

Visual Abstract

Keywords: cystic kidney disease, ADPKD, ARPKD, cysts, ductal plate, fibrocystin, mouse model, PKD1, PKHD1, polycystic liver, polycystin
Abstract
Background
A major difference between autosomal recessive polycystic kidney disease (ARPKD) and autosomal dominant polycystic kidney disease (ADPKD) lies in the pattern of inheritance, and the resultant timing and focality of cyst formation. In both diseases, cysts form in the kidney and liver as a consequence of the cellular recessive genotype of the respective disease gene, but this occurs by germline inheritance in ARPKD and somatic second hit mutations to the one normal allele in ADPKD. The fibrocystic liver phenotype in ARPKD is attributed to abnormal ductal plate formation because of the absence of PKHD1 expression during embryogenesis and organ development. The finding of polycystic liver disease in a subset of adult PKHD1 heterozygous carriers raises the question of whether somatic second hit mutations in PKHD1 in adults may also result in bile duct-derived cyst formation.
Methods
We used an adult-inducible Pkhd1 mouse model to examine whether Pkhd1 has a functional role in maintaining bile duct homeostasis after normal liver development.
Results
Inactivation of Pkhd1 beginning at 4 weeks of age resulted in a polycystic liver phenotype with minimal fibrosis at 17 weeks. Increased biliary epithelium, which lines these liver cysts, was most pronounced in female mice. We assessed genetic interaction of this phenotype with either reduced or increased copies of Pkd1, and found no significant effects on the Pkhd1 phenotype in the liver or kidney from altered Pkd1 expression.
Conclusions
Somatic adult inactivation of Pkhd1 results in a polycystic liver phenotype. Pkhd1 is a required gene in adulthood for biliary structural homeostasis independent of Pkd1. This suggests that PKHD1 heterozygous carrier patients can develop liver cysts after somatic mutations in their normal copy of PKHD1.
Introduction
PKHD1 is the disease gene for autosomal recessive polycystic kidney disease (ARPKD). ARPKD is a pediatric diagnosis with incidence 1:20,000 live births, characterized by diffusely cystic kidneys progressing to renal failure and congenital hepatic fibrosis (CHF) (1). CHF is a fully penetrant component of the diagnosis characterized by liver fibrosis and portal hypertension, whose consequences require liver transplant in 1%–7% of survivors of early childhood (2,3). CHF pathogenesis is attributed to malformation of the ductal plate during liver development, and ARPKD-related CHF can be distinguished from other forms of liver fibrosis by the occurrence of biliary-derived cysts. Patients with missense mutations tend to have a milder kidney phenotype compared with those with truncating mutations and in some cases are not diagnosed until liver abnormalities are identified in adulthood (4,5). Mouse models for ARPKD recapitulate the human liver phenotype with fibrosis and cysts (6–10). Pancreas phenotypes are variable. ARPKD mice lack a kidney phenotype at least until late in life when mild tubular dilations have been reported (7,8). The function of fibrocystin is not well understood, and this hinders attempts to identify ARPKD treatments.
The parents of ARPKD children are healthy, and there is no kidney or liver phenotype in heterozygous rodent models (6,9). Monoallelic loss of PKHD1 has been thought to be inconsequential. Nonetheless, 10% of asymptomatic carrier adults were found to have multiple cysts in their livers or kidneys, and a gene discovery analysis of individuals ascertained on the basis of isolated polycystic liver disease (PCLD) identified PKHD1 heterozygous carrier genotype as the causative gene mutation (11,12). Genetic studies and modeling of several other gene causes of PCLD have shown that these occur by somatic second hit mutations akin to the mechanisms underlying autosomal dominant polycystic kidney disease (ADPKD) (12–14). These findings allow for two possible genetic models for cyst pathogenesis in PKHD1 heterozygous mutation carriers. Bile duct cysts may form either because of haploinsufficiency or because of a focal cellular recessive genotype resulting from somatic loss of the single normal allele. This second possibility can only be true if the PKHD1 gene has an ongoing homeostatic role after liver development. We used an adult-inducible mouse knockout model of Pkhd1 to determine whether postdevelopmental inactivation of Pkhd1 results in a PCLD phenotype. We found that somatic inactivation of Pkhd1 is sufficient to cause bile duct cysts in adult livers.
Materials and Methods
Mouse Lines
All experiments were conducted in accordance with Yale University Institutional Animal Care and Use Committee guidelines and procedures. The Pkhd1flox3–4 (Pkhd1tm1Ggg (7)) allele was a generous gift from Dr. Gregory G. Germino (National Institute of Diabetes and Digestive and Kidney Diseases/National Institutes of Health). The Pkd1flox, Pkd1F/H-BAC, tamoxifen-inducible UBC-CreER, and doxycycline-inducible Pax8rtTA;TetO-Cre models have been previously described (13,15,16). The Pkd1F/H-BAC contains three copies of a bacterial artificial chromosome modified to express dual-epitope-tagged Pkd1. All mice are predominantly C57BL6. Littermates were used as controls.
Pharmacological Induction and Sacrifice
Gene inactivation using both the UBC-CreER and Pax8rtTA;TetO-Cre models began at postnatal day 28. Tamoxifen 20 mg/ml in sunflower seed oil was administered as a 0.1-ml intraperitoneal injection daily for 5 days. Doxycycline (2 mg/ml in a 5% sucrose solution) was provided in the drinking water for 2 weeks. UBC-CreER mice were sacrificed at age 17 weeks, and Pax8rtTA;TetO-Cre mice were sacrificed at age 26 weeks (6 months). Organs were perfused with 1× PBS then 4% paraformaldehyde, and stored in 4% paraformaldehyde before sectioning for histology.
Histology and Immunohistochemistry
Paraffin-embedded tissue sections were stained with hematoxylin and eosin (H&E), Masson Trichrome, Sirius Red, or left unstained. To perform anti-cytokeratin 19 (CK19) immunohistochemistry, unstained slides were treated with xylene and sequential ethanol dilutions to remove paraffin and rehydrate the tissue. Endogenous peroxidase was blocked with 10% hydrogen peroxide in methanol. After water wash, antigen retrieval was achieved with steaming in 1 mM EDTA pH 8 followed by 0.05% Tween 20 in PBS. Samples were then blocked with 1% BSA for 30 minutes before primary antibody application overnight at 4°C. Washes were completed with 0.05% Tween 20. Primary antibody: anti-CK19;TROMA-III (1:100; Developmental studies Hybridoma Bank, Iowa City, IA); secondary antibody: anti-rat horseradish peroxidase (1:500; Jackson ImmunoResearch, West Grove, PA). Horseradish peroxidase substrate was provided by the 3′-diaminobenzidine peroxidase substrate kit (Vector Laboratories, Burlingame, CA).
