

Abstract
Registry-based epidemiologic studies suggest associations between chronic inflammatory intestinal diseases and pancreatic ductal adenocarcinoma (PDAC). As genetic susceptibility contributes to a large proportion of chronic inflammatory intestinal diseases, we hypothesize that the genomic regions surrounding established genome-wide associated variants for these chronic inflammatory diseases are associated with PDAC. We examined the association between PDAC and genomic regions (+/− 500 kb) surrounding established common susceptibility variants for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, celiac disease, chronic pancreatitis, and primary sclerosing cholangitis. We analyzed summary statistics from genome-wide association studies data for 8,384 cases and 11,955 controls of European descent from two large consortium studies using the summary data-based adaptive rank truncated product method to examine the overall association of combined genomic regions for each inflammatory disease group. Combined genomic susceptibility regions for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, and chronic pancreatitis were associated with PDAC at P-values < 0.05 (0.0040, 0.0057, 0.011, and 3.4 × 10−6, respectively). After excluding the 20 PDAC susceptibility regions (+/− 500 kb) previously identified by GWAS, the genomic regions for ulcerative colitis, Crohn’s disease, and inflammatory bowel disease remained associated with PDAC (P-values = 0.0029, 0.0057, and 0.0098, respectively). Genomic regions for celiac disease (P-value = 0.22) and primary sclerosing cholangitis (P-value = 0.078) were not associated with PDAC. Our results support the hypothesis that genomic regions surrounding variants associated with inflammatory intestinal diseases, particularly, ulcerative colitis, Crohn’s disease, inflammatory bowel disease, and chronic pancreatitis are associated with PDAC.
Introduction
Pancreatic cancer, characterized by its increasing incidence and high fatality, is the 3rd leading cause of cancer-related mortality in the United States (US) (1). Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer (2). In addition to risk factors, such as cigarette smoking, excess weight and diabetes, genetic susceptibility contributes to the disease with an estimated heritability of up to 21% (3).
Chronic inflammation plays an important role in PDAC pathogenesis. The most direct link between inflammation and pancreatic cancer is derived from known associations with chronic pancreatitis, with risks being particularly strong for hereditary pancreatitis (4). Swedish registry-based studies have identified significant excess incidence of PDAC in patients with ulcerative colitis (5), Crohn’s disease (6), and primary sclerosing cholangitis (7) compared with the general Swedish population, albeit based on a relatively small number of PDAC cases. Ulcerative colitis and Crohn’s disease are characterized as idiopathic, immune-mediated recurrent or chronic inflammatory disorders of the gastrointestinal tract and are collectively classified as inflammatory bowel disease. Primary sclerosing cholangitis is a chronic autoimmune disease with progressive inflammation and fibrosis in the intrahepatic and extrahepatic biliary ducts and has also been associated with inflammatory bowel disease (8). Celiac disease, an autoimmune disorder characterized by chronic inflammation in small intestine, has been variably associated with PDAC (9,10). A limitation of registry-based studies and epidemiologic studies that rely on self-report is chronic inflammatory intestinal diseases may not be accurately ascertained (e.g., misdiagnosed with irritable bowel syndrome due to overlapping symptoms) (11,12), and thus affect risk estimates. Genetic susceptibility is known to contribute to each inflammatory disease mentioned above (13–16).
In this study, we examined the association between genomic regions surrounding common susceptibility variants identified from published genome-wide association studies (GWAS) of ulcerative colitis, Crohn’s disease, inflammatory bowel disease, celiac disease, chronic pancreatitis and primary sclerosing cholangitis, and risk of PDAC using GWAS data from two large PDAC consortia (17–21). We hypothesize that combined genomic susceptibility regions for each chronic inflammatory intestinal disease group will be associated with PDAC. The statistical approach we employed considers the joint association of multiple genomic regions with PDAC risk (22), and thus has the potential to detect associations that could be overlooked by traditional single-marker approaches.
Materials and Methods
Study sample
Our study was based on 9,038 primary PDAC cases (ICD-O-3 code C250-C259) and 12,389 controls of European ancestry from four GWAS conducted in the Pancreatic Cancer Cohort Consortium (PanScan I, II, III) and the Pancreatic Cancer Case Control Consortium (PanC4) (17–21). Participants with non-exocrine pancreatic tumors (histology types 8150, 8151, 8153, 8155 and 8240) were excluded because their etiologies are thought to be different. The details of the cases, controls and study design have been previously described (17–21). The three PanScan GWAS included participants from 16 cohorts [Alpha-Tocopherol, Beta-Carotene Study (ATBC, Finland), Agricultural Health Study (US), Give us a Clue to Cancer and Heart Disease Study (CLUE II, US), Cancer Preventions Study II (CPS-II, US), European Prospective Investigation into Cancer and Nutrition Study (EPIC, which comprises cohorts from Denmark, France, Germany, Great Britain, Greece, Italy, The Netherlands, Spain, and Sweden), Health Professional Follow-Up Study (HPFS, US), Multiethnic Cohort (US), Melbourne (MCCS, Australia), Nurse’s Health Study (NHS, US), New York University Women’s Health Study (US), Physician’s Health Study I (PHS, US), Prostate Lung Colorectal Ovarian Cancer Cohort (PLCO, US), Selenium and Vitamin E Cancer Prevention Trial (US), Vitamins and Lifestyle Cohort (US), Women Health Initiative Cohort (US), Women’s Health Study (WHS, US)] from the National Cancer Institute’s cohort consortium, 9 case-control studies and 1 case series (Gastrointestinal Cancer Clinic of Dana-Farber Cancer Institute, US). PanC4 included 9 case-control studies. The case-control studies included in either PanScan II or PanC4 were the Central Europe study coordinated by IARC (hospital-based, EU), Johns Hopkins Hospital (clinic-based, US), Mayo Clinic (clinic-based, US), MD Anderson Cancer Center (hospital-based, US), Memorial Sloan Kettering Cancer Center (clinic-based, US), PANDoRA- pancreatic cancer case-control study (clinic-based, EU), PACIFIC Study of Group Health and Northern California Kaiser Permanente (HMO, US), QIMR Berghofer Medical Research Institute (population-based, Australia), Spanish Pancreatic Cancer Study (hospital-based, Spain), University of California San Francisco (population-based, US), University of Toronto (population-based, Canada), and Yale University (population-based hospitals, US) (17–21). Controls for PanScan I and II and PanC4 were matched to cases by age, sex, self-reported race, area of residence (case-control studies) and/or smoking (HPFS, PHS, NHS, WHS cohorts only) and incidence density sampled within each respective cohort studies. PanScan Ⅲ used previously genotyped controls, mostly from cohort studies (ATBC, CPS-II, EPIC, HPFS, MCCS, MEC, NHS, PLCO, WHI). To facilitate stratified analyses by study design (cohort versus case-control) and evaluate potential survival bias, we excluded 654 cases and 434 controls from PanScan Ⅲ studies because they did not have comparable controls or cases by study designs because PanScan III used previously genotyped controls (e.g., exclusion of cases from case-control or case series studies that used cohort controls). We only included participants of European ancestry to avoid confounding by population stratification. Our final analytic data set included 8,384 (2,320 cohort, 6,064 case-control) PDAC cases and 11,955 (6,121 cohort, 5,834 case-control) controls.
