

Abstract
Immune responses to neonatal hypoxic ischemic encephalopathy (HIE) exacerbate brain injury. Phagocytes, including microglia, play a central role in the immune response, but how the activation of phagocytes is regulated remains elusive. Previously we have reported that interferon regulatory factor 5 (IRF5) signaling is closely correlated with a pro-inflammatory microglial phenotype in adult mice after stroke. The present study investigated IRF5’s regulatory role in post-HIE inflammation. Male IRF5 conditional knockout (CKO) and IRF5fl/fl postnatal day 10 (P10) pups were subjected to the Rice-Vannucci Model (RVM) to induce HIE. Outcomes including morphological and neurobehavioral changes were evaluated at day 7 after HIE. Microglia/macrophage phenotypes and inflammatory responses were evaluated by flow cytometry (FC), RT-PCR and multiplex cytokine assays. Lenti-IRF5 virus was administered in microglia-neuron co-cultures to evaluate the effects of microglial IRF5 upregulation in ischemic neurons exposed to oxygen glucose deprivation (OGD). Deletion of phagocytic IRF5 resulted in significantly decreased IRF5 expression, attenuated pro-inflammatory and enhanced anti-inflammatory responses to HIE, and improved outcomes compared to IRF5fl/fl control pups. In vitro lentivirus transfection experiments revealed that overexpression of IRF5 in microglia amplified pro-inflammatory signals and exacerbated OGD-induced neuronal apoptosis and neurite fragmentation. IRF5 signaling mediates microglial pro-inflammatory activation and also affects anti-inflammatory responses. Phagocytic IRF5 signaling is detrimental in HIE and is a potential therapeutic target for post-ischemic inflammation.
Keywords: Brain, Inflammation, Interferon regulatory factor 5 (IRF5), Neonatal Ischemia, Phagocyte
Introduction
HIE occurs in neonates when there is deprivation of oxygen supply, commonly known as intrapartum asphyxia. HIE is a major cause of neonatal mortality and long-term mental and motor disabilities, including cerebral palsy, seizmes, and developmental delay [1–3]. Immune responses to HIE is a fundamental pathological process and exerts secondary nemonal damage after the ischemic insult, in which phagocytic activation plays a central role. During ischemia, microglia and monocytes are activated through either classical or alternative pathways, with the former pro-inflammatory and the latter anti-inflammatory [4, 5]. Research efforts are focused on how to mitigate phagocytic pro-inflammatory responses, as phagocytes in this state release cytotoxic factors including iNOS, TNFα, IL-1ß and IL-6, etc.[6–8], leading to exacerbated immune responses [9, 10].
The interferon regulatory factor (IRF) family includes nine factors (IRF1-9) that are all involved in modulating the immune responses to various diseases. IRF5 plays a critical role in regulating pro-inflammatory responses in macrophages [11, 12]. However, whether IRF5 also regulates neonatal microglia activation is not clear. Om previous study has confirmed that another IRF, IRF4, is responsible for the anti-inflammatory responses of microglia to HIE. It has been suggested that in peripheral inflammatory diseases, IRF5 and IRF4 signaling acts in a balanced fashion to drive macrophage towards either pro- or anti-inflammatory polarization depending on the disease progression [11, 2]. In the present study, we hypothesized that IRF5 signaling regulates the pro-inflammatory activation of both microglia and macrophage, and that manipulation of phagocytic IRF5 signaling could impact HIE outcomes.
Materials and methods
Animals
To generate phagocytic IRF5 conditional knockout (CKO) mice, we crossed IRF5fl/fl with lysozyme M-Cre (LyzMCre) mice (Jackson Laboratory) in C57BL/6 background. All the breeding pairs were housed in pathogen-free rooms and had access to food and water ad libitum. Postnatal day 10 (P10) mice of IRF5 CKO and IRF5fl/fl (control) strains were used. All procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and were approved by the Institutional Animal care and use committee of the University of Texas Health Science Center.
Neonatal HIE model
The Rice-Vannucci Model (RVM) of neonatal hypoxia–ischemia was modified to induce HIE in P10 mice as described previously [13, 3]. Briefly, P10 pups were anesthetized with isoflurane (4% for induction and 1.5–2% for maintenance). A midline cervical incision was made and the right common carotid artery (CCA) was cauterized to occlude blood flow. After surgery, the pups were returned to their dams for two hours. To induce hypoxia, the CCA-occluded mice were put in a chamber containing 10% oxygen and 90% nitrogen for 45 minutes. After that, the animals were placed on a temperature-controlled blanket for 20 min and then returned to their dams. Mice were sacrificed at either 3d or 7d of surgery for histological, cellular, and molecular analysis. Sham mice underwent the same procedure except cauterization of the right CCA and induction of hypoxia in the chamber. In the present study, the HIE model led to 10-20% mortality in IRF5fl/fl pups and 10-30% mortality in IRF5 CKO cohorts by 7d.
Behavior testing
Seizure activity
Seizure activity was scored according to a seizure rating scale as previously reported [13, 3]. Every 5 min in 1 h at the same time of 1 day after HIE, the score corresponding to the highest level of seizure activity observed during that time period was recorded and summed to produce a total seizure score. Seizure behavior was scored as follows: 0 = normal behavior; 1 = immobility; 2 = rigid posture; 3 = repetitive scratching, circling, or head bobbing; 4 = forelimb clonus, rearing, and falling; 5 = mice that exhibited level four behaviors repeatedly; and 6 = severe tonic-clonic behavior.
Y-maze test
Spontaneous alternation using a Y-maze is a test for habituation and spatial working memory [14]. The symmetrical Y-maze consists of three white opaque plastic arms at a 120° angle from each other. After placing pups in the center, the animal is allowed to freely explore the three arms. Over the course of multiple arm entries, the subject should show a tendency to enter a less recently visited arm. The test consists of a single 5 min trial; spontaneous Alternation (%) is defined as consecutive entries in 3 different arms (arm A,B,C) divided by the number of total alternations (total arm entries minus 2) [15]. Mice with less than 8 arm entries during the 5-min trial were excluded from the analysis. An entry occurs when all four limbs are within the arm. All the arms were cleaned after each trial.