Cystic Parameters
Slides stained with H&E or immunohistochemistry were scanned on a Nikon Eclipse TE2000 microscope using MetaMorph software (Molecular Devices, San Jose, CA) to black and white TIFF files, respectively. For both hepatic cystic index (HCI) and the percentage of the two-dimensional liver area that stained positive for CK19 (CK19+%) calculations, a denominator was set with the manually outlined perimeter of the liver excluding large internal venous structures. Autothresholding within this space was performed by MetaMorph to determine white (cystic) from dark (noncystic) for HCI. HCI is defined as the percentage of cystic/total parenchymal area. CK19+% is defined as the percentage of CK19+ staining over the total parenchymal area.
Plots and Statistics
Two-way comparisons of sample means were calculated using the unpaired two-tailed t test, a parametric test using Prism version 7 (GraphPad Software, Inc., San Diego, CA). Fisher’s exact test was used to make pairwise comparisons of proportions. Sample size calculation was performed using online calculators (http://powerandsamplesize.com) for the Pkd1 dosage experiments to suggest a target of 15 mice of each genotype to have 80% power to detect a difference with 5% risk for type 1 error between a HCI of 12 and 20, with estimated SD of 7.8 from the initial experiment.
Results
Postdevelopmental Inactivation of Pkhd1 Results in Liver Cysts
Mouse liver achieves the mature portal triad morphology of bile ducts, hepatocytes, and vascular structures and an adult liver weight/body weight (LW/BW) ratio in the first 2–4 weeks of postnatal life (17,18). We inactivated the Pkhd1 gene at age 4 weeks using a tamoxifen-inducible, ubiquitously acting UBC-CreER that expresses in bile ducts and has previously been used to model PCLD with conditional Pkd1 alleles (16). Pkhd1fl/fl;UBC-CreER mice sacrificed at age 17 weeks showed bile duct cysts throughout the liver (Figure 1). This cystic phenotype was fully penetrant and, in most cases, grossly visible as a stippled texture of the organ upon perfusion (Supplemental Figure 1). Normal ductal plate formation is reported in mice with the germline heterozygous Pkhd1 genotype (6) and, indeed, tamoxifen-induced littermate mice with a monoallelic Pkhd1fl/+;UBC-CreER genotype had normal-appearing livers, including portal triads consisting of artery, vein, and bile duct, without cysts (Figure 1, A and B). Histologic analysis of livers from Pkhd1fl/fl;UBC-CreER mice showed clusters of cysts among normal-appearing hepatocytes (Figure 1, A and B). Pkhd1fl/fl;UBC-CreER mice (n=11) have an increased LW/BW ratio of 5.7% ± 0.3% compared with that of Pkhd1fl/+;UBC-Cre mice (n=10), 4.4% ± 0.2%, P=0.0016 (Figure 1C), without any effect of genotype on body weight (Supplemental Figure 2A). The female subgroup of Pkhd1fl/fl;UBC-CreER mice (n=4) had the most significant difference from the noncystic Pkhd1fl/+;UBC-CreER genotype female mice (n=3), P<0.001 (Figure 1C, right panel).
Figure 1.
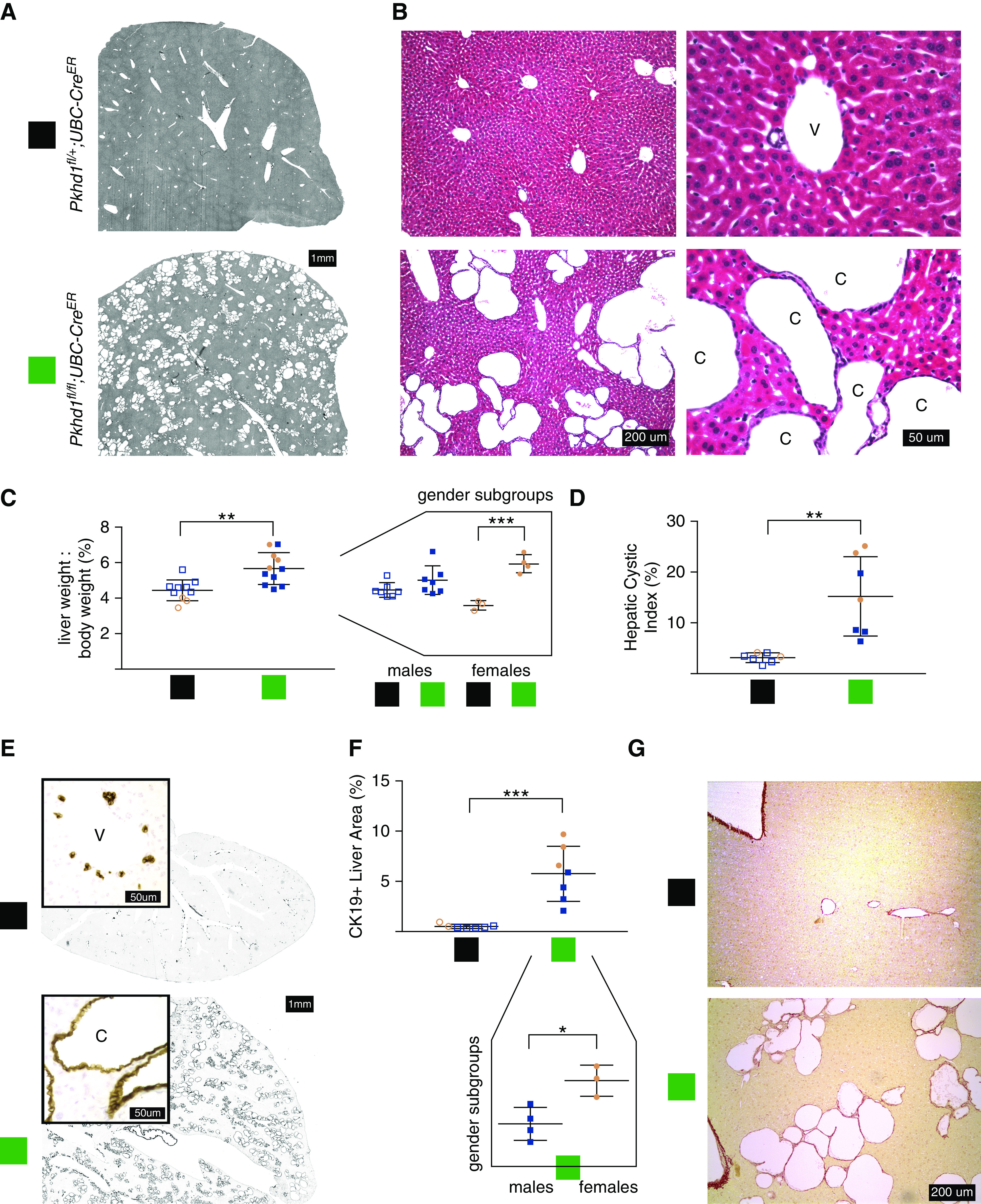
Biallelic loss of Pkhd1 with UBC-CreER results in diffuse liver cysts. (A) Representative liver sections of the indicated genotype at age 17 weeks. The colored boxes denote genotypes throughout the figure. (B) Hematoxylin and eosin-stained histology of Pkhd1fl/fl;UBC-CreER versus control monoallelic deletion (Pkhd1fl/+;UBC-CreER) at age 17 weeks. Representative structures marked as: v, vein; c, cyst. (C) Liver weight/body weight ratio of Pkhd1fl/fl;UBC-CreER versus Pkhd1fl/+;UBC-CreER mice. Right panel, sex subgroups visually separated. Blue shapes, male; pink shapes, female. (D) Hepatic cystic index, the percentage of liver section area that is background density (cysts+venous structures) rather than parenchyma on liver lobe sections scanned in greyscale as in (A). (E) Anti-cytokeratin 19 (CK19) immunohistochemistry localizes to the biliary epithelium, which lines all cysts, and not venous structures. (F) Percentage of liver section area positive with CK19 staining. Bottom panel, sex subgroups of Pkhd1fl/fl;UBC-CreER mice are visually separated. (G) Sirius Red stain at ×10 magnification.