Each participating study obtained written informed consent from participants and approval from their local Institutional Review Board. The National Cancer Institute’s Special Studies Institutional Review Board approved the consortia study.
GWAS summary statistics
Genotype imputation across the four study phases was based on the 1000 Genomes Project (Phase 3, v1) reference dataset (23) and IMPUTE2 (http://mathgen.stats.ox.ac.uk/impute/impute_v2.html) (24) as previously described (21). Due to the large overlap of variants on the arrays (Illumina HumanHap550 Infinium II, Human 610-Quad) used for PanScan I and II, these studies were combined and jointly analyzed, whereas PanScan III (OmniExpress, Omni1M, Omni2.5M and Omni5M) and PanC4 (Illumina HumanOmniExpressExome-8v1) were each analyzed separately. For quality control, SNPs with minor allele frequency (MAF) < 5% and low-quality imputation score (IMPUTE2 INFO score < 0.3) were excluded (21). For each GWAS phase, SNPTEST (http://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html) (25) was used to perform association analysis and generate summary statistics based on probabilistic genotype values from IMPUTE2. The analysis was adjusted for age (10-year categories, (⩽50, 51-60, 61-70, 71-80, and ⩾81)), sex, top eigenvectors for each study phase, study (PanScan), and geographic region (PanScan Ⅲ) as previously described (17–21). PanScan Ⅲ was adjusted for geographic region (US, central and northern Europe, and southern Europe) because of the use of the previously genotyped controls that were not necessarily from the same study or region as the cases (20).
SNP selection
We identified “index-” single-nucleotide polymorphisms (SNPs) associated with ulcerative colitis, Crohn’s disease, inflammatory bowel disease, celiac disease, chronic pancreatitis and primary sclerosing cholangitis at genome-wide association level (P-value < 5×10−8) using the most recent and largest studies curated by NHGRI-EBI Catalog of published GWAS (https://www.ebi.ac.uk/gwas/) as of October 9, 2017 (Supplementary Table S1). As some GWAS had inflammatory bowel disease as an outcome alone, we defined inflammatory bowel disease as a unique disease group rather than combining GWAS regions for ulcerative colitis and Crohn’s disease. We reviewed the original GWAS publications and added additional SNPs that met the significance level criterion (26). For SNPs that were not genotyped or imputed in our data set (i.e., genotyped on different platforms), we selected an alternative index-SNP in high linkage disequilibrium (LD, r2 ≥ 0.74) using LDlink (https://ldlink.nci.nih.gov) (27). We then included genomic regions +/− 500 kb surrounding each index-SNP and applied LD filtering to highly correlated SNP pairs (r2 > 0.80) in each disease group. In total, our analysis included the following number of index-SNP defined regions (SNPs in regions): 99 (728) for ulcerative colitis, 151 (997) for Crohn’s disease, 211 (1,403) for inflammatory bowel disease, 21 (148) for celiac disease, 11 (35) for chronic pancreatitis, and 15 (109) for primary sclerosing cholangitis. Fifty-five index-SNP defined regions associated with inflammatory bowel disease did not overlap with ulcerative colitis or Crohn’s disease.
Statistical analysis
We first conducted a meta-analysis combining SNP-level summary statistics from the four GWAS using an inverse-variance fixed-effects model. To eliminate the effect of population stratification, the square root of the genomic inflation factors for each study phase (λ = 1.02, 1.02, 1.01 and 1.06 for PanC4, PanScan I and II case-control, PanScan I and II cohort, and PanScan III cohort, respectively) was used to rescale the standard error of the estimated log odds ratio at each SNP before the meta-analysis. We then applied the summary data-based adaptive rank truncated product (sARTP) method (22) to the meta-analysis result. The genomic region surrounding an index-SNP was treated as a “gene”, and the disease-level analysis was the gene-set/pathway analysis by combining the signals across multiple regions defined by the index-SNPs for a given inflammatory intestinal disease group (22). The sARTP analysis selected up to five of the most significant outcome (PDAC)-associated SNPs within each index-SNP defined region and adjusted for multiple comparison through a resampling procedure. The disease-level sARTP adjusted for multiple comparison for tests done around all considered index-SNP/regions within a disease group through a similar resampling procedure. One-hundred million resampling steps were used to estimate the P-value of each index-SNP region and disease-level associations. A panel of 503 European subjects (population codes: CEU, TSI, FIN, GBR, IBS) in the 1000 Genomes Project (phase 3, v1) was used in sARTP to estimate the LD between SNPs. We considered a disease-level P-value less than or equal to 0.05 statistically significant. All statistical tests are two-sided.
We also conducted a sensitivity analysis excluding the 20 PDAC-associated risk signals identified by previous GWAS at 1q32.1 (NR5A2), 1p36.33 (NOC2L), 2p14 (ETAA1), 3q28 (TP63), 5p15.33 (CLPTM1L-TERT), 7p14.1 (SUGCT), 7q23.2 (LINC-PINT), 8q21.11 (HNF4G), 8q24.21 (MYC), 9q34.2 (ABO), 13q12.2 (PDX1), 13q22.1 (non-genic), 16q23.1 (BCAR1), 17q12 (HNF1B), 17q24.3 (LINC00673), 18q21.32 (GRP), and 22q12.1 (ZNRF3) (17–21,28) and genomic regions within +/− 500kb (22).
LD score regression
To estimate the genetic correlation between the disease groups and PDAC, we performed LD score regression (29) using summary statistics (excluding major histocompatibility complex regions) from PDAC GWAS (21) and GWAS for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, chronic pancreatitis and primary sclerosing cholangitis with comparable HapMap3 genotype imputation (30–32). Celiac disease was excluded from the analysis because the originating study did not have imputed genotyping data.
