

Abstract
Objective
MS is an autoimmune demyelinating disease of the CNS, which causes neurologic deficits in young adults and leads to progressive disability. The aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor, can drive anti-inflammatory functions in peripheral immune cells and also in CNS-resident cells. Laquinimod is a drug developed for the treatment of MS known to activate AHR, but the cellular targets of laquinimod are still not completely known. In this work, we analyzed the contribution of AHR activation in astrocytes to its beneficial effects in the experimental autoimmune encephalomyelitis (EAE) preclinical model of MS.
Methods
We used conditional knockout mice, in combination with genome-wide analysis of gene expression by RNA-seq and in vitro culture systems to investigate the effects of laquinimod on astrocytes.
Results
We found that AHR activation in astrocytes by laquinimod ameliorates EAE, a preclinical model of MS. Genome-wide RNA-seq transcriptional analyses detected anti-inflammatory effects of laquinimod in glial cells during EAE. Moreover, we established that the Delaq metabolite of laquinimod dampens proinflammatory mediator production while activating tissue-protective mechanisms in glia.
Conclusions
Taken together, these findings suggest that AHR activation by clinically relevant AHR agonists may represent a novel therapeutic approach for the treatment of MS.
MS is a chronic inflammatory disease of the CNS, which in most patients initially presents with a relapsing-remitting course (RRMS).1 This relapsing-remitting stage is often followed by a secondary progressive phase (SPMS) characterized by the progressive and irreversible accumulation of neurologic deficits. In primary progressive MS (PPMS), neurologic impairment progresses continuously without superimposed relapses.1 Most current immunomodulatory therapies in MS target the adaptive immune response, which is mostly relevant to the relapsing-remitting phase of MS.2 However, there is an unmet clinical need for therapeutic approaches to target CNS-intrinsic mechanisms of disease pathology driven by astrocytes and microglia, which are thought to promote disease progression in SPMS and PPMS.2
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor with important effects in inflammatory responses driven by CNS-resident and peripheral cells.3,4 Indeed, AHR activation by endogenous, microbial derived or therapeutically administered agonists has been shown to ameliorate experimental autoimmune encephalomyelitis (EAE), the animal model of MS.5–9 Moreover, AHR activation in CNS-resident glial cells including astrocytes and microglia mediates tissue-protective and regenerative effects relevant to progressive stages of MS.10–12 However, AHR agonistic activity in serum is reduced in patients with MS, potentially contributing to the dysregulation of proinflammatory and neurodegenerative mechanisms.10,13,14
Laquinimod (Laq) activates AHR and showed positive effects in some MS clinical trials.15–18 In addition to relapse reduction, Laq administration resulted in the reduction of white and gray matter atrophy and diminished permanent black hole formation in MS,18 suggesting that Laq acts on CNS-resident cells relevant to MS progression. However, clinical use of Laq is hampered due to an incomplete understanding of its modes of action, potential side effects, and limited clinical efficacy.
In this study, we report that AHR activation in astrocytes by the Laq metabolite Delaq ameliorates EAE. These findings may guide the development of future therapeutic approaches based on AHR activation in acute and chronic inflammation.
Methods
Mice
Female C57Bl/6 mice and 1–3-day-old pups from C57Bl/6J mice were obtained from the Jackson Laboratory and were kept in a pathogen-free facility at the Harvard Institutes of Medicine. Glial fibrillary acidic protein (GFAP)-AHR and LysM-AHR mice have been described before.10,11,19 All experiments were performed in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee at Harvard Medical School.
EAE Induction and Treatment
EAE was induced in 8-week-old mice by subcutaneous immunization with 150 μg MOG35–55 peptide emulsified in complete Freund adjuvant (Difco Laboratories, Detroit, MI) per mouse, followed by administration of 200 ng pertussis toxin (List biological Laboratories, Inc., Campbell, CA) on days 0 and 2 as described.10,20 Clinical signs of EAE were assessed as follows: 0, no signs of disease; 1, loss of tone in the tail; 2, hind limb paresis; 3, hind limb paralysis; 4, tetraplegia; and 5, moribund. All agents were purchased from Sigma-Aldrich (St. Louis, MO). Starting from day 2 after disease induction, mice were treaty daily with oral gavage of Laq (25 mg/kg) or vehicle, respectively.