We further characterized the cystic phenotype by two complementary measures. To quantify the area of the cysts as an HCI, we applied MetaMorph software thresholding to scanned H&E-stained liver section images (Figure 1A) to measure the two-dimensional cystic area as a percentage of the total area of the liver section defined within a perimeter that excluded large vascular structures. Mean HCI was 15.2 ± 2.9 in Pkhd1fl/fl;UBC-CreER mice (n=7) versus 3.14 ± 0.4 in Pkhd1fl/+;UBC-CreER mice (n=7), P=0.0015 (Figure 1D). Immunohistochemical staining of CK19, a marker of biliary epithelium, showed that the liver cysts were uniformly lined with a CK19-positive epithelium, indicating that they were derived from the bile ducts (Figure 1E). In Pkhd1fl/+;UBC-CreER mice, the CK19 staining highlighted the normal bile ducts, which represent a tiny minority of the cells in the liver (Figure 1E). Additional cells around the central veins were noted to be CK19-positive as has been defined in wild-type rodent liver CK19 immunostaining (19). To quantify the increased burden of biliary epithelium in our Pkhd1fl/fl;UBC-CreER mice, we again applied tissue perimeter outlining and color thresholding, this time to tissue sections stained with CK19. We calculated the CK19+%. Mean CK19+% was 5.8 ± 1 in Pkhd1fl/fl;UBC-CreER mice (n=7) versus 0.5 ± 0.1 in Pkhd1fl/+;UBC-CreER mice (n=7), P=0.0003 (Figure 1F).
Both HCI and CK19+% calculations were done by authors blinded to sample genotype on the first seven mice sacrificed for each genotype as only those had histologic sections. Nonzero values in the noncystic Pkhd1fl/+;UBC-CreER mice were expected because of the inability of the semiautomatic method to distinguish venous versus cystic structures on two-dimensional histology, and the expected presence of CK19 staining of native bile ducts. HCI and CK19+% were far superior to LW/BW at numerically quantifying the cystic phenotype distinguishing Pkhd1fl/fl;UBC-CreER and Pkhd1fl/+;UBC-CreER mice. For both HCI and CK19+%, the comparison between genotypes maintained statistical significance (P<0.05) even in the sex subgroups with small numbers. CK19+% was the one parameter by which the trend toward increased severity of liver cysts in cystic females versus cystic males met statistical significance (P=0.016) despite the small numbers once split by sex (Figure 1F). There was a robust positive correlation (r2=0.47), between the CK19+ area and the HCI values for each mouse showing that these measures were congruent with each other (Supplemental Figure 2B).
As the lesion in recessive polycystic kidney disease is CHF, we also sought to assess whether there was any significant fibrosis in our model. Pkhd1 null mice develop a significant burden of fibrosis as defined by Sirius Red staining between 3 and 9 months of age (6,20). We stained paraffin sections with Sirius Red stain and Masson’s trichrome stain (data not shown) to localize collagen. Sirius Red stained sections show positive staining outlining liver margins, central veins, and cysts (Figure 1G). There was no additional collagen staining beyond these structures on Sirius Red or Masson’s trichrome-stained sections, suggesting an absence of significant fibrosis in this model.