Flow cytometry and intracellular cytokines staining
Leukocytes from the brain tissue were prepared as previously described [3, 2, 11]. Briefly, animals were anesthetized with Avertin® (2,2,2-Tribromoethanol, Sigma) and transcardially perfused with phosphate-buffered saline (PBS) for 5 min. The brain was then divided along the interhemispheric fissure into two hemispheres. Ipsilateral brains were processed to isolate leukocytes. Isolated leukocytes were stained with antibodies of cell surface markers and intracellular cytokines as described in the online-only supplementary material. FlowJo (Tree Star Inc.) software was employed for data analysis.
mRNA extraction and gene expression analysis by real-time PCR
Total RNA was isolated from sorted microglia and monocytes using RNAqueous® Micro Total RNA Isolation Kit (Thermo Fisher Scientific, USA), and cDNA was then synthesized from 30 ng of total RNA with the Sensiscript RT Kit (Qiagen GmbH, Hilden, Gennany) in accordance with the manufacturer’s instructions. Real-time polymerase chain reaction (RT-PCR) was performed with a QuantiTect SYBR Green PCR Kit (Qiagen GmbH) using the BIO-RAD C1000 Thermal Cycler (BIO-RAD, Hercules, CA, USA). Results were normalized using the housekeeping gene GAPDH and the 2−ΔΔCt cycle threshold method. A complete list of primer is shown in Supplementary Table 1.
Immunohistochemistry and cresyl violet (CV) staining
At day 7, all the pups were anesthetized by Avertin® and then transcardially perfused with 0.1M sodium phosphate buffer (pH 7.4) followed by 4% paraformaldehyde for post-fixation of the brains. The brain sections were stained with rabbit anti-Iba1 (1:300, Wako, Japan) and goat anti-IRF5 (AP16060PU-T; 1:50, OriGene Technologies) for phagocyte staining. Brain images were obtained on Leica DMi8 confocal microscope. At day 7 of HIE, brain tissue loss was evaluated by performing CV staining as describes previously [15, 3, 2]. For details, see the online-only supplementary material.
Brain and serum cytokine levels by multiplex ELISA
Brain homogenate was prepared as describe previously [11]. Cytokine levels in plasma and in brain homogenates were measured by commercially available specific quantitative Bio-Plex Pro™ Mouse Cytokine 23-plex assay kit according to manufacturer’s instruction (# M60009RDPD, Bio-Rad Laboratories, Hercules, CA).
Primary mixed glial culture
Primary cortical mixed glial cultures were prepared from C57BL/6 mouse brains within 0-1 day (P0-P1) of birth as in [16]. Briefly, the cortices were dissociated via serial incubations with neuronal dissociation kit according to the manufacturer instruction (MACS Miltenyi Biotech.). The mixed cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fatal bovine serum (FBS) and 1% Penicillin/Streptomycin (P/S) for 10 days in an incubator at 37 °C and with 5% CO2 gas. The culture was boosted by replacing L929 conditioned media at day 3 and 5. After 10 days, flasks were shaken for 3 hours at 300rpm to remove loosely attached immature microglia. The collected microglia were further cultured in 8-well chamber slides for ICC, 96 well plate for viability assay and in 12 well cell culture insert (0.4 micron, 1.13 cm2 12W insert, catalog: 140652, ThermoFisher Scientific, USA) coated with poly D lysine (0.001%, Sigma) for coculture with neruons. The purity of these microglial cultures was 99% as determined by Iba1 immunoreactivity.
Primary cortical neuron culture
Cultures of cortical neurons from E15–16 embryonic C57BL/6 mouse (Jackson Laboratory) were prepared as described previously [17, 18]. Briefly, cortical tissue isolated from 8-10 E15-16 mice were dissected into small pieces and incubated at 37 °C with 5% trypsin/EDTA followed by trituration with siliconized Pastern pipettes. Trypsin activity was stopped by adding 1 mL of FBS. The cells were then passed through a 40μm filter to ensure single cell population. The cells were counted and plated on poly-d-lysine (50 μg/ml; Sigma)-coated round glass slide in 12W culture plates with 500mL of DMEM medium without FBS. After 24h, culture medium was replenished with Neurobasal media supplemented with 1% B27 (ThermoFisher Scientific) with L-glutamate and 1% PS. The microtubule-associated protein 2 (MAP2; Santa Cruz Biotechnology, USA)-immunocytochemical staining showed that the purity of primary neurons was over 97%. Half of the medium was replaced every 3 days. The neurons were used at 10 days of culture.
Lentivirus transfection
To investigate the transfection efficiency of the lentiviral vectors (conjugated with GFP) [19], microglia cells were seeded in 96-well plates at 2×103 cells per well and transfected at different multiplicity of infection (MOI) of the virus. After 3 days, nearly 90% of the transfected primary microglia were GFP positive at the MOI of 10 and 20, whereas 45% of the cells were GFP positive at the MOI of 3. On the 4th day of transfection, the GFP-positive microglia that had received lentivirus at MOI 10 and 30 began to aggregate and die by apoptosis; however, the microglia transfected with MOI 3 of lentivirus continued to expand and grow with a viability over 80%. The morphology of GFP positive microglia was homogeneous, similar to that of non-transfected primary microglia (Supp. Fig. S5). Therefore, the regimen of 4 days transfection with the virus of MOI 3 was selected for this study. The green fluorescence of GFP in transfected microglia was confirmed by ICC.