Isolation of Cells From Adult Mouse CNS
Mononuclear cells were isolated from the CNS as previously described, and astrocytes, monocytes, and microglia were sorted as described before.10,20 Isolated CNS cells were stained with fluorochrome-conjugated antibody to CD11b (M1/70, 1:100), CD45 (90, 1:100), Ly6C1 (HK1.4, 1:100), CD105 (N418, 1:100), CD140a (APA5, 1:100), CD11c (N418, 1:100), F4/80 (BM8, 1:50), O4 (O4, Miltenyi Biotec, 1:10), and CD19 (eBio1D3, 1:100). All antibodies were from eBioscience or BD Pharmingen, unless otherwise mentioned. Microglia were sorted as CD11b+ cells with low CD45 expression and low LY6C1 (CD11b+CD45lowLy6C1low), inflammatory monocytes were considered as CD45hiCD11b+Ly6C1hi. Astrocytes were sorted as CD11blowCD45low Ly6C1lowCD105lowCD140alowCD11blowF4/80lowO4lowCD19low after the exclusion of lymphocytes, microglia, oligodendrocytes, and monocytes. Sorted astrocytes were >85% GFAP+ as determined by fluorescence-activated cell sorting analysis and by quantitative PCR (qPCR) analysis of the expression of the astrocyte markers Gfap, Aldh1l1, and Aqp4.
Flow Cytometry Staining and Acquisition
Mononuclear cell suspensions were prepared as previously described.10 Antibodies for flow cytometry were purchased from eBioscience or BD Pharmingen and used at a concentration of 1:100 unless recommended otherwise by the manufacturer. Cells were then analyzed on an LSRII or MACSQuant flow cytometer (BD Biosciences, Franklin Lakes, NJ, and Miltenyi Biotec, Bergisch Gladbach, Germany, respectively).
Sequence Alignment and Quantification
The reads were aligned to the GRCm38 reference genome using STAR (v2.7.3a),21 and the aligned reads were quantified using RSEM (v1.3.1).22
Differential Expression Analysis and Downstream Analysis
The quantification results from RSEM were aggregated and analyzed using DESeq2,23 and the log2 fold changes of genes were shrunk using the Approximate posterior estimation for GLM coefficients (ApeGLM).24 The downstream analysis was performed using gene set enrichment analyses (GSEA) and ingenuity pathway analysis (IPA).
Ingenuity Pathway Analysis
To determine significant pathways, differentially expressed genes that passed false discovery rate <0.1 for vehicle vs Laq-treated mice were uploaded and analyzed using the IPA tool. p Values were calculated using the Fisher exact test.
Network Analysis
The network diagram was generated using IPA. The activation status of a pathway was predicted using IPA's upstream regulatory tool by an activation z score. The z score was calculated using the formula z = (Σiwixi)/(√Σiwi2), where wi is the weight of gene i and xi is the number of activating and inhibiting predictions of gene i. Positive z score indicated upregulated pathway, whereas negative z score indicated downregulated pathway as the overall effect.
Quantitative PCR
RNA was extracted with RNAeasy kit (Qiagen, Hilden, Germany), and complementary DNA was prepared and used for qPCR, with the results normalized to Gapdh levels. All primers and probes were from Applied Biosystems. Mouse: Ccl2 Mm00441242_m1, Cyp1b1 Mm00487229_m1, Gapdh Mm99999915_g1, Il10 Mm00439614_m1, Nos2 Mm00440502_m1, and Tnfa Mm00443258_m1.
Primary Astrocyte Cultures
Cerebral cortices from neonatal C57Bl/6J mice aged 1–3 days were dissected, stripped of their meninges, digested with 0.25% trypsin–ethylenediaminetetraacetic acid (EDTA) and DNAse I (1 mg/mL) for 15 minutes, and dispersed to single-cell level by passing through a cell strainer (70 μm). The cell suspension was then cultured at 37°C in humidified 5% CO2, 95% air on poly-l-lysine (Sigma) coated 175-cm2 cell culture flasks. The medium was replaced every 4–5 days. After 7–10 days, cells reached confluence, and astrocytes were isolated by mild trypsinization with trypsin-EDTA (0.06%) as previously described.10 Cells were >95% astrocytes as determined by staining with GFAP or GLAST, with less than 5% contamination of CD11b+ microglia cells After the isolation procedure, cells were further plated as required for the specific experiments. Astrocyte cultures were activated with tumor necrosis factor-α (TNFα) and interleukin (IL)-1β (5 ng/mL and 10 ng/mL, respectively, both R&D Systems) in the presence of Laq 100 ng/mL, Delaq 100 ng/mL, or vehicle. Unless otherwise indicated, RNA analysis was performed 24 hours after start of treatment.