Analysis of Postdevelopmental Pkhd1 Loss in the Kidney
Germline Pkhd1 null mouse kidneys have no phenotype for at least 6 months of life, or not at all (6,8,9). In keeping with this, the kidneys of the Pkhd1fl/fl;UBC-CreER mice at 17 weeks of age showed no gross, histologic, or kidney weight/body weight ratio abnormalities. Pkd1+/− mouse kidneys are noncystic other than a potential small number of discrete cysts if aged over 9 months (21). The combined Pkhd1-/-;Pkd1+/− genotype has diffusely cystic kidneys (7,13). In order to evaluate whether Pkhd1 plays a role after development in the kidney, we made use of this established genetic interaction between Pkhd1 and Pkd1. To make the conditional equivalent, we used Pkhd1fl and Pkd1fl alleles with the doxycycline-inducible Pax8rtTATetO-Cre system. This model has Cre activity in the majority of renal tubule segments including the collecting duct. We induced Pkhd1fl/fl;Pkd1fl/+;Pax8rtTATetO-Cre and Pkhd1fl/+;Pkd1fl/+;Pax8rtTATetO-Cre littermates with doxycycline for a 2-week duration from 4 to 6 weeks of age. We sacrificed the mice at age 6 months. Histologic analysis of the perfusion-fixed kidneys showed no apparent abnormalities in the renal tubules, interstitium, or glomeruli (Figure 2, A and B). There was no difference in the kidney weight/body weight ratio (Figure 2C). This ratio was higher in male mice (n=9) than female mice (n=9), P=0.0039, regardless of genotype. Scanned kidney sections from this experiment are shown in Supplemental Figure 3.
Figure 2.
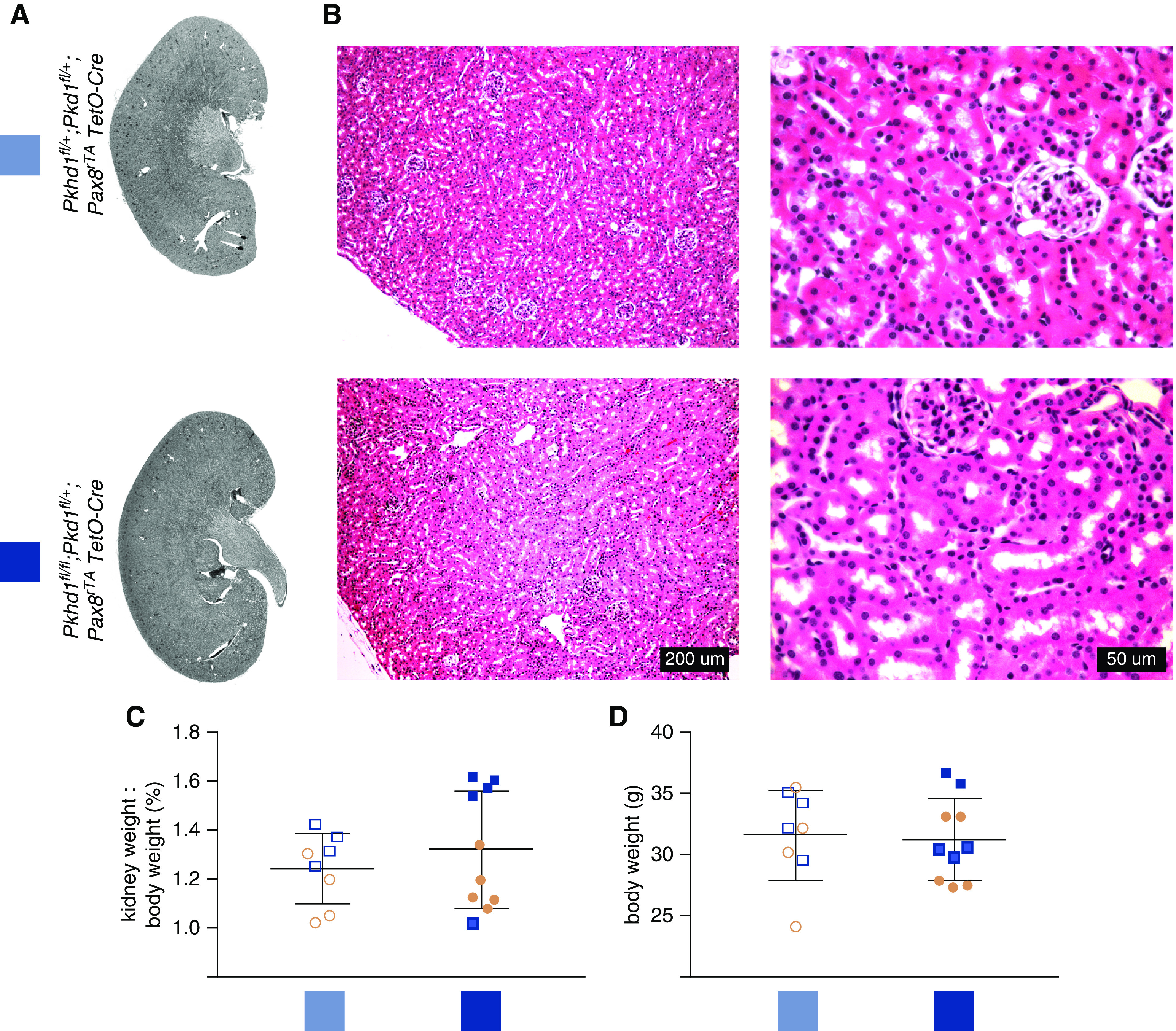
Pkhd1fl/fl;Pkd1fl/+;Pax8rtTA;TetO-Cre has no kidney phenotype. (A) Representative kidney sections of the indicated genotype at 6 months of age. (B) Hematoxylin and eosin-stained kidneys from Pkhd1fl/fl; Pkd1fl/+;Pax8rtTA;TetO-Cre mice at 6 months of age. (C) Kidney weight/body weight ratio and (D) body weight of Pkhd1fl/fl;Pkd1fl/+;Pax8rtTA;TetO-Cre mice. Genotypes indicated by color corresponding to labels in (A). Blue shapes, males; pink shapes, female.
Pkhd1fl/fl;UBC-Cre Model Liver Cyst Phenotype Is Independent of Pkd1 Dosage
ARPKD mouse liver is more severely affected when Pkd1 gene dosage is reduced (13). Specifically, the liver phenotype in Pkhd1del4/del4;Pkd1+/− is more severe than in Pkhd1del4/del4, even though the Pkd1+/− genotype itself has no liver phenotype. Increased Pkd1 dosage in the form of a three-copy Pkd1F/H-BAC rescued conditional mouse models of PCLD disease genes PRKCSH and SEC63, but had no effect on germline ARPKD mouse models (Pkhd1del4/del4;Pkd1F/H-BAC) (13). However, given that the Pkhd1del4/del4 mouse liver develops abnormally, this does not exclude the possibility that the postdevelopmental loss of Pkhd1 modeled in this study could be rescued by increased Pkd1 dosage. To assess the effect of Pkd1 dosage on our postdevelopmental Pkhd1 model, we generated Pkhd1fl/fl;Pkd1fl/+;UBC-CreER and Pkhd1fl/fl;Pkd1F/H-BAC;UBC-CreER mice to compare their littermates with the cystic Pkhd1fl/fl;UBC-CreER genotype. We induced these mice with tamoxifen injections at age 4 weeks, identical to our prior experiments, and followed them until 17 weeks of age. There was no appreciable difference in the cystic liver phenotype between these genotypes as assessed grossly by LW/BW or by HCI (Figure 3, A–C, Supplemental Figures 4 and 5). The body weight was also unchanged between these three genotypes (Figure 3D).