Oxygen-glucose deprivation
To model ischemia/reperfusion conditions in vitro, the microglia cultures were exposed to OGD as described previously [20]. The culture medium was replaced with serum-free, glucose-free Locke’s buffer (154 mM NaCl, 5.6 mM KCl, 2.3 mM CaCl2, 1 mM MgCl2, 3.6 mM NaHCO3, 5 mM HEPES and 5 mg/ml gentamicin, pH 7.2), and the cultures were incubated in an experimental hypoxia chamber in a saturated atmosphere of 95% N2 and 5% CO2 for 1 hour. The control (normoxia) cells were cultured in the presence of normal levels of glucose and were incubated in a humidified atmosphere of 95% air and 5% CO2. After 1 hours of OGD, cells were then supplemented with 10 mM glucose and incubated for 12 h at 37 °C.
Co-culture of neurons and microglia
Co-culture of neurons and microglia was performed as described previously [4]. Primary microglia were seeded in 96W and 12W polycarbonate cell culture insert (ThermoFisher Scientific, USA) coated with PDL (0.001%, Sigma). 48h later, microglia were transfected with either GFP only or GFP-IRF5 lentivirus as described above. Four days after the virus transfection, the cell culture insert containing microglia were transferred to corresponding neuronal cultures (96W plates for neuronal viability assays and 12W plates for immunocytochemistry (ICC) staining). 24h after the co-culture, cells were subjected to OGD [20]. After 2 hours of OGD, cells were then supplemented with 10 mM glucose and incubated for 12 h at 37 °C
Immunocytochemistry (ICC)
Cultured cells were fixed with 4% paraformaldehyde for 30 min at room temperature, washed with 0.1 M Tris-buffered solution (TBS; pH 7.5), blocked with 3% donkey serum (Sigma) in TBS containing 0.3% Triton X-100 at room temperature for 60 min, and incubated overnight with a unconjugated rabbit anti MAP2 antibody (sc-390543, Santa Cruz Biotechnology, Inc. Dallas, TX) for neuron and glial fibrillary acidic protein (GFAP) conjugated with Cy3 (# C9205, Millipore, Sigma) for astrocytes at 4°C. The cells were then washed with TBS and incubated with Alexa Fluor 488-conjugated secondary antibody (1:1000; Invitrogen, Carlsbad, CA) for 60 min. GFP signal was visualized in green channel (488). Finally, stained cells were mounted with Fluoroshield™ with DAPI (# F6057, Sigma) and visualized using a Leica DMi8 confocal microscopy (Buffalo Grove, IL, USA). Images were processed using ImageJ software (NIH, USA). The fluorescence intensity of IRF5 was measured in 21-24 random microscopic fields from 3 independent experiments by a blinded investigator.
Cell viability and apoptosis assay
Cell viability of cultured neurons was measured using MTS assay (Cell Titer 96® AQueous One Solution Cell Proliferation Assay kit, Promega, Madison, WI, USA) kit and Pierce LDH Cytotoxicity Assay Kit (# 88953, ThermoFisher Scentific, USA). Neuronal apoptosis was evaluated by TUNEL assay kit (In Situ Cell Death Detection Kit, Roche). All assays were performed according to the manufacturer’s protocols.
Statistical analysis
Data from individual experiments are presented as mean ± SD and assessed by Student t test or one-/two-way ANOVA with Tukey post-hoc test for multiple comparisons as appropriate (GraphPad Prism Software Inc., San Diego, CA, USA). P <0.05 was considered statistically significant. The ordinal data set for seizure behavioral scores were analyzed with Mann-Whitney U test. All the studies were performed by investigators blinded to the mouse strains for HIE surgery, behavioral testing, infarct, and inflammation analysis.
Results
Decreased expression of IRF5 in phagocytes of IRF5 CKO neonatal mice brains.
We first confirmed IRF5 deletion from phagocytes in our IRF5 CKO mouse model (IRF5fl/fl:LyzMCre). Colocalization of Iba1 and IRF5 immunofluorescent signals were detected in brain slices from IRF5 CKO pups. We found that almost none of the Iba1 positive cells express IRF5 in CKO groups; however, Iba1 and IRF5 were colocalized in control mice (IRF5fl/fl) (Supp. Fig. S1a). To further validate the deletion of IRF5, we performed RT-PCR in FC sorted microglia to examine IRF5 mRNA levels. Similar to the IHC results, microglia in the IRF5 CKO pups had significantly lower IRF5 mRNA levels compared to control mice (Supp. Fig. S1b). Since IRF5 and IRF4 counteract with each other [11], we also measured the IRF4 mRNA levels in sorted microglia of sham and HIE pups. The IRF5 CKO did not affect microglial IRF4 mRNA levels in either sham or HIE groups (Supp. Fig. S1c). The validation data confirmed efficient deletion of IRF5 by the LoxP-Cre system without affecting the baseline level of IRF4.
Attenuated immune responses to HIE in IRF5 CKO Vs. IRF5fl/fl pups.
IRF5 signaling is involved in post-stroke inflammation [11, 21]. To evaluate the effects of IRF5 CKO on the immune responses to HIE, we set out to examine microglial activation, peripheral leukocyte infiltration, and circulating cytokine levels in CKO mice.
Microglial activation
Microglial activation is a key triggering factor in the initiation and propagation of inflammatory responses to ischemia. We first performed FC to examine activation markers in microglia. Microglia were gated as CD45intermediateCD11b+, and the gating strategy is illustrated in Supp. Fig. S2. CD68 and CD206 are well-established cell membrane pro- and anti-inflammatory markers respectively [8, 22], whereas IL-1β/TNFα and IL-4/IL-10 are considered intracellular pro- and anti-inflammatory markers [23, 24]. We quantified the median fluorescence intensity (MFI) of CD68, CD206, IL-1β/TNFα and IL-4/IL-10 in microglia. 7 days after HIE, the MFI of CD68 was significantly decreased in IRF5 CKO microglia compared to controls (Fig. 1a). In contrast, the MFI of CD206 was significantly increased by IRF5 CKO (Fig. 1a). Intracellular staining revealed significantly lower levels of IL-1β in IRF5 CKO vs. control microglia (Supp. Fig. S3a&b), but no differences in TNFα, IL-4 and IL-10 MFI were found between two groups (Supp. Fig. S3c). However, we sorted microglia and measured pro- and anti-inflammatory cytokine mRNA levels with RT-PCR, and found IRF5 CKO microglia had significantly lower mRNA levels of pro-inflammatory markers (MHCII, TNFα) (Fig. 1c), and higher mRNA levels of anti-inflammatory markers (Arg1) compared to control microglia (Fig. 1d). We also examined FC-sorted monocytes and found IRF5 CKO decreased TNFα mRNA levels only in infiltrating monocytes (Supp. Fig. S4).
