

Abstract
We have developed a macromolecular prodrug platform based on poly (L-lysine succinylated) (PLS) that targets scavenger receptor A1 (SR-A1), a receptor expressed by myeloid and endothelial cells. We demonstrate the selective uptake of PLS by murine macrophage, RAW 264.7 cells, which was eliminated upon cotreatment with the SR-A inhibitor polyinosinic acid (poly I). Further, we observed no uptake of PLS in a SR-A1-deficient RAW 264.7 cell line, even after 24 h incubation. In mice, PLS distributed to lymphatic organs following i.v. injection, as observed by ex vivo fluorescent imaging, and accumulated in lymph nodes following both i.v. or i.d. administration, based on immunohistochemical analysis with high-resolution microscopy. As a proof-of-concept, the HIV antiviral emtricitabine (FTC) was conjugated to the polymer’s succinyl groups via ester bonds, with a drug loading of 14.2% (wt/wt). The prodrug (PLS-FTC) demonstrated controlled release properties in vitro with a release half-life of 15 h in human plasma and 29 h in esterase-inhibited plasma, indicating that drug release occurs through both enzymatic and non-enzymatic mechanisms. Upon incubation of PLS-FTC with human peripheral blood mononuclear cells (PBMC), released drug was converted to the active metabolite FTC triphosphate. In a pharmacokinetic study in rats, the prodrug achieved ~7 to 19-fold higher concentrations in lymphatic tissues compared to FTC control, supporting lymphatic-targeted drug delivery. We believe the SR-A1-targeted macromolecular PLS prodrug platform has extraordinary potential for treatment of infectious diseases.
Keywords: scavenger receptor A1 (SR-A1), prodrug, lymphatic distribution, HIV, FTC, emtricitabine, macromolecular prodrug, polymeric prodrug
INTRODUCTION
Recently, there has been extraordinary interest in utilizing immune cells, such as macrophages, as either carriers or cell targets for increasing drug exposure to tissues that are difficult to reach with traditional systemic therapies. For example, tumor-associated macrophages (TAMs) may represent a unique cellular target for novel cancer therapies, as well a means of infiltrating the dense stroma of pancreatic ductal adenocarcinoma.1,2 Macrophages have also been used as drug carriers to bypass the blood-brain-barrier for treatment of neurological diseases, such as Parkinson’s and Human Immunodeficiency Virus (HIV) infection of the central nervous system (CNS).3,4 Additionally, targeting macrophages, which were recently identified as a primary reservoir of active HIV in lymphoid tissues capable of sustaining the HIV replication cycle, may prove useful for improvement of HIV therapies.5,6,7 Class A scavenger receptors (SR-A) offer a unique receptor target since they are expressed on the surface of all myeloid cell types, including macrophages, monocytes, mast cells, dendritic cells, as well as endothelial cells8,9,10,11, and have been shown to mediate the uptake of negatively charged nanomaterials.12,13,14 Therefore, targeting SR-A may allow accumulation of drugs in the myeloid immune cells of lymphoid tissues. Few attempts have been made to target SR-A15,16, but the presence of these recycling receptors on the surface of myeloid and endothelial cells offers a potential mechanism for the selective delivery of drugs used to treat cancer, as well as infectious and neurological diseases, that are otherwise difficult to treat with traditional systemic therapies.
An example of an SR-A targeted therapy is succinylated human serum albumin (HSA) which was clinically investigated as an anti-HIV drug. Succinylated albumin is unique in that the succinyl modification of albumin is responsible for both its efficacy and lymphatic targeting. Early studies in rats showed rapid distribution to liver that appeared to be SR-A mediated, as uptake was reduced by competition with the SR-A inhibitor polyinosinic acid (poly I).17 SR-A is abundant on myeloid cells, such as macrophage, and Kupffer cells are the likely hepatic target of succinylated HSA. Interestingly, it has been shown that endothelial SR-A mediates lymphatic transcytosis of modified LDL, offering the potential for SR-A mediated targeting of the lymphatic system.18 In rats, succinylated HSA distributed to lymph much more rapidly and to a greater extent than unmodified albumin, and pre-administration of poly I, or the transcytosis inhibitor chloroquine, significantly reduced distribution of succinylated HSA, suggesting an endothelial transcytosis mechanism mediated by scavenger receptor.16 Despite its demonstrated safety and efficacy in rodent and non-human primate models, succinylated HSA ultimately failed in clinical trials due to hepatic toxicity, as evidenced by increased liver transaminase, at sub-efficacious doses.19,20 Nevertheless, succinylated HSA demonstrated how targeting endothelial SR-A can allow rapid lymphatic distribution.
There are many infectious agents that reside in lymphoid tissues such as tuberculosis21 and Epstein-Barr virus22, with HIV being the most prominent example23. Despite therapeutic advances, there is still not a viable cure for HIV infection, and acquired immunodeficiency syndrome (AIDS) remains a major cause of death worldwide. The current standard of care for HIV treatment consists of a combination of antiretroviral drugs referred to as highly active antiretroviral therapy (HAART), which is capable of reducing virus in the blood to undetectable levels.24 However, the inability of current HAART therapies to achieve efficacious drug concentrations within lymphatic tissues is hypothesized to result in the persistence of viral reservoirs that result in the rebound of blood virus levels when therapy is withdrawn.25 The HIV virus infects CD4+ lymphocytes, monocytes, macrophages, dendritic cells, and microglial cells that are present in various organs including liver, kidney, lung, and testes, but it is the lymphoid tissues and brain that remain the major sites of latent viral reservoirs that are difficult to access by traditional therapeutic strategies.5 Therefore, the eradication of these persistent viral reservoirs remains an unmet medical need, and targeted drug delivery strategies to increase lymphatic drug exposure may prove beneficial.
Nanoparticles are widely employed for drug delivery, and these delivery platforms often allow enhanced lymphatic drug distribution since many nanoparticle types are phagocytosed by immune cells and passively accumulate within lymphatic tissues. Indeed, several HIV antiviral formulations have achieved increased lymph node drug exposure.26,27,28,29 Additionally, a recently developed lipid nanosuspension capable of loading multiple anti-HIV drugs not only significantly increased drug concentrations in lymph node mononuclear cells (LNMCs) and peripheral blood mononuclear cells (PBMCs) compared to plasma, but also achieved persistent drug concentrations for five weeks from a single subcutaneous dose in a macaque model.30 However, the authors in these studies reported only the total drug concentration in these tissues and did not differentiate between encapsulated and unencapsulated (pharmacologically active) drug fractions. Simply increasing total drug concentrations in lymph nodes may not necessarily be effective against viral reservoirs if drug release is too slow. Nanomedicines must be stable enough to allow for drug transport to the target tissue, but also release sufficient drug at the target tissue to be efficacious.31
We have developed a novel macromolecular platform that achieves SR-A1 targeting and controlled drug release. The platform is based on poly(L-lysine succinylated) (PLS) which is a homopolymer of poly(L-lysine) of which all amine groups have been succinylated. The pendant carboxyl groups allow conjugation of dyes through stable amide bonds or conjugation of drugs through hydrolysable ester bonds, as well as selective interaction with scavenger receptor (Figure 1). Although this platform may have many applications, we selected an HIV drug, FTC, as a proof-of-concept to evaluate prodrug synthesis and lymphatic distribution. For the first time to our knowledge, we report the ability of PLS to target SR-A1. We further report its lymphatic distribution upon i.v. administration, as well as its accumulation in various lymph nodes following i.v. or i.d. administration. Additionally, in a rat model the FTC PLS prodrug achieved ~7 to 19-fold higher free FTC concentrations in lymphatic tissues compared to unformulated FTC, supporting rapid lymphatic-targeted drug delivery. Overall, this technology offers a unique drug delivery strategy for enhancing drug distribution to lymphatic tissues, with potential applications for infectious disease and cancer immunotherapy.
Figure 1.
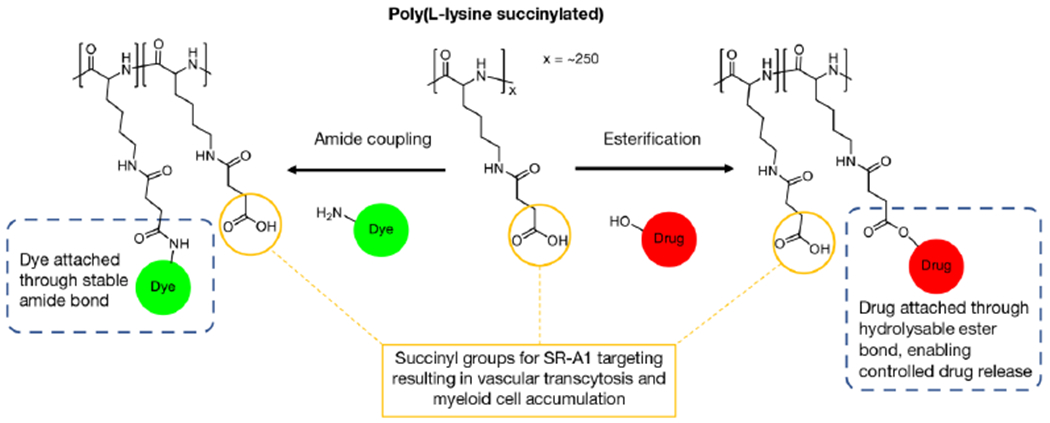
Schematic illustration of dye and drug conjugation to PLS through amide coupling and esterification, respectively.
EXPERIMENTAL SECTION
Materials.
Emtricitabine (FTC) and 2-(2-methoxyethoxy)ethanamine were purchased from Toronto Research Chemicals (Toronto, Canada). 13C15N2-FTC was purchased from Moravek, Inc (Brea, CA). Poly(L-lysine succinylated) (PLS, molecular weight 62,500 g/mol) was purchased from Alamanda Polymers (Huntsville, AL). Cy7.5 amine was purchased from Lumiprobe (Cockeysville, MD). N,N’-Diisopropylcarbodiimide (DIC), 4-(dimethylaminopyridine) (DMAP), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC-HCl), poly(inosinic acid) potassium salt (Poly I), poly(cytidylic acid) potassium salt (Poly C), anhydrous dimethylformamide (DMF), and anhydrous dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). N-hydroxysulfosuccinimide (sulfo-NHS) and 2-(n-morpholino) ethanesulfonic acid (MES) were purchased from ProteoChem (Hurricane, UT). Acetonitrile was purchased from VWR (Radnor, PA). Methanol (HPLC grade), acetonitrile (HPLC grade), water (HPLC grade), acetic acid (80% w/w), formic acid, and Alexa Fluor 488 cadaverine was purchased from Thermo Fisher Scientific (Waltham, MA). HyClone phosphate buffered saline (PBS) was purchased from GE Healthcare Life Sciences (Marlborough, MA). Spectra/Por 6 dialysis tubing (10 kDa molecular weight cut-off) was purchased from Spectrum Labs (Rancho Dominguez, CA). Amicon Ultra regenerated cellulose centrifugal filters (10 kDa molecular weight cut-off) were purchased from EMD Millipore (Burlington, MA). RAW 264.7 cells was purchased from ATCC (Old Town Manassas, VA). The RAW 264.7 derivative cell line (Clone 1/2) was developed as reported previously.
Synthesis of AlexaFluor488-labelled poly(L-lysine succinylated) (PLS-488).
Dye-labelled polymers were prepared using an amide coupling reaction based on a previously reported method 32. Briefly, PLS (11.0 mg) was dissolved in 1.0 mL MES buffer (0.1 M, pH 5.3). EDC-HCl (4.2 mg, 0.022 mmol) and sulfo-NHS (9.6 mg, 0.044 mmol) were added to the solution, sequentially. The reaction mixture was allowed to stir for 15 minutes. The pH was then adjusted to 7.2 by adding saturated sodium bicarbonate solution. AlexaFluor 488 cadaverine (90.2 μL, 10 mg/mL in 10X PBS) was added to the reaction mixture and allowed to stir at room temperature, protected from light. The reaction was quenched by raising the pH to 8.2 with saturated sodium bicarbonate solution. The product was isolated by spin filtering five times at 5,000 x g for 8 min each using Amicon Ultra centrifugal filters (10k molecular weight cut-off). The retentate was sterile filtered and lyophilized (11.0 mg). SEC analysis yielded a single product peak with an elution time similar to the unreacted polymer, and a strong absorption band at 493 nm in its UV spectrum (Figure S1). The SEC/photodiode array (PDA) system consisted of an LC-20AT pump, SIL-20AC auto injector, CTO-20AC column oven, SPD-M20A diode array detector, and C-R3A integrator (Shimadzu Scientific Instruments, Inc.). The SEC conditions were: 10 μL injection volume with a 50% acetonitrile in ammonium acetate (9 mM, pH 7.7) isocratic elution. Flow rate was 0.390 mL/min, and column temperature was 40 °C. PDA monitored at 488 nm. The column used was KW403-4F 4.6 mm id x 30 cm (Shodex).
Synthesis of Cy7.5-labelled poly(L-lysine succinylated) (PLS-Cy7.5).
PLS (11.0 mg) was dissolved in 1.0 mL PBS (pH 7.0). EDC-HCl (4.2 mg, 0.022 mmol) and sulfo-NHS (1.1 mg, 0.0049 mmol) were added to the solution, sequentially. The reaction mixture was allowed to stir for 15 minutes. Cy7.5 amine dye (115 μL, 10 mg/mL in DMSO) was added to the reaction mixture and allowed to stir for 45 minutes at room temperature, protected from light. The reaction was quenched by raising the pH to 8.2 with saturated sodium bicarbonate solution. The product was isolated by spin filtering five times at 5,000 x g for 8 min each using Amicon Ultra centrifugal filters (10k molecular weight cut-off). The retentate was sterile filtered and lyophilized (8.5 mg). The product was analyzed by an SEC/PDA system described above (Figure S9). PDA monitored at 786 nm. SEC analysis yielded a single product peak with an elution time similar to the unreacted polymer, and a strong absorption band at 785 nm in its UV spectrum.
Synthesis of partially capped poly(L-lysine succinylated) with 2-(2-methoxyethoxy)ethanamine.
PLS (40.0 mg) was dissolved in 4.0 mL MES buffer (0.1 M, pH 5.3). EDC-HCl (15.3 mg, 0.0800 mmol) and sulfo-NHS (9.7 mg, 0.045 mmol) were added to the solution, sequentially. The reaction mixture was allowed to stir for 15 minutes. The pH of the solution was adjusted to 7.2 using saturated sodium bicarbonate solution, if necessary. 2-(2-methoxyethoxy)ethanamine (1.6 μL, 0.013 mmol) was added to the reaction mixture, and the pH was adjusted to 7.2 using saturated sodium bicarbonate solution, if necessary. The reaction was allowed to stir for 2 hours at room temperature. The reaction was quenched by raising the pH to 8.2 with saturated sodium bicarbonate solution. The product was isolated by spin filtering seven times at 5,000 x g for 9 min each using Amicon Ultra centrifugal filters (10k molecular weight cut-off). The retentate was sterile filtered and lyophilized (35 mg). 1H NMR spectrum was obtained from a Bruker Nanobay 400 MHz instrument, using D2O as solvent. 1H NMR (400 MHz, D2O, ppm): Peaks attributed to polymer, δ: (4.30 (br, -C(O)CHNH-), 3.16 (br, -CH2CH2NHC(O)-), 2.45 (br, -C(O)CH2CH2C(O)-), 1.76 (br, -CHCH2CH2-), 1.52 (br, -CH2CH2NHC(O)-), 1.37 (br, -CHCH2CH2CH2-). Peaks attributed to 2-(2-methoxyethoxy)ethanamine, δ: 3.69 (br, -OCH2CH2O-), 3.61 (br, -NHCH2CH2O-), 3.40 (br, -OCH3), 3.16 (br, -NHCH2CH2O-) (Figure S4).