Figure 3.
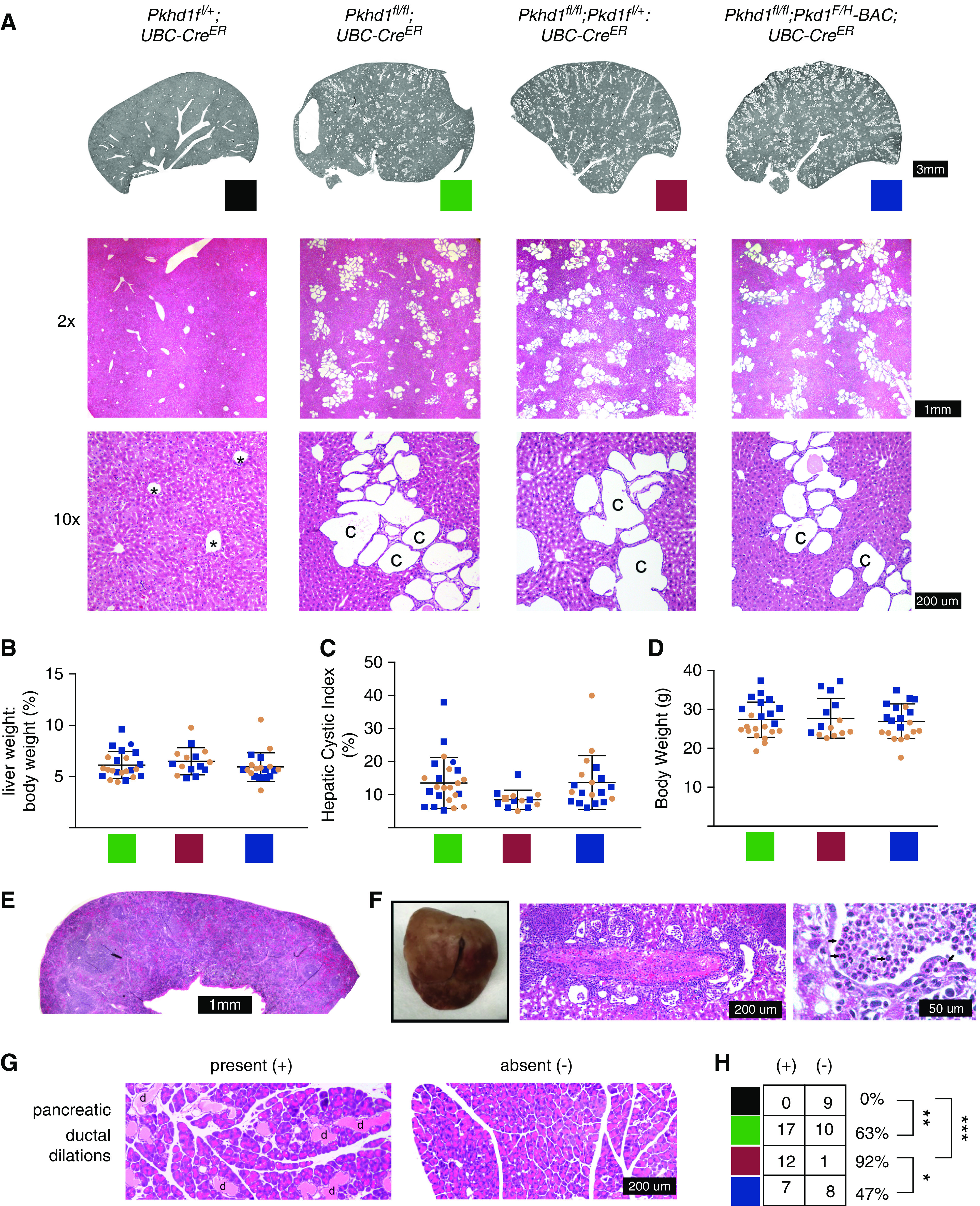
Liver phenotype does not change with alterations of Pkd1 in Pkhd1fl/fl;UBC-CreER mice. (A) Liver section scan and hematoxylin and eosin-stained histology for the indicated genotypes. Representative structures marked as: *, vein; c, cyst. Colors indicated for each genotype are used throughout the figure. (B) Liver weight/body weight ratio. (C) Hepatic cystic index as defined in Figure 1. (D) Body weight. (E) Hematoxylin and eosin-stained histology of spleen at ×2 magnification. (F) Patchy appearance of liver in one Pkhd1fl/fl;Pkd1fl/+;UBC-CreER mouse affected by cholangitis. Middle and right panels, hematoxylin and eosin-stained histology shows cystic biliary structures full of polymononuclear lymphocytes (arrows). (G) Pancreatic ductal dilations were seen in a subset of mice with Pkhd1fl/fl;UBC-CreER with or without alterations in Pkd1 genotype. Hematoxylin and eosin-stained histology of pancreas at ×10 magnification representative of presence or absence of ductal dilations. d, ductal dilation. (H) Quantification of number of mice of each genotype with pancreatic ductal dilations present or absent.