Figure 1.
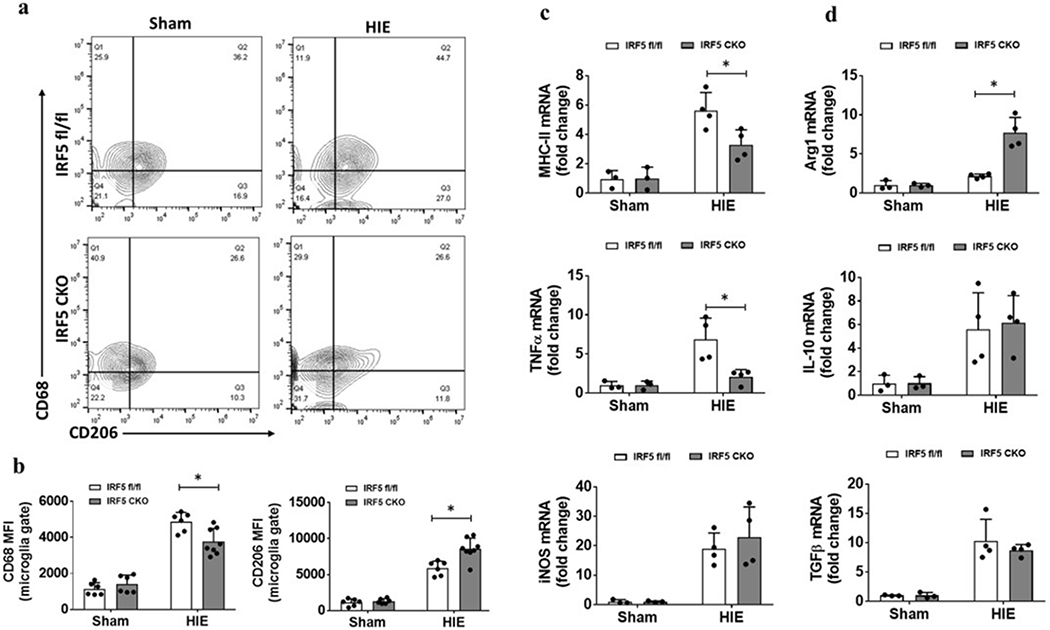
Microglial activation in IRF5 CKO vs. IRF floxed control mice assessed by flow cytometry at 7d after HIE. a, Representative contour plots and quantification of median fluorescence intensity (MFI) of cell surface markers CD68 and CD206 in microglia in both HIE and sham pups. b, Representative contour plots and quantification of MFI of intracellular cytokine TNFα and IL-1β in microglia in both HIE and sham pups. N=6 per sham and 6-8 per HIE group; *P < 0.05 (two-way ANOVA). c, mRNA level of MHCII, TNFα, and iNOS in sorted microglia, d, mRNA level of Arg1, IL-10, and TGFβ in sorted microglia. Data are expressed as fold change vs. sham-operated controls. N=3 per sham and 4 per HIE group; *P < 0.05 (two-way ANOVA).
Peripheral cell infiltration in HIE brains
We next quantified infiltrating peripheral leukocytes in HIE brains with FC. Total peripheral myeloid cells were gated as CD45highCD11b+, and lymphocytes as CD45highCD11b− (Supp. Fig. S2). Among total peripheral myeloid (pMyeloid) cells, monocytes were gated as CD45highCD11b+Ly6C+Ly6G−, and neutrophils as CD45highCD11b+Ly6C−Ly6G+. FC data showed IRF5 CKO mice had significantly fewer leukocytes (absolute counts/hemisphere), including total pMyeloid cells (Fig. 2b), neutrophils (Fig. 2c), and lymphocytes (Fig. 2d) in the ischemic brains compared with IRF5fl/fl pups 7 days after HIE. All sham mice had almost no leukocyte infiltration.
Figure 2.
