobility-mass spectrometry (IM-MS) is capable of revealing much that remains unknown within the structural proteome, promising such information on refractory protein targets. Here, we report the development of a unique drift tube IM-MS (DTIM-MS) platform, which combines high-energy source optics for improved collision induced unfolding (CIU) experiments and an electromagnetostatic cell for electron capture dissociation (ECD). We measured a series of high precision collision cross section (CCS) values for protein and protein complex ions ranging from 6–1600 kDa, exhibiting an average relative standard deviation (RSD) of 0.43 ± 0.20%. Furthermore, we compare our CCS results to previously reported DTIM values, finding strong agreement across similarly configured instrumentation (average RSD of 0.82 ± 0.73%), and systematic differences for DTIM CCS values commonly used to calibrate traveling-wave IM separators (−3% average RSD). Our CIU experiments reveal that the modified DTIM-MS instrument described here achieves enhanced levels of ion activation when compared with any previously reported IM-MS platforms, allowing for comprehensive unfolding of large multiprotein complex ions as well as interplatform CIU comparisons. Using our modified DTIM instrument, we studied two protein complexes. The enhanced CIU capabilities enable us to study the gas phase stability of the GroEL 7-mer and 14-mer complexes. Finally, we report CIU-ECD experiments for the alcohol dehydrogenase tetramer, demonstrating improved sequence coverage by combining ECD fragmentation integrated over multiple CIU intermediates. Further improvements for such native top-down sequencing experiments were possible by leveraging IM separation, which enabled us to separate and analyze CID and ECD fragmentation simultaneously.
Graphical Abstract
The emergence of native mass spectrometry (MS), which is concerned with the direct MS analysis of native proteins and multiprotein complexes, has been built upon many prior innovations in protein ionization1,2 and instrumentation.3 Over the past two decades, native MS has established itself as a bona fide tool for structural biology research, capable of analyzing large biomolecular ions such as chaperone assemblies,4,5 ribosomes,6–8 and intact viral particles.9–11 In addition, ion mobility-mass spectrometry (IM-MS), which separates protein ions according to their size and charge on the millisecond timescale, has been developed in combination with native MS to provide more detailed structures of a wide range of protein assemblies. Much of the utility of IM-MS for structural biology depends strongly on the accurate and precise measurement of ion collision cross section (CCS) values, the ion sizes reported by the technology.
Protein CCSs are especially difficult to measure with precision due to the inherent plasticity and dynamism adopted by such biomolecules. These challenges motivated the development of collision induced unfolding (CIU), which tracks the stability and unfolding of gas-phase protein ions through stepwise collisional heating in combination with native IM-MS.12 Such tools have enabled IM-MS to discern subtle changes in protein tertiary structures that remain obscured in ground-state protein CCS values, for systems ranging from protein kinases13 to biotherapeutics.14,15 Similarly, native proteomics16 seeks to leverage ion activation tools to merge protein structures and sequence information into a single rapid set of experiments. Current methods are, however, limited by the contemporary precision of protein CCS values (~3% RSD),17 the few number of intermediate features produced during typical CIU,13 and the low sequence coverages obtained for native protein ion precursors.18
Here, we describe and characterize a unique drift tube (DT) IM-MS platform for native protein analysis. In previous testing across multiple, geographically distinct instruments, an Agilent 6560 DTIM-MS platform has demonstrated CCS relative standard deviation (RSD) values of 0.29% for multiple classes of small and medium-sized biomolecules.19 Recent reports have expanded the use of this platform for native MS, although prior data included limited ion activation capabilities for large biomolecular ions.20,21 The DTIM-MS device disclosed here combines modified source optics to achieve high-energy collisional activation for CIU with an electromagnetostatic linear ExD device22 installed between the quadrupole and the collision cell of the standard platform. We use this instrument to perform the most comprehensive analysis of DTIM protein CCS data to date. We disclose evidence that precise protein size values can be recorded across a wide mass range using DTIM (6–1600 kDa), but systematic CCS differences are observed for values obtained from differently configured DTIM-MS platforms23–25 that likely relate to the different ion temperatures and lifetimes for the analytes interrogated in past experiments. Furthermore, we extensively characterize our high-energy source design for CIU, observing additional extended ion unfolding features, and demonstrating the reproducibility for CIU pathways across disparate IM-MS platforms. Using this higher energy source, we explore the limits of protein complex CIU. Finally, we leverage CIU and ECD to demonstrate the capability of IM-ECD to correlate changes in ECD fragmentation with gas-phase structural changes. Finally, the combination of unique fragments obtained at different levels of CIU enhanced native top-down sequencing of intact protein assemblies, illustrating the utility of such experiments for native proteomics workflows.
MATERIALS AND METHODS
Sample Preparation & IM-MS Measurements.
Lyophilized protein samples were purchased from Sigma Aldrich (St. Louis, Mo) (Table S3). GroEL was prepared following a previously published protocol.26 Samples were desalted before analysis using Bio-Rad BioSpin 6 and 30 columns (6 and 30 kDa MWCO). All IM-MS measurements were carried out at a protein concentration of 1–10 μM, prepared in 100–200 mM ammonium acetate solution, pH ~7.5, at room temperature. Ions were generated by static nanoelectrospray ionization (nESI) using an Agilent nESI source and gold-coated borosilicate nESI emitters, with a capillary voltage of 900–1400 V. The source drying gas was operated at 1.5 L/min (N2) or 2 L/min (SF6), at 25 °C. Source voltages were optimized for every protein to avoid activation.
All DTIM measurements were performed in high purity N2 with the front funnel (high-pressure funnel) and trap funnel operated at 4–4.50 torr and 3.80 torr, respectively, and the drift tube operated at 3.95 torr. The DTIM collision cross sections (DTCCSN2) reported were measured using multiple drift fields (MF, DT-MFCCSN2)27 or by the single-field method (SF, DT-SFCCSN2) calibrated with Agilent tune mix.19 The two approaches agree within 0.58% for a selection of ions from multiple classes.19
Instrumentation.
A modified Agilent 6560 drift tube ion mobility-quadrupole time-of-flight (DTIM-Q-TOF) (Santa Clara, CA) was used in most of the experiments reported. Instrument modifications include the addition of a new capillary exit (CE) lens immediately after the ion transfer capillary (Figure 1A). Elevating the voltage applied to the CE lens with respect to fragmentor (F) lens enables a ΔV of up to 450 V between CE and F, when F = 400 V (default F setting) (Figure 1B). Setting the F slightly lower to 360 V allows a ΔV of 490 V when activating larger protein complexes. This ΔV allows for increasingly faster acceleration of ions through the source, resulting in higher energy collisions with the background gas. For higher in-source activation (ΔV > 350 V), sulfur hexafluoride (SF6) was used as the drying gas, serving as an electron scavenger to prevent arcing between the high voltage CE/F lenses. The SF6 also contributes to ion activation by enabling larger center-of-mass frame energy collisions with the analyte ions. High purity N2 gas was used throughout the rest of the instrument.
Figure 1.