Pairwise LD analysis, functional annotation and eQTL analysis
We performed a pairwise LD analysis to evaluate haplotype patterns between alleles of the index-SNPs and sARTP-selected SNPs using LDlink (27). We conducted an exploratory analysis of expression quantitative trait locus (eQTL) data to assess the cis effects of the most statistically significant sARTP-selected SNPs (P-value ≤ 0.002) and their corresponding index-SNPs on gene expression in pancreas tissue and determined whether the same gene(s) were expressed in gastrointestinal tract, and whole blood tissues using data from the NIH Genotype-Tissue Expression (GTEx) v8 (33). We also examined the regulatory potential of these SNPs (and SNPs in LD) using experimental data and information from Ensembl (34), HaploReg v4.1 (35), and RegulomeDB v1.1 (36).
Data Availability
The majority of the data that support the findings of this study are available in dbGAP at https://www.ncbi.nlm.nih.gov/gap/, reference number [phs000206.v5.p3 and phs000648.v1.p1]. Biomedical research scientists from recognized research institutions can request data as bona fide researchers from dbGAP or by contacting the corresponding author.
Results
Table 1 shows the characteristics of our study population by genotyping phase and study design. The age and sex distribution of cases compared to controls within each study phase (PanScan I and II, PanC4) was similar except for PanScan Ⅲ which used previously genotyped controls. Overall, the majority of cases were diagnosed after age 60 years (74.1%), 39.1% of the cases were diagnosed after age 70 years, and 54.3% of the cases were men. A higher proportion of the cases from the cohort studies were diagnosed at older ages (87.7% > 60 years and 53.0% > 70 years) compared to the case-control studies (68.9% > 60 years and 33.8% > 70 years). The proportion of female cases was greater among the cohort studies (51.8%), while the proportion of male cases was greater in the case-control studies (56.6%).
Table 1.
Number of cases and controls, case diagnosis age, control enrollment age, and sex distribution by study phase and study design from PanScan and PanC4 studiesa
| Study Phase |
Total |
||||||
|---|---|---|---|---|---|---|---|
| Characteristic | PanScan I Case / control 1,746 / 1,812 |
PanScan II Case / control 1,768 / 1,841 |
PanScan III Case / control 937 / 4,651 |
PanC4 Case / control 3,933 / 3,651 |
Cohort Case / control 2,320 / 6,121 |
Case-control Case / control 6,064 / 5,834 |
Combined Case / control 8,384 / 11,955 |
| Case diagnosis age, > 60 years, n (%) | 1,419 (81.3) | 1,168 (66.0) | 880 (93.9) | 2,744 (69.7) | 2,034 (87.7) | 4,177 (68.9) | 6,211 (74.1) |
| Ageb, years, n (%) | |||||||
| ≤ 50 | 63 (3.6) / 44 (2.4) | 157 (8.9) / 181 (9.8) | 6 (0.6) / 104 (2.2) | 357 (9.1) / 389 (10.6) | 44 (1.9) / 125 (2.0) | 539 (8.9) / 593 (10.2) | 583 (6.9) / 718 (6.00) |
| 51-60 | 264 (15.1) / 226 (12.5) | 443 (25.1) / 422 (22.9) | 51 (5.5) / 687 (14.8) | 832 (21.2) / 910 (24.9) | 242 (10.4) / 849 (13.9) | 1,348 (22.2) / 1,396 (23.9) | 1,590 (19.0) / 2,245 (18.8) |
| 61-70 | 673 (38.6) / 724 (40.0) | 605 (34.2) / 599 (32.6) | 256 (27.3) / 2,450 (52.7) | 1,401 (35.6) / 1,225 (33.6) | 805 (34.7) / 3,053 (49.9) | 2,130 (35.1) / 1,945 (33.3) | 2,935 (35.0) / 4,998 (41.8) |
| ≥ 71 | 746 (42.7) / 818 (45.1) | 563 (31.8) / 639 (34.7) | 624 (66.6) / 1,410 (30.3) | 1,343 (34.1) / 1,127 (30.9) | 1,229 (53.0) / 2,094 (34.2) | 2,047 (33.8) / 1,900 (32.6) | 3,276 (39.1) / 3,994 (33.4) |
| Sex, n (%) | |||||||
| Male | 892 (51.1) / 925 (51.0) | 945 (53.45) / 965 (52.4) | 433 (46.2) / 3,422 (73.6) | 2,280 (57.97) / 2,038 (55.8) | 1,118 (48.2) / 4,154 (67.9) | 3,432 (56.6) / 3,196 (54.8) | 4,550 (54.3) / 7,350 (61.5) |
| Female | 854 (48.9) / 887 (49.0) | 823 (46.55) / 876 (47.6) | 504 (53.8) / 1,229 (26.4) | 1,653 (42.03) / 1,613 (44.2) | 1,202 (51.8) / 1,967 (32.1) | 2,632 (43.4) / 2,638 (45.2) | 3,834 (45.7) / 4,605 (38.5) |
The analysis (N=8,384 cases and 11,955 controls) was restricted to participants of European ancestry from the PanScan and PanC4 consortia.
Age at pancreatic cancer diagnosis or age when selected to be a control. PanScan III used previously genotyped controls.
Disease group and PDAC
Genetic susceptibility to the ulcerative colitis, Crohn’s disease, inflammatory bowel disease, and chronic pancreatitis groups (P-values = 0.0040, 0.0057, 0.011, and 3.4 × 10−6, respectively) was significantly associated with PDAC while that to the celiac disease and primary sclerosing cholangitis groups (P-values = 0.22 and 0.078) was not (Table 2). After excluding the previous PDAC GWAS risk signal regions, the associations for ulcerative colitis, Crohn’s disease, and inflammatory bowel disease remained (P-values = 0.0029, 0.0057, and 0.0098), but that for chronic pancreatitis did not (P-value = 0.073). Associations for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, chronic pancreatitis and primary sclerosing cholangitis (P-values = 0.022, 0.0069, 0.014, 3.2 × 10−6, and 0.018) were present in the case-control studies but not in the cohort studies (Supplementary Table S2).
Table 2.
