

Abstract
The 2011 discovery of the pathogenic hexanucleotide repeat expansion (HRE) in C9orf72, the leading genetic cause of both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), marked a breakthrough in the effort to unravel the etiology of these conditions. Ten years later, clinicians are still working to integrate the implications of this discovery into the care of individuals with ALS and/or FTD. Consensus management guidelines for ALS do not comprehensively address the issue of genetic testing, and questions remain about whom to test, what counseling should be provided before and after testing, laboratory methods, and test interpretation. These challenges have contributed to inconsistent clinical practices and present barriers to patients wishing to access testing. This review summarizes the clinical impact of the discovery of the C9orf72 HRE, outlines ongoing challenges, and provides recommendations for C9orf72 testing, counseling, and research.
Genetic studies as early as 2006 established that amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), 2 seemingly distinct neurodegenerative disorders, were linked to chromosome 9p21.1 In 2011, 2 groups independently identified a G4C2 hexanucleotide repeat expansion (HRE) in the first intron (or promoter region, depending on transcript) of C9orf72 and demonstrated that this expansion underlies a significant proportion of familial and sporadic ALS, FTD, and combined ALS-FTD. Normal alleles were reported to contain no more than 23 repeats, with pathogenic expansions having greater than 30 repeats. Intermediate alleles of 23–30 repeats were reported to have uncertain pathogenicity.2,3 The mechanism of disease is the focus of active investigation and may include loss of function of the C9orf72 protein and the formation of RNA foci and dipeptide repeat proteins. The mechanisms may act in concert and/or differ depending on repeat size.4 Antisense oligonucleotide, small molecule, and other therapies hold promise for slowing disease progression in patients with the C9orf72 HRE.
Most patients with the C9orf72 HRE present clinically with ALS or behavioral variant FTD, or a combination of both. The spectrum of phenotypes includes rapidly and slowly progressive disease, and clinical presentations may differ from classic ALS or FTD.5,6 In families, the C9orf72 HRE is transmitted in an autosomal dominant manner, with age-dependent and incomplete penetrance. Age at onset and presenting symptomotology is variable, even among members of the same family.7 Notably, some individuals with a HRE in the clearly pathogenic range remain symptom free, even in old age.8 The C9orf72 HRE is by far the most common known genetic cause of ALS and FTD, far exceeding the prevalence of pathogenic variants in any other gene, although frequencies vary greatly by geoancestry. The highest HRE frequencies are observed in Europe, reported in 40% of familial and 6% of sporadic cases of ALS and in 18% of familial and 6% of sporadic cases of FTD in European patient cohorts. The HRE is rarely found in Asia.9–13
C9orf72 in the Clinic: Progress and Challenges
Current Status of Testing and Counseling
Clinical testing for the C9orf72 HRE has been available since 2012.14 Currently available commercial assays include repeat-primed PCR (RP-PCR), fluorescent amplicon length analysis, and Southern blot. United States ALS management guidelines do not address the offer of genetic testing, whereas European guidelines specify that testing be offered to patients with familial disease.15,16 Several published clinician surveys suggest a growing consensus to offer genetic testing to patients with familial ALS, but the offer of testing to the typical ALS patient with sporadic disease remains inconsistent.17 Table 1 summarizes published clinician practice surveys.
Table 1.