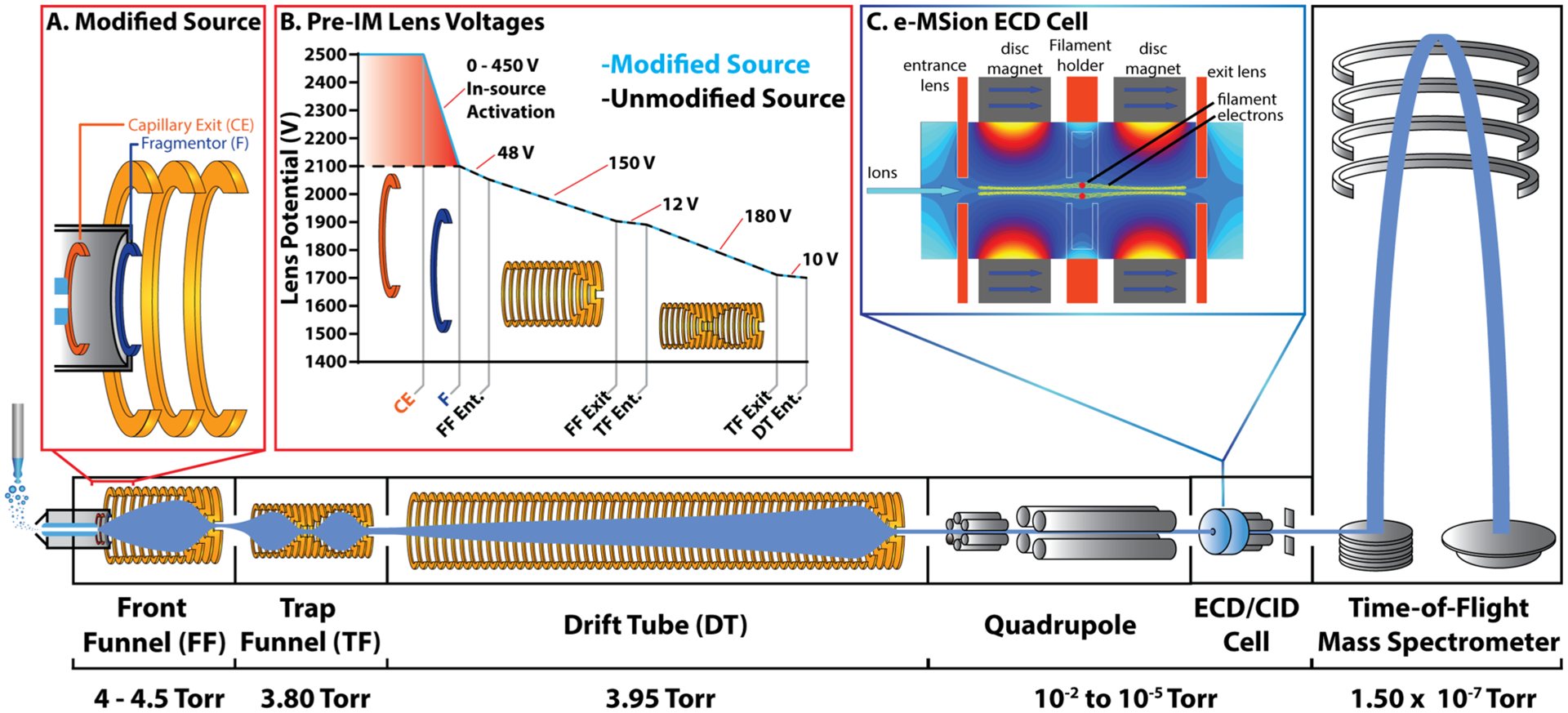
Agilent 6560 DTIM-QTOF modified for native mass spectrometry: (A) Agilent 6560 DTIM-QTOF mass spectrometer was retrofitted with a custom desolvation assembly equipped with one additional high voltage lens (CE), which enables a ΔV of up to 450 volts relative to the fragmentor (F). (B) Source voltages in the modified high energy source. Voltages labeled are ΔV values between the two adjacent lenses. (C) e-MSion electromagnetostatic linear ExD cell was installed before the post-IM collision cell, to enable IM-resolved electron capture dissociation experiments. The instrument is also upgraded for an extended mass range, which allows analysis up to 20,000 m/z. The instrument was operated at the indicated pressures, and pure N2 was used as the IM separation gas. The 78.2 cm drift tube was operated at <18.5 V/cm.
The Agilent 6560 was also modified with the addition of an e-MSion electromagnetostatic linear electron capture dissociation cell (Corvallis, OR) positioned between the quadrupole and the collision cell, to enable post-IM electron capture dissociation (ECD) experiments while retaining the IM-separation (Figure 1C). Finally, the instrument was upgraded to enable acquisition up to 20,000 m/z in the TOF region, and the optics were tuned in sensitivity mode to optimize transmission of larger ions by adjusting the top and bottom slit potentials before the TOF entrance.
CIU.
CIU experiments were conducted by increasing the CE lens to +490 V relative to the fragmentor. The time segment feature was used in the Agilent Mass Hunter Acquisition software to collect all activation steps in a single file. The MIDAC Agilent data extractor (packaged with CIUSuite2) was used to extract the arrival time distribution (ATD) of the ions of interest from each time segment. The ATDs were plotted against the applied ΔV, referred to as collision voltage (CV), using CIUSuite2.28 All IM-MS data were analyzed using Agilent Mass Hunter IM-MS Browser software 10.0 and mMass.29
Some CIU experiments were conducted using a Waters Synapt G2 (Milford, MA) travelling wave ion mobility (TWIM) mass spectrometer to assess the feasibility of comparing CIU across various instrument platforms. All CIU experiments were conducted by increasing the trap voltage from 5 V to 200 V in 5 V steps. The TWIM cell was operated with a wave velocity of 500 m/s and a wave height of 30 V. A recently developed calibration framework30 was used to convert all ATDs to collision cross sections (TWCCSN2) using polyalanine, in combination with TTR, BSA, ConA, or GDH as calibrants. All TWIM data were extracted for analysis using TWIMExtract31 and analyzed as previously described using CIUSuite 2.
CCSN2 of the unfolding intermediate features and the change in CCSN2 between two adjacent features (ΔCCSN2) were used to compare the CIU performance between these two instruments. This approach was chosen due to the different ion activation energy scales employed by the two instruments, which result from very different instrument designs and pressure regimes in the pre-IM activation region. Specifically, in the Synapt G2, the ion activation occurs in the trap cell filled with argon gas where the pressure is ~25 mTorr. In contrast, ion activation in the Agilent 6560 occurs in the ~4.5 Torr source region, with a mixture of nitrogen and sulfur hexafluoride gas as the collision gas. Thus, it is currently not possible to directly compare the ion activation voltages required to unfold proteins on the two instruments.
ECD.
ECD experiments were conducted using an e-MSion linear ExD cell without quadrupole selection. The ECD filament was operated at 1.2 μA to generate a magnetically confined cloud of electrons. The parent ions were fragmented by passing through the electron cloud. In some cases, supplemental activation was applied in the collision cell after the ECD cell, to release ECD fragments that were not ejected from the parent ion. Data were extracted in .csv format using Agilent MassHunter IM-MS Browser 10.0. Mascot generic format (MGF) files were produced from CSV files by an in-house python script. Next, MGF files were analyzed by MASH Explorer.32 eTHRASH was used for deconvolution with a minimum fit of 90%, and TopPIC was used for peak matching with a MIScore Threshold of 0.90. Other parameters for eTHRASH and TopPIC were default values. Mass tolerance for peak matching was 10 ppm. Sequence coverage was calculated by dividing observed fragment sites by the total possible number of fragment sites.