Synthesis of fully capped poly(L-lysine succinylated) with 2-(2-methoxyethoxy)ethanamine.
PLS (40.0 mg) was dissolved in 4.0 mL MES buffer (0.1 M, pH 5.3). EDC-HCl (92.0 mg, 0.480 mmol) and sulfo-NHS (104 mg, 0.480 mmol) were added to the solution, sequentially. The reaction mixture was allowed to stir for 15 minutes. 2-(2-methoxyethoxy)ethanamine (59.6 μL, 0.480 mmol) was added to the reaction mixture, and the pH was adjusted to 7.2 using saturated sodium bicarbonate solution, if necessary. The reaction was allowed to stir for 2 hours at room temperature. The reaction was quenched by raising the pH to 8.2 with saturated sodium bicarbonate solution. The product was isolated by spin filtering seven times at 5,000 x g for 9 min each using Amicon Ultra centrifugal filters (10k molecular weight cut-off). The retentate was sterile filtered and lyophilized (42 mg). 1H NMR spectrum was obtained from a Bruker Nanobay 400 MHz instrument, using D2O as solvent. 1H NMR (400 MHz, D2O, ppm): Peaks attributed to polymer, δ: (4.25 (br, -C(O)CHNH-), 3.13 (br, -CH2CH2NHC(O)-), 2.43 (br, -C(O)CH2CH2C(O)-), 1.76 (br, -CHCH2CH2-), 1.49 (br, -CH2CH2NHC(O)-), 1.35 (br, -CHCH2CH2CH2-). Peaks attributed to 2-(2-methoxyethoxy)ethanamine, δ: 3.67 (br, -OCH2CH2O-), 3.61 (br, -NHCH2CH2O-), 3.38 (br, -OCH3), 3.16 (br, -NHCH2CH2O-) (Figure S5).
Synthesis of poly(L-lysine succinylated) FTC prodrug (PLS-FTC).
PLS was converted to the free acid form by dissolving 1000 mg PLS into ~80 mL cold water and adding 4.4 mL 1N HCl. The resulting precipitant (PLS-COOH) was pelleted by centrifugation, washed several times with water, and lyophilized (890 mg yield). PLS-COOH (800 mg, 3.51 mmol acid) and FTC (174 mg, 0.702 mmol) were weighed and added to an oven-dried 100 mL round-bottom flask equipped with stir bar. The flask was capped with a rubber septum and purged with nitrogen for 5 minutes. Anhydrous DMF (20.0 mL) was added to the flask followed by sonication until dissolution was completed. In an oven-dried vial, DMAP (429 mg, 3.51 mmol) was added, and the vial was capped and purged with nitrogen for 5 minutes. The DMAP was then dissolved with 8.00 mL anhydrous DMSO under nitrogen. The DMAP solution was transferred to the PLS-COOH/FTC reaction flask under nitrogen via syringe. DIC (272 μL, 1.75 mmol) was added to the reaction flask dropwise via microsyringe, and the reaction was allowed to stir at room temperature. The reaction was monitored using HPLC for approximately 7 h until unreacted FTC was undetectable. The reaction was then diluted with 100 mM sodium acetate buffer (pH 5.8) and dialyzed in Spectra/Por 6 regenerated cellulose dialysis tubing (10k molecular weight cut-off) against acetone overnight. In order to completely remove the DMAP without cleaving the polymer prodrug product, dialysis proceeded in different solvents in the following order: 50% acetone in water → sodium acetate buffer pH 5.8 → 100% water. Next, the product was converted to the sodium salt by raising the pH inside the dialysis bags above pH 6 using saturated sodium bicarbonate solution. Raising the pH above 7 risked cleaving the ester linkage of the prodrug. Several rounds of dialysis against 100% water were performed at 4 °C to remove bicarbonate salts. Finally, the product was sterile filtered and lyophilized to yield a fluffy, white material (961 mg). 1H NMR spectrum of PLS-FTC was obtained from a Bruker Nanobay 400 MHz instrument, using D2O as solvent. PLS-FTC 1H NMR (500 MHz, D2O, ppm): Peaks attributed to polymer, δ: (4.25 (br, -C(O)CHNH-), 3.13 (br, -CH2CH2NHC(O)-), 2.43 (br, -C(O)CH2CH2C(O)-), 1.76 (br, -CHCH2CH2-), 1.49 (br, -CH2CH2NHC(O)-), 1.35 (br, -CHCH2CH2CH2-). Peaks attributed to FTC, δ: 8.04 (br, -NCHCF-), 6.22 (br, -OCHCH2S-), 5.45 (br, -OCHS-), 4.56 (br, -HOCH2CH-), 3.57 (br, -SCH2CH-), 3.12 (br, -SCH2CH-) (Figure 8). SEC/PDA analysis was performed using a system consisting of an LC-20AT pump, SIL-20AC auto injector, CTO-20AC column oven, SPD-M20A diode array detector, and C-R3A integrator (Shimadzu Scientific Instruments, Inc.). The SEC conditions were: 10 μL injection volume with a 50% acetonitrile in ammonium acetate (9 mM, pH 7.4) isocratic elution. Flow rate was 0.390 mL/min, and column temperature was 40 °C. PDA monitored at 280 nm. The column used was KW403-4F 4.6 mm id x 30 cm (Shodex) (Figure 6B). Molecular weight and PDI were analyzed by an SEC/MALS system consisting of Agilent 1260 Infinity II pump, DAWN HELEOS II multiangle light scattering detector (Wyatt), and Optilab T-rEX infrared detector (Wyatt) (Figure 6C,D). The SEC conditions were: 100 μL injection volume with a 90% methanol in water (containing 50 mM LiCl) isocratic elution. Flow rate was 1.00 mL/min. The column used was TOSOH Bioscience Alpha-6000, 7.8 mm id x 30 cm, 7 micron particle size with guard column TOSOH Bioscience Guard Alpha, 6.0 mm id x 4.0 cm, 13 μm particle size. Normalization constants were determined using PLS. Measured dn/dc values for PLS and PLS-FTC were 0.193 and 0.184 mL/g, respectively.
Figure 8.
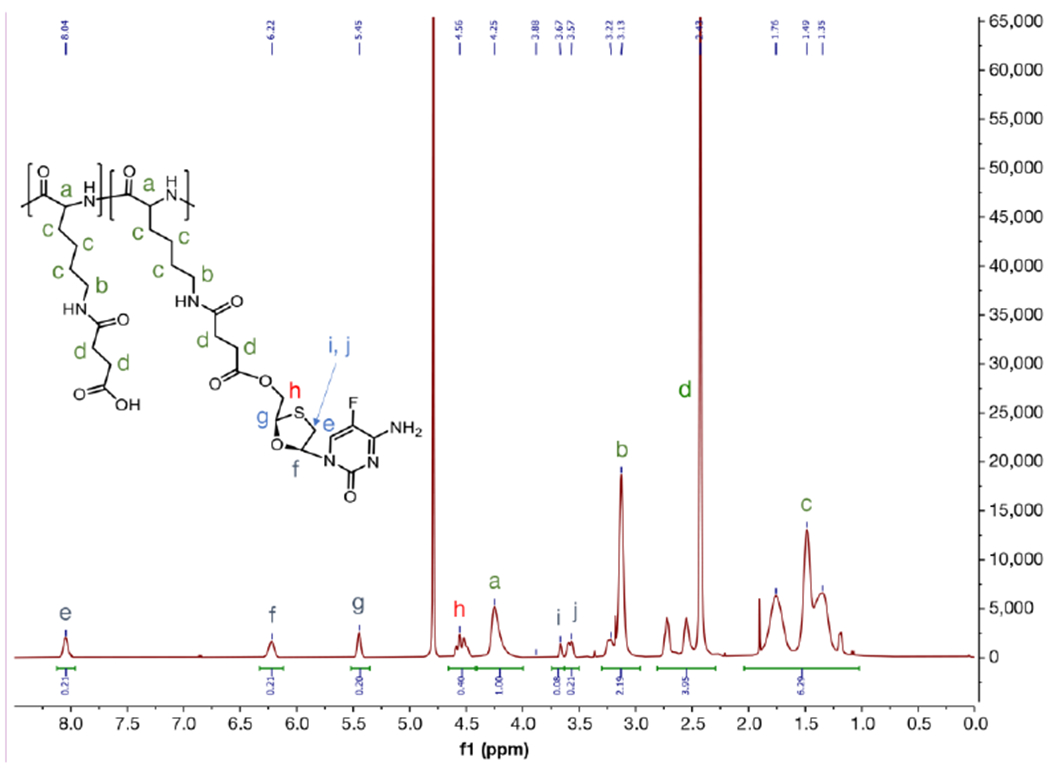
1H NMR of PLS-FTC in D2O (400 MHz)
Figure 6.
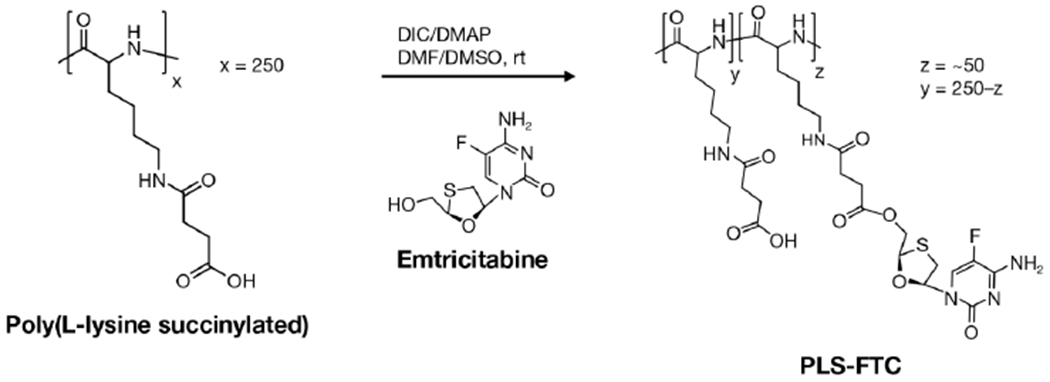
Synthesis of poly(L-lysine succinylated) FTC prodrug via carbodiimide esterification.
PLS-FTC drug loading analysis.
FTC content in PLS-FTC was determined using a sodium hydroxide degradation sample preparation and HPLC/PDA analytical method (Figure 9; Figure S14,15; Table S1). PLS-FTC and FTC standards were prepared in 50% acetonitrile in water. 150 μL of each sample was treated with 10 μL 1N NaOH for 1 h to completely hydrolyze all esters, followed by neutralization with 10 μL 1N HCl. A set of non-degraded FTC standards were prepared by taking 150 μL of each standard and adding 20 μL water. The samples were analyzed by an HPLC/PDA system consisting of an LC-20AT pump, SIL-20AC auto injector, CTO-20AC column oven, SPD-M20A diode array detector, and C-R3A integrator (Shimadzu Scientific Instruments, Inc.). The HPLC conditions for the NaOH-treated samples were: 10 μL injection volume with a 50% acetonitrile in ammonium acetate (20 mM, pH 5) isocratic elution. Flow rate was 0.600 mL/min, and column temperature was 35 °C. PDA monitored at 280 nm. The column used was Zorbax SB C18 4.6 mm id x 15 cm.
Figure 9.
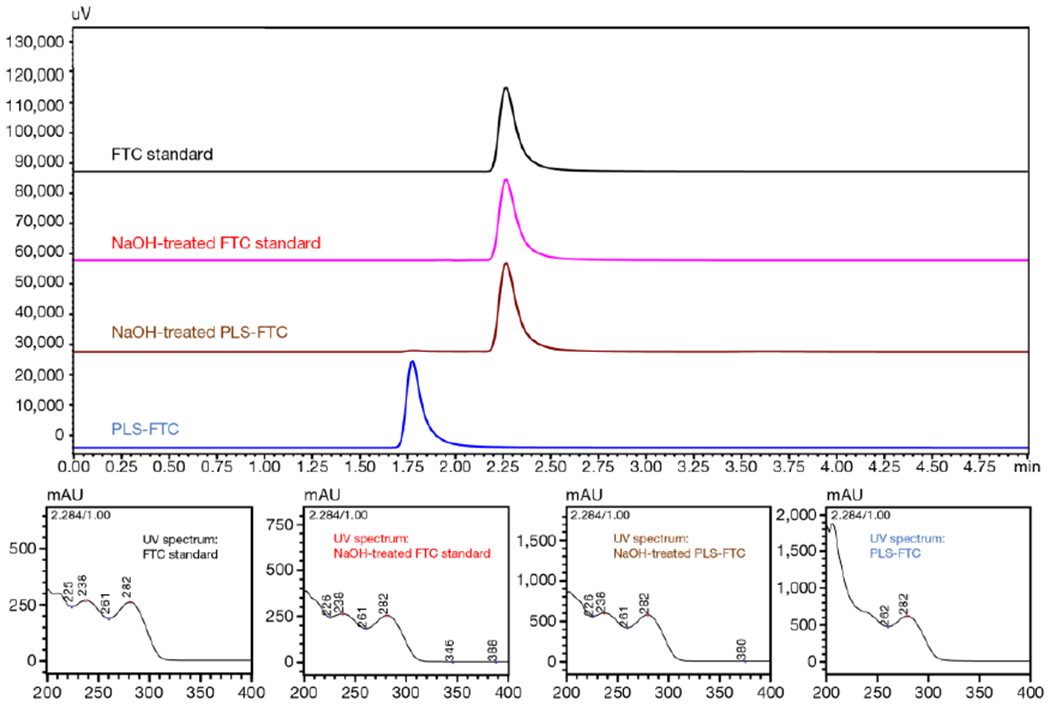
HPLC chromatograms of FTC (top), FTC degraded with sodium hydroxide for 1 h (2nd from top), PLS-FTC treated with sodium hydroxide for 1 h (3rd from top), and PLS-FTC (bottom). HPLC/UV conditions were: 10 μL injection volume, isocratic elution solvent consisting of 50% acetonitrile and 50% ammonium acetate (20 mM, pH 5), flow rate of 0.600 mL/min, column temperature of 35 °C, and UV detection set at 280 nm. The UV spectra of each peak are shown below the chromatograms.
Scavenger receptor A competitive inhibition studies.