Statistical Analysis
Statistical analyses were performed with Prism software (GraphPad, San Diego, CA), using the statistical tests indicated in the individual figure legends. No samples were excluded. The investigators were blinded as to the treatment of mice in individual experiments. p Values of <0.05 were considered significant. All error bars represent SEM or SD as noted in the individual figure legends. Unless otherwise stated, 3 independent experiments were used for all assays, and displayed figures are representative.
Data Availability
Anonymized data that are not published in this article will be made available on request from any qualified investigator.
Results
Laq Ameliorates Autoimmune CNS Inflammation Independent of AHR in Myeloid Cells
To analyze the cell populations involved in the AHR-dependent therapeutic effects of Laq on EAE, we used LysMCre AHRfl/fl mice (LysM-AHR mice) in which AHR is deleted in myeloid cells including monocytes, macrophages, granulocytes, and some microglia. Starting 2 days after EAE induction by immunization with MOG35-55, mice were treated with Laq or vehicle administered by gavage. Laq ameliorated EAE both in control and LysM-AHR mice as indicated by a reduction of the clinical score (figure 1, A and B) and the number and fractions of CNS-infiltrating effector T cells (figure 1, C–E).
Figure 1. Laquinimod Acts on Dendritic Cells to Ameliorate EAE.

EAE was induced in control (A) and LysM-AHR (LysMCre AHRfl/fl, B) mice. Animals were treated with daily oral doses of laquinimod or vehicle starting from day 2 after immunization. Clinical scores are mean ± SEM and representative of 2 independent experiments. **p < 0.01 by 2-way analysis of variance (ANOVA). (C and D) Absolute numbers and (E) relative fractions of CNS-infiltrating T cells were determined by fluorescence-activated cell sorting staining for CD3, CD4, IFN-γ, and IL-17A. Data are mean ± SEM. *p < 0.05. AHR = aryl hydrocarbon receptor; EAE = experimental autoimmune encephalomyelitis; IFN = interferon; IL = interleukin; n.s. = not significant as determined by 1-way analysis of variance followed by the Tukey post hoc test.
AHR in Astrocytes Contributes to the Protective Effects of Laq in Late-Stage EAE
AHR in astrocytes limits CNS inflammation by suppressing nuclear factor kappa B (NF-κB) activation and the expression of NF-κB-driven transcriptional modules that promote inflammation and neurodegeneration.4,10 Thus, to evaluate the role of AHR activation in astrocytes in the therapeutic effects of Laq in EAE, we used GFAPCre Ahrfl/fl mice (GFAP-AHR mice) in which AHR has been deleted from astrocytes.10
Laq administration delayed EAE onset in GFAP-AHR mice, but failed to ameliorate EAE in GFAP-AHR mice during the late stage of the disease (figure 2A). Moreover, Laq administration failed to reduce the number of CNS-infiltrating proinflammatory monocytes and number and fractions of effector T cells in GFAP-AHR mice (figure 2, B–D). Indeed, Laq administration in GFAP-AHR mice exacerbated proinflammatory monocyte influx into the CNS (figure 2B), potentially due to the pronounced upregulation of CCL2 following AHR deletion in astrocytes and a dysregulation of the astrocyte to microglia crosstalk in GFAP-AHR mice, which we have previously demonstrated.10 Taken together, these findings suggest that AHR activation in astrocytes contributes to the beneficial effects of Laq during chronic CNS inflammation.
Figure 2. Laquinimod Acts on Astrocytes to Ameliorate Late Stage EAE.