Association of genetic susceptibility to inflammatory intestinal disease groups and risk of PDAC in participants of European ancestry from PanScan and PanC4 studiesa
| Index-SNP defined region |
Excluding previous PDAC GWAS locic |
|||||
|---|---|---|---|---|---|---|
| Disease group | n | SNPb, n | P-value | n | SNP, n | P-value |
| Ulcerative colitis | 99 | 728 | 0.0040 | 96 | 716 | 0.0029 |
| Crohn’s disease | 151 | 997 | 0.0057 | 151 | 997 | 0.0057 |
| Inflammatory bowel disease | 211 | 1,403 | 0.011 | 208 | 1,393 | 0.0098 |
| Celiac disease | 21 | 148 | 0.22 | 21 | 148 | 0.22 |
| Chronic pancreatitis | 11 | 35 | 3.4 × 10−6 | 10 | 32 | 0.073 |
| Primary sclerosing cholangitis | 15 | 109 | 0.078 | 15 | 109 | 0.078 |
Abbreviations: GWAS, genome-wide association study; PDAC, pancreatic ductal adenocarcinoma; SNP, single-nucleotide polymorphism.
The analysis (N=8,384 cases and 11,955 controls) was adjusted for age, sex, study, geographic region, and the top eigenvectors for the PanScan studies and age, sex, and the top eigenvectors for the PanC4 studies.
These SNPs are within +/− 500kb genomic regions around index-SNPs and correlated with index-SNPs at r2 > 0.80.
Results were obtained after excluding +/− 500kb genomic regions of 20 PDAC GWAS risk signals.
The PDAC associations for the index-SNP defined regions, and for SNPs selected by sARTP within each region are shown in Supplementary Table S3 and Supplementary Table S4, respectively. The index-SNPs were highly correlated with their sARTP-selected SNPs, with LD r2 value ≥ 0.78 among all SNP pairs.
In total, 32 out of 99 index-SNP defined regions for ulcerative colitis, 28 out of 151 regions for Crohn’s disease and 30 out of 211 regions for inflammatory bowel disease were selected by sARTP and contributed to the associations between disease groups and PDAC (Table 3). There was some overlap of the regions across these disease groups. For example, 6 index-SNP defined regions were in common for ulcerative colitis and Crohn’s disease (rs2872507, rs12946510, rs2413583, rs7517847, rs9868809, rs3766606) (Figure 1). The most significant index-SNP defined regions associated with PDAC (P-value < 0.01) and corresponding gene (location) contributing to the associations included rs9858542 (BSN, 3p21.31; Crohn’s disease, inflammatory bowel disease), rs6651252 (LINC00824, 8q24.21; Crohn’s disease, inflammatory bowel disease), rs2872507 (intergenic variant, 17q21.1; ulcerative colitis, Crohn’s disease, inflammatory bowel disease), rs10883365 (LINC01475, 10q24.2; Crohn’s disease, inflammatory bowel disease), rs9988642 (IL23R, 1p31.3; Crohn’s disease, inflammatory bowel disease), rs4256018 (FERMT1, 20p12.3; inflammatory bowel disease), rs7809799 (KPNA7, 7q22.1; ulcerative colitis, inflammatory bowel disease), rs9822268 (APEH, 3p21.31; ulcerative colitis), rs4409764 (intergenic variant, 10q24.2; ulcerative colitis), rs80174646 (IL23R, 1p31.3; ulcerative colitis), and rs568617 (FIBP, 11q13.1; Crohn’s disease).
Table 3.
Expression quantitative trait loci (eQTL) for select index-SNPsa in normal tissues of pancreas, traverse colon and small intestine from GTExb
| GTEx pancreas (n=305) |
GTEx colon-transverse (n=368) |
GTEx small intestine-terminal ileum (n=174) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Index-SNP | Location | Positionc | Gene | r2d | eQTL gene | P-value | Effect size (β)e | P-value | Effect size (β) | P-value | Effect size (β) |
| rs9858542 ‖# | 3p21.31 | 49701983 | BSN | Ref. | UBA7 | 2.4 × 10−9 | 0.29 | 1.2 × 10−12 | 0.26 | 5.5 × 10−6 | 0.17 |
| rs9822268 ╪ | 3p21.31 | 49719729 | APEH | 1.00 | UBA7 | 7.4 × 10−10 | 0.30 | 6.9 × 10−13 | 0.26 | 6.0 × 10−6 | 0.16 |
| rs3197999 ¶ | 3p21.31 | 49721532 | MST1 | 0.95 | UBA7 | 2.7 × 10−9 | 0.29 | 1.2 × 10−12 | 0.26 | 3.9 × 10−6 | 0.17 |
| rs8055167 § | 16q23.1 | 75254889 | CTRB1, CRTB2 | — | CTRB2 | 3.4 × 10−17 | −0.50 | — | — | — | — |
| rs2872507 ‖╪# | 17q21.1 | 38040763 | — | — | PGAP3 | 5.6 × 10−10 | −0.23 | 2.8 × 10−8 | −0.11 | 2.5 × 10−7 | −0.16 |
Abbreviations: Chr., chromosome; eQTL, expression quantitative trait loci; PDAC, pancreatic ductal adenocarcinoma; sARTP, summary data-based adaptive rank truncated product; SNP, single-nucleotide polymorphism; TSS, transcription start site.
Only index-SNPs whose corresponding sARTP-selected SNPs show the strongest association with PDAC (P-value ≤ 0.002) are presented in the table.
cis-acting eQTLs are examined by using +/− 1 Mb cis-window around a gene’s TSS.
SNP position according to NCBI Human Genome Build 37.
Linkage disequilibrium r2 values are obtained from NCI LDlink EUR population data.
Effect size (β) denotes the directional effect for alternative allele relative to the reference allele. The magnitude has no direct biological interpretation.
Crohn’s disease
Ulcerative colitis
Inflammatory bowel disease
Chronic pancreatitis
Primary sclerosing cholangitis
Figure 1.