Raw 264.7 cells were cultured in phenol-free RPMI 1640 media with 10% heat inactivated fetal bovine serum and 50 μM beta-mercaptoethanol at 37 °C in a humidified atmosphere at 5% CO2. In the first experiment, various concentrations of PLS-488 were co-incubated with poly I or poly C during cell treatment based on a previously reported method 14. Raw 264.7 cells were seeded into 96-well plates (100 μL of 400,000 cells/mL) and incubated overnight at 37 °C in a 5% CO2 humidified atmosphere. Stocks of PLS-488, poly I, and poly C were prepared using cell media. Cells were treated with PLS-488 at concentrations ranging from 4.22 ng/mL to 1.11 mg/mL along with 200 μg/mL poly I, 200 μg/mL poly C, or no inhibitor. After 3 h incubation at 37 °C, the cells were rinsed 3x with phenol-free media and fluorescence measured at excitation/emission, 493/516 nm, on a SpectraMax M5 (Molecular Devices). In the second experiment, various concentrations of either poly I or poly C were co-incubated with PLS-488 during cell treatment. Raw 264.7 cells were seeded into 96-well plates as described above. Stocks of PLS-488 and poly C were prepared in cell media. Poly I stock was prepared in DMSO and diluted into cell media to achieve the high concentrations needed for this experiment. Cells were treated with 69.3 μg/mL PLS-488 and co-treated with either poly I or poly C at concentrations ranging from 4.51 μg/mL to 2.31 mg/mL. After 3 h incubation at 37 °C, the cells were rinsed 6x with phenol-free media and fluorescence (excitation/emission, 493/516 nm) measured on a SpectraMax M5 (Molecular Devices). Fluorescence values were normalized to cells receiving PLS-488 treatment alone. In the third experiment, PLS-488 was co-incubated with fully capped PLS, partially capped PLS, poly C, or poly I during cell treatment. Raw 264.7 cells were seeded into 24-well plates (500 μL of 500,000 cells/mL) and incubated overnight at 37 °C in a 5% CO2 humidified atmosphere. Stocks of fully conjugated and partially conjugated PLS were prepared in cell media, and stocks of PLS-488, poly C, and poly I were prepared as described above. Cells were treated with 5.00 μg/mL PLS-488 and co-treated with either fully conjugated PLS, partially conjugated PLS, or poly C at 2.00 mg/mL or poly I at 100 μg/mL. After 3 h incubation at 37 °C, the cells were rinsed 3x with phenol-free media and fluorescence (excitation/emission, 493/516 nm) measured on a SpectraMax M5 (Molecular Devices). Fluorescence values were normalized to cells receiving PLS-488 treatment alone.
Flow cytometry analysis of SR-A1-deficient cells treated with PLS-488.
Cells of both wild-type Raw 264.7 cells and SR-A1 knockout clone 33 were seeded into 24-well plates at a concentration of 1x105 cells per well, and incubated overnight at 37 °C in a 5% CO2 humidified atmosphere. Stock of PLS-488 was prepared in complete cell culture media. Cells were treated with 1.00 g/mL PLS-488 and incubated for 24 h at 37 °C in a 5% CO2 humidified atmosphere. Untreated cells were used as controls. After 24 h incubation, cells were harvested from each well using trypsin, washed 3x with PBS, resuspended in FACS buffer (PBS supplemented with 2% FBS), and analyzed using a Becton Dickson FACSCalibur flow cytometer (excitation/emission, 495/519 nm). Data was analyzed using FCS Express5 software (De Novo Software).
Confocal microscopy of SR-A1-deficient and wild type RAW 264.7 cells treated with PLS-488.
Wild type and SR-A1 knockout Raw 264.7 cells (clone 1/2) were seeded into an 8-well chambered coverslip slides (ibidi) at a concentration of 4x104 cells per well and incubated overnight at 37 °C in a humidified atmosphere with 5% CO2. Both the cell lines were treated in triplicate with PLS-488 at a concentration of 1 μg/mL or control media, and incubated for 0.5 and 24 h at 37 °C in a 5% CO2 humidified atmosphere. After the incubation period, cells were washed twice with media and stained with 200 μL of 50 nM Lysotracker Red (ThermoFisher Scientific). After 30 min, cells were washed 4 times with phenol-free media and nuclei were stained with 250 μL of fresh phenol-free media containing Hoechst 33342 (ThermoFisher Scientific). Fluorescence images of both cell lines were taken using a Nikon A1R confocal microscope equipped with 4 lasers (405 nm, 488 nm, 561 nm, and 640nm), with capture conditions: Lysotracker Red ex./em. 561/590 nm; Hoechst 33342 ex./em. 405/461 nm; PLS-488 ex./em. 488/519 nm. Image processing was carried out using NIS-elements (Nikon) to separate images taken with each individual laser. In vitro drug release studies. Healthy human volunteer blood specimens were drawn under NCI at Frederick Protocol OH99-C-N046 and collected in lithium heparin tubes. Plasma was obtained by centrifugation of the blood at 2,500 x g for 10 minutes. Human plasma was collected and pooled from six donors, followed by supplementation with HEPES buffer to a concentration of 25 mM, and either 5 mM PMSF or DMSO control (equivalent to 5 mM PMSF). PLS-FTC and FTC stocks were prepared at 50.0 μg/mL in water. 50 μL of either PLS-FTC or FTC control were added to 450 μL plasma in triplicate, vortexed, and incubated at 37 °C with agitation. At each time point, 50 μL sample was removed and added to 200 μL ice cold methanol containing 625 ng/mL 13C15N2-FTC internal standard. The sample was placed in −80 °C for 10 minutes, and then thawed at room temperature. The thawed sample was then centrifuged at 14,000 x g for 20 minutes at 4 °C. The supernatant was transferred to a glass tube and dried under nitrogen gas in a turbo vap at 45 °C. The dried residue was resuspended in 150 μL 10% ACN in water. The extracted sample was transferred to a 1.5 mL Eppendorf tube and spun at 14,000 x g for 5 minutes at 4 °C. The supernatant was transferred to 1.5 mL amber glass screw HPLC vials with fixed insert and cap and placed in HPLC vial rack. FTC calibration standards were prepared in plasma with concentrations ranging from 10 to 6000 ng/mL. FTC quality controls (QC) were also prepared in plasma at concentrations 25, 500, and 5000 ng/mL. A plasma blank was also run with the calibration curve.
The liquid chromatography/mass spectrometry (LC/MS) system consisted of an LC-20AT pump, SIL-20AC auto injector, CTO-20AC column oven, LC/MS/MS 8040 triple quad, C-R3A integrator (Shimadzu Scientific Instruments, Inc.). The HPLC/MS/MS conditions were: 5 μL injection volume, water-acetonitrile gradient: (Hold at 3% ACN/0.1% formic acid for 1 min, 3% ACN/0.1% formic acid linear increase to 10% ACN/0.1% formic acid from 1-2 min, 10% ACN/0.1% formic acid linear increase to 95% ACN/0.1% formic acid from 2-4 min, hold at 95% ACN/0.1% formic acid from 4-4.5 min, and linear decrease to 3% ACN/0.1% formic acid from 4.5-4.6 min, column regeneration time between injections of 7.6 min), flow rate of 0.35 mL/min, and column temperature of 35 °C. The column used was a Zorbax SB C18 2.1 mm id x 10 cm (Agilent Technologies, Inc.) with a Sunfire C18 2.1 mm id x 10 mm guard column (Waters, Inc.). FTC and 13C15N2-FTC elution times were both 3.7 min, and m/z ions monitored by MRM transitions were 248.10 → 130.05 and 251.10 → 133.00, respectively. The MS instrument used an electrospray ionization source in positive ion mode. Detector voltage was 4.5 kv and a DL and heat block temperature were 300 °C and 200 °C, respectively. High pressure liquid nitrogen was used as the drying gas at a rate of 15 L/min. Argon was used as the collision gas at a pressure of 230 kPa.
Conversion of FTC to FTC triphosphate (FTC-TP) in human PBMCs.
Frozen human PBMCs were purchased from PPA Research Group. Upon thawing, PBMCs were grown in RPMI 1640 20% heat-inactivated FBS for 24 hours. The cells were resuspended at 3 x 106 cells/mL and plated on 6-well plates (2 mLs/well). FTC and PLS-FTC were added to wells in triplicate at 10 ug/mL FTC and incubated at 37 °C with 5% CO2 for 24 h. Cells were washed twice with ice-cold serum free media and centrifuged at 800 x g for 5 min. The cell pellet was lysed with 400 μL ice-cold 60% methanol in water containing 200 ng/mL 13C15N2-FTC as an internal standard and frozen at −80 °C. The samples were then thawed at room temperature and centrifuged at 18,000 x g for 20 min at 4 °C. An aliquot of the supernatant was analyzed for protein concentration using micro BCA protein assay kit (Thermo Fisher Scientific) by incubating the supernatant with micro BCA reagent at a 1:1 ratio at 37 °C for 2 h followed by absorbance measurement at 562 nm on a SpectraMax i3x (Molecular Devices). The remainder of the supernatant was transferred to 1.5 mL amber glass screw HPLC vials with fixed insert and cap and placed in a HPLC vial rack. FTC and FTC-TP calibration standards were prepared in 60% methanol with concentrations ranging from 100 to 50,000 ng/mL, and 10 to 500 ng/mL, respectively. FTC and FTC-TP quality controls (QC) were also prepared in 60% methanol at concentrations 500, 5000, and 20,000 ng/mL, and 25 and 500 ng/mL, respectively.
The LC analytical system consisted of a VH-P10-A binary pump, VH-A10-A Vanquish autosampler HT, VH-C10-A column compartment, Q Exactive basic mass spectrometer, and VH-S01-A Vanquish system base (Thermo Fisher Scientific). The following HPLC conditions were used: Gradient elution consisting of (A) pH 10.1, 750 mM ammonium acetate and (B) 75% 5 mM ammonium in 25% acetonitrile: 0-0.25 min 20% B, 0.25-1 min linear increase to 100% B, hold at 100% B 1-3.25 min, 3.25-3.5 min linear decrease to 20% B, column regeneration time of 4.5 min; flow rate of 0.30 mL/min; column temperature of 35 °C. The column used was an Acclaim Trinity P2 HILIC/WCX/SAX mixed mode 3 um 3.0 mm id x 10 cm (Thermo Fisher Scientific). The elution times for FTC, 13C15N2-FTC, and FTC-TP were 1.9, 1.9, and 1.8 min, respectively. PRM transitions were 487.949 → 130.041, 230.039, 258.917 for FTC-TP, 248.10 → 130.05 for FTC, and 251.21 → 133.04 for 13C15N2-FTC. The MS instrument used an electrospray ionization source in positive ion mode.
Husbandry.
Animal rooms were kept at 50% relative humidity, 68-72 °F with 12 h light/dark cycles. Rats were housed with two animals/cage (Rat polycarbonate cage type), with ¼″ corncob bedding. Rats were allowed ad libitum access to Purina 18% NIH Block and chlorinated tap water. Balb/c and C57BL/6 mice were housed in Thoren polycarbonate caging and allowed ad libitum access to Purina 5L79 and chlorinated tap water. NCI-Frederick is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the Guide for Care and Use of Laboratory Animals (National Research Council, 1996; National Academy Press, Washington, D.C.)
Biodistribution of PLS-Cy7.5.
Female Balb/c mice, aged between 7 and 9-weeks-old, were purchased from Charles River Laboratories (Raleigh, N.C.). Mice, 5 animals per treatment group, were treated with PLS-Cy7.5 (equivalent to 1.5 nmol dye) by intravenous tail-vein injection under anesthesia using 3% isoflurane. At each time point, mice were transferred to the imaging chamber where isoflurane was reduced to 2% and O2 was used as a carrier at 1 liter/minute flow rate. Imaging was performed at 0.5, 1, 2, 4 and 6 h after injection using IVIS Spectrum (PerkinElmer) and the following parameters: excitation 675 nm and 745 nm, emission at 800, 820, and 840 nm, and exposure time set to auto (1-120 seconds per wavelength). Images were unmixed using the vendor specified protocol to separate skin autofluorescence from the Cy7.5 signal. An unmixed component image of Cy7.5 was used for analysis. After the 6-h whole body image, mice were euthanized using CO2 exposure followed by thoracotomy as per ACUC guidelines. Mice were dissected, and lymph nodes, brain, liver, lung, pancreas, spleen, and muscle were harvested, weighed, and placed on a black imaging plate. Ex vivo imaging was performed using the same parameters described above. To quantify the organ fluorescence, a fitted region of interest (ROI) was drawn over each organ and total radiance efficiency within the ROI was measured. To eliminate organ size dependence on quantification, total radiance efficiency values were normalized by the respective ROI area (cm2). Lymph node distribution of PLS-488. Male and female C57BL/6 mice, aged between 8 and 12-weeks-old, were purchased from Charles River Laboratories (Raleigh, N.C.). Mice, 3 males and 3 females per treatment group, were treated with PLS-Alexa488 (equivalent to 0.25 nmol of dye) or with PBS vehicle by either tail-vein injection or intra-dermal injection in the foot pads. Mice were euthanized by CO2 exposure followed by thoracotomy as per ACUC guidelines at 6 and 24 h. Liver, spleen, and lymph nodes (popliteal, axillary, mesenteric, and inguinal) were collected and fixed with 4% paraformaldehyde at room temperature for 18-24 h followed by paraffin embedding. Immunohistochemistry to detect PLS-Alexa488 was performed using a biotin-free polymer approach (Golden Bridge International, Inc.) on 5-μm tissue sections mounted on Fisherbrand™ Superfrost™ Plus Microscope Slides (Thermo Fisher Scientific), which were dewaxed and rehydrated with double-distilled H2O. Heat induced epitope retrieval (HIER) was performed by heating sections in 0.01% citraconic anhydride containing 0.05% Tween-20 in a pressure cooker set at 122-125°C for 30 s. Slides were incubated with blocking buffer (TBS with 0.05% Tween-20 and 0.25% casein) for 10min then incubated with rabbit anti-Alexa488 antibody (1:400; Cat. No. A-11094, Invitrogen) diluted in blocking buffer over night at 4°C. Slides were washed in 1X TBS with 0.05% Tween-20 and endogenous peroxidases blocked using 1.5% (v/v) H2O2 in TBS (pH 7.4) for 10min. Slides were incubated Rabbit Polink-1 HRP for 30 min at room temperature, washed and incubated with Impact™ DAB (3,3′-diaminobenzidine; Vector Laboratories) for 2-5 min. Slides were washed in ddH2O, counterstained with hematoxylin, mounted in Permount (Thermo Fisher Scientific), and whole tissue sections were scanned at high magnification (x200) using the ScanScope AT2 System (Aperio Technologies) yielding high-resolution data from the entire tissue section. Representative high-resolution images were extracted from these whole-tissue scans.
Pharmacokinetics of PLS-FTC.