(A) EAE was induced in control and GFAP-AHR (GFAPCre AHRfl/fl). Animals were treated with daily oral doses of laquinimod or vehicle starting from day 2 after immunization. Clinical scores are mean ± SEM and representative of 2 independent experiments. *p < 0.05. **p < 0.01. ***p < 0.001 by 2-way analysis of variance (ANOVA). (B) Absolute numbers of CNS-infiltrating proinflammatory monocytes as determined by fluorescence-activated cell sorting (FACS) staining for CD11b, CD45, and Ly6C. Data are mean ± SEM. (C) Absolute numbers and (D) relative fractions of CNS-infiltrating T cells were determined by FACS staining for CD3, CD4, IFN-γ, and IL-17A. Data are mean ± SEM of n = 5 mice per group and representative of 3 independent experiments. AHR = aryl hydrocarbon receptor; EAE = experimental autoimmune encephalomyelitis; GFAP = glial fibrillary acidic protein; IFN = interferon; IL = interleukin; n.s. = not significant as determined by 1-way ANOVA followed by the Tukey post hoc test.
Laq Suppresses Disease-Promoting Transcriptional Modules in Astrocytes via AHR
To further investigate the beneficial effects of AHR activation by Laq during EAE, we analyzed by RNA-seq the transcriptional response of control and AHR-deficient astrocytes in Laq-treated mice. Laq treatment had significant effects in the transcriptional response of astrocytes in control, but not GFAP-AHR mice, suggesting that the effects of Laq on astrocytes are mediated by AHR (figure 3, A and B). Indeed, in agreement with the reported ability of AHR to suppress NF-κB activation in astrocytes during the course of EAE,10,11,25 ingenuity pathway analyses detected a significant downregulation of NF-κB-driven proinflammatory mechanisms (table). In agreement with these findings, we detected the downregulation of NF-κB–driven proinflammatory mediators such as IL-6 and iNOS (table).11,26–28 Moreover, we also detected a Laq-induced regulation of transcriptional programs associated with the production of chemokines, neurotrophin, and axonal guidance signaling, as well as pathways relevant for transendothelial migration (figure e-1, links.lww.com/NXI/A382). Collectively, these findings suggest that AHR activation induced in astrocytes by Laq treatment downmodulates transcriptional responses that promote CNS pathogenesis.
Figure 3. Laquinimod Acts on Astrocytes by Modulating the Production of Proinflammatory Cytokines, Chemokines, and Neurotoxic Mediators.

(A) Heat map of expressed (detected at level 0.1 in at least 2 of 3 samples) genes (signal:noise ratio) of astrocytes fluorescence-activated cell sorting sorted from animals as in (figure 2A). Data represent means of n = 3 mice. Gene expression levels are row centered and log2 transformed and saturated at levels −0.5 and +0.5 for visualization satisfying a false discovery rate <0.1. (B) Principal component analysis plot of RNA sequencing data isolated from microglia of mice as in (A). AHR = aryl hydrocarbon receptor; GFAP = glial fibrillary acidic protein.
Table.
Ingenuity Pathway Analysis of Pathways Regulated in Astrocytes from Control Laquinimod vs Control Vehicle-Treated Mice

Laq Acts on Astrocytes to Reduce Microglia Proinflammatory Pathways
Astrocytes and microglia play important roles in the CNS both during health and disease.12,29 Indeed, the crosstalk between astrocytes and microglia controls their proinflammatory and disease-promoting responses in EAE and MS.30 The inefficient activity of LysM-Cre in microglia limits our ability to evaluate the effects of direct microglial AHR activation by Laq treatment in EAE. However, astrocytes can secrete granulocyte-macrophage colony-stimulating factor, IL-33, and additional factors to modulate microglial responses.31 Thus, we evaluated the effects of AHR activation in astrocytes by Laq administration on microglial transcriptional response during EAE. Laq-triggered AHR activation in astrocytes during EAE had significant effects on the transcriptional response of microglia as determined by RNA-seq (figure 4). Indeed, GSEA detected the upregulation of proinflammatory pathways in microglia from Laq-treated GFAP-AHR compared with control mice (figure e-2, links.lww.com/NXI/A382), supporting a role of AHR-driven transcriptional modules in astrocytes on the control of microglial responses during EAE.
Figure 4. Laq Acts on Astrocytes to Indirectly Modulate Microglial Proinflammatory Pathways.