RESULTS & DISCUSSION
Native IM-MS and CIU of E. coli GroEL.
The upgrades to our DTIM-QTOF instrument enable analyses of significantly larger ions than previously possible on this platform. The higher energy source improves desolvation and ion transmission, while the extended mass range of the TOF enables detection up to 20,000 m/z. To evaluate the instrument for large ion analysis, we selected GroEL, a 14-subunit protein complex with a molecular weight of ~801 kDa, and a standard for native MS measurements. GroEL consists of two stacked 7-subunit rings with molecular weights of ~400 kDa each.
In our data, we observe that the 14-subunit complex accounts for the majority of signal intensity; however, smaller subcomplexes are also observed at lower intensities. We also observed a charge state distribution at ~15,800–16,500 m/z, which corresponds to a 28-subunit complex (~1.6 MDa) (Figure 2A,B). CCSs were measured and are reported for all GroEL ions observed (Table S1), some for the first time. High IM resolution values (~40 t/Δt) are observed for all of the GroEL assemblies detected.
Figure 2.
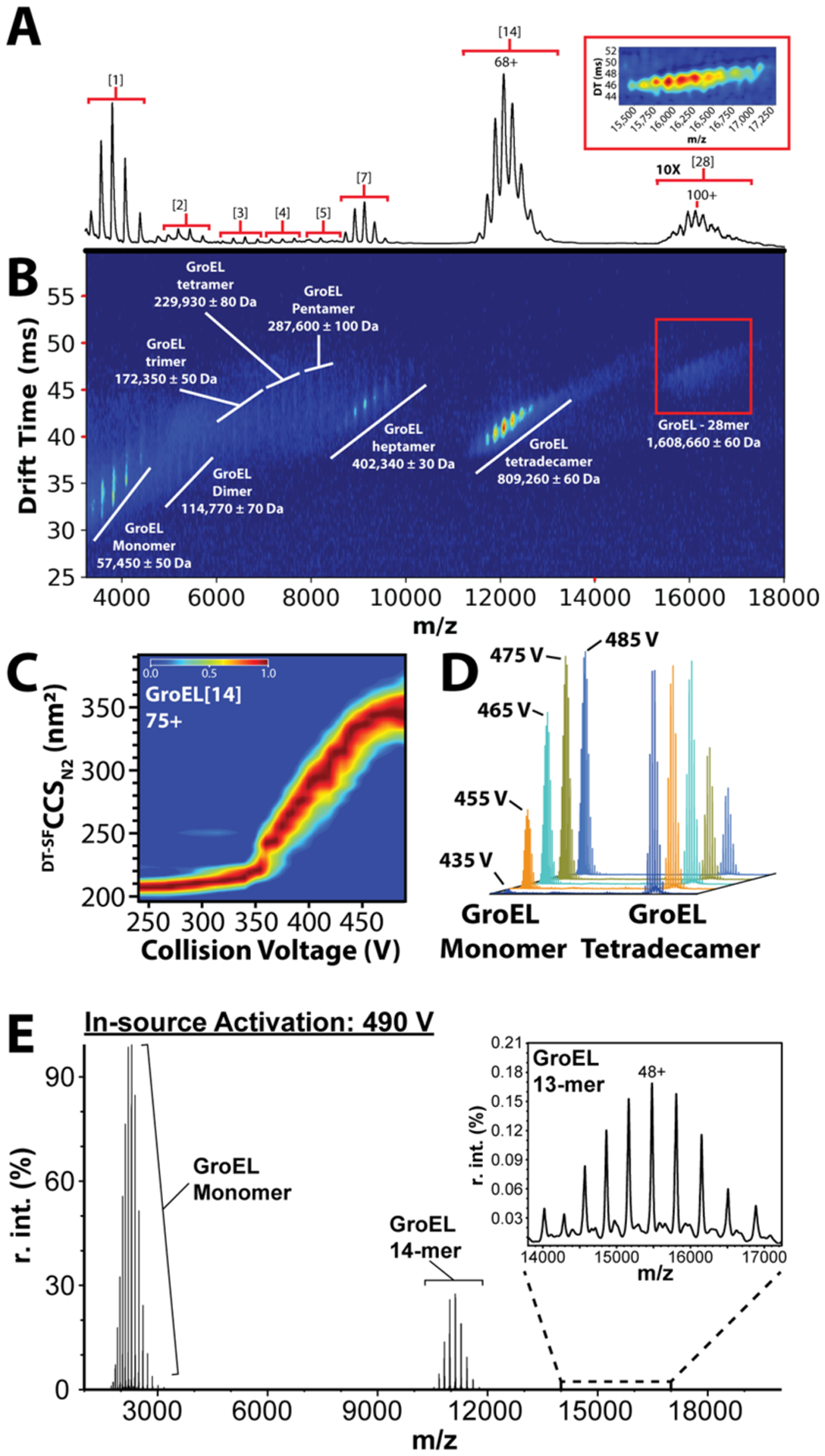
Native MS and CIU of E. coli GroEL: (A) native mass spectrum of chaperonin 60 from E. coli (groEL). Charge state distributions of 1-, 2-, 3-, 4-, 5-, 7-, 14-, and 28-mer complexes of GroEL are labeled (bracketed numbers). The signal intensity for the 28-mer complex is magnified 10x. The inset shows an enlarged IMMS plot of the 28-mer complex. (B) Corresponding IM-MS plot of GroEL with complexes labeled. (C) CIU of the 75+ charge state of GroEL. (D) Stacked mass spectra that show CID of GroEL 14-mer as in-source activation is increased (collision voltages labeled). Spectral intensity values are normalized relative to the most intense signal per collision voltage step. (E) Extracted mass spectrum of GroEL 14-mer CID products at 490 V in-source activation, demonstrating the stability of the 14-mer complex, and the presence of the stripped 13-mer complex (48+ charge state labeled).
Our high-resolution IM measurements extend to the GroEL 28-mer, which exhibits CCSs of approximately 330–366 nm2, some of the largest protein CCSs reported to date. Currently, there is no evidence for a physiological role for such a large GroEL complex; however, the observation of these ions is noteworthy as it indicates the high transmission efficiency possible for such megadalton assemblies on our modified DTIM platform. To investigate the potential structure adopted by GroEL 28-mers in our experiments, we constructed two archetypal models (Figure S1) and computed their CCSs using IMPACT.33 Since the computed CCSs of these structures are within 3.5% of each other, it is difficult to confidently assign a specific structure to the 28mer based on the CCS alone. We note that the relative increase in CCS between 14-mer and average 28-mer model structures, ~75%, is similar to that observed experimentally (~72%). These results stand in contrast to CCS comparisons made between GroEL 7-mers and 14-mers, where models predict a 53% increase, while a 43% increase is observed experimentally. Previous reports have discussed the apparent gas-phase compaction of the GroEL 14-mer,26 and these results indicate that such compaction does not extend to GroEL subcomplexes or higher-order assemblies.