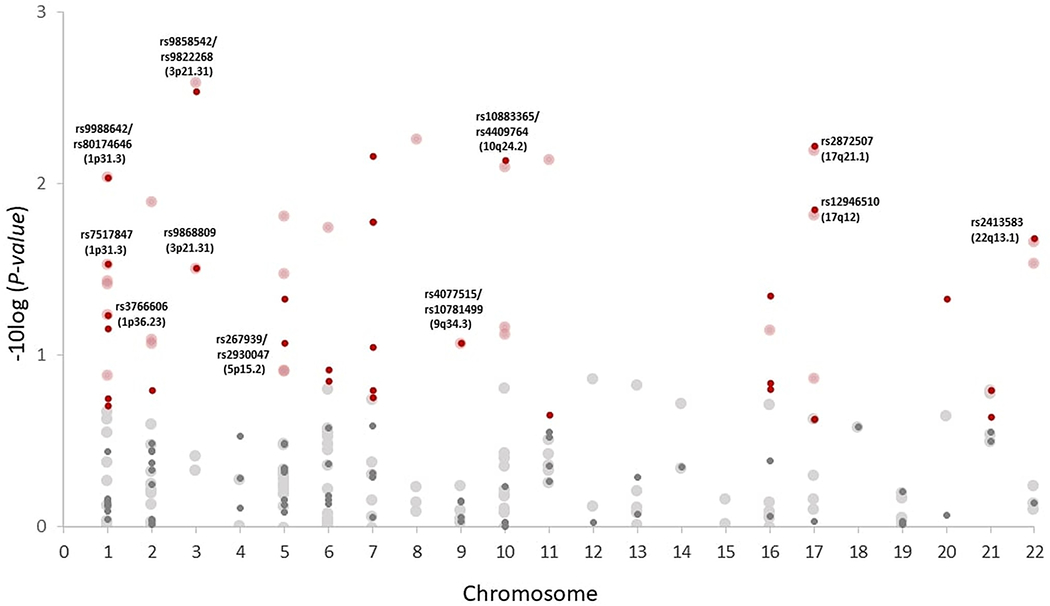
Index-SNP defined regions contributing to the associations of Crohn’s disease and ulcerative colitis with PDAC. Index-SNP defined regions associated with PDAC in ulcerative colitis (small circle) and Crohn’s disease (large circle). Circles highlighted in red are regions contributing to each disease group-PDAC association. Circles labeled with SNP IDs and locations represent index-SNP defined regions that are in common between Crohn’s disease and ulcerative colitis. The analysis (N=8,384 cases and 11,955 controls) is adjusted for age, sex, study, geographic region, and the top eigenvectors for the PanScan studies and age, sex, and the top eigenvectors for the PanC4 studies. All statistical tests are two-sided. PDAC = pancreatic ductal adenocarcinoma; sARTP = summary data-based adaptive rank truncated product.
Five out of 21 index-SNP defined regions for celiac disease, 1 out of 11 regions for chronic pancreatitis, and 1 out of 15 regions for primary sclerosing cholangitis contributed to the associations. The index-SNP defined region for chronic pancreatitis that contributed to the chronic pancreatitis-PDAC association was rs8055167 (CTRB1, CTRB2, 16q23.1, P-value = 1.0 × 10−7). The index-SNP defined region that contributed to the primary sclerosing cholangitis-PDAC association was rs3197999 (MST1, 3p21.31, P-value = 0.0028).
LD score regression
The genetic correlation rg (standard error, se) of each disease group with PDAC was 0.08 (P-value = 0.42, se = 0.10) for ulcerative colitis, −0.16 (P-value = 0.055, se = 0.08) for Crohn’s disease, −0.07 (P-value = 0.39, se = 0.08) for inflammatory bowel disease, 0.28 (P-value = 0.49, se = 0.41) for chronic pancreatitis, and −0.13 (P-value = 0.25, se = 0.12) for primary sclerosing cholangitis.
eQTL and functional annotation
The exploratory eQTL results for select index-SNPs and their corresponding sARTP-selected SNPs with the strongest evidence of association with PDAC are shown in Table 3 and Supplementary Table S5. Their functional annotations were shown in Supplementary Table S6 and Supplementary Table S7. In three regions defined by correlated index-SNPs (r2 ≥ 0.95) associated with ulcerative colitis, Crohn’s disease, inflammatory bowel disease, or primary sclerosing cholangitis, variants rs9858542-A, rs9822268-A, rs3197999-A were associated with increased UBA7 expression in normal tissues of pancreas, transverse colon, and small intestine (P-values ≤ 6.0 × 10−6). These SNPs were associated with increased UBA7 expression in esophageal mucosa, stomach, and whole blood (Supplementary Table S5). In the region defined by the chronic pancreatitis index-SNP rs8055167, variant rs8055167-C was associated with lower CTRB2 expression and increased CTRB1 expression in normal pancreatic tissue (P-values ≤ 2.0 × 10−6). In the region defined by ulcerative colitis/Crohn’s disease/inflammatory bowel disease index-SNP rs2872507, variant rs2872507-A was associated with decreased PGAP3 expression in normal pancreatic, transverse colon and small intestine tissues (P-values ≤ 2.5 × 10−7). Similar eQTL effects were present in the esophagus-gastroesophageal junction, stomach, and whole blood.
Discussion
Our analysis of GWAS data showed that the joint effects of common variants in genomic regions containing susceptibility loci for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, and chronic pancreatitis were associated with PDAC. After excluding the previously established PDAC-associated regions, the genomic regions for ulcerative colitis, Crohn’s disease, and inflammatory bowel disease remained associated with PDAC, but the association for chronic pancreatitis was no longer significant. No significant associations were observed for genetic susceptibility to celiac disease or primary sclerosing cholangitis. Our results are consistent with the previous Swedish registry-based studies of patients previously diagnosed with ulcerative colitis and Crohn’s disease that found positive associations with PDAC compared with the general population (5,6), and the known association between chronic pancreatitis and PDAC (4).
Although previous GWAS suggest overlap of genetic susceptibility between ulcerative colitis and Crohn’s disease (37), a connection with PDAC remains to be elucidated. In this study, we observed that multiple common regions between ulcerative colitis and Crohn’s disease are associated with PDAC, suggesting some shared underlying biology between the two inflammatory diseases that contributes to the risk of PDAC. Of note, index-SNP rs7517847 (ulcerative colitis, Crohn’s disease) resides in IL23R (Supplementary Table S3), which encodes a subunit of the receptor for interleukin-23 (IL-23). The IL-23 signaling pathway mediates intestinal inflammation (38) and promotes inflammation-induced tumorigenesis in colon in murine models (39). In addition, expression of IL-23 was significantly higher in pancreatic tumor compared with normal adjacent tissue (40).