Percentage of Clinicians Offering ALS Genetic Testing, by Family History Indication, in Practice Surveys (Adapted from Klepek et al., 2019)
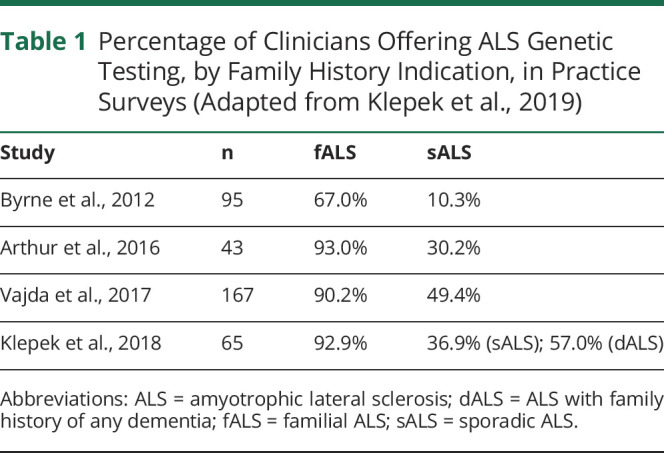
Patient experience and attitudes toward genetic testing in ALS have been investigated in several studies. In a 2015 survey of US ALS patients, 33.4% of respondents (n = 449) reported being offered genetic testing, including 68.9% of those with familial and 27.5% of those with sporadic ALS. Respondents expressed positive attitudes toward genetic testing and perceived benefits to ALS patients, families, the medical community, and society; this was most pronounced in those who reported a positive family history of ALS. Among those who had testing, those who received positive and negative results reported equally favorable experiences, suggesting that those who get “bad news” maintained a favorable outlook about the benefits of testing.18,19 Attitudes toward presymptomatic ALS testing have also been studied; in a study of family members of persons with ALS, 60% indicated they would have presymptomatic testing if offered.20
Challenges in Laboratory Detection and Interpretation of the HRE
PCR-based assays for detection of the HRE are widely used in commercial laboratories, but their accuracy has been questioned. In a 2014 blinded study, only 5/14 laboratories reported PCR-based C9orf72 results in complete concordance with the reference Southern blot result, and both false negative and false positive results were identified.21 Insertions and deletions adjacent to the repeat have been shown to interfere with detection of the expansion using PCR-based assays.22 Southern blot has been advocated as the gold standard for clinical testing.6 Currently, it is the only commercial test capable of sizing large repeats, but few laboratories offer it, and it may not detect intermediate repeats or small expansions.23 In the future, alternative methods for detecting and sizing the HRE may be commercially available, such as short- and long-read sequencing techniques that are currently used in the research setting.
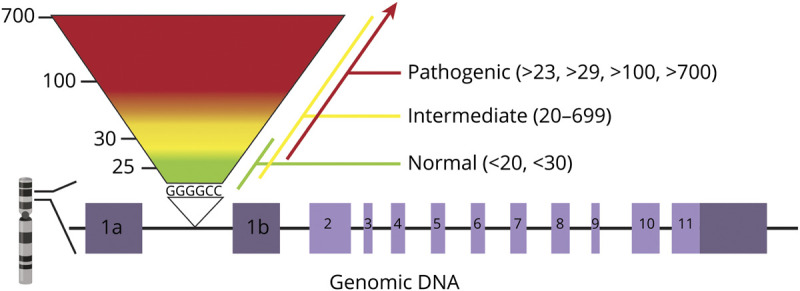
In addition, laboratory interpretation and reporting of the HRE is inconsistent. Published surveys of commercial laboratory tests reveal that cutoffs for normal, intermediate, and expanded alleles varied widely. Cutoffs for intermediate alleles ranged from 20 to several hundred; cutoffs for pathogenic alleles ranged from >23 to >700 (figure). Intermediate or small expansions could therefore be interpreted differently by different laboratories, and such cases have been reported.23,24 There is currently no validated cutoff that differentiates between pathogenic and nonpathogenic alleles.
Figure. Variable Interpretations of the C9orf72 Hexanucleotide Repeat Number Among Commercial Laboratories.
Finally, the C9orf72 HRE shows somatic and germline instability further complicating result interpretation. For example, expansion sizes as measured in blood may differ from those in relevant neural tissues.25 Intermediate alleles identified in patients with ALS may be incidental or could be associated with larger, pathogenic expansions in the brain.6 The size of the HRE as measured in blood may change over time in the same individual,26 although this has not been observed to increase from the normal to pathogenic range. Repeat instability also occurs in the germline, but evidence does not support the common occurrence of genetic and clinical anticipation as seen in other repeat expansion disorders such as myotonic dystrophy type 1 and Fragile X syndrome.26 Contraction of the HRE in successive generations has recently been demonstrated, particularly with paternal transmission.26,27
Unanswered Questions Regarding Disease Expression
When patients with C9orf72 HRE present with symptoms of motor neuron disease, the phenotype is often indistinguishable from classic ALS. Although the most common cognitive phenotype is behavioral variant FTD, a range of cognitive-behavioral presentations are observed, including psychotic features such as delusions and hallucinations. Patients presenting with neuropsychiatric changes may not be recognized and diagnosed with C9orf72-related disease.6 Repeat number has not been robustly shown to correlate with age at onset, disease duration, or clinical presentation (ALS and/or FTD),25,28 although this remains an active area of investigation.