Using the higher in-source activation available on our DTIM platform, we monitored the CIU of GroEL ions. In our experiment, we were able to clearly observe the unfolding of both GroEL 7-mer and 14-mer ions. For example, CIU data for the 75+ GroEL 14-mer begin in a compact state and unfold to produce a wide range of intermediate unfolding steps of varying stabilities, until a more stable putatively terminally unfolded structure is achieved at ~490 V (Figure 2C). CIU of such a large complex has never been studied in detail before, and thus, a structural interpretation for these intermediate features remains unclear. Interestingly, our GroEL experiments reveal that a 75% CCS increase is achieved by the ions at the end of the CIU experiment. This value is significantly larger than what has been observed previously for the CIU of smaller protein complexes.34–36 For example, DTIM CIU data reported here for BSA and IgG result in CCS increases of 40–45% over the course of an experiment (Figure 3; Figure S4).
Figure 3.
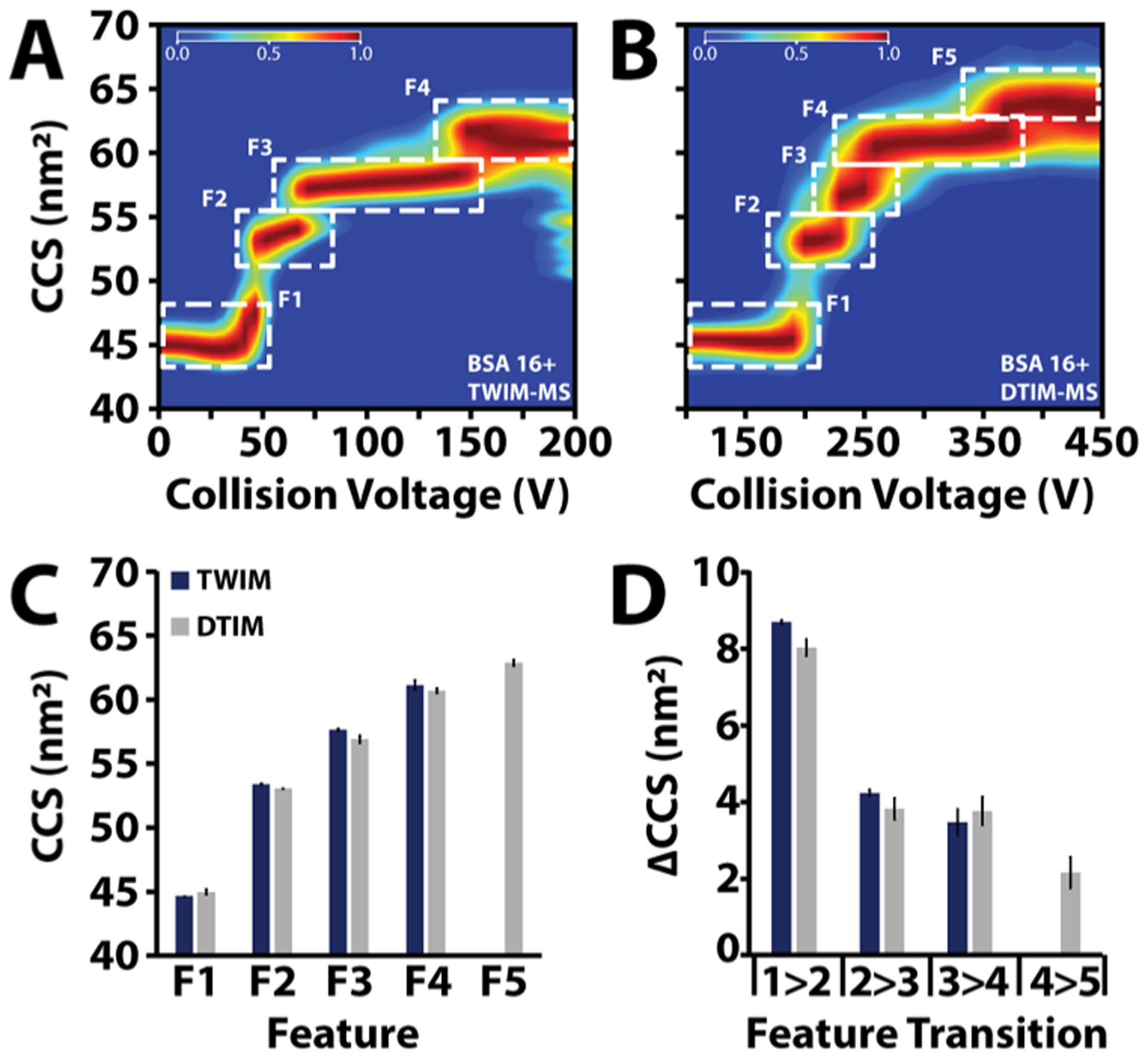
Enhanced collision induced unfolding comparison of bovine serum albumin (BSA) CIU from a (A) TWIM-MS vs a (B) DTIMMS. Higher pre-IM activation is achievable on the DTIM-MS as evidenced by the fifth feature at higher CCS observed on the DTIMMS. (C) Median CCS of each feature from TWIM and DTIM CIU experiments. (D) ΔCCS between features from the TWIM and DTIM CIU experiments.
In parallel with our CIU experiments, we observe collision induced dissociation (CID) of the 14-mer complex. Five mass spectra are shown stacked in order of increasing in-source activation, demonstrating the decrease of the GroEL 14-mer signal and the concomitant increase in the GroEL monomer signal (Figure 2D). As previously described,37,38 CID of the 14-mer complex produces highly charged ejected GroEL monomers at low m/z and the resultant stripped 13-mer complex at higher m/z (Figure 2E). The relative abundance of each complex was extracted and plotted versus the in-source activation, to monitor the conversion of 14-mer into a monomer and 13-mer complex (Figure S2). Although the 7-mer complex was observed in these experiments, Figure S2 demonstrates that it was not a product of 14-mer CID, since its abundance does not increase upon 14-mer activation. Finally, it is noteworthy that even at a maximum in-source activation voltage of 490 V, the 14-mer complex signal was not completely depleted indicating the overall stability of the 14-mer complex in our experiments (Figure 2E).
Integration across the entire activation regime shows four 7-mer (47+ to 50+) and seven 14-mer charge states (69+ to 75+) simultaneously undergoing CIU (Figure S3A), and we extracted high-quality fingerprint data comprehensively across these ions (Figure S3B−K). As observed previously, GroEL 7-mer is a common subcomplex observed upon disruption of the complex in solution, and the 7-mer signals observed here are likely the result of a similar process.39 A comparison of the GroEL 7-mer and 14-mer CIU data reveals that the lab frame energy required to reach the first feature transition for the 14-mer complex is ~50% greater than that for the 7-mer complex. Though this difference in overall stability is expected, we were surprised to observe that the ΔCCS observed for the first CIU transition is substantially greater for GroEL 7-mer. Specifically, we find that the ΔCCS associated with the first CIU transition is approximately 9 nm2 in the 14-mer complex and 35 nm2 in the 7-mer complex. Interestingly, the second transition for the 14-mer complex accounts for a ~30 nm2 increase in CCS. Prior results have demonstrated that CIU data for low charge state proteins can be correlated with the domain structure40 and that protein complex CIU is likely dominated by the unfolding of single subunits within assemblies. The results presented here clearly illustrate the challenges, however, in extracting subunit domain information from protein complex CIU data and suggest that the gas-phase compaction of the GroEL 14-mer is not linked to its aberrant stability in the absence of the solvent.