With regard to the three inflammatory bowel disease groups, susceptibility variants residing at loci 3p21.31 and 17q12-q21 are of interest due to their associations with PDAC (Supplementary Table S4), and significant cis-eQTL effects in pancreas, transverse colon and small intestine (Table 3). Index-SNPs rs9858542 (Crohn’s disease, inflammatory bowel disease), rs9822268 (ulcerative colitis) and rs3197999 (primary sclerosing cholangitis) reside at 3p21.31 (Supplementary Table S3), a region in which multiple susceptibility loci for chronic inflammatory intestinal diseases have been identified (26,31,37,41). The three index SNPs and their sARTP-selected SNPs are eQTLs for UBA7. Previous studies suggested a tumor suppressive effect of UBE1L (the protein encoded by UBA7) in both lung cancer and acute promyelocytic leukemia (42,43). Index-SNP rs2872507 is an intergenic variant located at 17q12-q21, a locus found to be associated with many immune-mediated diseases (44). rs2872507 and its sARTP-selected SNPs are associated with decreased PGAP3 expression in pancreas, transverse colon and small intestine. PGAP3 is frequently coamplified with the oncogene ERBB2 in breast and gastric cancer (45,46), and co-silencing of PGAP3 with ERBB2 in vitro results in an additive inhibition of cell viability and induced apoptosis (47). In an agnostic pathway analysis, we previously identified PGAP3 as a top contributing gene associated with PDAC within the Nikolsky breast cancer chr17q11-q21 amplicon gene set with four SNPs associated with decreased PGAP3 expression (48). In a separate transcriptome-wide association study, genetically predicted expression of PGAP3 was found to be associated with PDAC risk (49). Our findings based on eQTL analysis are suggestive and further colocalization analysis may help identify a shared etiology between inflammatory bowel disease and PDAC.
Our sensitivity analyses suggest that the associations we observe between ulcerative colitis, Crohn’s disease, inflammatory bowel disease groups and PDAC are not driven by previously identified PDAC GWAS loci. The chronic pancreatitis group, however, was no longer associated with PDAC. The association observed in the primary analysis was driven by the PDAC-associated SNP rs8051363, which is in close proximity to the PDAC risk signal rs7190458 (BCAR1/CTRB1/CTRB2) at 16q23.1 (20,21).
Our analysis is based on regions surrounding established susceptibility variants from GWAS of each inflammatory intestinal disease. As a result, we may not be able to observe an association for disease groups with only few established GWAS loci (e.g., primary sclerosing cholangitis). In contrast to Mendelian randomization and polygenic risk scores that sum trait-associated single SNPs from GWAS weighted by their effect sizes to provide an overall measure of an individual’s genetic risk to develop disease (50), our approach uses genomic regions surrounding established gastrointestinal disease SNPs from GWAS and selects only SNPs associated with PDAC in those regions to examine trait-disease associations. These aforementioned approaches rely on previous GWAS and may overlook a significant portion of heritability, given that heritability is often distributed over thousands of genetic variants with small effects in complex traits (29). To complement our current approach, we performed LD score regression which took the effect of all SNPs into account, regardless of their genome-wide significance level. The LD regression analyses did not show significant genome-wide shared heritability between inflammatory intestinal diseases and PDAC, which might be due to limited number of participants in the originating studies and lack of power to observe associations, particularly for pancreatitis and primary sclerosing cholangitis. Our results suggest that only selective regions surrounding established susceptibility variants for inflammatory intestinal diseases are associated with PDAC.
Strengths of the study include the large number of PDAC cases and controls, and our statistical approach using GWAS data. Our associations are based on genomic regions identified from GWAS of inflammatory intestinal diseases, which provides the opportunity to identify susceptibility to chronic intestinal inflammation, beyond that of questionnaires or registry data with variable accuracy. sARTP allowed us to discover disease group-PDAC associations that would not be detected by studies using traditional single-marker approaches. One limitation of the study is that although we highlighted individual PDAC-associated SNPs that contributed to the overall associations of inflammatory intestinal diseases with PDAC, none of them alone was statistically significant after correction for multiple comparisons. We observed more significant associations in the case-control studies compared with cohort studies. This is most likely due to the larger number of cases in the case-control compared to the cohort studies (n = 6,064 versus n = 2,320), and thus increased power to detect associations. Alternatively, the differences might be related to selection bias of either the cases (e.g., referral bias, IBD-related, survival bias) or controls (e.g., participation bias) from the case-control studies particularly as many of the studies were clinic or hospital based. Lastly, our study lacks clinical diagnosis of inflammatory intestinal diseases and information on important environmental factors (e.g., gluten exposure for celiac disease) that play a role in the etiology of these diseases.
In conclusion, our results support the hypothesis that variants within genomic regions surrounding susceptibility variants for ulcerative colitis, Crohn’s disease, inflammatory bowel disease, and chronic pancreatitis are associated with PDAC. Further investigations are warranted to replicate our findings and examine these associations in populations of non-European ancestry.
Supplementary Material
Significance:
The joint effects of common variants in genomic regions containing susceptibility loci for inflammatory bowel disease and chronic pancreatitis are associated with PDAC and may provide insights to understanding pancreatic cancer etiology.
Acknowledgements