Early family studies suggested that nearly all HRE carriers develop disease by the age of 80 years.9,29 However, the identification of the HRE in 0.2%–0.6% of unaffected controls and the presence of elderly, neurologically healthy obligate HRE carriers in affected families support incomplete penetrance.5,25,29 Penetrance has not been established to correlate with repeat size.26 A recent study modeled HRE penetrance in a large cohort drawn from published literature, concluding that lifetime penetrance is incomplete (estimated at ∼90% by the age of 80 years).29 Oligogenic inheritance has been proposed to contribute to the variable penetrance of the HRE and other pathogenic ALS variants; however, evidence in support of this model is lacking.30
Incomplete Knowledge of C9orf72 HRE Prevalence
HRE frequencies are highest in Scandinavian countries9,31 and lowest in Asian populations.9,10,12,13,31 HRE frequencies in other populations, however, including non-White or admixed populations in the United States and Europe have not been studied extensively. Tables e-1 and e-2 (links.lww.com/NXG/A345) summarize published frequencies by geoancestry.
Practice Recommendations
All Persons With ALS and/or FTD of European Geoancestry Are Candidates for HRE Testing
Many persons with ALS and/or FTD wish to know as much as possible about why they developed the condition and the risk that family members could also be affected. A genetic diagnosis enables clinicians to provide personalized, specific answers to these questions, and affected persons and their relatives may feel empowered by the knowledge and the opportunity to act, whether for reproductive planning, presymptomatic testing, or research participation. Testing of persons with ALS and/or FTD, but without a known family history of either condition, identifies the HRE in up to 10%.32 HRE testing should be offered to any patient who meets clinical diagnostic criteria for either condition.33 The current availability of clinical trials targeting HRE pathology in ALS underscores the importance of HRE testing in ALS management. Although pathogenic variation in more 30 other genes has been established to cause ALS and/or FTD, these account for a smaller proportion of cases in populations of European descent.34 In addition, uncertain or indeterminate results are rare with C9orf72 HRE testing, occurring in less than 1% cases; most patients get a clear positive or negative result.
Amplicon Length Analysis With Bidirectional RP-PCR Is Suitable for Large Scale HRE Testing
The RP-PCR assays widely used in commercial testing for the HRE are cost-effective, rapid, and identify patients with an expanded allele by revealing a “saw-tooth pattern” when a significant repeat expansion is present. However, as noted previously, interpretation of C9orf72 RP-PCR results is not always straightforward. Amplicon length analysis, however, is a reliable method for sizing intermediate and smaller HREs and may be helpful in resolving cases in which sequence variants interfere with RP-PCR results. A combination of amplicon-length analysis and bidirectional 5′ and 3′ RP-PCR is recommended to obtain the highest level of sensitivity and specificity.21,22 The use of 2 RP-PCR assays from both the 3′ and 5′ end of the repeat region and 2 nonoverlapping primer sets for amplicon length analysis buffers against potential allele drop out due to neighboring sequence variants and aids in determining zygosity in patients with HRE (Luke Drury, PhD, written communication, August 20, 2020). Although this approach cannot size large expansions, there are currently no established correlations between expansion size and disease expression. This combined technique currently represents the best approach to the clinical detection of the C9orf72 HRE on a large scale.
Southern blot or advanced sequencing techniques may be necessary in rare cases, such as those in which RP-PCR results are discrepant. Southern blot may also be considered if PCR detects only one allele, particularly with a rare repeat size, to determine whether the individual is truly homozygous or whether an HRE is present that failed to amplify.35 Recently published testing guidelines for myotonic dystrophy, type 2, include performing Southern blot in all apparently homozygous cases.36 Although up to 34% of individuals are expected to have homozygous C9orf72 repeat sizes, rare homozygous repeats (3, 7, 11, and 12) are found in less than 1% (Matthew Harms, MD, written communication, April 1, 2020); such cases could be targeted for additional testing. Southern blot and other methods capable of sizing the HRE remain important tools in the research setting and may be clinically necessary in the future if HRE size is clearly shown to affect disease expression.
Genetic Counseling Should Be Provided to All Affected Persons
All persons with ALS or FTD should be provided genetic counseling, irrespective of whether they have a positive family history of ALS or FTD or are seeking genetic testing. A discussion of ALS and/or FTD genetics should introduce genetic heterogeneity, dominant transmission, disease expression, and penetrance, leading to the offer of C9orf72 HRE testing for persons with European geoancestry.