Bile Duct and Extrahepatic Findings
We performed gross and histologic analysis of the spleen and pancreas, in addition to the liver and kidney, in a subset of Pkhd1fl+;UBC-CreER, Pkhd1fl/fl;UBC-CreER, Pkhd1fl/fl;Pkd1fl/+;UBC-CreER, and Pkhd1fl/fl;Pkd1F/H-BAC;UBC-CreER mice. Germline Pkhd1 null mouse models have an age-dependent penetrance of common bile duct dilation, cholangitis, pancreatic cysts, and splenomegaly in the setting of portal hypertension (7–9). In our postdevelopmental Pkhd1fl/fl;UBC-CreER model, we did not find common bile duct dilation or splenomegaly. Spleen histology appeared normal (Figure 3E). Considering liver histology on 67 Pkhd1fl/fl;UBC-CreER mice with or without concurrent alterations in Pkd1 dosage, we noted findings consistent with varying degrees of cholangitis in four mice. This ranged from very mild focal pericystic and intracystic infiltrates including PMNs (n=3) to more diffuse histologic findings and grossly visible patchy erythema on the liver surface (n=1, Figure 3F). The four mice in which this was noted each had biallelic Pkhd1 loss, but included at least one mouse of each sex and Pkd1 dosage tested.
Histology of the pancreas in Pkhd1fl/fl;UBC-CreER mice demonstrated microscopic ductal dilations containing eosinophilic material, albeit with incomplete penetrance. The gross appearance of the pancreas was unremarkable. Figure 3G shows representative histologic images of mouse pancreatic tissue with or without pancreatic ductal dilations. This phenotype was either notable throughout the sampled tissue or it was absent. This pancreatic ductal dilation was seen in 17 of 27 (63%) of Pkhd1fl/fl;UBC-CreER mice, but was never seen in Pkhd1fl/+;UBC-CreER pancreas (n=7), P=0.007 (Figure 3H). The pancreatic ductal dilations were seen in a significantly higher percentage of the mice with reduced Pkd1 compared with those who had extra copies of Pkd1: 12 of 13 (92%) Pkhd1fl/fl;Pkd1fl/+; UBC-CreER mice versus 7 of 15 (47%) Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER mice, P=0.016.
Discussion
This study is the first to evaluate whether Pkhd1, and thus its protein product fibrocystin, continue to play a critical role in biliary homeostasis in the developed liver. We show that grown mice that lose Pkhd1 after normal liver development have a dramatic and fully penetrant cystic liver phenotype. Whereas ductal plate malformation is known to be involved in the pathogenesis of CHF, we now show that it is not required for the Pkhd1-related liver cysts themselves. This finding could serve to distinguish the pathogenesis of cysts that are seen in variable degrees in patients with ARPKD from the fibrotic phenotype of CHF. CHF accompanied by cysts is a distinguishing feature of ARPKD unique from other genetic causes of such hepatic fibrosis. Further, this finding demonstrates that the development of the recessive genotype in adulthood, as would be the case in a heterozygous individual who has a somatic mutation to the normal allele, likely results in cysts as is known to occur in PCLD. One limitation to conditional mouse models of dominantly inherited polycystic kidney disease and PCLD is that all biliary epithelial cells lose the disease gene simultaneously, in contrast to human organs in which somatic mutations occur in individual cells. Given this, we cannot yet definitively conclude that a single cell affected with PKHD1 loss is sufficient to initiate a cyst as is known to be the case in patients with PKD1 and PKD2. Nonetheless, a recent study followed Pkhd1 heterozygous mice by magnetic resonance imaging and although they showed no abnormalities at 10 months of age, by 1.5 years when somatic mutations could have occurred with time for subsequent cyst growth, the mice had cysts analogous to those found in human heterozygotes (22). This finding in mice, together with the focal nature of some cysts seen in PKHD1 mutation carriers, supports the idea that recessive loss in individual cells is sufficient to form cysts.
Our discovery of PKHD1 as an autosomal dominant PCLD disease gene prompted the investigations in this study. Unlike all other established PCLD disease genes, Pkhd1 does not cause PCLD by affecting polycystin-1 (PC1) maturation (12,23). Biallelic loss of Prkcsh, Sec63, Alg8, Ganab, Sec61b, Dnajb11, or Alg9 results in significant reduction of the expression of mature PC1, whereas biallelic loss of Pkhd1 does not (12,13,24–26). Instead, we asked the question of whether Pkhd1 loss affected the PC1 functional dosage without affecting PC1 protein levels of trafficking. We found that in contrast to mouse models of Prkcsh and Sec63, in which the presence of extra genomic copies of Pkd1 in the form of the Pkd1F/H-BAC rescues the liver cystic phenotype, the extra copies of PC1 had no effect on bile duct cysts because of loss of Pkhd1. Similarly, whereas reduced Pkd1 dosage worsens PCLD because of Prkcsh and Sec63, it has no effect on Pkhd1 liver cysts resulting from adult inactivation. This shows that the Pkhd1-related liver cystic phenotype is independent of PC1 dosage and thus likely occurs via a non-PC1-dependent mechanism.
Adult-inducible inactivation of Pkhd1 does not recapitulate the genetic interaction between Pkhd1 and Pkd1 that occurs with germline Pkhd1-/-;Pkd1+/− mice. The loss of one Pkd1 allele was not able to bring out a kidney phenotype in our Pkhd1fl/fl;Pkd1fl/+;Pax8rtTATetO-Cre system, and the cystic liver phenotype was no worse in Pkhd1fl/fl;Pkd1fl/+;UBC-CreER mice than those with two functional copies of Pkd1. This suggests that the previously described genetic interaction between the Pkhd1-/- and Pkd1+/− genotypes in both the kidney and liver is not involved in adult tissue homeostasis, but rather requires abnormal organogenesis as likely occurs with the germline null Pkhd1 alleles. Both patients with ADPKD and mice, and ARPKD mice, develop pancreatic cysts with incomplete penetrance (7–9,27,28). Pancreatic ductal dilation in our postdevelopmental Pkhd1 model occurred with significantly higher frequency in mice with Pkhd1fl/fl;Pkd1fl/+;UBC-Cre versus Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER genotypes, even though there was no appreciable difference in severity. This was the only parameter in the Pkhd1fl/fl;UBC-CreER mice that differed with PC1 dosage in adult inactivation of Pkhd1.