This research is supported by the Intramural Research Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health. The content of the article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Laurie Burdett, Aurelie Vogt, Belynda Hicks, Amy Hutchinson, Meredith Yeager and other staff at the National Cancer Institute’s Division of Epidemiology and Genetics (DECG) Cancer Genomics Research Laboratory (CGR) for GWAS genotyping. The IARC/Central Europe study was supported by a grant from the US National Cancer Institute at the National Institutes of Health (R03 CA123546-02) and grants from the Ministry of Health of the Czech Republic (NR 9029-4/2006, NR9422-3, NR9998-3, MH CZ-DRO-MMCI 00209805). Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer /World Health Organization. The work at Johns Hopkins University was supported by the NCI Grants P50CA062924, R01CA97075, and P30 CA006973. Additional support was provided by the Lustgarten Foundation, Susan Wojcicki and Dennis Troper and the Sol Goldman Pancreas Cancer Research Center. This work was supported by RO1 CA154823 and federal funds from the National Cancer Institute (NCI), US National Institutes of Health (NIH) under contract number HHSN261200800001E. The Mayo Clinic Biospecimen Resource for Pancreas Research study is supported by the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701). The Memorial Sloan Kettering Cancer Center Pancreatic Tumor Registry is supported by P30CA008748, the Geoffrey Beene Foundation, the Arnold and Arlene Goldstein Family Foundation, and the Society of MSKCC. We acknowledge the contribution of Dr. Irene Orlow to the study. The Queensland Pancreatic Cancer Study was supported by a grant from the National Health and Medical Research Council of Australia (NHMRC) (Grant number 442302). RE Neale is supported by a NHMRC Senior Research Fellowship (#1060183). The UCSF pancreas study was supported by NIH-NCI grants (R01CA1009767, R01CA109767-S1 and R01CA059706) and the Joan Rombauer Pancreatic Cancer Fund. Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program; the NCI’s SEER Program under contract HSN261201000140C awarded to CPIC; and the CDC’s National Program of Cancer Registries, under agreement #U58DP003862-01 awarded to the California Department of Public Health. The Yale (CT) pancreas cancer study was supported by National Cancer Institute at the U.S. National Institutes of Health, grant 5R01CA098870. The cooperation of 30 Connecticut hospitals, including Stamford Hospital, in allowing patient access, is gratefully acknowledged. The Connecticut Pancreas Cancer Study was approved by the State of Connecticut Department of Public Health Human Investigation Committee. Certain data used in that study were obtained from the Connecticut Tumor Registry in the Connecticut Department of Public Health. The authors assume full responsibility for analyses and interpretation of these data. Studies included in PANDoRA were partly funded by: the Czech Science Foundation (No. P301/12/1734), the Internal Grant Agency of the Czech Ministry of Health (IGA NT 13 263); the Baden-Württemberg State Ministry of Research, Science and Arts (Prof. H. Brenner), the Heidelberger EPZ-Pancobank (Prof. M.W. Büchler and team: Prof. T. Hackert, Dr. N. A. Giese, Dr. Ch. Tjaden, E. Soyka, M. Meinhardt; Heidelberger Stiftung Chirurgie and BMBF grant 01GS08114), the BMBH (Prof. P. Schirmacher; BMBF grant 01EY1101), the “5x1000” voluntary contribution of the Italian Government, the Italian Ministry of Health (RC1203GA57, RC1303GA53, RC1303GA54, RC1303GA50), the Italian Association for Research on Cancer (Prof. A. Scarpa; AIRC n. 12182), the Italian Ministry of Research (Prof. A. Scarpa; FIRB - RBAP10AHJB), the Italian FIMP-Ministry of Health (Prof. A. Scarpa; 12 CUP_J33G13000210001), and by the National Institute for Health Research Liverpool Pancreas Biomedical Research Unit, UK. We would like to acknowledge the contribution of Dr Frederike Dijk and Prof. Oliver Busch (Academic Medical Center, Amsterdam, the Netherlands). Assistance with genotype data quality control was provided by Cecelia Laurie and Cathy Laurie at University of Washington Genetic Analysis Center. The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II cohort. The authors express sincere appreciation to all Cancer Prevention Study-II participants, and to each member of the study and biospecimen management group. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries and cancer registries supported by the National Cancer Institute’s Surveillance Epidemiology and End Results Program. Cancer incidence data for CLUE were provided by the Maryland Cancer Registry, Center for Cancer Surveillance and Control, Department of Health and Mental Hygiene, 201 W. Preston Street, Room 400, Baltimore, MD 21201, http://phpa.dhmh.maryland.gov/cancer, 410-767-4055. We acknowledge the State of Maryland, the Maryland Cigarette Restitution Fund, and the National Program of Cancer Registries of the Centers for Disease Control and Prevention for the funds that support the collection and availability of the cancer registry data.” We thank Kathy J. Helzlsouer for her contribution in CLUE. We also thank all the CLUE participants. The Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. The NYU Women’s Health Study (AZJ and AAA) was funded by NIH UM1 CA182934 and center grants P30 CA016087 and P30 ES000260. The PANKRAS II Study in Spain was supported by research grants from Instituto de Salud Carlos III-FEDER, Spain; Fondo de Investigaciones Sanitarias (FIS) (#PI13/00082 and #PI15/01573) and Red Temática de Investigación Cooperativa en Cáncer, Spain (#RD12/0036/0050); and European Cooperation in Science and Technology (COST Action #BM1204: EU_Pancreas), Ministerio de Ciencia y Tecnología (CICYT SAF 2000-0097), Fondo de Investigación Sanitaria (95/0017), Madrid, Spain; Generalitat de Catalunya (CIRIT - SGR); ‘Red temática de investigación cooperativa de centros en Cáncer’ (C03/10), ‘Red temática de investigación cooperativa de centros en Epidemiología y salud pública’ (C03/09), and CIBER de Epidemiología (CIBERESP), Madrid. The Physicians’ Health Study was supported by research grants CA-097193, CA-34944, CA-40360, HL-26490, and HL-34595 from the National Institutes of Health, Bethesda, MD USA. The Women’s Health Study was supported by research grants CA-047988, CA-182913, HL-043851, HL-080467, and HL-099355 from the National Institutes of Health, Bethesda, MD USA. Health Professionals Follow-up Study is supported by NIH grant UM1 CA167552 from the National Cancer Institute, Bethesda, MD USA. Nurses’ Health Study is supported by NIH grants UM1 CA186107, P01 CA87969, and R01 CA49449 from the National Cancer Institute, Bethesda, MD USA. Multiethnic Cohort Study was supported by NIH grants CA63464, CA54281, CA098758, CA132839 and CA164973 from the National Cancer Institute, Bethesda, MD USA. Additional support from the Hale Center for Pancreatic Cancer Research, U01 CA21017 from the National Cancer Institute, Bethesda, MD USA, and the United States Department of Defense CA130288, Lustgarten Foundation, Pancreatic Cancer Action Network, Noble Effort Fund, Peter R. Leavitt Family Fund, Wexler Family Fund, and Promises for Purple to B.M. Wolpin; Broman Family Fund for Pancreatic Cancer Research to K. Ng; and K07 CA222159 from the National Cancer Institute, Bethesda, MD USA, and Bob Parsons Fellowship to A. Babic. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201600018C, HHSN268201600001C, HHSN268201600002C, HHSN268201600003C, and HHSN268201600004C. The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf. SELECT is funded by the National Cancer Institute of the National Institutes of Health under Award Numbers U10 CA37429 (CD Blanke), and UM1 CA182883 (CM Tangen/IM Thompson). The authors thank the site investigators and staff and, most importantly, the participants from SELECT who donated their time to this trial. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH, Bethesda, MD, USA (http://biowulf.nih.gov).The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on September 24, 2019.