Provide Genetic Risk Assessment Informed by Family History
The family history is a fundamental aspect of genetic risk assessment and genetic counseling for persons with ALS or FTD. Clinicians should document a 3-generation pedigree, at minimum, that ascertains ALS, FTD, other dementias, parkinsonism, ataxia, psychiatric disorders, and suicide.33 The pedigree should be reviewed for evidence of dominant transmission. However, many circumstances may obscure a clear pattern of dominant transmission in families with the C9orf72 HRE,37 and data from ALS clinic cohorts suggest that only about half of those who carry the HRE have a family history of ALS.38,39 Family history information may be used to provide HRE risk assessment for affected persons who are considering HRE testing. See table 2 for HRE incidence estimates by diagnosis and family history.
Table 2.
Incidence of C9orf72 HRE in Patients With ALS, FTD, and ALS/FTD of European Geoancestry

Empower Affected Persons to Make an Informed Decision About HRE Testing
Pretest counseling should prepare individuals for the possible impact of testing on themselves and their family. Cognitively impaired persons should be assisted by a legal guardian or health care proxy throughout the counseling and testing process. All affected persons and their families should be informed that testing for the most common genetic cause of ALS (or FTD) is available and that this is a personal/family choice. The availability of clinical trials should be discussed. The clinical heterogeneity and incomplete penetrance of the HRE should be emphasized. Patients and families should be informed of the limitations of HRE testing, including that (1) a negative result does not exclude a genetic basis or contribution to the proband's condition, (2) rarely, an intermediate HRE of uncertain significance may be found, or the laboratory test may not be able to provide a result, and (3) positive results do not allow prediction of disease course.14,37 Post-test counseling points for patients with positive, negative, and intermediate HRE test outcomes are outlined in table 3.
Table 3.
C9orf72 HRE Post-test Counseling Considerations for Persons With ALS, ALS-FTD, and FTD

Prepare Unaffected Family Members Who Seek Presymptomatic Testing
Presymptomatic testing for the C9orf72 HRE may be offered to adult relatives of persons with a documented, pathogenic HRE. This clinical service requires extensive genetic counseling and is best provided by a certified genetic counselor and/or at a tertiary ALS or FTD clinic. Individuals seeking presymptomatic testing should be assessed for psychosocial readiness to receive the news of their genetic status and motivations for seeking testing explored. Pretest counseling should review risks and legal protections pertaining to genetic discrimination. Most importantly, the limitations of testing must be emphasized. A positive result does not resolve uncertainty regarding disease expression, age at onset, or whether disease will actually occur. Most patients seek testing to determine whether they will develop ALS or FTD, but we currently cannot predict whether a healthy HRE carrier will develop ALS, FTD, both, or neither. Although some studies have suggested that penetrance approaches 90%–100% by age 80, other evidence suggests a far lower penetrance.9,29,32 This uncertainty has affected clinical practice, and family members undergoing presymptomatic testing at different centers have reported being told widely different disease risks. Providing specific risk figures conveys a degree of certainty that does not exist for the individual patient at this time and is not recommended. Many HRE carriers will die from other causes; thus, stating penetrance figures based on a theoretical 80- or 90-year lifespan may be misleading. Until penetrance is better understood, at-risk persons should be told that testing can determine whether they inherited their family's predisposition to ALS and/or FTD, and the magnitude of that risk is not precisely known but increases with age. HRE carriers should also know that they have a 50% chance to pass on the HRE to each child whether or not they themselves develop disease.
Goals for Future Research
Develop Evidence-based Guidelines for ALS and FTD Genetic Testing and Counseling
There is currently no consistent approach to the offer of testing for the typical ALS or FTD patient, who has no family history of the disease. Many patients who desire testing and counseling are not offered these services.18 A recent survey of ALS clinicians revealed that 74% would be more likely to offer ALS genetic testing if testing guidelines were published.17 Genetic testing guidelines have been published for other neurodegenerative diseases such as Huntington and Alzheimer disease40,41 and successfully incorporated into patient care. Consistent genetic testing practices would also support the identification of appropriate patients for gene-targeted trials and therapies.