Double-jugular catheterized and non-catheterized 10-week-old male Sprague Dawley rats (approx. weight of 250 grams) were purchased from Charles River Laboratories (Raleigh, N.C.). Rats were treated intravenously either by left jugular catheter or tail vein injection (for non-catheterized tissue collection animals) with 10 mg FTC equivalent/10 mL/kg of either PLS-FTC or FTC. Blood samples (200 μL) were collected from the right jugular catheter and placed in K2EDTA tubes at 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, and 96 h post dose. Blood samples were centrifuged at 2,500 x g for 10 min to collect plasma, and the collected plasma was snap-frozen and stored at −80 °C until analysis. Plasma from the untreated control animals was also collected and used to prepare a matrix standard curve and quality control standards. For tissue collection animals, tissues were collected at 4, 24, and 96 h post dose. Liver, spleen, lymph nodes (mesenteric, axillary, popliteal, and inguinal), and plasma were collected. The tissues were weighed, and half the tissue (left node) was snap frozen in liquid nitrogen (for LC-MS/MS analysis) while the other half (right node) was weighed and frozen by methyl butane dry ice slush on foil (for IR-MALDESI analysis) and stored at −80 °C. Tissues from the untreated control animals were also collected and used to prepare matrix standard curves and quality control standards.
LC-MS/MS analysis of FTC in plasma.
The extraction of FTC from rat plasma was performed using a protein precipitation process with isotopically-labeled internal standard. In this assay, 25 μL of plasma was mixed with 75 μL of methanol containing 20 ng/mL of the internal standard, 13C15N2-FTC (FTC-IS). Following vortex and centrifugation steps, 60 μL of the supernatant was transferred to a clean tube and evaporated to dryness at 50 °C. Extracts were reconstituted with 75 μL of water prior to LC-MS/MS analysis.
The following LC-MS/MS conditions were used. 10 μL injection volume, gradient mobile phase consisting of (A) 0.1% acetic acid in water and (B) methanol: hold at 0% methanol for 0.25 min, linear increase to 3% methanol at 0.5 min, 3% methanol linear increase to 10% methanol at 1 min, hold at 10% methanol from 1-1.5 min, ramp to 18% methanol at 2.25 min, with a final ramp to 65% methanol at 3.25 min, and linear decrease to 0% methanol at 3.80 min and held for the complete run time of 6 min, with a flow rate of 0.45 mL/min on a Waters Atlantis T3 (2.1 x 50 mm, 3 μm particle size) analytical column. FTC and 13C15N2-FTC elution times were both 2.7 min, and m/z ions monitored by MRM transitions were 248.0 → 112.0 and 251.0 → 133.00, respectively. The MS instrument used an electrospray ionization source in positive ion mode with an IonSpray voltage of 4500V at 400 °C. The instrumentation included Shimadzu SIL 20-AC Auto sampler, LC-20AD (binary pump) solvent delivery system, DGU-20A Inline Degasser, CTO-20A Column Oven, AB Sciex API-5000 Triple Quadrupole Mass Spectrometer System, AB Sciex Analyst LC-MS/MS Software. Data was collected using Sciex Analyst Chromatography Software from the AB Sciex API-5000 triple quadrupole mass spectrometer.
LC-MS/MS analysis of FTC in tissues.
Tissue samples were transferred from the freezer into pre-labeled Precellys Lysing Kit (2 mL reinforced tube with metal beads) tubes. This process was performed quickly so as not to allow the unhomogenized tissue to come to room temperature. Tissue weights were recorded, and 1.00 mL of ice-cold 70:30 acetonitrile:1 mM ammonium phosphate was added to each tube. Tissues were homogenized on Precellys 24 using settings of: 5500 RPM, 3 cycles: 60 sec/cycle, pause 15 sec. 300 μL of the homogenate was mixed with 50 μL of FTC-IS (20 ng/mL) in acetonitrile. Following vortex and centrifugation steps, 60 μL of the samples were evaporated to dryness at 50 °C. Extracts were reconstituted with 100 μL of 1mM ammonium phosphate (pH 7.4) prior to LC-MS/MS analysis.
The following LC-MS/MS conditions were used. 8 μL injection volume, gradient mobile phase consisting of (A) 0.1% formic acid in water and (B) acetonitrile with 0.1 % formic acid: (Hold at 3% ACN/0.1% formic acid for 1 min, 3% ACN/0.1% formic acid linear increase to 10% ACN/0.1% formic acid from 1-2 min, 10% ACN/0.1% formic acid linear increase to 95% ACN/0.1% formic acid from 2-4 min, hold at 95% ACN/0.1% formic acid from 4-4.5 min, and linear decrease to 3% ACN/0.1% formic acid from 4.5-4.6 min, and held for a total run time of 6 min), with a flow rate of 0.35 mL/min on a Waters Atlantis T3 (2.1 mm id x 10 cm, 3 μm particle size) analytical column. FTC and 13C15N2-FTC elution times were both 3.9 min, and m/z ions monitored by MRM transitions were 248.0 → 130.0 and 251.0 → 133.00, respectively. The MS instrument used an electrospray ionization source in positive ion mode with an IonSpray voltage of 5500V at 500 °C. The instrumentation included Shimadzu SIL 20-AC Auto sampler, LC-20AD (binary pump) solvent delivery system, DGU-20A Inline Degasser, CTO-20A Column Oven, AB Sciex API-5000 Triple Quadrupole Mass Spectrometer System, AB Sciex Analyst LC-MS/MS Software. Data are collected using Sciex Analyst Chromatography Software from the AB Sciex API-5000 triple quadrupole mass spectrometer.
IR-MALDESI lymph node analysis.
Spatial distribution of FTC drug concentrations was measured by infrared matrix-assisted laser desorption-electrospray ionization (IR-MALDESI) mass spectrometry imaging (MSI) 34,35 in rat axillary lymph nodes collected at 4h post dose. Lymph node sections (10 μm) of samples were prepared in a cryotome, thaw-mounted on glass microscope slides, and maintained at −10 °C on the sample stage of the IR-MALDESI source chamber prior to analysis. The stage translated the sample step-wise across the focused beam of an IR laser (λ=2.94 μm, IR-Opolette 2371; Opotek, Carlsbad, CA, USA), which desorbed sample material from adjacent 100 μm diameter sampling locations. An electrospray (50/50 mixture of methanol/water (v/v) with 0.2% formic acid) ionized the desorbed neutral molecules, and resulting ions were sampled into a high resolving power Thermo Fisher Scientific Q Exactive Plus (Bremen, Germany) mass spectrometer for synchronized analysis. The mass spectrometer was operated in positive ion mode from m/z 200 to 800, with resolving power of 140,000FWHM at m/z 200. With high mass measurement accuracy (MMA) within 5 ppm maintained using protonated and sodiated adducts of diisooctyl phthalate as two internal lock masses at m/z 391.28428 and 413.26623, FTC was identified by its protonated molecular ion [M+H+]+ at m/z 248.049969. To generate images from mass spectrometry data, raw data from each voxel was converted to the mzXML format using MSConvert software.36 These mzXML files were interrogated using MSiReader37, a free software developed for processing MSI data.
Pharmacokinetic analysis.
Noncompartmental pharmacokinetic parameters were determined using Phoenix WinNonlin version 8.0 software (Certara, L.P., Princeton, NJ): the area under the time concentration curve including all time points (AUCall) was calculated using the linear up/log down trapezoidal rule; the Cmax term is the maximum measured concentration; the MRT term is the mean residence time, calculated using the following equation: AUC/AUMC; The AUMC term is the area under the first moment curve; The Vss term is the volume of distribution steady state, calculated using the following equation: CLxMRT; C0 is the extrapolated concentration time zero; T1/2 is the half-life calculated using the following equation: half-life=ln2/kel. All values below the limit of quantiation were imputed at ½ the lower limit of quantification for graphing purposes.
Statistics.
Statistical differences were identified by Student’s t test, with level of significance, p ≤ 0.05.
RESULTS
PLS targets scavenger receptor A1.
SR-mediated binding and uptake of PLS by macrophage was verified using a competitive binding assay with the SR competitive inhibitor, polyinosinic acid (poly I), or the non-inhibitor control, polycytidylic acid (poly C). A model murine macrophage cell line, RAW 264.7, was incubated with various concentrations of AlexaFluor488-labelled PLS (PLS-488, Figure S1) in the presence or absence of either poly I or poly C. The results presented in Figure 2A demonstrate inhibition of PLS-488 binding to macrophages in the presence of the SR-A substrate poly I, and not with the control polymer poly C, which is consistent with SR-A selective uptake.38 Poly I co-treatment required higher concentrations of PLS-488 to detect fluorescence due to competition for the SR-A receptor. The results were further confirmed in a similar study in which increasing concentrations of poly I or poly C were co-incubated with a constant concentration of PLS-488 (Figure 2B). As expected, concentration-dependent inhibition was observed with poly I co-treatment, but no inhibition was observed even with very high concentrations of poly C. Since SR-A are known to bind negatively charged species, we hypothesized the free succinyl groups of PLS were responsible for its interactions with SR-A. To support this theory, we performed a competitive binding study between PLS-488 and PLS partially capped (95% free succinyl groups) or fully capped (no free succinyl groups) with 2-(2-methoxyethoxy)ethanamine (Figure S2–5). The partially capped PLS demonstrated inhibition of PLS-488 binding to macrophages, whereas the fully capped PLS was equivalent to poly C treatment, indicating no competition for SR-A (Figure S6). This data suggests the free succinyl groups of PLS are responsible for its ability to target SR-A. To further explore SR-A selective binding and uptake of PLS by macrophages, we utilized a RAW 264.7 derivative cell line (Clone 1/2) which is SR-A1 deficient.33 The abundant expression of SR-A1 on macrophages, and receptor preference for polyanionic substrates, made SR-A1 our primary receptor candidate to test.39 Both the wild-type RAW 264.7 cells and the SR-A1 deficient Clone 1/2 were incubated with PLS-488 for 24 h and analyzed by flow cytometry while media-treated cells were used as controls (Figure 2C; Figure S7). Remarkably, there was uniform uptake of PLS-488 by the wild-type cells and no uptake by the SR-A1 deficient cell line, demonstrating the importance of SR-A1 for binding and uptake of PLS. Collectively, these studies strongly suggest a selective interaction between PLS and SR-A1. Based on these conclusive findings we did not continue with evaluation of other SR superfamily or alternative receptor candidates.
Figure 2.
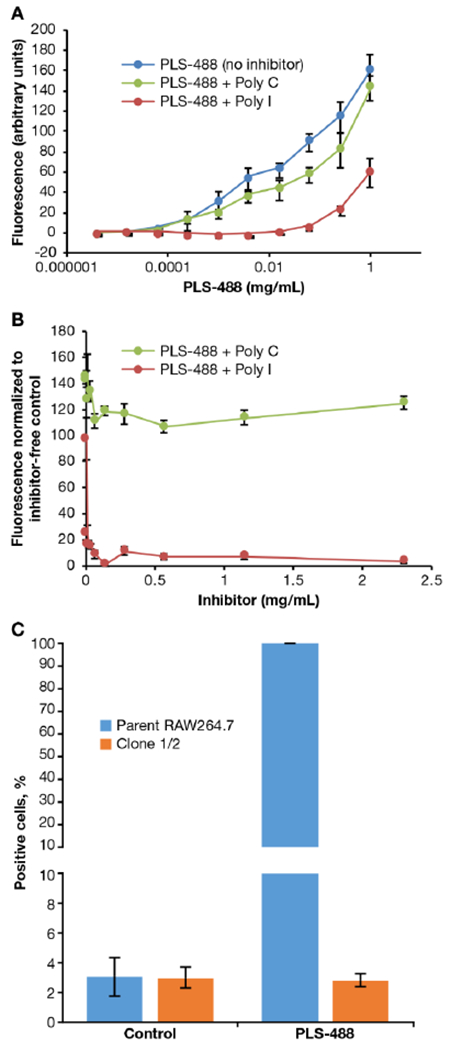
Selective uptake studies in murine Raw 264.7 cells by flow cytometry. (A) Binding and uptake of PLS-488 in RAW 264.7 cells after 3 h co-treatment with 4.22 ng/mL to 1.11 mg/mL PLS 488 and 200 μg/mL poly I or poly C. (B) Binding and uptake of PLS-488 in RAW 264.7 cells after 3 h co-treatment with increasing concentrations of poly I or poly C (0.00451 - 2.31 mg/mL). (C) Selective uptake of PLS-488 in RAW 264.7 and Clone 1/2 cells after 24 h incubation with 1.00 μg/mL PLS-488. Data are expressed as mean ± SD (n=3). PLS-488 ex./em. 493/516 nm.
SR-A1-mediated PLS cellular uptake was further confirmed from a time course PLS-488 uptake study carried out using confocal laser scanning microscopy (Figure 3; Figure S8). PLS-488 uptake was observed at the earliest 0.5 h time point only in the wild type Raw 264.7 cells. The PLS-488 fluorescence signal colocalized with Lysotracker Red, suggesting PLS-488 was located within lysosome and consistent with SR-A1 receptor-mediated phagocytosis. Following the 24 h treatment period, PLS-488 fluorescence signal was markedly greater in the wild type Raw 264.7 cells than the SR-A1 deficient Raw cells, similar to results obtained from the flow cytometry study above and confirming the PLS specificity for the SR-A1 receptor.
Figure 3.
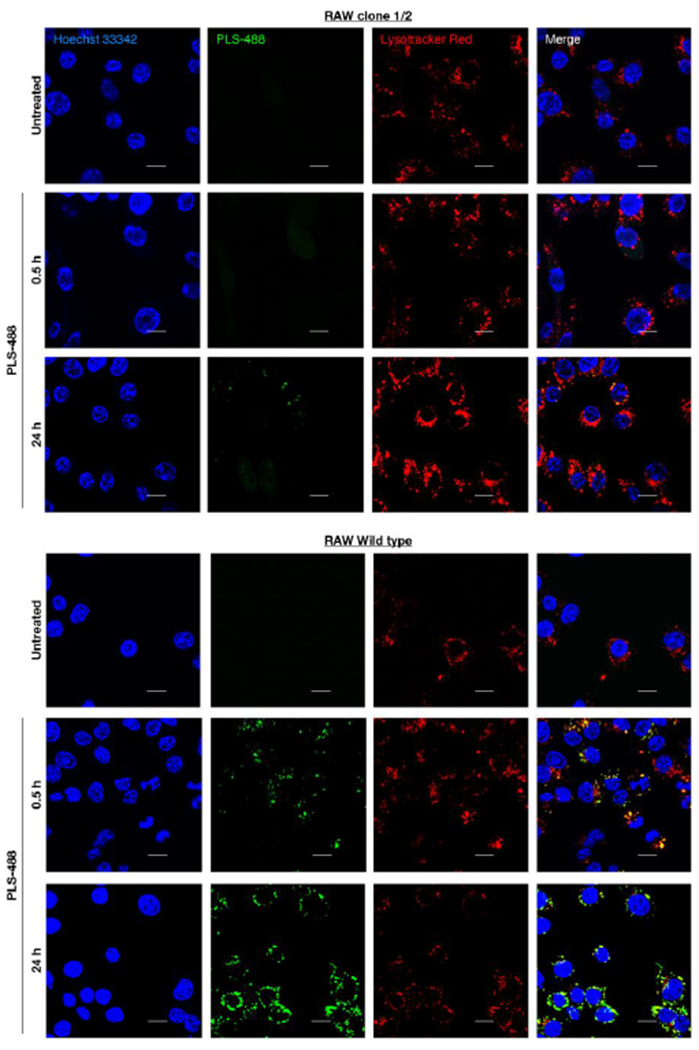
Selective uptake studies in murine Raw 264.7 cells by confocal microscopy. (top- clone ½ cells) Representative confocal images of untreated (top row) and PLS-488 treated clone 1/2 cells, 0.5 h (middle row) and 24 h (bottom row) post treatment. (bottom- wild type Raw 264.7 cells) Representative confocal images of untreated (top row) and PLS-488 treated Raw 264.7 cells, 0.5 h (middle row) and 24 h (bottom row) post treatment. (Lyosomes in red were stained with Lysotracker Red, ex./em. 561/590 nm; Nuclei in blue were stained with Hoechst 33342, ex./em. 405/461 nm; PLS-488 tagged with Alexa Fluor 488 is shown in green, ex./em. 488/519 nm). Scale bar represents 10 μm.