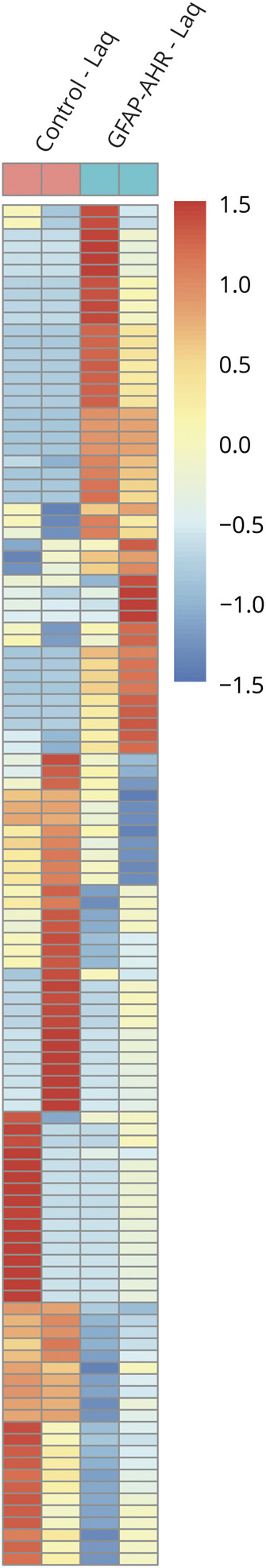
Heat map of expressed genes (signal:noise ratio) of microglia fluorescence-activated cell sorting sorted from Laq-treated control and GFAP-AHR animals. Gene expression levels are row centered and log2 transformed and saturated at levels −0.5 and +0.5 for visualization satisfying a false discovery rate <0.1. AHR = aryl hydrocarbon receptor; GFAP = glial fibrillary acidic protein; Laq = laquinimod.
Laq Metabolite Delaq Activates AHR-Driven Protective Mechanisms in EAE
Hepatic metabolism leads to the breakdown of Laq to its de-ethylated metabolite Delaq (de-ethylated Laq), a potent AHR agonist.32 However, it is still unclear whether Laq or its metabolite Delaq activates AHR to limit CNS inflammation. To address this point, we treated primary murine astrocyte cultures with equimolar concentrations of Laq or Delaq. Delaq, but not Laq, induced the expression of the AHR transcriptional target Cyp1b1 both in resting astrocytes and in astrocytes activated with the proinflammatory stimuli TNFα and IL-β (figure 5A). Indeed, Delaq reduced the expression of genes associated with disease-promoting astrocyte activities during EAE, such as immune cell recruitment (Ccl2) and neurotoxicity and microglia activation (Nos2 and Tnfa), while it enhanced the expression of anti-inflammatory Il10 (figure 5B). These findings suggest that Delaq, but not Laq, activates AHR in astrocytes to induce anti-inflammatory and tissue protective mechanisms that limit CNS pathology.
Figure 5. Delaq Is the Active Metabolite of Laquinimod and Dampens Proinflammatory Activation of Primary Astrocytes.

(A) Primary astrocytes from neonatal mice were activated with TNFα and IL-1β in the presence of vehicle, laquinimod, or Delaq. RNA was isolated, transcribed, and subjected to qPCR for the AHR responsive gene Cyp1b1. Data are mean ± SEM and representative of 2 independent experiments. (B) Primary astrocytes were activated as in (A) in the presence of vehicle or Delaq. RNA was isolated, transcribed, and subjected to qPCR for the indicated genes. Data are mean ± SEM and representative of 2 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. IL = interleukin; n.s. = not significant as determined by 1-way analysis of variance followed by the Tukey post hoc test; qPCR = quantitative PCR; TNFα = tumor necrosis factor-α.