Although further study is needed regarding the minimum HRE size necessary to cause disease, commercial laboratories are in urgent need of guidance to ensure consistent interpretations of HRE sizes. Currently, intermediate or smaller expansions could be interpreted differently at different laboratory tests, potentially leading to discrepant and/or inaccurate results for patients or family members undergoing diagnostic or presymptomatic testing.23,24 Laboratories, clinicians, and patients could benefit from guidelines establishing consistent HRE cutoffs, which will likely require updating as new information becomes available. Given that some affected HRE carriers have been found to carry pathogenic variants in other genes, it is unclear whether additional genetic should be offered after a patient tests positive for the HRE. Guidance is also needed to arm genetic counselors and other clinicians with a consistent message regarding penetrance of the C9orf72 HRE. The development of evidence-based guidelines that encompass HRE test indication, methodology, interpretation, and genetic counseling would support clinicians and laboratories in providing consistent and equitable testing and care.
Prioritize C9orf72 as a Target for Clinical Trials
Recent data from clinic-based cohorts has shown that the relative contribution of the C9orf72 HRE, as compared to other ALS genes, is even greater than initial estimates suggested. Among more than 500 ALS patients tested via the ALS Genetic Access program of the Northeast ALS Consortium, the HRE accounted for 75% of all pathogenic or likely pathogenic variants identified in the 6 genes tested (unpublished data). Similarly, the HRE comprised 79% of pathogenic or likely pathogenic variants detected in a Midwestern clinic cohort; testing included comprehensive multigene panel testing for familial or early onset cases. The incidence of pathogenic or likely pathogenic variants in SOD1 and in genes other than C9orf72 in clinic-based ALS cases in the United States may be lower than that reported in research cohorts.39 Accordingly, research and industry should prioritize the C9orf72 HRE as a target for therapy. Carriers of the HRE are present in every ALS and FTD clinic, and with consistent testing practices, they will be increasingly identified. They represent a rich opportunity to study the natural history of the condition, identify early biomarkers of disease, and ultimately, develop gene-targeted therapies and neuroprotective interventions. With coordinated effort from the scientific community, the next generation of HRE carriers may stand to benefit.
Engage Diverse Patients in Research
Further study is needed to better understand the incidence of the HRE in diverse populations. Our current understanding of the familial and genetic basis of ALS and FTD is based on the study of White individuals, and data thus derived (such as published empiric recurrence risks) may not be applicable to other populations.37 Although the HRE has been identified in persons with ALS in Europe, North and South America, and East and South Asia,9 incidence has not been extensively studied in many populations. Data on the genetic profile of sporadic and familial ALS and FTD in persons of diverse geoancestry are critical to determine the need for population-specific testing approaches. It is currently unclear, for example, whether the universal offer of HRE testing is warranted in affected persons of non-European descent. Likewise, for familial ALS or FTD cases of non-European descent, it is uncertain whether the first step of testing should be multigene panel testing or HRE testing. The success of ALS and FTD research hinges on successfully recruiting and retaining patients of diverse backgrounds. Table 4 summarizes practice recommendations and goals for future research.
Table 4.
Summary of Practice Recommendations and Goals for Research

Conclusion
The discovery of the C9orf72 HRE marked the finish line in the long search for the underlying cause of chromosome 9-linked FTD and ALS. The work of applying the many clinical implications of this discovery then began, and much progress has been made in the 10 years since. Commercial testing for the HRE is now widely available, and many clinicians are offering testing to persons with familial ALS. Going forward, several important steps will continue to advance patient care, research, and therapy. This includes the development of consensus guidelines for the genetic testing and counseling of persons with ALS and FTD, including the offer of HRE testing to all affected persons of European geoancestry, recommendations for consistent repeat size cutoffs and interpretation, and guidance in providing counseling regarding penetrance and other issues. Given that the HRE accounts for a significant majority of pathogenic variants identified in clinical testing in the North America and Europe, C9orf72 should be prioritized as a target for gene-specific therapies. Finally, researchers should recruit and engage affected persons of diverse backgrounds to understand the genetic profile of ALS and FTD in all populations. Achieving these goals represents the next milepost in the work of understanding and treating the C9orf72 HRE.
Acknowledgment
The author thanks Dr. Stephen Kolb for helpful comments on the manuscript, Courtney Fleming for assistance with the figure, and Leah Vicini for assistance with the supplementary tables.
Glossary
- ALS
amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- HRE
hexanucleotide repeat expansion
- RP-PCR
repeat-primed PCR
Author Contribution
J. Roggenbuck: Drafting/revision of the manuscript for content, including medical writing for content; major role in the acquisition of data.
Study Funding
No targeted funding reported.