Biodistribution of fluorescently-labelled PLS.
Cy7.5-labelled poly(L-lysine succinylated) (PLS-Cy7.5, Figure S9) was administered to mice by intravenous tail vein injection. At various time points, whole-body images were taken, and at 6 h post injection animals were euthanized and organs imaged ex vivo (Figure 4; Figure S10–11). Liver had the greatest PLS-Cy7.5 fluorescent signal, consistent with Kupffer cell uptake, followed in descending order by spleen, lymph node, lungs and kidneys. The inguinal lymph nodes were imaged intact with the skin, and lymph node distribution is clearly visible. No PLS-Cy7.5 signal was detected in the brain, pancreas, or muscle tissues collected.
Figure 4.
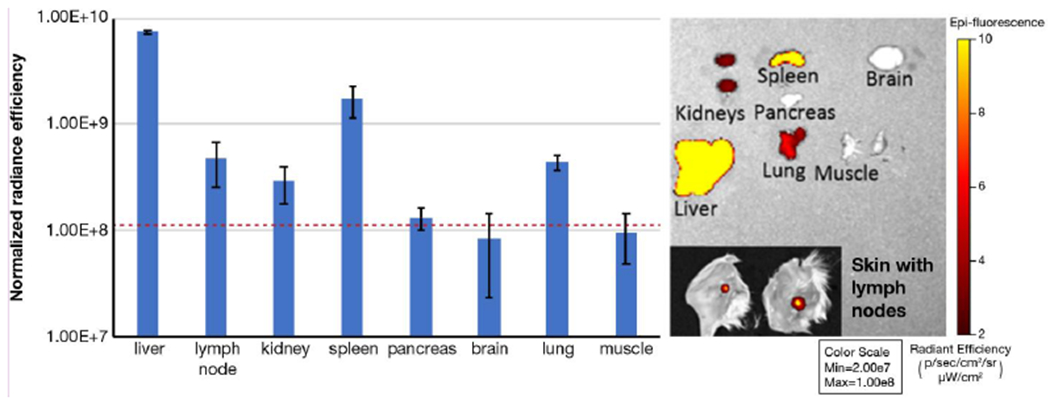
Biodistribution of PLS-Cy7.5 in mice after IV administration. Representative ex vivo images of organs harvested 6 h post-injection are shown. Subset image displays inguinal lymph node intact with skin. Data are presented as mean ± SD (n=3). Dashed red line represents the average background (autofluorescence) level for all organs (mean +/−SD, 1.15E+08 +/− 0.08E+08 normalized radiance efficiency). Organs are displayed at the same colorscale to demonstrate relative intensity levels.
Lymph node distribution of fluorescently-labelled PLS.
To determine the cellular distribution of PLS into lymphoid tissues in vivo, PLS-488 or PBS was administered to mice by either intravenous tail vein injection or intradermal injection into the foot pads. Mice were euthanized 6 h and 24 h post administration and organs harvested, fixed and PLS-488 distribution determined by immunohistochemistry. Importantly, control mice stained for Alex488 showed no staining in any tissue examined (Figure 5; Figure S12). Both routes of administration resulted in abundant PLS-488 accumulation into macrophage-rich liver sinusoids and spleen red pulp by 6 h that was still evident 24 h post administration, albeit at lower levels (Figure 5). As expected, intradermal administration resulted in robust accumulation of PLS-488 in regions of the draining (popliteal) and peripheral (axillary and inguinal) lymph nodes consistent with myeloid cells that was still evident 24 h post administration. Intravenous administration resulted in early accumulation of PLS-488 into myeloid-rich regions of the deep mesenteric lymph nodes by 6 h, with later accumulation in peripheral lymph nodes. Regardless of the route of administration, PLS-488 accumulation in lymph nodes occurred in macrophage-rich subcapsular and medullary regions, and at 24 h even into the B cell follicles.
Figure 5.
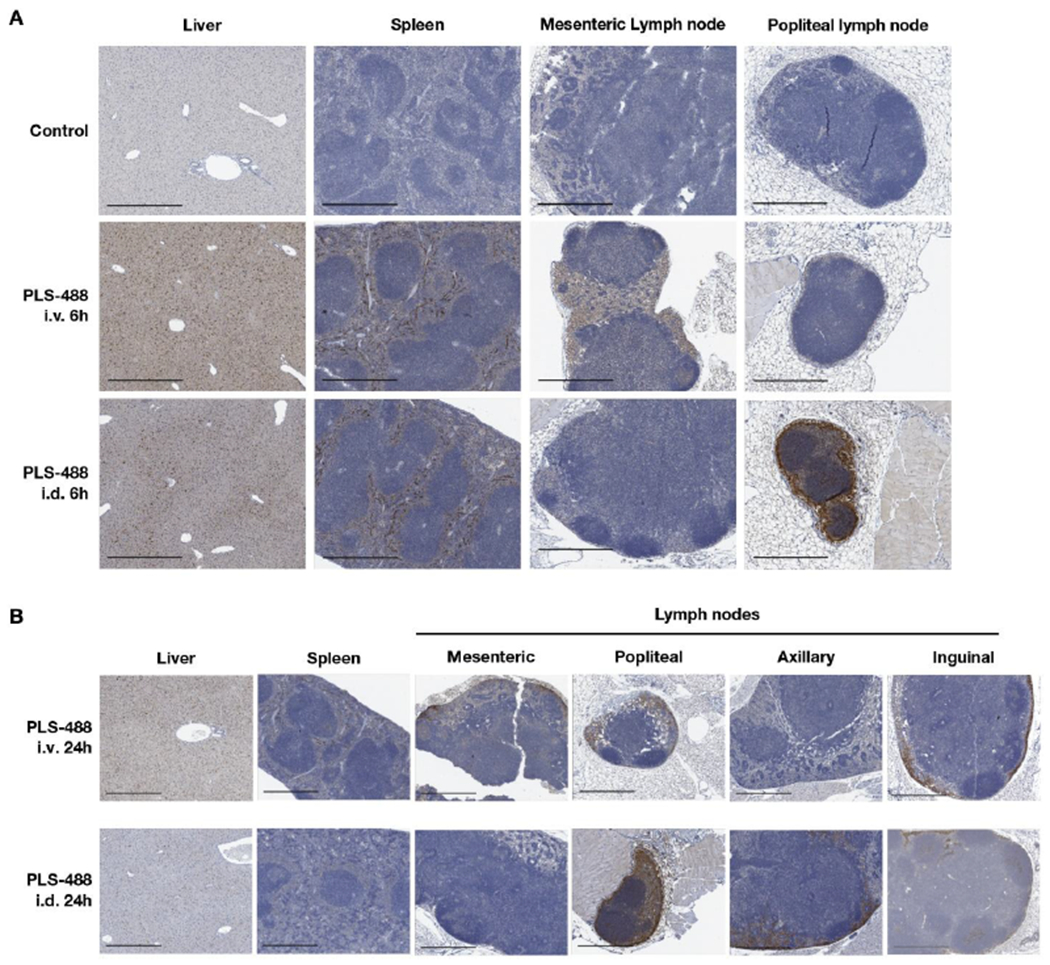
(A) Displayed are the anti-Alexa488 immunohistochemistry images for liver, spleen, mesenteric lymph node and popliteal lymph node from mice 6 h after PBS vehicle control treatment and i.v. and i.d. injection of PLS-488. (B) Displayed are the anti-Alexa488 IHC images for liver, spleen, mesenteric lymph node, popliteal lymph node, axillary lymph node, and inguinal lymph node from mice 24 h after i.v. and i.d. PLS-488 treatment. The scale bars in the images are 500 um.
Synthesis of PLS-FTC.
Emtricitabine (FTC) was conjugated to the pendant carboxyl groups of PLS using 4-dimethylaminopyridine (DMAP)-catalyzed carbodiimide esterification (Figure 6). The resulting polymer prodrug product was characterized by size-exclusion chromatography (SEC), proton nuclear magnetic resonance (1H NMR), and high-performance liquid chromatography (HPLC) to confirm conjugation and determine drug loading. SEC/ultra-violet (UV) analysis yielded a single macromolecular peak at roughly the same elution time as the starting polymer, but the extracted UV spectrum showed strong absorption at 280 nm (λmax for FTC) which is indicative of successful conjugation since the starting PLS absorbs poorly at this wavelength (Figure 7A,B). SEC/multi-angle light scattering (MALS) analysis confirmed a single macromolecular peak with a polydispersity (PDI) of 1.22 and a molecular weight of 109,000 g/mol, which is larger than that of the polymer starting material due to the conjugation of FTC molecules and is above the molecular weight cut-off for renal clearance (Figure 7C,D).40 1H NMR analysis in D2O further suggested successful conjugation, as it showed proton peaks for both PLS and FTC as well as proton peak changes corresponding to esterification. Specifically, the disappearance of the peaks at 3.92 and 4.01 ppm, which represent the CH2 protons adjacent to the hydroxyl of FTC, and the emergence of the corresponding peak at 4.56 ppm indicate ester formation (Figure 8; Figure S2,13). Integration of the 1H NMR spectrum shows there are approximately 0.2 FTC molecules per succinylated lysine unit, corresponding to approximately 50 FTC molecules per polymer on average.
Figure 7.
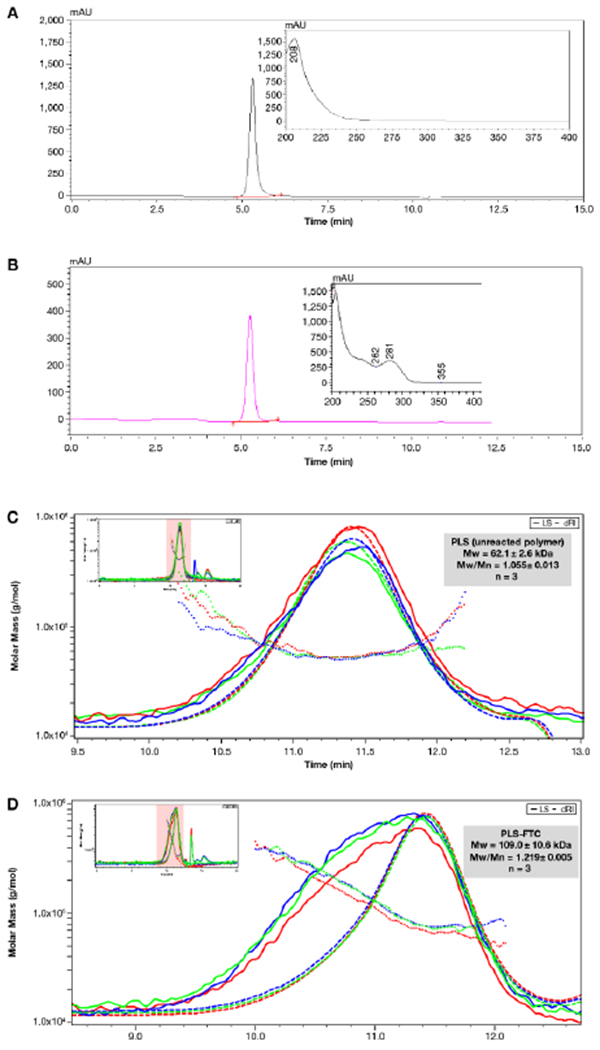
(A,B) SEC chromatograms of PLS starting material (A) and PLS-FTC (B). SEC/UV conditions were: 10 μL injection volume, isocratic elution solvent consisting of 50% acetonitrile and 50% ammonium acetate (9 mM, pH 7.7), flow rate of 0.39 mL/min, column temperature of 40 °C, and UV detection set at 210 nm (A) or 280 nm (B). The UV spectra of the sample peaks are shown in the subset images. (C,D) SEC chromatograms and molecular weight distributions of PLS starting material (C) and PLS-FTC (D). SEC/MALS conditions were: 100 μL injection volume, isocratic elution solvent consisting of 90% methanol and 10% water containing 50 mM LiCl, flow rate of 1.00 mL/min, and detecting with DAWN HELEOS II multiangle light scattering detector (Wyatt) and Optilab T-rEX infrared detector (Wyatt). Greed, red, and blue traces represent individual runs. Mw and Mw/Mn (PDI) are presented as average ± SD (n=3).
Drug loading determination, as a weight percent, was performed using a sodium hydroxide (NaOH) degradation method followed by HPLC analysis. Prodrug samples were treated with 1N NaOH for 1 h to completely hydrolyze the linker ester and release all the FTC from the prodrug (Figure 9). A standard curve of NaOH-treated FTC was generated and used to quantify the FTC content in PLS-FTC after NaOH treatment (Figure S14,15; Table S1). Excellent drug FTC loading at 14.2% (wt/wt) was achieved. No free FTC or other contaminants in the prodrug sample were detected by HPLC.
In vitro drug release in human plasma.
The stability of PLS-FTC was assessed by incubation in fresh human plasma, with or without the esterase inhibitor phenylmethylsulfonyl fluoride (PMSF) to determine the extent of enzymatic hydrolysis. PLS-FTC demonstrated surprising plasma stability, releasing the drug at a log-linear rate (first-order). After 24 h incubation in plasma, ~65% of the FTC had been released compared to 43% released over the same period in the esterase-inhibited plasma (Figure 10). The drug release plot was linearized by plotting the natural log of % drug remaining on the y-axis, and the slope of the linear least squares regression was used to determine the first-order drug release half-life of 15 h in plasma and 29 h in esterase-inhibited plasma, indicating that drug release occurs through both enzymatic and non-enzymatic mechanisms.
Figure 10.