Discussion
In clinical trials conducted in patients with MS, Laq reduced MS relapse rates and slowed the accumulation of white and gray matter atrophy,16,17,33 suggesting that in addition to its effects on peripheral immune cells,34–38 AHR activation interferes with mechanisms of disease pathogenesis driven by CNS-resident cells. However, it remains unclear whether these suppressive effects of Laq on brain atrophy are an indirect result from its effects on peripheral immune cells and/or reflect direct effects of Laq on CNS-residents cells. In this context, our study identifies neuroprotective and anti-inflammatory effects of Laq mediated by AHR activation in CNS-resident glial cells. Indeed, Laq administration led to the activation of AHR in astrocytes, which was associated with clinical improvement during late stages of EAE, reduction of CNS-infiltrating proinflammatory monocytes and T-cell numbers, and polarization. Transcriptional analyses identified astrocyte-intrinsic effects of Laq, which included dampening of proinflammatory pathways and enhancement of anti-inflammatory and tissue-regenerative mechanisms. Moreover, the astrocyte responses induced by Laq also decreased proinflammatory responses in microglia indirectly, suggesting that Laq affects the modulation of microglial responses by astrocytes. Finally, our in vitro studies on primary astrocytes in culture suggest that these beneficial effects of Laq administration are mediated by its metabolite Delaq. Taken together, these results underline the potential role of AHR-targeted therapies for the management of neurodegenerative diseases.
Several studies analyzed the effects of Laq on the peripheral immune compartment. One of the first studies testing the effects of Laq in EAE determined dose-dependent reduction of clinical symptoms in MOG35-55-induced EAE: although peripheral immune cell counts in secondary lymphoid organs were unaltered, immune cell infiltration into the CNS was greatly reduced in Laq-treated animals.38 Moreover, Laq treatment initiated after the first relapse also proved effective in reducing relapse number and severity in a chronic EAE model.39 More recently, Laq was also shown to reduce autoimmune inflammatory damage to the optic nerve and retina, which are usually affected in MS.40 These beneficial effects were attributed in part to a shift in the phenotype and migratory properties of T cells.37,38 Moreover, Laq showed beneficial effects in B cell–dependent spontaneous chronic EAE models, where Laq reduced the activation of dendritic, T follicular helper, and T memory cells, while expanding the compartment of regulatory T cells.41 In addition, Laq inhibited the development of meningeal B-cell aggregates and myelin-specific antibody secretion.41 Finally, Laq was also shown to enhance immunomodulatory functions of natural killer cells in their interaction with CD155 positive dendritic cells due to alterations in dendritic cell antigen presentation function.42 Together with previous observations, our findings suggest that the anti-inflammatory effects of Laq in the peripheral immune compartment target the interactions of dendritic cells with components of the adaptive immune system.
Laq administration has been shown to trigger AHR activation in vivo,43,44 most likely via its active metabolite Delaq, as demonstrated for glial cells here. Of note, Delaq itself cannot be administered in vivo because of its low solubility and short half-life, suggesting that Laq is metabolized into Delaq in the local microenvironment.32 Using complete AHR-deficient mice, 2 independent groups determined that the suppression of EAE by Laq administration is AHR dependent, although AHR-independent mechanisms may also play a role.43,44 Indeed, the induction of brain-derived natriuretic factor, a glial neuroprotective factor, is driven by Laq in an AHR-independent manner, suggesting that Laq engages additional pathways in glial cells in addition to AHR to promote the production of neuroprotective factors and regulators of excitotoxic neurotransmitters including glutamate.44–46 Although our transcriptional analyses corroborate profound alterations in astrocyte transcriptional profiles during EAE induced by astrocyte AHR deficiency,10 they also point to the existence of AHR-independent mechanisms induced by Laq in AHR-deficient astrocytes in GFAP-AHR mice. In these lines, the expression of neuroprotective factors induced by Laq in astrocytes was only partially suppressed in GFAP-AHR mice. In this context, Tapinarof is a novel AHR agonist developed for the treatment of skin inflammation.47 Of interest, the therapeutic effects of Laq are mediated by AHR and the transcription factor NFR2,47 which promotes the production of neutrotrophic and anti-oxidant factors in astrocytes and is the target of dimethyl fumarate.48 Thus, Laq may potentially activate NRF2 and other transcription factors in addition to AHR to modulate the transcriptional response of astrocytes during CNS inflammation. These pathways are of great relevance for disease progression in chronic stages of MS, which are not properly modeled by the C57Bl/6 model of EAE. Hence, additional studies should investigate the effects of Laq on additional preclinical models, such as the chronic progressive EAE model in nonobese diabetic mice or the cuprizone-induced model of demyelination and neurodegeneration, which recapitulates recapitulate some aspects of the progressive MS.