We describe a novel mouse model of Pkhd1/fibrocystin inactivation after liver development. We learn that Pkhd1 plays a crucial ongoing role in biliary homeostasis. This may suggest that the pathogenesis of ARPKD liver cysts is not intertwined with that of CHF. Further, our findings support the possibility that the polycystic liver phenotype in PKHD1 heterozygous carriers could occur through somatic mutation, and the lack of Pkhd1/Pkd1 genetic interaction in this model lends support to a hypothesis that liver cyst formation in ARPKD and ADPKD occur through different mechanistic pathways. Such investigations are relevant to understanding whether future targeted treatments for ADPKD should be expected to be relevant to ARPKD. The robust liver cystic phenotype in our Pkhd1fl/fl;UBC-CreER mice makes this a valuable model for future studies to compare the functional pathways of Pkhd1 and Pkd1 and their treatment.
Disclosures
S. Somlo is a founder, shareholder, and consultant for Goldfinch Bio, and reports personal fees from Otsuka Pharmaceuticals and Goldfinch Bio, outside the submitted work. All remaining authors have nothing to disclose.
Funding
This study was supported by grants from the Polycystic Kidney Disease Founation (217G18a) and National Institutes of Health (K08DK119642) to W. Besse, and R01DK54053 and R01DK100592 to S. Somlo.
Supplemental Material
This article contains the following supplemental material online at http://kidney360.asnjournals.org/lookup/suppl/doi:10.34067/KID.0002522020/-/DCSupplemental.
Liver histology of all Pkhd1fl/fl;UBC-CreER and Pkhd1fl/+;UBC-CreER mice evaluated by this method. Download Supplemental Figure 1, PDF file, 6.2 MB (6.1MB, pdf)
Additional information for the UBC-CreER model. Download Supplemental Figure 2, PDF file, 6.2 MB (6.1MB, pdf)
Kidney histology of all Pkhd1fl/fl;Pkd1fl/+;Pax8rtTATetO-Cre mice in the study. Download Supplemental Figure 3, PDF file, 6.2 MB (6.1MB, pdf)
Males from contemporaneous mouse cohort of Pkhd1fl/fl;UBC-CreER Pkhd1fl/fl;Pkd1fl/+;UBC-CreER and Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER. Download Supplemental Figure 4, PDF file, 6.2 MB (6.1MB, pdf)
Females from contemporaneous mouse cohort of Pkhd1fl/fl;UBC-CreER Pkhd1fl/fl;Pkd1fl/+;UBC-CreER and Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER. Download Supplemental Figure 5, PDF file, 6.2 MB (6.1MB, pdf)
Acknowledgments
The authors would like to thank Dr. Gregory G. Germino for the Pkhd1flox3-4 mouse allele.
Some of the data in this manuscript were presented at American Society of Nephrology Kidney Week.
Author Contributions
W. Besse, S. Ghosh Roy, C. Roosendaal, and L. Tuccillo were responsible for investigation; W. Besse was responsible for formal analysis, funding acquisition, methodology, visualization, and wrote the original draft; A. Gallagher was responsible for data curation; S. Somlo was responsible for funding acquisition and methodology; W. Besse, A. Gallagher, and S. Somlo conceptualized the study, and reviewed and edited the manuscript.
References
- 1.Guay-Woodford LM: Autosomal recessive polycystic kidney disease: The prototype of the hepato-renal fibrocystic diseases. J Pediatr Genet 3: 89–101, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guay-Woodford LM, Desmond RA: Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics 111: 1072–1080, 2003. Available at: 10.1542/peds.111.5.1072 [DOI] [PubMed] [Google Scholar]
- 3.Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani KT, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler NG, Roque A, Douek DC, Graf J, Huizing M, Bryant JC, Mohan P, Gahl WA, Heller T: Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology 144: 112–121.e2, 2013. Available at: 10.1053/j.gastro.2012.09.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furu L, Onuchic LF, Gharavi A, Hou X, Esquivel EL, Nagasawa Y, Bergmann C, Senderek J, Avner E, Zerres K, Germino GG, Guay-Woodford LM, Somlo S: Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J Am Soc Nephrol 14: 2004–2014, 2003. Available at: 10.1097/01.ASN.0000078805.87038.05 [DOI] [PubMed] [Google Scholar]
- 5.Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC: Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore) 85: 1–21, 2006. Available at: 10.1097/01.md.0000200165.90373.9a [DOI] [PubMed] [Google Scholar]
- 6.Moser M, Matthiesen S, Kirfel J, Schorle H, Bergmann C, Senderek J, Rudnik-Schöneborn S, Zerres K, Buettner R: A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 41: 1113–1121, 2005. Available at: 10.1002/hep.20655 [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Gonzalez MA, Menezes LF, Piontek KB, Kaimori J, Huso DL, Watnick T, Onuchic LF, Guay-Woodford LM, Germino GG: Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum Mol Genet 16: 1940–1950, 2007. Available at: 10.1093/hmg/ddm141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woollard JR, Punyashtiti R, Richardson S, Masyuk TV, Whelan S, Huang BQ, Lager DJ, vanDeursen J, Torres VE, Gattone VH, LaRusso NF, Harris PC, Ward CJ: A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int 72: 328–336, 2007. Available at: 10.1038/sj.ki.5002294 [DOI] [PubMed] [Google Scholar]
- 9.Gallagher AR, Esquivel EL, Briere TS, Tian X, Mitobe M, Menezes LF, Markowitz GS, Jain D, Onuchic LF, Somlo S: Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am J Pathol 172: 417–429, 2008. Available at: 10.2353/ajpath.2008.070381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams SS, Cobo-Stark P, James LR, Somlo S, Igarashi P: Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr Nephrol 23: 733–741, 2008. Available at: 10.1007/s00467-007-0735-4 [DOI] [PubMed] [Google Scholar]