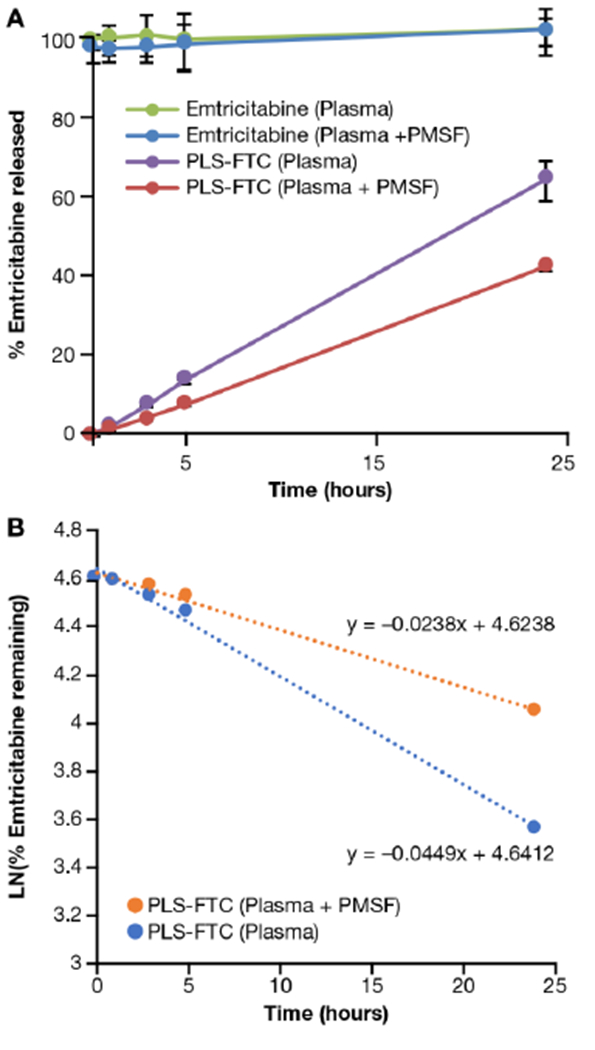
In vitro drug release of PLS-FTC in human plasma. (A) Drug release of PLS-FTC in human plasma matrix at 37 °C, with or without 5 mM PMSF, was compared to free FTC control. (B) The plot of the natural log of PLS-FTC concentration remaining vs. time was used to determine the first-order release kinetics. Data in (A) are presented as mean ± SD (n=3), data in (B) are presented as mean (n=3). Dotted lines and accompanying equations in (B) are the linear least squares regression plots fit to the data. Phenylmethylsulfonyl fluoride (PMSF) is an esterase inhibitor used to assess enzyme dependency of hydrolysis.
Conversion of FTC to FTC triphosphate (FTC-TP) in human PBMCs.
To verify that the FTC released from the prodrug can undergo conversion to its active triphosphate metabolite, liquid chromatography-mass spectrometry (LC-MS) was used to measure FTC and FTC-TP concentrations in human PBMCs following 24 h incubation with PLS-FTC prodrug or FTC free drug control, and normalized to soluble protein concentration in each sample as determined by micro BCA assay. FTC and FTC-TP were detected for both the prodrug (40.6 ± 1.7 ng/mg protein and 52.7 ± 14.3 ng/mg protein, respectively) and free drug control (182 ± 18 ng/mg protein and 79.7 ± 9.0 ng/mg protein, respectively), confirming the FTC released from the prodrug can undergo conversion to its active triphosphate form. Of note, the ratio of the active metabolite to precursor, FTC-TP/FTC, was greater for the prodrug than free drug control, 1.3 vs. o.4, respectively, supporting the more efficient conversion of the prodrug. This increased prodrug FTC-TP conversion is likely the result of prodrug-mediated cellular concentration of FTC relative to free FTC.
Pharmacokinetics of PLS-FTC.
The pharmacokinetics of the FTC prodrug was assessed in male Sprague-Dawley rats and compared to FTC free drug control. Following i.v. bolus administration of 10 mg FTC equivalents/kg of either PLS-FTC or FTC control, plasma and tissue samples were collected at specified time points and analyzed for FTC concentrations by liquid chromatography with tandem mass spectrometry (LC-MS/MS). The resulting plasma pharmacokinetic profiles are displayed in Figure 11. Pharmacokinetic parameters differed markedly between the prodrug and FTC control groups, with the prodrug having a 2-fold lower concentration time zero, and half-life, and 2-fold higher mean residence time and area under the concentration-time curve (Table 1). The significance of these findings is considered in the discussion section
Figure 11.
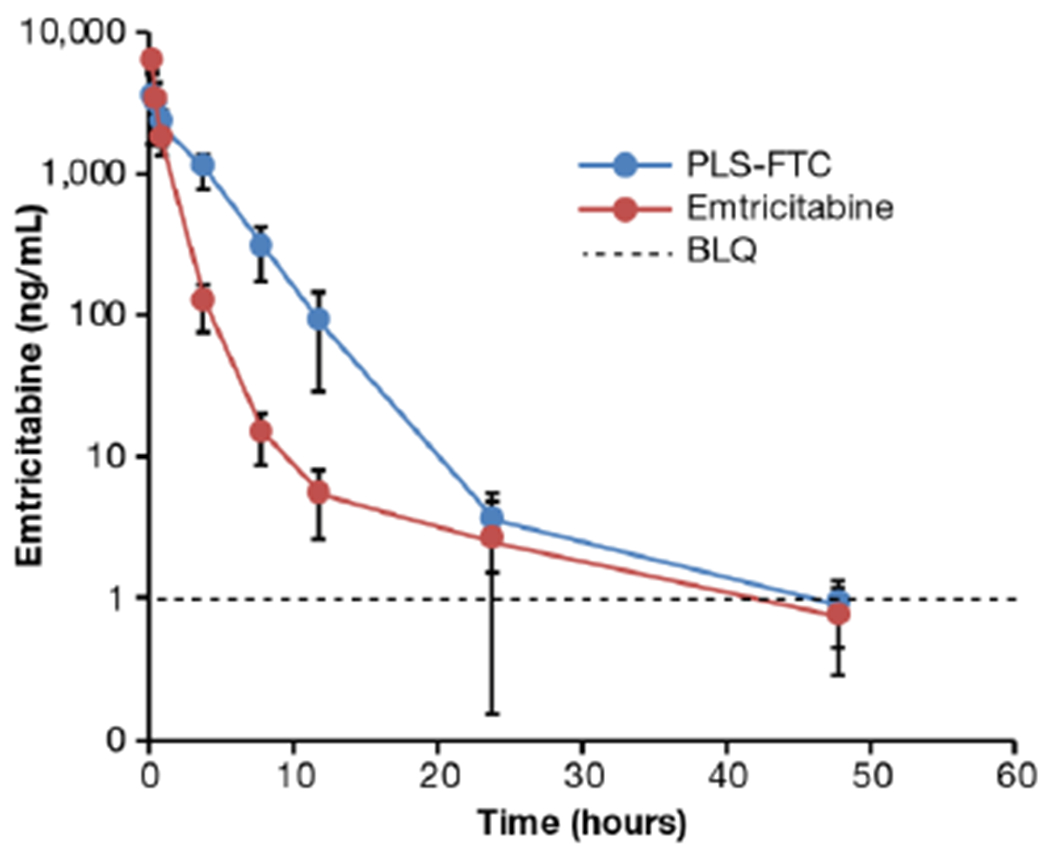
FTC concentrations in plasma after i.v. bolus administration of 10 mg FTC equivalents/kg PLS-FTC or free FTC control. Data are presented as mean ± SD (n=8). Dashed line represents lower limit of quantitation (1.00 ng/mL).
Table 1.
FTC plasma pharmacokinetic parameters. Displayed are the FTC PK parameters for rats administered an i.v. bolus dose of 10 mg FTC equivalents/kg of PLS-FTC or FTC.
| Parameter | PLS-FTC | Emtricitabine |
|---|---|---|
| C0 (ng/mL) | 4901 ± 3621* | 10610 ± 2947 |
| Cmax (ng/mL) | 3839 ± 1734* | 5850 ± 853 |
| T1/2 (h) | 6.99 ± 2.62* | 14.10 ± 5.17 |
| AUCall (h*ng/mL) | 11100 ± 3281* | 6396 ± 1014 |
| MRT (h) | 3.64 ± 0.47* | 1.80 ± 0.32 |
| Cl (mL/h/kg) | 1053 ± 620* | 1600 ± 287 |
| Vss (mL/Kg) | 3816 ± 2254 | 2937 ± 1095 |
C0, concentration extrapolated to time zero; Cmax, maximum measured concentration; T1/2, elimination half-life; AUCall, area under the curve from time zero to the last measurable time point; MRT, mean residence time; Cl, clearance; Vss, volume of distribution at steady state. Data are presented as mean ± SD (n=8).
Asterisk (*) represents statistical significance, two-sided t-test, p < 0.05.
Remarkably, the prodrug achieved ~7 to 19-fold higher concentrations in lymphatic tissues at 4 h post injection compared to the free FTC control (mesenteric lymph node 850 ± 540 vs. 75 + 35 ng/g; axillary lymph node 811 ± 65 vs. 109 ± 37 ng/g; inguinal lymph node 587 ± 56 vs. 87 ± 51 ng/g; ; popliteal lymph node 864 ± 330 vs. 105 ± 64 ng/g; liver 2,257 ± 143 vs. 119 ± 51 ng/g; spleen 1,788 ± 164 vs. 106 ± 44 ng/g (mean ± SD, n=3)), supporting lymphatic-targeted drug delivery (Figure 12).
Figure 12.
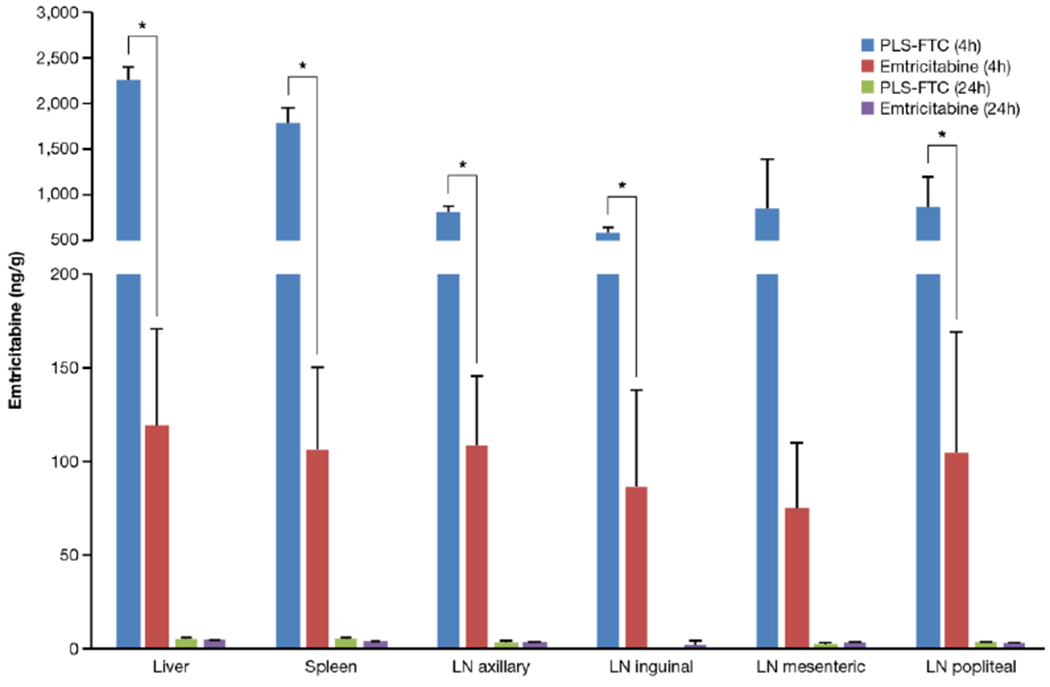
FTC concentrations in tissues at 4 and 24 h after i.v. bolus administration of PLS-FTC or FTC control. Data are presented as mean ± SD (n=3 for all tissues at 4 h and PLS-FTC at 24 h (LN axillary), n=2 for PLS-FTC 24 h (liver, spleen, and LN mesenteric) and FTC 24 h (LN inguinal), n=1 for PLS-FTC 24 h (LN popliteal) and FTC 24 h (liver, spleen, LN axillary, mesenteric, popliteal)). All PLS-FTC 24 h LN inguinal samples were below limit of quantitation. Asterisk (*) represents statistical significance, two-sided t-test, p < 0.05.
Infrared matrix-assisted laser desorption electrospray ionization mass spectrometry imaging (IR-MALDESI MSI) ion maps of FTC spatial distribution in axillary lymph node tissues are shown in Figure 13, overlaid on the ion map for endogenous cholesterol to delineate tissue section shape. These results indicated greater signal abundance (158.0 ± 96.0 vs. 18.5 ± 16.6, arbitrary units) and frequency of detection (11.1 ± 2.2 vs. 2.9 ± 2.8 % of sampling locations with measurable drug) for FTC prodrug compared to the free FTC control, further supporting the FTC-PLS lymphatic drug delivery.
Figure 13.
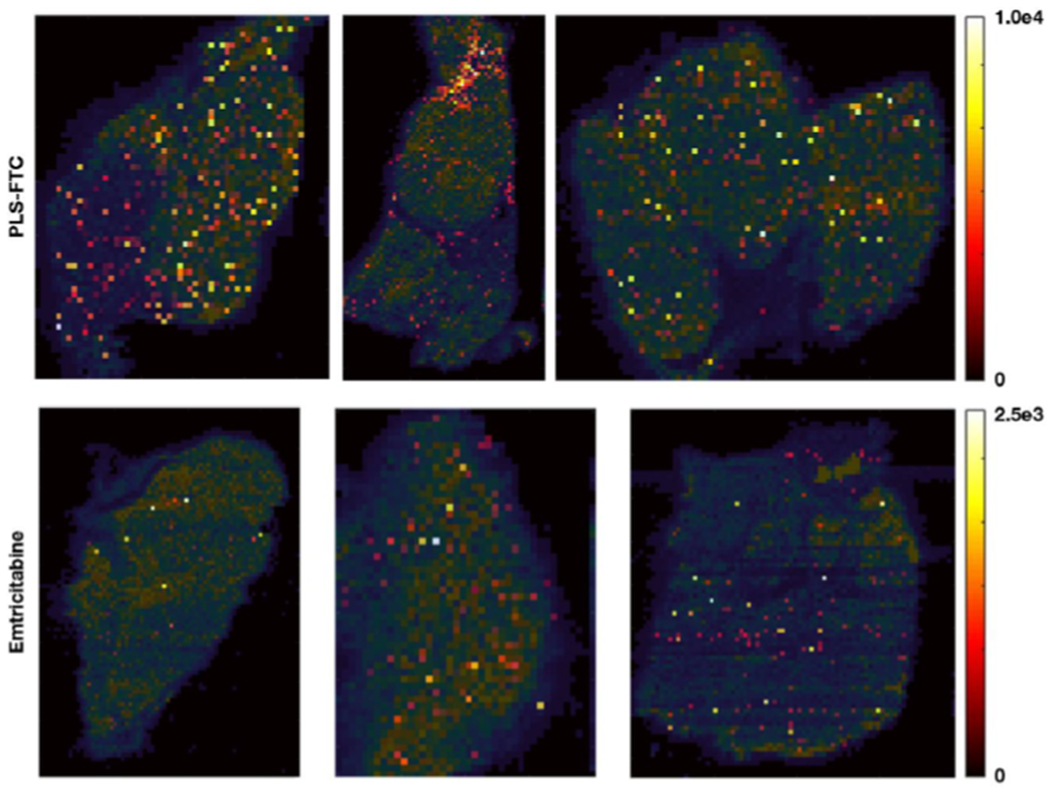
Spatial distribution of FTC in axillary lymph nodes 4 h post dose measured by IR-MALDESI MSI. FTC signal abundance, shown using a color map ranging from dark/red for areas of low concentration to yellow for areas of high concentration, is overlaid on the ion map for cholesterol (shown in blue-green color map) to indicate section shape and extent.