Enhanced CIU.
In order to cross-compare CIU fingerprint data across disparate IM-MS platforms, we selected bovine serum albumin (BSA) as a model system, as previous results suggest a detailed domain-correlated CIU mechanism.40 Specifically, previous results for low charge state ions indicated four total states, three total transitions, for the CIU data collected for BSA, where each feature correlates with the gas-phase unfolding of a specific albumin domain. CIU fingerprints were measured across both TWIM and DTIM platforms, and we observe very strong correlations between the CCS values recorded for CIU features across both instruments (Figure 3A,B). Furthermore, we detect an additional feature, possessing a larger CCS than those features observed in previous BSA CIU data for this charge state, suggesting that our modified DTIM source is able to achieve a higher level of activation than that achieved in our previous TWIM CIU measurements. We have observed similar results for other large protein ions (Figure S4), paving the way for CIU fingerprinting that incorporates a greater number of features than previously possible. Despite the strong correlations in CCS noted above, we note that the measured median CCSN2 values recorded for individual TWIM features exhibit a systematic offset of 1.1 ± 0.6% (Figure 3C) from those recorded by DTIM. Such differences likely result from the different kinetic and ion temperature conditions experienced by ions analyzed by the two platforms probed in this report. Interestingly, a comparison of the ΔCCS values recorded for CIU features reveals a significantly greater level of agreement, indicating that although median CCS values may vary between platforms, CIU may provide a more precise method for cross-platform comparisons of intact protein IM-MS data (Figure 3D).
ECD.
The combination of our enhanced energy ion source and an ExD cell situated between the quadrupole and collision cell enables unique protein analysis options that combine electron-based fragmentation IM separation and CIU. We assessed this combination in the context of native top down mass spectrometry experiments by focusing on the fragmentation of the alcohol dehydrogenase (ADH) tetramer.
Fragmentation data for ADH were recorded both with 100 V and 400 V in-source activation to evaluate the impact of CIU on ECD fragment ion production (Figure 4A). Exposing ADH tetramer ions to free electrons following 100 V of in-source collisional activation results in no discernable ECD fragment ion current (Figure S5), and this observation is in line with previous reports that have indicated that threshold collisional activation is necessary to liberate potential ECD fragments from the protein surface.41 Upon the addition of 80 V post-ECD activation in the collision cell, 64 unique fragments (Table S2) were identified, corresponding to 20% sequence coverage (Figure 4B). Based on past reports where increased fragment ion production is linked to extent of solvent accessibility of the exposed surface,42–44 we exposed ADH ions to free electrons following CIU using 400 V of in-source activation (Figure 4A), resulting in 44 unique fragments and 13% sequence coverage (Figure 4B). Unlike our data collected at following 100 V of in-source activation, these ions required no post-ECD activation to release fragment ions following electron capture. Conversely, we observe that the addition of post-ECD activation following CIU at 400 V results in fewer ECD fragment ions from the ADH tetramer, yet many were uniquely observed under such conditions (Figure S6).
Figure 4.
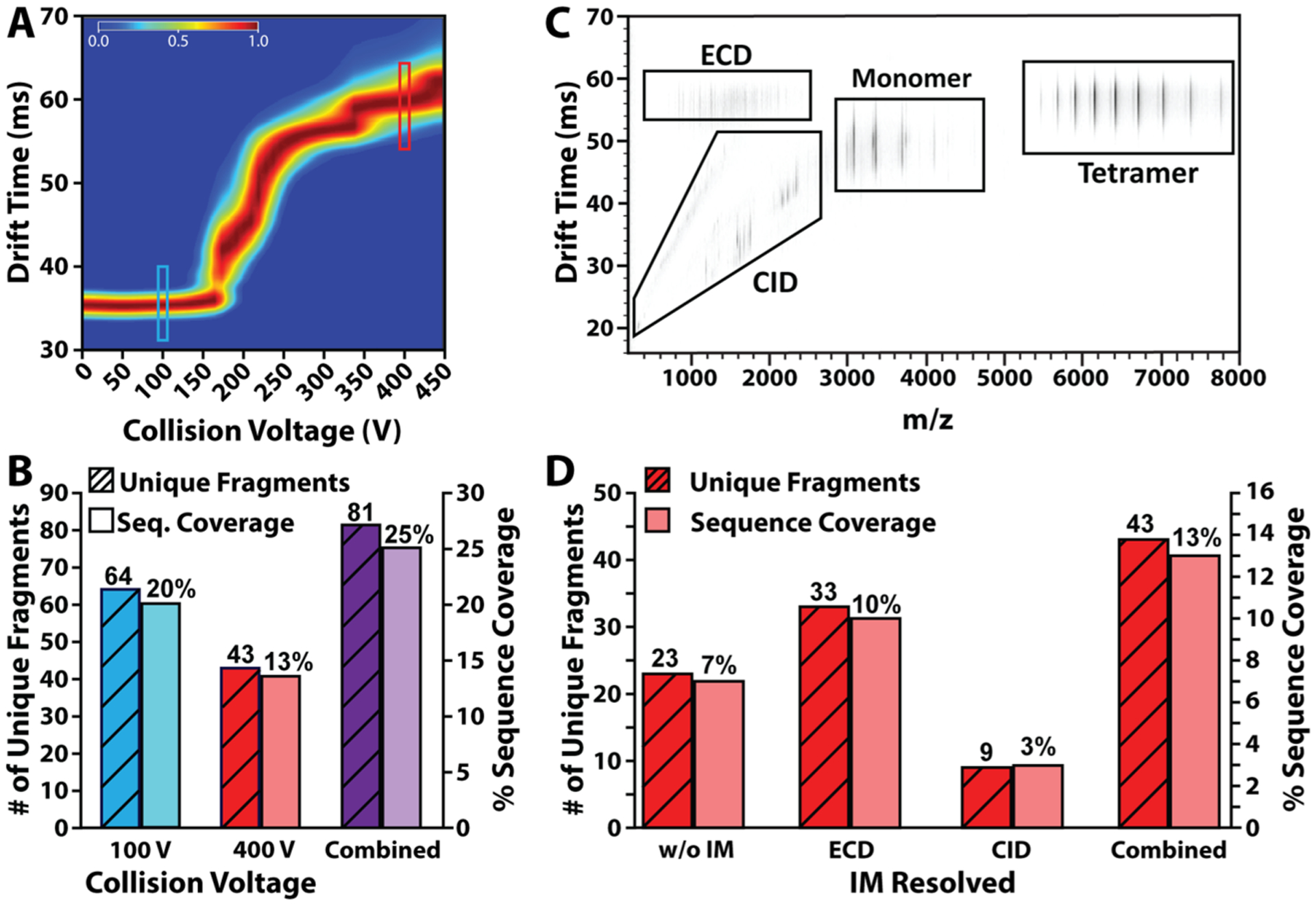
IM-ECD of alcohol dehydrodgenase. (A) All ion CIU fingerprint of ADH 23+ to 26+ charge states. CIU fingerprints of the 23+, 24+, 25+, and 26+ charge states are combined into one fingerprint representative of all ions unfolding in parallel, since all ions undergo ECD simultaneously. Turquoise box marks 100 V; red box marks 400 V. (B) Histogram showing number of unique fragments and sequence coverage for each collision voltage sampled for ADH. The colors correspond to the regions indicated in the CIU fingerprint in (A). (C) IM-MS spectrum of ADH IM-ECD with 400 V in-source activation. (D) Histogram showing a number of unique fragments and sequence coverage for collision voltage of 400 V without and without IM separation. The combination of both fragmentation techniques is also shown.