In summary, our study suggests that AHR activation in astrocytes is a potential therapeutic approach for neurologic disorders. Our findings also suggest that Delaq, a short-lived metabolite of Laq produced in the liver, mediates the therapeutic effects of Laq. Thus, a second generation of AHR activators aimed at achieving increased AHR activation within the CNS may provide novel therapeutic approaches for MS and other neurologic disorders.
Glossary
- AHR
aryl hydrocarbon receptor
- EAE
experimental autoimmune encephalomyelitis
- EDTA
ethylenediaminetetraacetic acid
- GFAP
glial fibrillary acidic protein
- GSEA
gene set enrichment analyses
- IL
interleukin
- IPA
ingenuity pathway analysis
- Laq
laquinimod
- NF-κB
nuclear factor kappa B
- PPMS
primary progressive MS
- qPCR
quantitative PCR
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- TNFα
tumor necrosis factor-α
Appendix. Authors

Contributor Information
Veit Rothhammer, Email: veit.rothhammer@tum.de.
Jessica E. Kenison, Email: jessica.kenison@antolrx.com.
Zahorong Li, Email: zli37@bwh.harvard.edu.
Emily Tjon, Email: emilyctjon@gmail.com.
Maisa C. Takenaka, Email: mcstakenaka@gmail.com.
Chun-Cheih Chao, Email: u911636@gmail.com.
Kalil Alves de Lima, Email: kalil.delima@gmail.com.
Davis M. Borucki, Email: boruckid@musc.edu.
Joel Kaye, Email: joel.k@ayalapharma.com.
Study Funding
This work was supported by grants AI075285 and AI093903 from the NIH and RG4111A1 and JF2161-A-5 from the National Multiple Sclerosis Society to F.J. Quintana. V. Rothhammer received support from an educational grant from Mallinkrodt Pharmaceuticals (A219074), a Heisenberg fellowship from the German Research Foundation (DFG RO4866 1/1, RO4866 3/1) and the European Research Council (ERC Starting Grant 851693 HICI). Teva provided research support and laquinimod material for the study.
Disclosure
Teva provided research support and laquinimod material for the study. Go to Neurology.org/NN for full disclosures.
References
- 1.Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 2015;14:406–419. [DOI] [PubMed] [Google Scholar]
- 2.Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018;97:742–768. [DOI] [PubMed] [Google Scholar]
- 3.Gutierrez-Vazquez C, Quintana FJ. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018;48:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 2019;19:184–197. [DOI] [PubMed] [Google Scholar]
- 5.Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008;453:65–71. [DOI] [PubMed] [Google Scholar]
- 6.Apetoh L, Quintana FJ, Pot C, et al. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol 2010;11:854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quintana FJ, Murugaiyan G, Farez MF, et al. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 2010;107:20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu HY, Quintana FJ, da Cunha AP, et al. In vivo induction of Tr1 cells via mucosal dendritic cells and AHR signaling. PLoS One 2011;6:e23618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quintana FJ, Jin H, Burns EJ, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol 2012;13:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rothhammer V, Mascanfroni ID, Bunse L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 2016;22:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rothhammer V, Borucki DM, Tjon EC, et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018;557:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler MA, Quintana FJ. Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb Perspect Med 2018;9:a029009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rothhammer V, Borucki DM, Kenison JE, et al. Detection of aryl hydrocarbon receptor agonists in human samples. Sci Rep 2018;8:4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rothhammer V, Borucki DM, Garcia Sanchez MI, et al. Dynamic regulation of serum aryl hydrocarbon receptor agonists in MS. Neurol Neuroimmunol Neuroinflamm 2017;4:e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorensen PS, Comi G, Vollmer TL, et al. Laquinimod safety profile: pooled analyses from the ALLEGRO and BRAVO trials. Int J MS Care 2017;19:16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vollmer TL, Sorensen PS, Selmaj K, et al. A randomized placebo-controlled phase III trial of oral laquinimod for multiple sclerosis. J Neurol 2014;261:773–783. [DOI] [PubMed] [Google Scholar]
- 17.Filippi M, Rocca MA, Pagani E, et al. Placebo-controlled trial of oral laquinimod in multiple sclerosis: MRI evidence of an effect on brain tissue damage. J Neurol Neurosurg Psychiatry 2013;85:851–858. [DOI] [PubMed] [Google Scholar]
- 18.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med 2012;366:1000–1009. [DOI] [PubMed] [Google Scholar]