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Ward E, Sherman RL, Henley SJ, Jemal A, Siegel DA, Feuer EJ, et al. Annual Report to the Nation on the Status of Cancer, 1999-2015, Featuring Cancer in Men and Women ages 20-49. J Natl Cancer Inst 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014;371:1039–49 [DOI] [PubMed] [Google Scholar]
- 3.Chen F, Childs EJ, Mocci E, Bracci PM, Gallinger S, Li D, et al. Analysis of heritability and genetic architecture of Pancreatic Cancer: A PanC4 Study. Cancer Epidemiol Biomarkers Prev 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013;144:1252–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemminki K, Li X, Sundquist J, Sundquist K. Cancer risks in ulcerative colitis patients. Int J Cancer 2008;123:1417–21 [DOI] [PubMed] [Google Scholar]
- 6.Hemminki K, Li X, Sundquist J, Sundquist K. Cancer risks in Crohn disease patients. Ann Oncol 2009;20:574–80 [DOI] [PubMed] [Google Scholar]
- 7.Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Loof L, Danielsson A, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36:321–7 [DOI] [PubMed] [Google Scholar]
- 8.Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013;382:1587–99 [DOI] [PubMed] [Google Scholar]
- 9.Askling J, Linet M, Gridley G, Halstensen TS, Ekstrom K, Ekbom A. Cancer incidence in a population-based cohort of individuals hospitalized with celiac disease or dermatitis herpetiformis. Gastroenterology 2002;123:1428–35 [DOI] [PubMed] [Google Scholar]
- 10.Ilus T, Kaukinen K, Virta LJ, Pukkala E, Collin P. Incidence of malignancies in diagnosed celiac patients: a population-based estimate. Am J Gastroenterol 2014;109:1471–7 [DOI] [PubMed] [Google Scholar]
- 11.Card TR, Siffledeen J, Fleming KM. Are IBD patients more likely to have a prior diagnosis of irritable bowel syndrome? Report of a case-control study in the General Practice Research Database. United European Gastroenterol J 2014;2:505–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Salhy M, Hatlebakk JG, Gilja OH, Hausken T. The relation between celiac disease, nonceliac gluten sensitivity and irritable bowel syndrome. Nutr J 2015;14:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon H, Trier Moller F, Andersen V, Harbord M. Heritability in inflammatory bowel disease: from the first twin study to genome-wide association studies. Inflamm Bowel Dis 2015;21:1428–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuja-Halkola R, Lebwohl B, Halfvarson J, Wijmenga C, Magnusson PK, Ludvigsson JF. Heritability of non-HLA genetics in coeliac disease: a population-based study in 107 000 twins. Gut 2016;65:1793–8 [DOI] [PubMed] [Google Scholar]
- 15.Witt H, Apte MV, Keim V, Wilson JS. Chronic pancreatitis: challenges and advances in pathogenesis, genetics, diagnosis, and therapy. Gastroenterology 2007;132:1557–73 [DOI] [PubMed] [Google Scholar]
- 16.Jiang X, Karlsen TH. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat Rev Gastroenterol Hepatol 2017;14:279–95 [DOI] [PubMed] [Google Scholar]
- 17.Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet 2009;41:986–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet 2015;47:911–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet 2010;42:224–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet 2014;46:994–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein AP, Wolpin BM, Risch HA, Stolzenberg-Solomon RZ, Mocci E, Zhang M, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Commun 2018;9:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Wheeler W, Hyland PL, Yang Y, Shi J, Chatterjee N, et al. A Powerful Procedure for Pathway-Based Meta-analysis Using Summary Statistics Identifies 43 Pathways Associated with Type II Diabetes in European Populations. PLoS Genet 2016;12:e1006122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009;5:e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007;39:906–13 [DOI] [PubMed] [Google Scholar]
- 26.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47:979–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015;31:3555–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang M, Wang Z, Obazee O, Jia J, Childs EJ, Hoskins J, et al. Three new pancreatic cancer susceptibility signals identified on chromosomes 1q32.1, 5p15.33 and 8q24.21. Oncotarget 2016;7:66328–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017;49:256–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji SG, Juran BD, Mucha S, Folseraas T, Jostins L, Melum E, et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat Genet 2017;49:269–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosendahl J, Kirsten H, Hegyi E, Kovacs P, Weiss FU, Laumen H, et al. Genome-wide association study identifies inversion in the CTRB1-CTRB2 locus to modify risk for alcoholic and non-alcoholic chronic pancreatitis. Gut 2018;67:1855–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The GTEx Consortium. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res 2018;46:D754–D61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res 2012;40:D930–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 2012;22:1790–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maxwell JR, Zhang Y, Brown WA, Smith CL, Byrne FR, Fiorino M, et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015;43:739–50 [DOI] [PubMed] [Google Scholar]
- 39.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 2009;15:1016–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Y, Wu X, Wang J. Correlation of miR-425-5p and IL-23 with pancreatic cancer. Oncol Lett 2019;17:4595–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet 2011;43:246–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pitha-Rowe I, Petty WJ, Feng Q, Koza-Taylor PH, Dimattia DA, Pinder L, et al. Microarray analyses uncover UBE1L as a candidate target gene for lung cancer chemoprevention. Cancer Res 2004;64:8109–15 [DOI] [PubMed] [Google Scholar]
- 43.Kitareewan S, Pitha-Rowe I, Sekula D, Lowrey CH, Nemeth MJ, Golub TR, et al. UBE1L is a retinoid target that triggers PML/RARalpha degradation and apoptosis in acute promyelocytic leukemia. Proc Natl Acad Sci U S A 2002;99:3806–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stein MM, Thompson EE, Schoettler N, Helling BA, Magnaye KM, Stanhope C, et al. A decade of research on the 17q12-21 asthma locus: Piecing together the puzzle. J Allergy Clin Immunol 2018;142:749–64 e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Singh RR, Patel KP, Routbort MJ, Aldape K, Lu X, Manekia J, et al. Clinical massively parallel next-generation sequencing analysis of 409 cancer-related genes for mutations and copy number variations in solid tumours. Br J Cancer 2014;111:2014–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kwon MJ, Kim RN, Song K, Jeon S, Jeong HM, Kim JS, et al. Genes co-amplified with ERBB2 or MET as novel potential cancer-promoting genes in gastric cancer. Oncotarget 2017;8:92209–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sahlberg KK, Hongisto V, Edgren H, Makela R, Hellstrom K, Due EU, et al. The HER2 amplicon includes several genes required for the growth and survival of HER2 positive breast cancer cells. Mol Oncol 2013;7:392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walsh N, Zhang H, Hyland PL, Yang Q, Mocci E, Zhang M, et al. Agnostic Pathway/Gene Set Analysis of Genome-Wide Association Data Identifies Associations for Pancreatic Cancer. J Natl Cancer Inst 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhong J, Jermusyk A, Wu L, Hoskins JW, Collins I, Mocci E, et al. A Transcriptome-Wide Association Study (TWAS) Identifies Novel Candidate Susceptibility Genes for Pancreatic Cancer. J Natl Cancer Inst 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat Rev Genet 2018;19:581–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The majority of the data that support the findings of this study are available in dbGAP at https://www.ncbi.nlm.nih.gov/gap/, reference number [phs000206.v5.p3 and phs000648.v1.p1]. Biomedical research scientists from recognized research institutions can request data as bona fide researchers from dbGAP or by contacting the corresponding author.