Disclosure
J. Roggenbuck received support from the OSUWMC Neurological Research Institute and the Julie Bonasera Fund for ALS and Neuromuscular Diseases. Go to Neurology.org/NG for full disclosures.
References
- 1.Vance C, Al-Chalabi A, Ruddy D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 2006;129:868–876. 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 2018;14:544–558. 10.1038/s41582-018-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beck J, Poulter M, Hensman D, et al. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am J Hum Genet 2013;92:345–353. 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohrer JD, Isaacs AM, Mizielinska S, et al. C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol 2015;14:291–301. 10.1016/S1474-4422(14)70233-9. [DOI] [PubMed] [Google Scholar]
- 7.Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783. 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langenhove TV, van der Zee J, Gijselinck I, et al. Distinct clinical characteristics of C9orf72 expansion carriers compared with GRN, MAPT, and nonmutation carriers in a Flanders-Belgian FTLD cohort. JAMA Neurol 2013;70:365–373. 10.1001/2013.jamaneurol.181. [DOI] [PubMed] [Google Scholar]
- 9.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai CP, Soong BW, Tu P-H, et al. A hexanucleotide repeat expansion in C9ORF72 causes familial and sporadic ALS in Taiwan. Neurobiol Aging 2012;33:2232.e11–2232.e18. 10.1016/j.neurobiolaging.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Jang JH, Kwon MJ, Choi WJ, et al. Analysis of the C9orf72 hexanucleotide repeat expansion in Korean patients with familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:1311.e7–1311.e9. 10.1016/j.neurobiolaging.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Konno T, Shiga A, Tsujino A, et al. Japanese amyotrophic lateral sclerosis patients with GGGGCC hexanucleotide repeat expansion in C9ORF72. J Neurol Neurosurg Psychiatry 2013;84:398–401. 10.1136/jnnp-2012-302272. [DOI] [PubMed] [Google Scholar]
- 13.Zou ZY, Li XG, Liu MS, Cui LY. Screening for C9orf72 repeat expansions in Chinese amyotrophic lateral sclerosis patients. Neurobiol Aging 2013;34:1710.e5–1710.e6. 10.1016/j.neurobiolaging.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 14.Fong JC, Karydas AM, Goldman JS. Genetic counseling for FTD/ALS caused by the C9ORF72 hexanucleotide expansion. Alzheimers Res Ther 2012;4:27 10.1186/alzrt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller RG, Jackson CE, Kasarskis EJ, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73:1227–1233. 10.1212/WNL.0b013e3181bc01a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen PM, Borasio GD, Dengler R, et al. EFNS task force on management of amyotrophic lateral sclerosis: guidelines for diagnosing and clinical care of patients and relatives. Eur J Neurol 2005;12:921–938. 10.1111/j.1468-1331.2005.01351.x. [DOI] [PubMed] [Google Scholar]
- 17.Klepek H, Nagaraja H, Goutman SA, Quick A, Kolb SJ, Roggenbuck J. Lack of consensus in ALS genetic testing practices and divergent views between ALS clinicians and patients. Amyotroph Lateral Scler Frontotemporal Degener 2019;20:216–221. 10.1080/21678421.2019.1582670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner KN, Nagaraja H, Allain DC, Quick A, Kolb S, Roggenbuck J. Patients with amyotrophic lateral sclerosis have high interest in and limited access to genetic testing. J Genet Couns 2017;26:604–611. 10.1007/s10897-016-0034-y. [DOI] [PubMed] [Google Scholar]
- 19.Wagner KN, Nagaraja HN, Allain DC, Quick A, Kolb SJ, Roggenbuck J. Patients with sporadic and familial amyotrophic lateral sclerosis found value in genetic testing. Mol Genet Genomic Med 2018;6:224–229. 10.1002/mgg3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fanos JH, Gelinas DF, Miller RG. “You have shown me my end”: attitudes toward presymptomatic testing for familial amyotrophic lateral sclerosis. Am J Med Genet A 2004;129A:248–253. 10.1002/ajmg.a.30178. [DOI] [PubMed] [Google Scholar]
- 21.Akimoto C, Volk AE, van Blitterswijk M, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 2014;51:419–424. 10.1136/jmedgenet-2014-102360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nordin A, Akimoto C, Wuolikainen A, et al. Sequence variations in C9orf72 downstream of the hexanucleotide repeat region and its effect on repeat-primed PCR interpretation: a large multinational screening study. Amyotroph Lateral Scler Frontotemporal Degener 2017;18:256–264. 10.1080/21678421.2016.1262423. [DOI] [PubMed] [Google Scholar]