- 19.Takenaka MC, Gabriely G, Rothhammer V, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 2019;22:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayo L, Trauger SA, Blain M, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med 2014;20:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 2019;35:2084–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeste A, Takenaka MC, Mascanfroni ID, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal 2016;9:ra61. [DOI] [PubMed] [Google Scholar]
- 26.Wheeler MA, Jaronen M, Covacu R, et al. Environmental control of astrocyte pathogenic activities in CNS inflammation. Cell 2019;176:581–596.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler MA, Clark IC, Tjon EC, et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020;578:593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chao CC, Gutierrez-Vazquez C, Rothhammer V, et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell 2019;179:1483–1498.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giovannoni F, Quintana FJ. The role of astrocytes in CNS inflammation. Trends Immunol 2020;41:805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron 2020;108:608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vainchtein ID, Chin G, Cho FS, et al. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 2018;359:1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahiout S, Tagliabue SG, Nasri A, et al. In vitro toxicity and in silico docking analysis of two novel selective AH-receptor modulators. Toxicol In Vitro 2018;52:178–188. [DOI] [PubMed] [Google Scholar]
- 33.Comi G, Pulizzi A, Rovaris M, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet 2008;371:2085–2092. [DOI] [PubMed] [Google Scholar]
- 34.Toubi E, Nussbaum S, Staun-Ram E, et al. Laquinimod modulates B cells and their regulatory effects on T cells in multiple sclerosis. J Neuroimmunol 2012;251:45–54. [DOI] [PubMed] [Google Scholar]
- 35.Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 2012;251:14–24. [DOI] [PubMed] [Google Scholar]
- 36.Schulze-Topphoff U, Shetty A, Varrin-Doyer M, et al. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS One 2012;7:e33797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wegner C, Stadelmann C, Pfortner R, et al. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 2010;227:133–143. [DOI] [PubMed] [Google Scholar]
- 38.Yang JS, Xu LY, Xiao BG, Hedlund G, Link H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, modulates the Th1/Th2 balance and induces the Th3 cytokine TGF-beta in Lewis rats. J Neuroimmunol 2004;156:3–9. [DOI] [PubMed] [Google Scholar]
- 39.Brunmark C, Runstrom A, Ohlsson L, et al. The new orally active immunoregulator laquinimod (ABR-215062) effectively inhibits development and relapses of experimental autoimmune encephalomyelitis. J Neuroimmunol 2002;130:163–172. [DOI] [PubMed] [Google Scholar]
- 40.Wilmes AT, Reinehr S, Kuhn S, et al. Laquinimod protects the optic nerve and retina in an experimental autoimmune encephalomyelitis model. J Neuroinflammation 2018;15:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varrin-Doyer M, Pekarek KL, Spencer CM, et al. Treatment of spontaneous EAE by laquinimod reduces Tfh, B cell aggregates, and disease progression. Neurol Neuroimmunol Neuroinflamm 2016;3:e272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ott M, Avendano-Guzman E, Ullrich E, et al. Laquinimod, a prototypic quinoline-3-carboxamide and aryl hydrocarbon receptor agonist, utilizes a CD155-mediated natural killer/dendritic cell interaction to suppress CNS autoimmunity. J Neuroinflammation 2019;16:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaye J, Piryatinsky V, Birnberg T, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci USA 2016;113:E6145–E6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berg J, Mahmoudjanlou Y, Duscha A, et al. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J Neuroimmunol 2016;298:9–15. [DOI] [PubMed] [Google Scholar]
- 45.Mishra MK, Wang J, Keough MB, et al. Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Ann Clin Transl Neurol 2014;1:409–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gentile A, Musella A, De Vito F, et al. Laquinimod ameliorates excitotoxic damage by regulating glutamate re-uptake. J Neuroinflammation 2018;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith SH, Jayawickreme C, Rickard DJ, et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest Dermatol 2017;137:2110–2119. [DOI] [PubMed] [Google Scholar]
- 48.Montes Diaz G, Hupperts R, Fraussen J, Somers V. Dimethyl fumarate treatment in multiple sclerosis: recent advances in clinical and immunological studies. Autoimmun Rev 2018;17:1240–1250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data that are not published in this article will be made available on request from any qualified investigator.