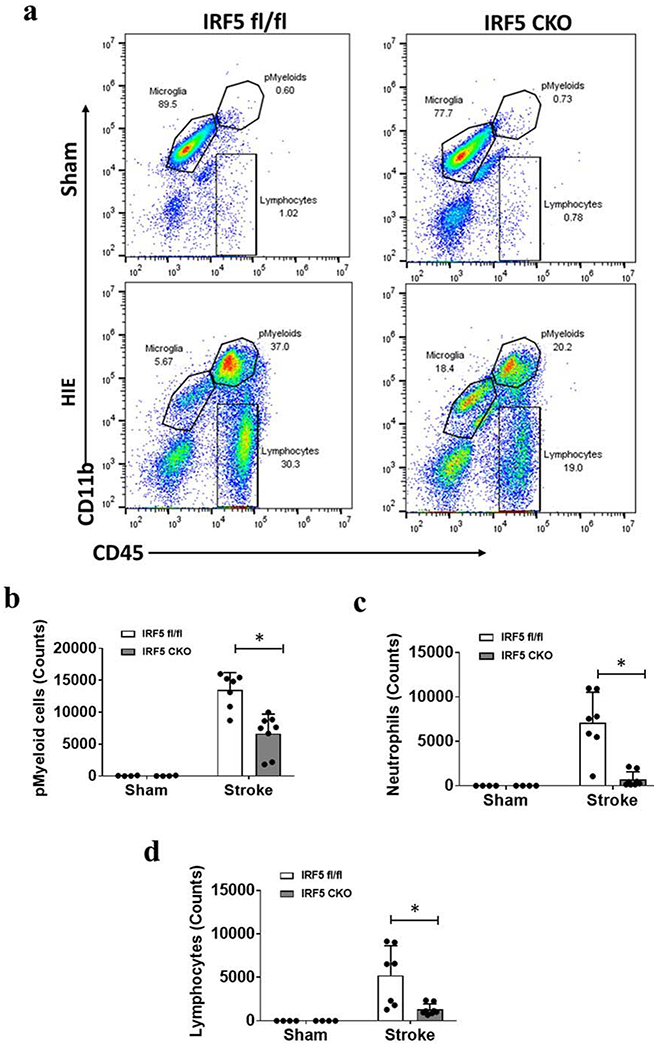
Infiltration of peripheral immune cells in the brain 7d after HIE. a, Representative dot plots depict the gating strategy to gate monocytes as CD45highCD11b+Ly6C+Ly6G−, and neutrophils as CD45highCD11b+Ly6G+Ly6C− from peripheral myeloid cells (pMyeloids). Lymphocyte gating strategy is shown in Supp. Fig. S2 Fig. b-d, Quantification (absolute counts) of pMyeloids, neutrophils, and lymphocytes in IRF5 CKO vs. control mice. N=4 per sham and 7-8 per HIE group; *P < 0.05 (two-way ANOVA).
Plasma and brain cytokine levels
We next examined plasma and brain cytokine levels at 3d and 7d after HIE, as cytokine levels are reflective of the level of immune response to ischemic injury [11, 25]. A same panel of pro-/anti-inflammatory cytokines and chemokines were examined with multiplex in both brain and plasma samples. We observed a decrease in pro-inflammatory (IL-6), and an increase in anti-inflammatory (IL-10) cytokine level in the plasma and the HIE brains of IRF5 CKO HIE pups compared to IRF5 fl/fl at 3d after HIE, (Fig. 3a, b). No differences were found at this time point in the levels of other inflammatory mediators (TNFα, IL-1β, IL-4, monocyte chemoattractant protein-1 (MCP-1), and Eotaxin) between CKO and the control groups (Supp. Fig. S5). At 7d after HIE, plasma IL-1β was significantly decreased and IL-10 increased in IRF5 CKO compared to IRF5 fl/fl strain (Fig. 3c); in the brain, the levels of pro-inflammatory mediators, i.e. IL-1β, IL-1α, IL-12, IL-6, Eotaxin, and MCP-1, were all significantly decreased in IRF5 CKO groups compared to the control (Fig. 3d), and no differences were seen in the levels of anti-inflammatory cytokines (IL-4, IL-10) (Supp. Fig. S6). Plasma levels of IL-1α, IL-12, IL-6, Eotaxin, MCP-1, and IL-4 were not different between CKO vs. control groups at 7d (Supp. Fig. S6a). These immune response data indicated an attenuated pro-inflammatory response in the brains of IRF5 CKO pups after HIE, and suggested IRF5 CKO favors an anti-inflammatory response.
Figure 3.
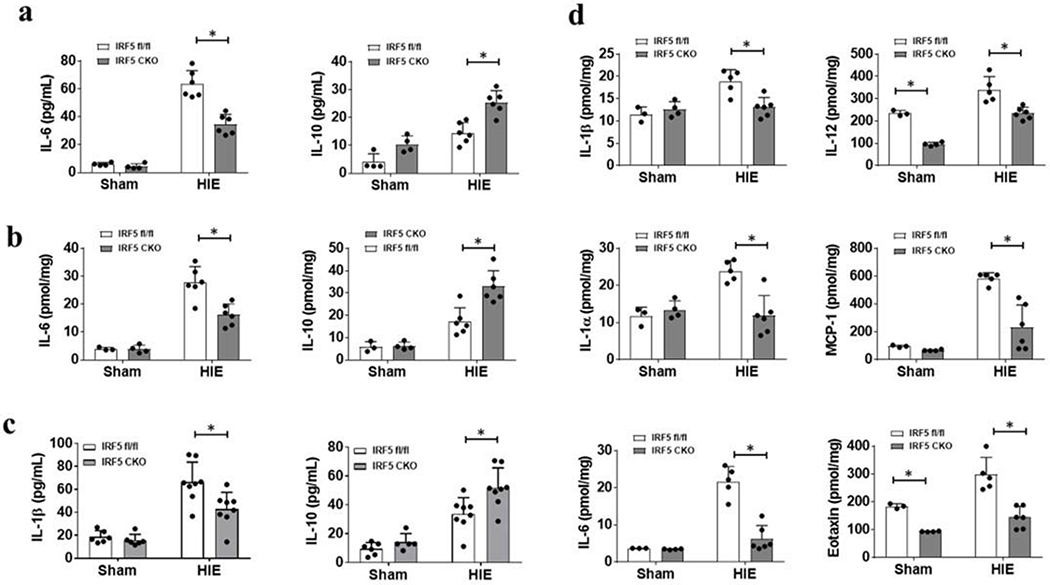
Cytokine levels in plasma and brain homogenates at 3d and 7d after HIE. a, Plasma pro-inflammatory (IL-6) and anti-inflammatory (IL-10) cytokine levels at 3d. b, pro-inflammatory (IL-6) and anti-inflammatory (IL-10) cytokine levels at 3d. N=3-4 per sham and 5-6 per HIE group; *P < 0.05. c, Pro-inflammatory (IL-1β) and anti-inflammatory (IL-10) cytokine levels in plasma at 7d. N=6 per sham and 8 per HIE group; *P < 0.05 d, Pro-inflammatory (IL-1β, IL-1α, IL-6, IL-12, Eotaxin, and MCP-1) cytokine levels in the ipsilateral hemispheres of sham and HIE brain. N=3-4 per sham and 5-6 per HIE group; *P < 0.05. Two-way ANOVA for a-d.