A key advantage of IM-MS methodologies for native top-down protein sequencing is the ability of IM to segregate peptide fragment ions produced during in-source CID from those generated via post-IM fragmentation methods (e.g., ECD), increasing the total peak capacity of the methodology over those relying on MS alone (Figure 4C).45–48 Integrating the ADH fragmentation data collected across all of the above experimental conditions (ECD and CID, vide infra) results in a total of 81 unique fragments, generating a total sequence coverage of 25% (Figure 4B), a significant improvement over the previous reports.49,50,43 As observed previously,43,44,49–52 identified ECD fragments originate from the N-terminus of ADH monomers (Figure S7).
In order to evaluate and quantify the impact of IM separation on the sequencing data gathered during our CID/ECD experiments, we began by extracting MS data across the entire IM drift time range collapsing all species into a single mass spectrum. This resulted in the identification of only 23 unique fragments (19 c- and 4 y-ions) accounting for 7% coverage of the ADH sequence (Figure 4D). In contrast, when MS data were extracted based on discrete IM drift time ranges (boxes within Figure 4C), the number of unique sequence informative fragments identified more than doubled to 43 (33 c-, 2 b-, and 8 y-ions), improving sequence coverage to 13% (Figure 4D), indicating that the increased peak capacity available in IM-MS mode increased the sequence coverage obtained in our native top-down experiments by approximately a factor of two.
CCSs of Standard Protein Ions.
CCSs of several native IM-MS standards were measured to benchmark the precision and accuracy of our modified DTIM-QTOF for such native protein measurements. The measurements reported in this work represent the most comprehensive survey of native protein standards measured on this commercially available DTIM platform to date. The DT-SFCCSN2 values reported in Figure 5A and Table S3 are the average of at least 3 or more technical replicates and include those resulting from single field (SF) and multifield (MF) approaches for CCS measurement. DT-SFCCSN2 measurements of large native proteins exhibit an average percent difference of 1.61 ± 0.98%, versus respective DT-MFCCSN2 measurements (Table S4).
Figure 5.
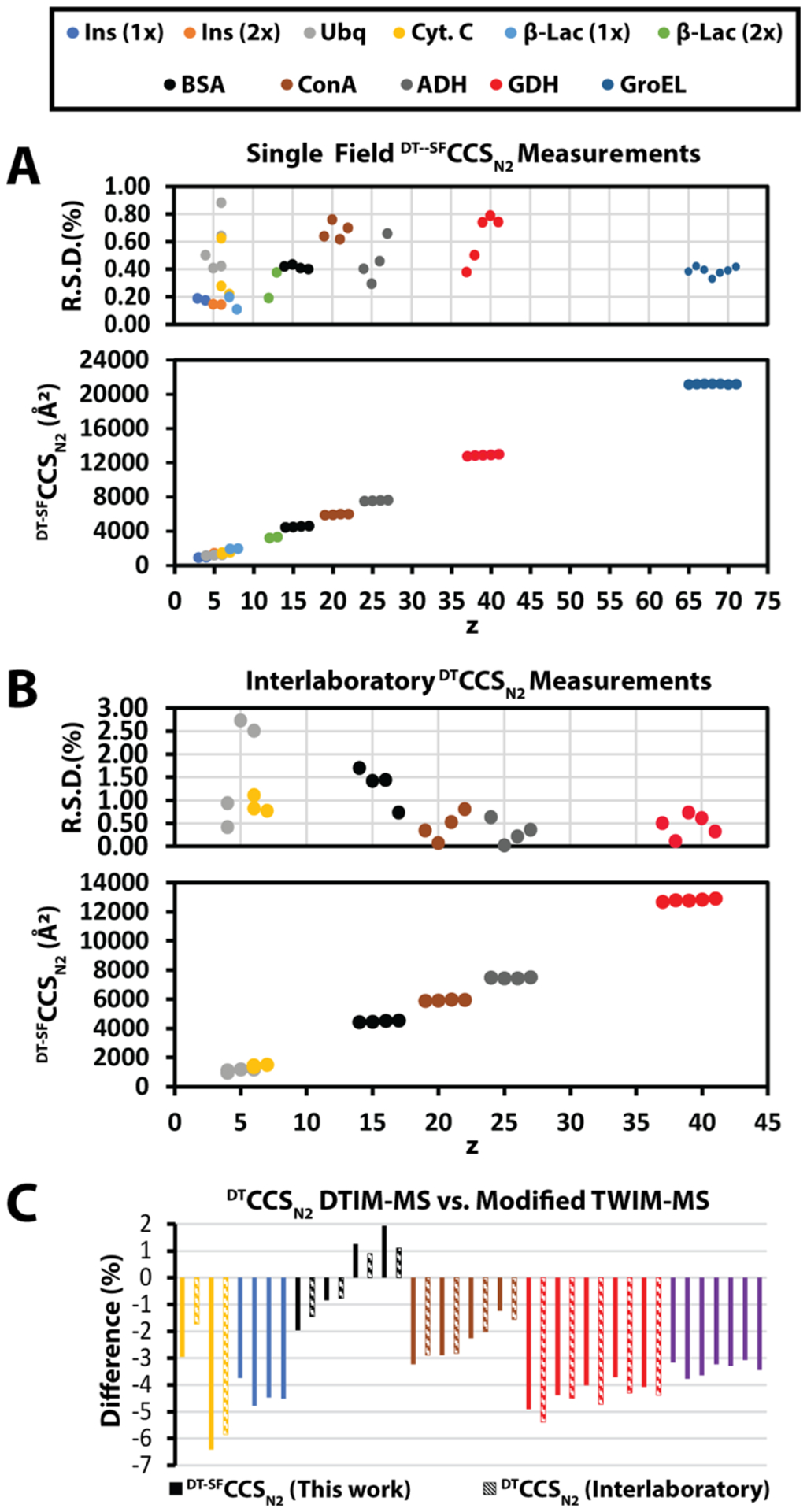
Collision cross sections of native protein ions. (A) Collision cross sections of 11 native proteins were measured on our modified DTIM-QTOF (1x, monomer; 2x, dimer). The CCS’s are plotted versus charge state (z). The average RSD for all measurements is 0.43 ± 0.20%. (B) Collision cross section averages from the interlaboratory comparison are plotted vs charge state (z). The average RSD is 0.82 ± 0.73%. (C) Comparison of our measurements and the interlaboratory measurements vs the measurements reported by Bush et al. These measurements were conducted using a modified TWIM instrument retrofitted with an RF-confining drift cell.