- 11.Gunay-Aygun M, Turkbey BI, Bryant J, Daryanani KT, Gerstein MT, Piwnica-Worms K, Choyke P, Heller T, Gahl WA: Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol Genet Metab 104: 677–681, 2011. Available at: 10.1016/j.ymgme.2011.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, Gallagher AR, Huang EB, Gulati A, Knight J, Mane S, Tahvanainen E, Tahvanainen P, Sanna-Cherchi S, Lifton RP, Watnick T, Pei YP, Torres VE, Somlo S: Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest 127: 3558, 2017. Available at: 10.1172/JCI96729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fedeles SV, Tian X, Gallagher AR, Mitobe M, Nishio S, Lee SH, Cai Y, Geng L, Crews CM, Somlo S: A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat Genet 43: 639–647, 2011. Available at: 10.1038/ng.860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen MJ, Salomon J, Cnossen WR, Bergmann C, Pfundt R, Drenth JP: Somatic loss of polycystic disease genes contributes to the formation of isolated and polycystic liver cysts. Gut 64: 688–690, 2015. Available at: 10.1136/gutjnl-2014-308062 [DOI] [PubMed] [Google Scholar]
- 15.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, Igarashi P, Somlo S: Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet 17: 1505–1516, 2008. Available at: 10.1093/hmg/ddn039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S: Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet 45: 1004–1012, 2013. Available at: 10.1038/ng.2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swartley OM, Foley JF, Livingston DP 3rd, Cullen JM, Elmore SA: Histology atlas of the developing mouse hepatobiliary hemolymphatic vascular system with emphasis on embryonic days 11.5-18.5 and early postnatal development. Toxicol Pathol 44: 705–725, 2016. Available at: 10.1177/0192623316630836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fausto N, Laird AD, Webber EM: Liver regeneration. 2. Role of growth factors and cytokines in hepatic regeneration. FASEB J 9: 1527–1536, 1995. Available at: 10.1096/fasebj.9.15.8529831 [DOI] [PubMed] [Google Scholar]
- 19.Paku S, Dezso K, Kopper L, Nagy P: Immunohistochemical analysis of cytokeratin 7 expression in resting and proliferating biliary structures of rat liver. Hepatology 42: 863–870, 2005. Available at: 10.1002/hep.20858 [DOI] [PubMed] [Google Scholar]
- 20.Locatelli L, Cadamuro M, Spirlì C, Fiorotto R, Lecchi S, Morell CM, Popov Y, Scirpo R, De Matteis M, Amenduni M, Pietrobattista A, Torre G, Schuppan D, Fabris L, Strazzabosco M: Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology 63: 965–982, 2016. Available at: 10.1002/hep.28382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu G, Tian X, Nishimura S, Markowitz GS, D’Agati V, Park JH, Yao L, Li L, Geng L, Zhao H, Edelmann W, Somlo S: Trans-heterozygous Pkd1 and Pkd2 mutations modify expression of polycystic kidney disease. Hum Mol Genet 11: 1845–1854, 2002. Available at: 10.1093/hmg/11.16.1845 [DOI] [PubMed] [Google Scholar]
- 22.Shan D, Rezonzew G, Mullen S, Roye R, Zhou J, Chumley P, Revell DZ, Challa A, Kim H, Lockhart ME, Schoeb TR, Croyle MJ, Kesterson RA, Yoder BK, Guay-Woodford LM, Mrug M: Heterozygous Pkhd1C642* mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am J Physiol Renal Physiol 316: F463–F472, 2019. Available at: 10.1152/ajprenal.00181.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olson RJ, Hopp K, Wells H, Smith JM, Furtado J, Constans MM, Escobar DL, Geurts AM, Torres VE, Harris PC: Synergistic genetic interactions between Pkhd1 and Pkd1 result in an ARPKD-like phenotype in murine models. J Am Soc Nephrol 30: 2113–2127, 2019. Available at: 10.1681/ASN.2019020150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR, Banks CJ, Baheti S, Reddy B, Herrero JI, Banales JM, Hogan MC, Tasic V, Watnick TJ, Chapman AB, Vigneau C, Lavainne F, Audrezet MP, Ferec C, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group, HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Mutations in GANAB, encoding the glucosidase IIα subunit, cause autosomal-dominant polycystic kidney and liver disease. Am J Hum Genet 98: 1193–1207, 2016. Available at: 10.1016/j.ajhg.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornec-Le Gall E, Olson RJ, Besse W, Heyer CM, Gainullin VG, Smith JM, Audrézet M-P, Hopp K, Porath B, Shi B, Baheti S, Senum SR, Arroyo J, Madsen CD, Férec C, Joly D, Jouret F, Fikri-Benbrahim O, Charasse C, Coulibaly J-M, Yu AS, Khalili K, Pei Y, Somlo S, Le Meur Y, Torres VE, Harris PC; Genkyst Study Group; HALT Progression of Polycystic Kidney Disease Group; Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease : Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am J Hum Genet 102: 832–844, 2018. Available at: 10.1016/j.ajhg.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besse W, Chang AR, Luo JZ, Triffo WJ, Moore BS, Gulati A, Hartzel DN, Mane S, Torres VE, Somlo S, Mirshahi T; Regeneron Genetics Center : ALG9 mutation carriers develop kidney and liver cysts. J Am Soc Nephrol 30: 2091–2102, 2019. Available at: 10.1681/ASN.2019030298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J: Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet 17: 179–181, 1997. Available at: 10.1038/ng1097-179 [DOI] [PubMed] [Google Scholar]
- 28.McNicholas BA, Kotaro Y, Martin W, Sharma A, Kamath PS, Edwards ME, Kremers WK, Chari ST, Torres VE, Harris PC, Takahashi N, Hogan MC: Pancreatic cysts and intraductal papillary mucinous neoplasm in autosomal dominant polycystic kidney disease. Pancreas 48: 698–705, 2019. Available at: 10.1097/MPA.0000000000001306 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Liver histology of all Pkhd1fl/fl;UBC-CreER and Pkhd1fl/+;UBC-CreER mice evaluated by this method. Download Supplemental Figure 1, PDF file, 6.2 MB (6.1MB, pdf)
Additional information for the UBC-CreER model. Download Supplemental Figure 2, PDF file, 6.2 MB (6.1MB, pdf)
Kidney histology of all Pkhd1fl/fl;Pkd1fl/+;Pax8rtTATetO-Cre mice in the study. Download Supplemental Figure 3, PDF file, 6.2 MB (6.1MB, pdf)
Males from contemporaneous mouse cohort of Pkhd1fl/fl;UBC-CreER Pkhd1fl/fl;Pkd1fl/+;UBC-CreER and Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER. Download Supplemental Figure 4, PDF file, 6.2 MB (6.1MB, pdf)
Females from contemporaneous mouse cohort of Pkhd1fl/fl;UBC-CreER Pkhd1fl/fl;Pkd1fl/+;UBC-CreER and Pkhd1fl/fl;Pkd1F/H-BAC UBC-CreER. Download Supplemental Figure 5, PDF file, 6.2 MB (6.1MB, pdf)