DISCUSSION
In this study, a SR-A1-targeted macromolecular platform was developed to enhance drug delivery to lymphatic tissues. One of the most successful examples of a SR-A-targeted formulation is succinylated HSA which distributes rapidly to lymphatic tissues through scavenger receptor-mediated endothelial transcytosis.16 Similar to succinylated HSA, poly(L-lysine succinylated) is a highly succinylated macromolecule composed of amino acids. Therefore, we hypothesized PLS would also interact with scavenger receptors and demonstrate similar biodistribution to succinylated HSA. Indeed, PLS binding and uptake in RAW 264.7 macrophage cells were inhibited by the SR-A competitive inhibitor poly I, but not the negative control poly C, supporting SR-A mediated uptake (Figure 2). Uptake of the PLS-488 by RAW 264.7 is consistent with a recent study demonstrating uptake of PLS-coated silica nanoparticles by RAW 264.7 cells.41 These researchers, however, did not identify SR-A receptor involvement in the PLS-coated silica nanoparticle uptake mechanism. In SR-A1-deficient RAW cells, there was very little observed uptake or binding of PLS even after 24 h incubation (Figures 2 and 3). Our focus on the SR-A1 member of the SR superfamily was driven by the current knowledge regarding expression level, abundance and ligand specificity of these receptors.39 Considering the numerous potential receptor-mediated mechanisms of macrophage phagocytosis42, the absence of any significant binding or uptake in SR-A1 deficient cells suggests remarkable selectivity of PLS for SR-A1.
SR-A1 is known to have affinity for polyanionic molecules, supporting the theory that PLS is a suitable substrate. The broad specificity of SR-A1 for polyanionic substrates is due to interaction of the polyanion with cationic collagenous domains of the receptor. The binding of substrates with SR-A1 on macrophage results in internalization through clathrin-dependent and -independent processes, resulting in entry into the endosome-lysosome pathway. Alternatively, SR-A1 on endothelial cells has been shown to be involved in transcytosis of substrates. Consistent with cellular uptake of PLS in the murine macrophage RAW cells occurring through a SR-A1-dependent receptor-mediated endocytosis mechanism, confocal studies demonstrated colocalization of the PLS-488 signal with the lysosomal marker Lysotracker red. By contrast, greatly reduced PLS-488 uptake was observed in the SR-A1 deficient RAW cell line.
Following i.v. administration, Cy7.5-tagged PLS distributed primarily to liver, likely due to Kupffer cell uptake, and lymphatic organs, including spleen and lymph nodes (Figure 4). Lymph node distribution was further confirmed by high resolution IHC analysis (Figure 5). This high resolution IHC imaging to the AlexaFluor488 tag is benefit over other distribution techniques such as radiolabel, which is more quantitative but fails to give cellular tissue distribution. High resolution microscopy images of anti-488 stained tissues from mice administered Alexa488-tagged PLS i.v. or i.d. identified distribution to subcapsular, medullary and follicular regions of lymph nodes consistent with myeloid cells. The distribution to myeloid cell populations within lymph nodes was expected due to the high expression of SR-A1 on both myeloid and endothelial cells, allowing the polymer to undergo endothelial transcytosis into the lymphatic system similar to the transcytosis mechanism described for succinylated HSA.16 Interestingly, even though brain microvessel endothelial cells have been shown to take up succinylated proteins via a scavenger receptor-dependent mechanism, we did not observe significant brain uptake in our imaging studies.43 The ability of PLS to undergo lymphatic transcytosis following intravenous administration appears to be a common attribute to scavenger receptor A1 ligands and may have tremendous therapeutic implications for infectious diseases.16
Traditional HIV anti-retroviral therapies achieve efficacious concentrations systemically, but do not distribute sufficiently to lymphatic tissues where latent viral reservoirs primarily reside. To determine if the PLS platform could improve anti-retroviral lymphatic distribution, FTC was selected as a model anti-HIV drug, and the prodrug was synthesized using established carbodiimide esterification chemistry. Although FTC contains an amine group which could potentially react to form an amide, aromatic amines are generally unreactive under the conditions used. Indeed, 1H NMR analysis provided evidence of esterification (Figure 8), and treatment with NaOH completely released the FTC from the prodrug, which would be expected for ester prodrugs and not for amide prodrugs due to the differences in hydrolytic stability between ester and amide bonds (Figure 9). Integration of the 1H NMR spectrum showed there are approximately 0.2 FTC molecules per succinylated lysine unit, which corresponded with the initial feed ratio in the synthesis reaction, indicating excellent ester conversion. SEC/MALS analysis of PLS-FTC showed a single macromolecular peak with a molecular weight of 109,000 g/mol, which was larger than expected if only ~50 FTC molecules were added. This larger-than-expected molecular weight could be due to secondary structures forming during analysis or interactions between the column and PLS-FTC. Further, molecular weight determination by SEC/MALS is highly dependent on selection of the peak range and accurate concentration and dn/dc values. Therefore, we developed a NaOH degradation method followed by HPLC analysis as a more accurate method to determine drug loading as a weight percent. Prodrug samples treated with NaOH released all its FTC from the prodrug as evidenced by the retention time shift from 1.8 min of PLS-FTC prior to NaOH treatment to 2.3 min after treatment, which matches the retention time of FTC with or without NaOH treatment (Figure 9). The stability of the FTC prodrug was evaluated in human plasma and was found to have controlled release properties with a half-life of approximately 15 h (Figure 10). This release half-life was prolonged ~2-fold in PMSF-treated plasma, suggesting drug release can occur through enzymatic hydrolysis, as well as non-enzymatic hydrolysis.
The proximity of the ester linkage to the polymer backbone may have a dramatic influence on enzymatic hydrolysis, as evidenced by the 119 h release half-life of Xyotax, a structurally similar poly(glutamic acid) ester prodrug of paclitaxel, which reportedly releases the drug nearly exclusively through a non-enzymatic hydrolysis mechanism.44 Of note, this polyanionic prodrug was also found to distribute to the lymphatic system, and may also undergo SR-A1-mediated uptake although this has not been evaluated. Despite achieving significant tumor distribution in pre-clinical models, Xyotax was unable to demonstrate superiority in clinical trials, with its slow drug release being one potential contributor to the lack of efficacy observed.44,45 A moderately stable formulation which is sufficiently stable to reach its intended tissue or target, but not overly stable where drug release is too slow to attain therapeutic concentrations, may prove to be a superior strategy. Given that our FTC prodrug reaches the lymph nodes as quickly as 4 h, our earliest time point for tissue collection, its ~15 h release half-life is sufficiently stable to deliver and release the majority of the drug within the target tissues. Moreover, the detection of FTC-TP in human PBMCs following prodrug treatment confirmed that the released FTC is available to undergo conversion to its active triphosphate metabolite.
The plasma pharmacokinetic profile of FTC released from the prodrug following i.v. administration showed a ~2-fold increase in AUC compared to the free drug control. For controlled release formulations, generally released drug plasma AUC is the same or lower than the free drug control due to the sustained or sometimes incomplete release of drug over time.31,46 With renal clearance being the primary route of elimination for FTC47, one possible explanation for the higher AUC is that the polymer backbone of the prodrug, or components of backbone, diminished renal clearance of the released FTC. This reduced clearance may also explain the difference in MRT which was 2-fold higher in the prodrug group. However, similar to orally administered drugs, MRT of controlled release formulations reflects both drug input and elimination rates.48 Since we were unable to directly measure the prodrug release rate in vivo, the calculated MRT and Vss (which is derived from MRT) are likely overestimated for the prodrug group. The lack of an observed absorption phase relating to prodrug release most likely reflects the rapid SR-A1-mediated uptake and hydrolysis of the prodrug in myeloid cells.
It is believed that the lymph node distribution of the PLS platform is the result of a combination of endothelial SR-A1 mediated transcytosis and SR-A1-mediated myeloid cell uptake within the lymph node due to the interactions between the receptor and the succinyl groups of PLS. Even with high FTC loading of 14% by weight, there remains approximately 200 free succinyl groups per prodrug molecule available for SR-A1 targeting, which resulted in ~7-19-fold higher concentrations of FTC in spleen, liver, and lymph nodes. Previous studies utilizing liposomes also successfully increased total drug concentrations in lymph nodes. For example, pegylated liposomes (immunoliposomes) targeting HLA-DR, an MHC II receptor that is found not only on the surface of macrophages and activated CD4+ T cells, but also within the envelope of HIV-1 virus particles in chronically infected patients, were shown to achieve 21 to 126-fold higher indinavir drug exposure to lymphatic tissues and plasma.49, 50, 51 One major caveat to these liposome studies is that the authors only reported the total drug concentrations and did not differentiate between encapsulated and unencapsulated (pharmacologically active) drug fractions. Liposomes are generally very stable, with drugs loaded in the aqueous core releasing very slowly, as in the case of Doxil, the most well-known example.52 Therefore, achieving increased total drug concentrations in lymph nodes may not be efficacious if drug release is too slow. Instead, optimal drug release rates must be coordinated with lymphatic-targeted delivery to achieve and maintain efficacious drug concentrations in tissues harboring the HIV viral reservoirs.31
Our PLS platform targets SR-A1, which is primarily found on endothelial cells and myeloid cells, such as macrophages.39,,43 Although myeloid cells within the lymph node are targeted by PLS, it is the CD4+ T cells that are recognized as the primary HIV reservoir within this tissue.5 Regardless, the PLS-FTC prodrug was able to raise lymph node FTC concentrations dramatically in comparison to FTC control, suggesting that CD4+ T cell exposure to FTC within the lymph node also increased. It is also important to note, however, that recent studies suggest that the macrophage may play a much more important role in HIV transmission, viral latency and viral rebound than previously recognized.53 For example, macrophages have been shown to be the primary HIV reservoir within the urethral mucosa of patients under suppressive antiretroviral therapy and may play a dominant role in both initial infection and transmission.54 In antiretroviral suppressed humanized mice, macrophage were identified as rare transcription/translation competent cells within the lymphatic reservoir, highlighting their importance in any type of “kick and kill” eradication strategy.55
Overall, these findings support a novel, macromolecular prodrug strategy to selectively deliver drugs to the lymphatic system. Although the FTC prodrug significantly increased FTC exposure in lymphatic tissues, efficacy studies in non-human primate simian immunodeficiency virus (SIV) or humanized mouse models will be required to determine if the prodrug has utility as an HIV therapy. Due to its ability to increase drug exposure to lymphatic tissues and therefore potentially offer cells within these tissues better protection from infection, the prodrug may be useful as an HIV prevention strategy or as a combination therapy with “kick and kill” strategies. The PLS platform is versatile, and various other PLS-prodrug versions can be envisioned that may prove useful in other infectious disease indications where lymphatic organs or myeloid cells are the therapeutic target. For example, alveolar macrophages are involved in early infection and lung dissemination of mycobacterium tuberculosis, and our platform has demonstrated both lung distribution and macrophage targeting.56 Another area that is potentially amenable to PLS-mediated macrophage targeting includes the depletion or reprogramming of immunosuppressive tumor associated macrophage that is a recent focus of cancer immunotherapy.57
CONCLUSION
In summary, this study demonstrates the SR-A1 targeting and controlled drug release properties of PLS, and its potential use as a macromolecular prodrug platform. In vitro assays confirmed SR-A1-mediated cell uptake, and in vivo studies demonstrated dramatic lymph node distribution, consistent with previously reported SR-A targeted constructs. As a proof-of-concept, an FTC-PLS prodrug was synthesized, although it is important to note that virtually any drug with a primary or secondary hydroxyl group is amenable to this esterification chemistry. The FTC-PLS prodrug was found to have controlled drug release properties in vitro that occurred through both enzymatic and non-enzymatic mechanisms. Most importantly, in rats the prodrug achieved significantly higher FTC concentrations in lymph nodes in comparison to the free drug control, supporting the use of PLS as a prodrug carrier to increase lymphatic drug exposure. Overall, we believe the PLS prodrug platform has tremendous potential for the treatment of infectious diseases and cancer.
Supplementary Material
ACKNOWLEDGMENTS
The study was supported in whole or in part by federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E, and by the the University of North Carolina at Chapel Hill Center for AIDS Research (CFAR), an NIH funded program P30 AI50410. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The authors would like to acknowledge Drs. Yingwen Hu and Jeffrey Clogston (Nanotechnology Characterization Lab, Frederick National Laboratory for Cancer Research sponsored by the National Cancer Institute, Frederick, Maryland) for their assistance performing SEC/MALS analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org
SEC chromatograms of PLS-488 and PLS-Cy7.5; 1H NMR spectra of PLS, partially capped PLS, fully capped PLS, FTC, 2-(2-methoxyethoxy)ethanamine, and FTC; binding and uptake of PLS-488 in RAW 264.7 cells following co-treatment with partially capped PLS, fully capped PLS, poly C, or poly I; flow cytometry plots of RAW 264.7 and clone 1/2 cells following 24 h incubation with PLS-488; confocal images of PLS-488 uptake in RAW 264.7 and clone ½ cells; whole-body images of PLS-Cy7.5 treated mice; ex vivo images of organs harvested 6 h after PLS-Cy7.5 i.v. injection; IHC staining controls; standard curves of FTC with and without NaOH hydrolysis.