Previous reports have disclosed percent differences ranging between 0.5 and 0.58% for small molecule, metabolite, and peptide DTIM CCS measurements measured using SF and MF methods.19 The larger difference between SF and MF CCS values reported here is expected due to the inherent flexibility of the analytes in question.
The RSD for all our SF CCS measurements is <0.88% with an average RSD of 0.43 ± 0.20%. For comparison, the RSD of our MF CCS measurements is 0.25 ± 0.21%. The higher average RSD for DT-SFCCSN2 measurements compared to DT-MFCCSN2 can be attributed to the inherent error introduced by using an external calibration. Both datasets represent the highest precision assessments of large, native-like proteins and complexes reported to date.
Comparisons to Literature CCS Values.
In order to assess the accuracy and robustness of protein CCS measurements in general, we begin by comparing our DT-SFCCSN2 measurements to an existing database commonly used for TWIM CCS calibration.53 These prior DTIM CCS measurements were conducted using a modified TWIM instrument where the IM separator was replaced with an RF confined drift cell.53 The DT-SFCCSN2 measurements reported here exhibit an average difference of −3.1 ± 1.8% (abs. Avg. diff.: 3.4 ± 1.2%) relative to these prior measurements (Figure 5C; Table S5) across the 26 ions compared, with individual datasets exhibiting percent CCS differences ranging from 6.3 to 1.9%. It is likely that these differences stem directly from the different lifetimes and internal temperatures experienced by ions analyzed on these differently configured IM-MS platforms.
Notably, the proteins that we observe to possess the largest net negative DTCCSN2 difference between our DTIM CCS values and TWIM reference DTIM CCS values are the two smallest proteins compared in our analysis, cytochrome C and β-lactoglobulin, with percent difference values of −4.7 and −4.2%, respectively. In order to confirm that our DTIM CCS measurements were not skewed due to unintentional ion activation in the ion funnel region prior to the IM separator, we monitored the arrival time of cytochrome C 7+ ions as a function of the confinement RF potentials operative on both the front and trapping ion funnels within our DTIM platform, which previous reports have identified as capable of ion activation.20 We found that the front funnel RF potential has no impact on the structure of cytochrome C 7+ under our conditions. Conversely, we discovered that trap funnel RF confinement potentials above 160 V led to ion activation and CIU (Figure S8). As such, we adjusted both potentials so as to avoid such collisional activation during the CCS measurements reported here.
We continued our comparative analysis of our DTIM DTCCSN2 measurements by analyzing their agreement with prior reports containing CCS values acquired on similarly configured Agilent 6560 DTIM platforms.23–25 As this was not a coordinated effort, our measurements are compared to those made independently by the referenced research groups, using independently sourced samples. Across four total DTIM CCS datasets, we were able to extract measurements for six proteins, which were collected by at least two of the groups represented (Table S6). When possible, only the DT-MFCCSN2 measurements were compared; however, DT-SFCCSN2 measurements were also used for comparison if a multifield measurement was not available. Based on 24 ions, from 6 proteins, we computed an excellent interlaboratory reproducibility for DTCCSN2 of 0.82 ± 0.73% (Figure 5B; Table S6). This level of reproducibility is rendered even more impressive by the fact that each study carried out ionization and sample preparation differently, yet the precision of the resulting CCS values remains within those reported previously for a controlled interlaboratory study focusing on smaller biomolecules. Furthermore, the precision of our interlaboratory analysis of DTIM CCS data allows us to more confidently assign the systematic differences observed between protein CCS values recorded using differently configured DTIM platforms as related to ion temperature and lifetime factors, rather than stemming from issues associated with sample preparation and ionization (Figure 5C).
CONCLUSIONS
In this report, we have extensively characterized a uniquely modified Agilent 6560 DTIM-QTOF MS system for the analysis of nativelike proteins. Our results demonstrate the improved in-source activation capabilities of our modified source optics for CIU experiments. Our results extend across a broader range of ion masses and CCS values than previous DTIM studies. Specifically, our results for the GroEL 14-mer offer high IM resolution as well as new insights into the structure and stability of this important standard for native MS. Furthermore, we demonstrate the capabilities of DTIM for native top-down protein sequencing efforts, reporting IM-enabled combined CID and ECD fragmentation data that produce the largest sequence coverage yet reported for the ADH tetramer.
Finally, our comprehensive CCS measurements of several native IM-MS protein and protein complex standards demonstrate both a systematic difference (~−3%) from a database of DTIM CCS values commonly used to calibrate TWIM CCS values, as well as a remarkable level of agreement (~0.8% RSD) with CCS values recorded on other similarly configured DTIM platforms. Our results also confirm the reproducibility of CIU features across differently configured platforms and indicate that such measurements may provide a more confident route for the cross-comparison of protein IM-MS data for future applications ranging from biotherapeutic quality control to structural proteomics.
Supplementary Material
ACKNOWLEDGMENTS
CIU method development in the Ruotolo group is supported through an Agilent Technologies Applications and Core Technology University Research (ACT-UR) Grant. Additional support for this project was provided by the Agilent Technologies Thought Leader Award and University Relations grant programs. C.R.R. was supported by grant T32 CA140044 from the National Institutes of Health.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03372.