Ameliorated HIE outcomes in IRF5 CKO vs. IRF5fl/fl pups.
Inflammatory responses shape HIE outcomes [26]. To investigate how changes in the immune response impacts HIE outcomes, morphological changes and functional deficits after HIE were examined. Brain atrophy was quantified at 7 days of HIE, as the ischemic infarct is less visible due to the tissue loss at the sub-acute stage [2]. Quantitative data showed IRF5 CKO pups had significantly less tissue loss than IRF5fl/fl pups (Fig. 4a). We also assessed Y-maze performance at 7 days after HIE to examine working memory in HIE pups. Corresponding with the observed tissue loss, IRF5 CKO pups had more spontaneous alternate entries in the Y-maze compared to control mice (Fig. 4b), indicating the CKO pups had less cognitive deficits after HIE. Seizures reflect acute brain damage in neonatal disease models [27, 28]. We performed seizure scoring in both CKO and flox control pups at either 5 min or 24h after HIE. At both time points, IRF5 CKO pups had significantly lower seizure scores than their control counterparts (Fig. 4c&d). The outcome data clearly showed that IRF5 signaling in phagocytes has detrimental effects after HIE.
Figure 4.
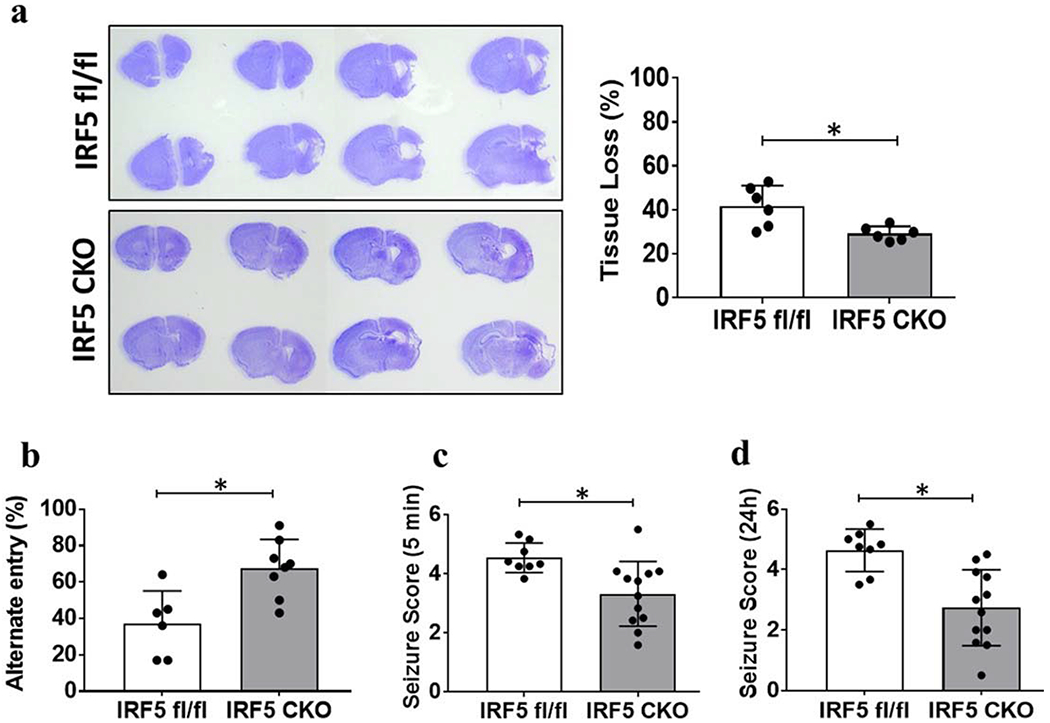
HIE outcomes. a, Representative images of brain slices stained with cresyl violet (CV) and quantification of brain tissue loss at 7d after HIE, N=6-8/group, *P < 0.05 (Student’s t-test). b, Functional outcomes evaluated by Y-maze for working memory, N=6-8/group, *P < 0.05 (Student’s t-test). c&d, Seizure scores were significantly different between IRF5 fl/fl and IRF5 CKO pups either at 5 min (C) and 24 h (D) of HIE. N=8-12/group; *P < 0.05 (Mann-Whitney U test).
Overexpression of IRF5 with lentivirus in primary microglia exacerbates inflammation after HIE.
We further investigated the role of IRF5 signaling by overexpression of IRF5 using lentivirus in primary microglial cultures. Cultured microglia were transfected with lenti-IRF5, a highly efficient GFP tagged virus for gene overexpression. The overexpression was performed under the control of CX3CR1 promoter for microglia [29, 30]. 3 MOI viruses were used for transfection based on our efficiency gradient test (Supp. Fig. S7a&b), and the virus specifically transfected microglia (Fig. 5a) but not astrocytes (Supp. Fig. S7c). After lenti-IRF5 transfection, both mRNA and protein levels of IRF5 were increased in lenti-IRF5 vs. lenti-GFP treated cells (Fig. 5b&c). We next subjected the transfected microglia to OGD, an in vitro ischemia model, to analyze the pro-inflammatory state demonstrated by TNFα signaling. Lenti-IRF5 itself did not increase TNFα expression in the culture supernatant under normoxic conditions; however, after OGD, both lenti-GFP and lenti-IRF5 induced significantly higher level of TNFα compared to the normoxic controls (Fig. 5d). Furthermore, TNFα levels in the culture supernatant after OGD were significantly higher in lenti-IRF5 vs. lenti-GFP treated cells (Fig. 5d). These data suggest that IRF5 overexpression amplifies pro-inflammatory signaling in microglia after ischemia.