References
- 1.Hu H; Hang JJ; Han T; Zhuo M; Jiao F; Wang LW, The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumor Biol 2016, 37 (7), 8657–8664. [DOI] [PubMed] [Google Scholar]
- 2.Ries CH; Cannarile MA; Hoves S; Benz J; Wartha K; Runza V; Rey-Giraud F; Pradel LP; Feuerhake F; Klaman I; Jones T; Jucknischke U; Scheiblich S; Kaluza K; Gorr IH; Walz A; Abiraj K; Cassier PA; Sica A; Gomez-Roca C; de Visser KE; Italiano A; Le Tourneau C; Delord JP; Levitsky H; Blay JY; Ruttinger D, Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25 (6), 846–859. [DOI] [PubMed] [Google Scholar]
- 3.Zhao YL; Haney MJ; Gupta R; Bohnsack JP; He ZJ; Kabanov AV; Batrakova EV, GDNF-Transfected Macrophages Produce Potent Neuroprotective Effects in Parkinson’s Disease Mouse Model. Plos One 2014, 9 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dou HY; Grotepas CB; McMillan JM; Destache CJ; Chaubal M; Werling J; Kipp J; Rabinow B; Gendelman HE, Macrophage Delivery of Nanoformulated Antiretroviral Drug to the Brain in a Murine Model of NeuroAIDS. J Immunol 2009, 183 (1), 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blankson JN; Persaud D; Siliciano RF, The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med 2002, 53, 557–593. [DOI] [PubMed] [Google Scholar]
- 6.Honeycutt JB; Wahl A; Baker C; Spagnuolo RA; Foster J; Zakharova O; Wietgrefe S; Caro-Vegas C; Madden V; Sharpe G; Haase AT; Eron JJ; Garcia JV, Macrophages sustain HIV replication in vivo independently of T cells. J Clin Invest 2016, 126 (4), 1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan L; Pooyan S; Hu P; Leibowitz MJ; Stein S; Sinko PJ, Peritoneal macrophage uptake, pharmacokinetics and biodistribution of macrophage-targeted PEG-fMLF (N-formyl-methionyl-leucyl-phenylalanine) nanocarriers for improving HIV drug delivery. Pharm Res 2007, 24 (11), 2110–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingersoll MA; Spanbroek R; Lottaz C; Gautier EL; Frankenberger M; Hoffmann R; Lang R; Haniffa M; Collin M; Tacke F; Habenicht AJR; Ziegler-Heitbrock L; Randolph GJ, Comparison of gene expression profiles between human and mouse monocyte subsets. (vol 115, pg e10, 2010). Blood 2010, 116 (5), 857–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draude G; Von Hundelshausen P; Frankenberger M; Ziegler-Heitbrock HWL; Weber C, Distinct scavenger receptor expression and function in the human CD14(+)/CD16(+) monocyte subset. Am J Physiol-Heart C 1999, 276 (4), H1144–H1149. [DOI] [PubMed] [Google Scholar]
- 10.Loboda A; Jazwa A; Jozkowicz A; Molema G; Dulak J, Angiogenic transcriptome of human microvascular endothelial cells: Effect of hypoxia, modulation by atorvastatin. Vasc Pharmacol 2006, 44 (4), 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apostolov EO; Shah SV; Ray D; Basnakian AG, Scavenger Receptors of Endothelial Cells Mediate the Uptake and Cellular Proatherogenic Effects of Carbamylated LDL. Arterioscl Throm Vas 2009, 29 (10), 1622–U532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao Y; Karmali PP; Mukthavaram R; Kesari S; Kouznetsova VL; Tsigelny IF; Simberg D, Direct Recognition of Superparamagnetic Nanocrystals by Macrophage Scavenger Receptor SR-AI. Acs Nano 2013, 7 (5), 4289–4298. [DOI] [PubMed] [Google Scholar]
- 13.Segers FME; den Adel B; Bot I; van der Graaf LM; van der Veer EP; Gonzalez W; Raynal I; de Winther M; Wodzig WK; Poelmann RE; van Berkel TJC; van der Weerd L; Biessen EAL, Scavenger Receptor-AI-Targeted Iron Oxide Nanoparticles for In Vivo MRI Detection of Atherosclerotic Lesions. Arterioscl Throm Vas 2013, 33 (8), 1812–1819. [DOI] [PubMed] [Google Scholar]
- 14.Wang HY; Wu LX; Reinhard BM, Scavenger Receptor Mediated Endocytosis of Silver Nanoparticles into J774A.1 Macrophages Is Heterogeneous. Acs Nano 2012, 6 (8), 7122–7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Basu S; Mukhopadhyay B; Basu SK; Mukhopadhyay A, Enhanced Intracellular Delivery of Doxorubicin by Scavenger Receptor-Mediated Endocytosis for Preferential Killing of Histiocytic Lymphoma-Cells in Culture. Febs Lett 1994, 342 (3), 249–254. [DOI] [PubMed] [Google Scholar]
- 16.Swart PJ; Beljaars L; Kuipers ME; Smit C; Nieuwenhuis P; Meijer DKF, Homing of negatively charged albumins to the lymphatic system - General implications for drug targeting to peripheral tissues and viral reservoirs. Biochem Pharmacol 1999, 58 (9), 1425–1435. [DOI] [PubMed] [Google Scholar]
- 17.Jansen RW; Olinga P; Harms G; Meijer DKF, Pharmacokinetic Analysis and Cellular-Distribution of the Anti-Hiv Compound Succinylated Human Serum-Albumin (Suc-Hsa) in-Vivo and in the Isolated-Perfused Rat-Liver. Pharm Res 1993, 10 (11), 1611–1614. [DOI] [PubMed] [Google Scholar]
- 18.Apostolov EO; Shah SV; Ray D; Basnakian AG, Scavenger receptors of endothelial cells mediate the uptake and cellular proatherogenic effects of carbamylated LDL. Arterioscler Thromb Vasc Biol 2009, 29 (10), 1622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swart PJ; Kuipers ME; Schutten M; Proost JH; vanAmerongen G; Osterhaus ADME; Meijer DKF, Pharmacokinetics of succinylated serum albumin in mice, rats and monkeys: Implications for dosage regimens in the therapy of HIV-infection and anti-HIV efficacy in mice. Crs Bui Nat 1996, 103–104. [Google Scholar]
- 20.Vermeulen JN; Meijer DKF; Over J; Lange JMA; Proost JH; Bakker HI; Beljaars L; Wit FWNM; Prins JM, A Phase I/IIa study with succinylated human serum albumin (Suc-HSA), a candidate HIV-1 fusion inhibitor. Antivir Ther 2007, 12 (2), 273–278. [PubMed] [Google Scholar]
- 21.Ganchua SKC; Cadena AM; Maiello P; Gideon HP; Myers AJ; Junecko BF; Klein EC; Lin PL; Mattila JT; Flynn JL, Lymph nodes are sites of prolonged bacterial persistence during Mycobacterium tuberculosis infection in macaques. PLoS Pathog 2018, 14 (11), e1007337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.G.R.F K, Pathology of Epstein-Barr Virus (EBV)-Associated Disease (The Lymphatic System) In Epstein-Barr Virus and Associated Diseases. Developments in Medical Virology, Levine PH, A. D V, Pearson GR, Kottaridis SD , Ed. Springer: Boston, MA, 1985; Vol. 1. [Google Scholar]
- 23.Lorenzo-Redondo R; Fryer HR; Bedford T; Kim EY; Archer J; Pond SLK; Chung YS; Penugonda S; Chipman J; Fletcher CV; Schacker TW; Malim MH; Rambaut A; Haase AT; McLean AR; Wolinsky SM, Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530 (7588), 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarmati L; D’Ettorre G; Parisi SG; Andreoni M, HIV Replication at Low Copy Number and its Correlation with the HIV Reservoir: A Clinical Perspective. Curr Hiv Res 2015, 13 (3), 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorenzo-Redondo R; Fryer HR; Bedford T; Kim EY; Archer J; Pond SLK; Chung YS; Penugonda S; Chipman JG; Fletcher CV; Schacker TW; Malim MH; Rambaut A; Haase AT; McLean AR; Wolinsky SM, Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530 (7588), 51-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur CD; Nahar M; Jain NK, Lymphatic targeting of zidovudine using surface-engineered liposomes. J Drug Target 2008, 16 (10), 798–805. [DOI] [PubMed] [Google Scholar]
- 27.Kaur A; Jain S; Tiwary AK, Mannan-coated gelatin nanoparticles for sustained and targeted delivery of didanosine: In vitro and in vivo evaluation. Acta Pharmaceut 2008, 58 (1), 61–74. [DOI] [PubMed] [Google Scholar]
- 28.Garg M; Jain NK, Reduced hematopoietic toxicity, enhanced cellular uptake and altered pharmacokinetics of azidothymidine loaded galactosylated liposomes. J Drug Target 2006, 14 (1), 1–11. [DOI] [PubMed] [Google Scholar]
- 29.Garg M; Dutta T; Jain NK, Reduced hepatic toxicity, enhanced cellular uptake and altered pharmacokinetics of stavudine loaded galactosylated liposomes. Eur J Pharm Biopharm 2007, 67 (1), 76–85. [DOI] [PubMed] [Google Scholar]
- 30.McConnachie LA; Kinman LM; Koehn J; Kraft JC; Lane S; Lee W; Collier AC; Ho RJY, Long-Acting Profile of 4 Drugs in 1 Anti-HIV Nanosuspension in Nonhuman Primates for 5 Weeks After a Single Subcutaneous Injection. J Pharm Sci 2018, 107 (7), 1787–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stern ST; Hall JB; Yu LL; Wood LJ; Paciotti GF; Tamarkin L; Long SE; McNeil SE, Translational considerations for cancer nanomedicine. Journal of Controlled Release 2010, 146 (2), 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grabarek Z; Gergely J, Zero-Length Crosslinking Procedure with the Use of Active Esters. Anal Biochem 1990, 185 (1), 131–135. [DOI] [PubMed] [Google Scholar]
- 33.Franca A; Aggarwal P; Barsov EV; Kozlov SV; Dobrovolskaia MA; Gonzalez-Fernandez A, Macrophage scavenger receptor A mediates the uptake of gold colloids by macrophages in vitro. Nanomedicine-Uk 2011, 6 (7), 1175–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robichaud G; Barry JA; Muddiman DC, IR-MALDESI mass spectrometry imaging of biological tissue sections using ice as a matrix. J Am Soc Mass Spectrom 2014, 25 (3), 319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bokhart MT; Rosen E; Thompson C; Sykes C; Kashuba AD; Muddiman DC, Quantitative mass spectrometry imaging of emtricitabine in cervical tissue model using infrared matrix-assisted laser desorption electrospray ionization. Anal Bioanal Chem 2015, 407 (8), 2073–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kessner D; Chambers M; Burke R; Agus D; Mallick P, ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 2008, 24 (21), 2534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robichaud G; Garrard KP; Barry JA; Muddiman DC, MSiReader: an open-source interface to view and analyze high resolving power MS imaging files on Matlab platform. J Am Soc Mass Spectrom 2013, 24 (5), 718–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice PJ; Kelley JL; Kogan G; Ensley HE; Kalbfleisch JH; Browder IW; Williams DL, Human monocyte scavenger receptors are pattern recognition receptors for (1 -> 3)-beta-D-glucans. J Leukocyte Biol 2002, 72 (1), 140–146. [PubMed] [Google Scholar]
- 39.Zani IA; Stephen SL; Mughal NA; Russell D; Homer-Vanniasinkam S; Wheatcroft SB; Ponnambalam S, Scavenger Receptor Structure and Function in Health and Disease. Cells 2015, 4 (2), 178–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jorgensen KE; Moller JV, Use of Flexible Polymers as Probes of Glomerular Pore-Size. Am J Physiol 1979, 236 (2), F103–F111. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen CT; Webb RI; Lambert LK; Strounina E; Lee EC; Parat MO; McGuckin MA; Popat A; Cabot PJ; Ross BP, Bifunctional Succinylated epsilon-Polylysine-Coated Mesoporous Silica Nanoparticles for pH-Responsive and Intracellular Drug Delivery Targeting the Colon. ACS applied materials & interfaces 2017, 9 (11), 9470–9483. [DOI] [PubMed] [Google Scholar]
- 42.Aderem A; Underhill DM, Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 1999, 17, 593–623. [DOI] [PubMed] [Google Scholar]
- 43.Tokuda H; Masuda S; Takakura Y; Sezaki H; Hashida M, Specific Uptake of Succinylated Proteins Via a Scavenger Receptor-Mediated Mechanism in Cultured Brain Microvessel Endothelial-Cells. Biochem Bioph Res Co 1993, 196 (1), 18–24. [DOI] [PubMed] [Google Scholar]
- 44.Singer JW; Shaffer S; Baker B; Bernareggi A; Stromatt S; Nienstedt D; Besman M, Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anti-Cancer Drug 2005, 16 (3), 243–254. [DOI] [PubMed] [Google Scholar]
- 45.Zhao J; Koay EJ; Li T; Wen X; Li C, A hindsight reflection on the clinical studies of poly(l-glutamic acid)-paclitaxel. Wiley Interdiscip Rev Nanomed Nanobiotechnol 2018, 10 (3), e1497. [DOI] [PubMed] [Google Scholar]
- 46.Stern ST; Zou P; Skoczen S; Xie S; Liboiron B; Harasym T; Tardi P; Mayer LD; McNeil SE, Prediction of nanoparticle prodrug metabolism by pharmacokinetic modeling of biliary excretion. J Control Release 2013, 172 (2), 558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.FDA, Application Number 21-500, Emtriva. Center for Drug Evaluation and Research, Clnical Pharmacology and Biopharmaceutics Review 2003. [Google Scholar]
- 48.Bialer M; Look ZM; Silber BM; Yacobi A, The relationship between drug input and mean residence time in the body. Biopharm Drug Dispos 1986, 7 (6), 577–83. [DOI] [PubMed] [Google Scholar]
- 49.Cantin R; Fortin JF; Lamontagne G; Tremblay M, The presence of host-derived HLA-DR1 on human immunodeficiency virus type 1 increases viral infectivity. J Virol 1997, 71 (3), 1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawn SD; Butera ST, Incorporation of HLA-DR into the envelope of human immunodeficiency virus type 1 in vivo: Correlation with stage of disease and presence of opportunistic infection. J Virol 2000, 74 (21), 10256–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gagne JF; Desormeaux A; Perron S; Tremblay MJ; Bergeron MG, Targeted delivery of indinavir to HIV-1 primary reservoirs with immunoliposomes. Bba-Biomembranes 2002, 1558 (2), 198–210. [DOI] [PubMed] [Google Scholar]
- 52.Barenholz Y, Doxil(R)--the first FDA-approved nano-drug: lessons learned. J Control Release 2012, 160 (2), 117–34. [DOI] [PubMed] [Google Scholar]
- 53.Wong ME; Jaworowski A; Hearps AC, The HIV Reservoir in Monocytes and Macrophages. Front Immunol 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ganor Y; Real F; Sennepin A; Dutertre CA; Prevedel L; Xu L; Tudor D; Charmeteau B; Couedel-Courteille A; Marion S; Zenak AR; Jourdain JP; Zhou ZC; Schmitt A; Capron C; Eugenin EA; Cheynier R; Revol M; Cristofari S; Hosmalin A; Bomsel M, HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol 2019, 4 (4), 633–644. [DOI] [PubMed] [Google Scholar]
- 55.Maidji E; Moreno ME; Rivera JM; Joshi P; Galkina SA; Kosikova G; Somsouk M; Stoddart CA, Cellular HIV Reservoirs and Viral Rebound from the Lymphoid Compartments of 4 ′-Ethynyl-2-Fluoro-2 ′-Deoxyadenosine (EFdA)-Suppressed Humanized Mice. Viruses-Basel 2019, 11 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen SB; Gern BH; Delahaye JL; Adams KN; Plumlee CR; Winkler JK; Sherman DR; Gerner MY; Urdahl KB, Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24 (3), 439–446 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guerriero JL, Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol Med 2018, 24 (5), 472–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.