Computationally measured CCS of GroEL complexes, CID and CIU of GroEL complexes, additional CIU comparisons between TWIM and DTIM platforms, IM-ECD supplemental information regarding analysis, collision cross section measurement tables, and IM-ECD fragment count tables (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c03372
REFERENCES
- (1).Wilm M; Mann M Anal. Chem 1996, 68, 1–8. [DOI] [PubMed] [Google Scholar]
- (2).Tahallah N; Pinkse M; Maier CS; Heck AJR Rapid Commun. Mass Spectrom 2001, 15, 596–601. [DOI] [PubMed] [Google Scholar]
- (3).Sobott F; Hernández H; McCammon MG; Tito MA; Robinson CV Anal. Chem 2002, 74, 1402–1407. [DOI] [PubMed] [Google Scholar]
- (4).Rostom AA; Robinson CV J. Am. Chem. Soc 1999, 121, 4718–4719. [Google Scholar]
- (5).Van Duijn E; Simmons DA; Van Den Heuvel RHH; Bakkes PJ; Van Heerikhuizen H; Heeren RMA; Robinson CV; Van Der Vies SM; Heck AJR J. Am. Chem. Soc 2006, 128, 4694–4702. [DOI] [PubMed] [Google Scholar]
- (6).Benjamin DR; Robinson CV; Hendrick JP; Hartl FU; Dobson CM Proc. Natl. Acad. Sci. U. S. A 1998, 95, 7391–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rostom AA; Fucini P; Benjamin DR; Juenemann R; Nierhaus KH; Hartl FU; Dobson CM; Robinson CV Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).McKay AR; Ruotolo BT; Ilag LL; Robinson CV J. Am. Chem. Soc 2006, 128, 11433–11442. [DOI] [PubMed] [Google Scholar]
- (9).Tito MA; Tars K; Valegard K; Hajdu J; Robinson CV J. Am. Chem. Soc April 12, 2000, 3550–3551. [Google Scholar]
- (10).Uetrecht C; Barbu IM; Shoemaker GK; Van Duijn E; Heck AJR Nat. Chem 2011, 3, 126–132. [DOI] [PubMed] [Google Scholar]
- (11).van de Waterbeemd M; Snijder J; Tsvetkova IB; Dragnea BG; Cornelissen JJ; Heck AJR J. Am. Soc. Mass Spectrom 2016, 27, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dixit SM; Polasky DA; Ruotolo BT Curr. Opin. Chem. Biol 2018, 42, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Rabuck JN; Hyung SJ; Ko KS; Fox CC; Soellner MB; Ruotolo BT Anal. Chem 2013, 85, 6995–7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Tian Y; Han L; Buckner AC; Ruotolo BT Anal. Chem 2015, 87, 11509–11515. [DOI] [PubMed] [Google Scholar]
- (15).Vallejo DD; Polasky DA; Kurulugama RT; Eschweiler JD; Fjeldsted JC; Ruotolo BT Anal. Chem 2019, 91 (13), 8137–8146. [DOI] [PubMed] [Google Scholar]
- (16).Hyung SJ; Ruotolo BT Proteomics 2012, 12, 1547–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT Proc. Natl. Acad. Sci. U.S.A 2010, 39, 31. [Google Scholar]
- (18).Reid GE; Wu J; Chrisman PA; Mitchell Wells J; McLuckey SA; Biol CS J. Am. Soc. Mass Spectrom 1999, 17, 12. [Google Scholar]
- (19).Stow SM; Causon TJ; Zheng X; Kurulugama RT; Mairinger T; May JC; Rennie EE; Baker ES; Smith RD; McLean JA; Hann S; Fjeldsted JC Anal. Chem 2017, 89, 9048–9055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gabelica V; Livet S; Rosu FJ Am. Soc. Mass Spectrom 2018, 29, 2189–2198. [DOI] [PubMed] [Google Scholar]
- (21).Zheng X; Kurulugama RT; Laganowsky A; Russell DH Anal. Chem 2020, 92, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Shaw JB; Malhan N; Vasil’ev YV; Lopez NI; Makarov A; Beckman JS; Voinov VG Anal. Chem 2018, 90, 10819–10827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).May JC; Jurneczko E; Stow SM; Kratochvil I; Kalkhof S; McLean JA Int. J. Mass Spectrom 2018, 427, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Harrison JA; Kelso C; Pukala TL; Beck JL J. Am. Soc. Mass Spectrom 2019, 30, 256–267. [DOI] [PubMed] [Google Scholar]
- (25).France AP; Migas LG; Sinclair E; Bellina B; Barran PE Anal. Chem 2020, 92 (6), 4340–4348. [DOI] [PubMed] [Google Scholar]
- (26).Hogan CJ; Ruotolo BT; Robinson CV; Fernandez de la Mora J J. Phys. Chem. B 2011, 115, 3614–3621. [DOI] [PubMed] [Google Scholar]
- (27).Mason EA; McDaniel EW Transport Properties of Ions in Gases; 1988.
- (28).Polasky DA; Dixit SM; Fantin SM; Ruotolo BT Anal. Chem 2019, 91, 3147. [DOI] [PubMed] [Google Scholar]
- (29).Strohalm M; Hassman M; Košata B; M K Rapid Commun. Mass Spectrom 2008, 22, 905–908. [DOI] [PubMed] [Google Scholar]
- (30).Richardson K; Langridge D; Dixit SM; Giles K An Improved Calibration Approach for Travelling Wave Ion Mobility Spectrometry: Robust, High-Precision Collision Cross Sections. In Proceedings of the American Society of Mass Spectrometry Annual Conference; 2019; p THP319. [DOI] [PubMed] [Google Scholar]
- (31).Haynes SE; Polasky DA; Dixit SM; Majmudar JD; Neeson K; Ruotolo BT; Martin BR Anal. Chem 2017, 89, 5669–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cai W; Guner H; Gregorich ZR; Chen AJ; Ayaz-Guner S; Peng Y; Valeja SG; Liu X; Ge Y Mol. Cell. Proteomics 2016, 15, 703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Marklund EG; Degiacomi MT; Robinson CV; Baldwin AJ; Benesch JLP Structure 2015, 23 (4), 791–799. [DOI] [PubMed] [Google Scholar]
- (34).Wang H; Eschweiler J; Cui W; Zhang H; Frieden C; Ruotolo BT; Gross ML J. Am. Soc. Mass Spectrom 2019, 30, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hong S; Bush MF J. Am. Soc. Mass Spectrom 2020, 30, 2430–2437. [DOI] [PubMed] [Google Scholar]
- (36).Shirzadeh M; Poltash ML; Laganowsky A; Russell DH Biochemistry 2020, 59, 1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Benesch JLP; Aquilina JA; Ruotolo BT; Sobott F; Robinson CV Chem. Biol 2006, 13, 597–605. [DOI] [PubMed] [Google Scholar]
- (38).Schwartz BL; Bruce JE; Anderson GA; Hofstadler SA; Rockwood AL; Smith RD; Chilkoti A; Stayton PS J. Am. Soc. Mass Spectrom 1995, 6, 459–465. [DOI] [PubMed] [Google Scholar]
- (39).Freeke J; Robinson CV; Ruotolo BT Int. J. Mass Spectrom 2010, 298, 91–98. [Google Scholar]
- (40).Eschweiler JD; Martini RM; Ruotolo BT J. Am. Chem. Soc 2017, 139, 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lermyte F; Valkenborg D; Loo JA; Sobott F Mass Spectrom. Rev 2018, 37, 750–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Horn DM; Breuker K; Frank AJ; McLafferty FW J. Am. Chem. Soc 2001, 123, 9792–9799. [DOI] [PubMed] [Google Scholar]
- (43).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML Anal. Chem 2011, 83, 5598–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lermyte F; Konijnenberg A; Williams JP; Brown JM; Valkenborg D; Sobott FJ Am. Soc. Mass Spectrom 2014, 25, 343–350. [DOI] [PubMed] [Google Scholar]
- (45).Hoaglund CS; Valentine SJ; Sporleder CR; Reilly JP; Clemmer DE Anal. Chem 1998, 70, 2236–2242. [DOI] [PubMed] [Google Scholar]
- (46).Hoaglund-Hyzer CS; Li J; Clemmer DE Anal. Chem 2000, 72, 2737–2740. [DOI] [PubMed] [Google Scholar]
- (47).Merenbloom SI; Koeniger SL; Valentine SJ; Plasencia MD; Clemmer DE Anal. Chem 2006, 78, 2802–2809. [DOI] [PubMed] [Google Scholar]
- (48).Zinnel NF; Pai PJ; Russell DH Anal. Chem 2012, 84, 3390–3397. [DOI] [PubMed] [Google Scholar]
- (49).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML J. Am. Soc. Mass Spectrom 2010, 21, 1966–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Williams JP; Morrison LJ; Brown JM; Beckman JS; Voinov VG; Lermyte F Anal. Chem 2020, 3674. [DOI] [PMC free article] [PubMed]
- (51).Zhou M; Liu W; Shaw JB Anal. Chem 2020, 92, 1788–1795. [DOI] [PubMed] [Google Scholar]
- (52).Lermyte F; Sobott F Proteomics 2015, 15, 2813–2822. [DOI] [PubMed] [Google Scholar]
- (53).Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT Anal. Chem 2010, 82, 9557–9565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.