- 23.Crook A, McEwen A, Fifita JA, et al. The C9orf72 hexanucleotide repeat expansion presents a challenge for testing laboratories and genetic counseling. Amyotroph Lateral Scler Frontotemporal Degener 2019;20:310–316. 10.1080/21678421.2019.1588904. [DOI] [PubMed] [Google Scholar]
- 24.Klepek H, Goutman SA, Quick A, Kolb SJ, Roggenbuck J. Variable reporting of C9orf72 and a high rate of uncertain results in ALS genetic testing. Neurol Genet 2019;5:e301 doi: 10.1212/NXG.0000000000000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12:978–988. 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fournier C, Barbier M, Camuzat A, et al. Relations between C9orf72 expansion size in blood, age at onset, age at collection and transmission across generations in patients and presymptomatic carriers. Neurobiol Aging 2019;74:234.e1–234.e8. 10.1016/j.neurobiolaging.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Jackson JL, Finch NA, Baker MC, et al. Elevated methylation levels, reduced expression levels, and frequent contractions in a clinical cohort of C9orf72 expansion carriers. Mol Neurodegener 2020;15:7 10.1186/s13024-020-0359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gendron TF, Belzil VV, Zhang YJ, Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol 2014;127:359–376. 10.1007/s00401-013-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chiò A, Traynor BJ. Age-related penetrance of the C9orf72 repeat expansion. Sci Rep 2017;7:2116 10.1038/s41598-017-02364-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross JP, Leblond CS, Laurent SB, et al. Oligogenicity, C9orf72 expansion, and variant severity in ALS. Neurogenetics 2020;21:227–242. 10.1007/s10048-020-00612-7. [DOI] [PubMed] [Google Scholar]
- 31.van der Zee J, Gijselinck I, Dillen L, et al. A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 2013;34:363–373. 10.1002/humu.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turner MR, Al-Chalabi A, Chio A, et al. Genetic screening in sporadic ALS and FTD. J Neurol Neurosurg Psychiatry 2017;88:1042–1044. 10.1136/jnnp-2017-315995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roggenbuck J, Fong JC. Genetic testing for amyotrophic lateral sclerosis and frontotemporal dementia. Clin Lab Med 2020;40:271–287. ClinicalKey. Availabe at: clinicalkey.com/#!/content/playContent/1-s2.0-S0272271220300342?returnurl=null&referrer=null. Accessed July 20, 2020. [DOI] [PubMed] [Google Scholar]
- 34.Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol 2017;13:96–104. 10.1038/nrneurol.2016.182. [DOI] [PubMed] [Google Scholar]
- 35.Cleary EM, Pal S, Azam T, et al. Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol Cell Probes 2016;30:218–224. 10.1016/j.mcp.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoser B, Montagnese F, Bassez G, et al. Consensus-based care recommendations for adults with myotonic dystrophy type 2. Neurol Clin Pract 2019;9:343–353. 10.1212/CPJ.0000000000000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roggenbuck J, Quick A, Kolb SJ. Genetic testing and genetic counseling for amyotrophic lateral sclerosis: an update for clinicians. Genet Med 2017;19:267–274. 10.1038/gim.2016.107. [DOI] [PubMed] [Google Scholar]
- 38.Umoh ME, Fournier C, Li Y, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016;87:1024–1030. 10.1212/WNL.0000000000003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roggenbuck J, Palettas M, Vicini L, Patel R, Quick A, Kolb SJ. Incidence of pathogenic, likely pathogenic, and uncertain ALS variants in a clinic cohort. Neurol Genet 2020;6:e390 doi: 10.1212/NXG.0000000000000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nance MA Genetic testing of children at risk for Huntington's disease. US Huntington Disease Genetic Testing Group. Neurology 1997;49:1048–1053. 10.1212/wnl.49.4.1048. [DOI] [PubMed] [Google Scholar]
- 41.Goldman J, Farmer J, Deerlin VV, Wilhelmsen K, Miller B, Grossman M. Frontotemporal dementia: genetics and genetic counseling dilemmas. Neurologist 2004;10:227–234. 10.1097/01.nrl.0000138735.48533.26. [DOI] [PubMed] [Google Scholar]