Figure 5.
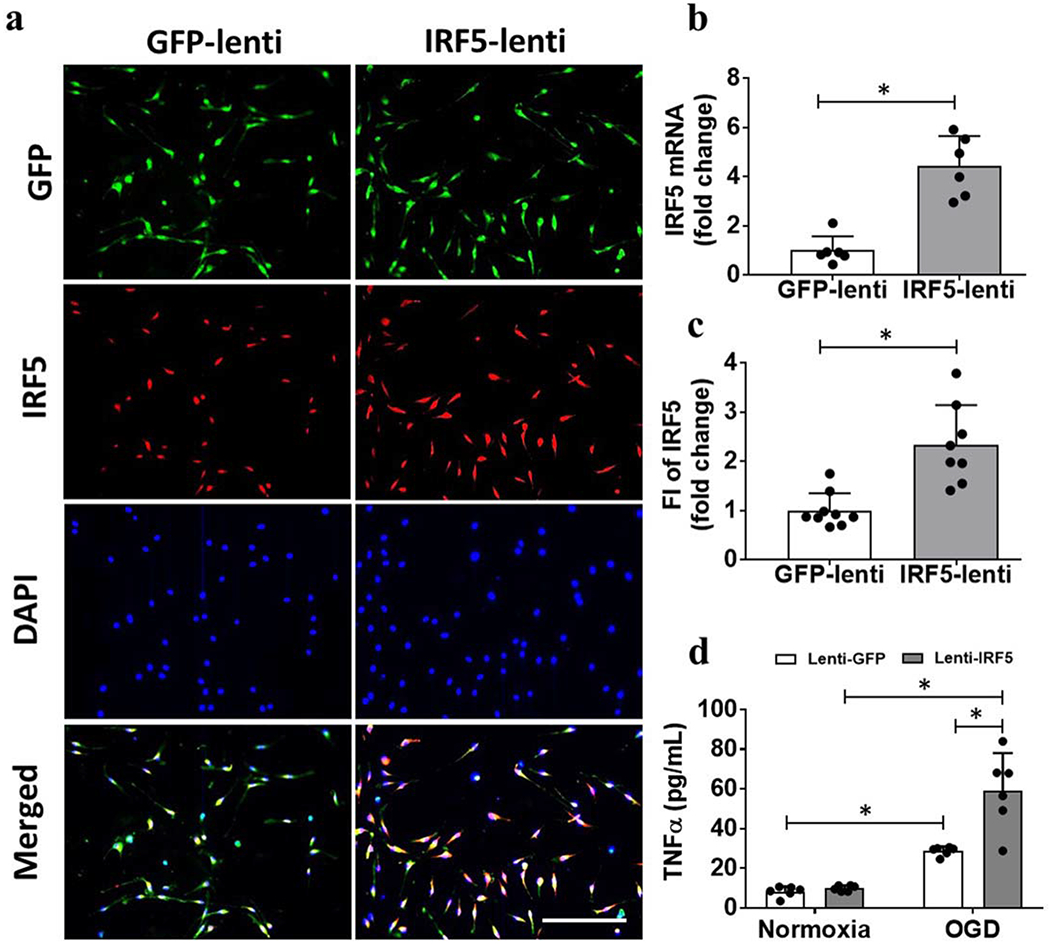
Over expression of IRF5 in primary microglial cultures. a, Representative ICC images of microglial IRF5 overexpression by GFP-tagged lenti-IRF5 transfection. b, IRF5 mRNA levels in cell homogenates. Each dot represents a replicate value from 3 independent experiment. *P < 0.05 (Student’s t-test). c, IRF5 fluorescence intensity (FI) in the ICC images; ; Scale bar=100μm. N=3 independent experiments with duplicate treatments. *P < 0.05 (Student’s t-test). D, TNFα levels in culture supernatant after GFP/IRF5-lenti virus treatment. N=3 independent experiments with duplicate treatments. *P < 0.05 (two-way ANOVA).
Upregulation of microglial IRF5 signaling compromises neuronal survival after OGD.
Finally, we evaluated the effects of microglial overexpression of IRF5 on neuronal survival after in vitro ischemia. We first transfected microglial cultures with lenti-IRF5, and then subjected the microglia co-cultured with primary neurons to a 2 hour OGD exposure. Neuronal survival and morphological changes were examined 48 hours after the co-culture by LDH/MTS assays, TUNEL and MAP2 staining. Over-expression of IRF5 in microglia exacerbated OGD-induced neuronal death compared to lenti-GFP treated cultures, as manifested by increased LDH release and decreased viability by MTS assay (Fig. 6a). TUNEL positive cells were also significantly increased by lenti-IRF5 treatment (Fig. 6b), indicating more neuronal apoptosis after IRF5 overexpression in microglia. MAP2 staining revealed that neuronal processes were more fragile when co-cultured with lenti-IRF5 treated microglia, demonstrated by scattered pieces of broken neuronal axons and dendrites (Fig. 6c). Quantitative data showed the neurons co-cultured with lenti-IRF5 treated microglia had significantly shorter neurites compared to the lenti-GFP counterparts (Fig. 6d). Together, these data suggested a cytotoxic effect of IRF5 on ischemic neurons.
Figure 6.
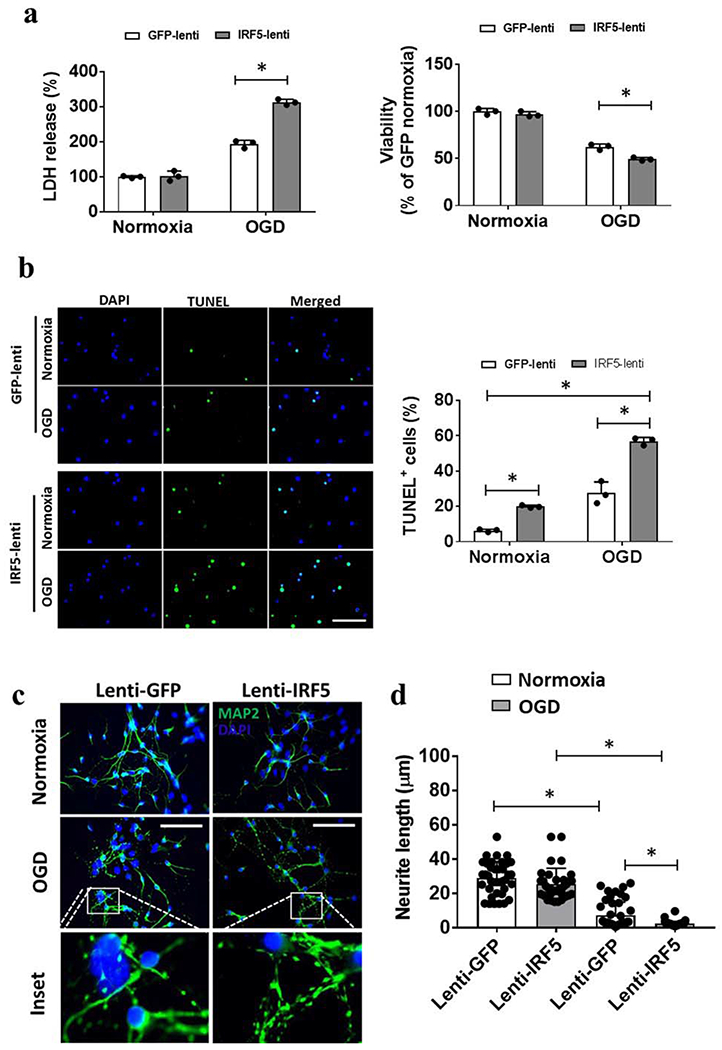
Co-culture of neurons with IRF5 overexpressed microglia. a, Viability of neurons measured by LDH release in the culture medium and by MTS assay after OGD. b, Representative immunofluorescence images of TUNEL positive cells and the quantification for each treatment. Each dot represents the average value of each independent experiment. N=3 independent experiments with duplicate treatments. c, Representative immunofluorescence images of MAP2 staining. d, Quantification of neurite length measured by ImageJ. Each dot represents an average value from randomly chosen 36-48 images (40x image) from 3 independent experiments with at least duplicate assays of each treatment; scale bar=100μm. *P < 0.05 (two-way ANOVA).
Discussion
Previously we have found that microglial IRF5 signaling was activated after stroke in young adult mice and that this was accompanied by a pro-inflammatory response [11, 31]. In the present study, we examined IRF5 signaling in a neonatal HIE model. We performed genetic manipulations to mechanistically examine the role of IRF5 in HIE. Several important findings were revealed. Firstly, IRF5 signaling in phagocytes plays a regulatory role in mediating pro-inflammatory responses after HIE, as demonstrated by the significantly reduced immune cell infiltration into the ischemic brain and the lower levels of pro-inflammatory mediators in both the brain and plasma samples from IRF5 CKO mice (Fig. 2&3). Secondly, deletion of IRF5 in phagocytes primes microglia towards alternative activation, as IRF5 deficiency increased the expression of anti-inflammatory cytokines (Arg1, CD206) in microglia (Fig. 1) and in the blood (IL-10) (Fig. 3). Thirdly, phagocytic IRF5 signaling impacts HIE outcomes, evidenced by histological and behavioral data (Fig. 4). Lastly, microglial IRF5 is cytotoxic to neurons after ischemia, by increasing neuronal apoptosis and inducing fragmentation of neuronal processes (Fig. 6). To our knowledge, this is the first study on IRF5 signaling in neonatal HIE that highlights the importance of regulation of phagocytes in cerebral ischemia.
Phagocytes play a central role in inflammatory responses [11, 32–34]. The present study provides clear evidence that IRF5 signaling not only regulates the activation of microglia, but also shapes the entire immune response to HIE. The responses of microglia to stimuli are characterized as either classical activation (pro-inflammatory; M1) or alternative activation (anti-inflammatory; M2) [32]. Although this binary classification has been increasingly recognized as an over-simplified characterization, as the two activation states overlap during disease progress, one of these two predominates at different stages of the disease. In the past decade, mounting evidence has indicated that IRF5 directly induces the expression of pro-inflammatory cytokines such as IL-6, IL-12b, and IL-23a while repressing transcription of anti-inflammatory cytokines such as IL-4 and IL-10 in peripheral macrophages. This accordingly up-regulates M1 activation and down-regulates M2 activation in a robust inflammatory process [35, 36]. We have previously demonstrated that the expression of IRF5 in microglia after stroke exhibited a “low-high-low” parabolic pattern, coinciding with the states of inflammatory responses at the acute and chronic stages of stroke in young male mice [11]. The present study further demonstrates that the IRF5 signaling is not simply a result of post-stroke inflammation; instead, it is a driving force that regulates the immune response to ischemic injury, and interacts with other inflammatory signals. Our findings are consistent with a previous study that showed that inhibition of IRF5 signaling in peripheral macrophages resulted in a reduction in inflammation and lower TNFα and IL-1β, and increased infarct repair after myocardial infarction [37]. Similarly, Li et al. reported that IRF5 gene silencing in macrophages conferred long lasting protection by decreasing pro-inflammatory markers (i.e: TNFα IL-6, IL-10, and IL-12.) and increased expression of M2 macrophage markers such as Arg1, and CD206 after spinal cord injury [38]. As the resident immune cells in CNS, microglia appear to share the same IRF5 signaling mechanisms as that seen in peripheral macrophages.
Both our in vivo and in vitro data revealed that IRF5 manipulation alone is not sufficient to modulate cytokine expression, and can only function under ischemic conditions, as IRF5 CKO and lenti-IRF5 treatment had less effect in sham mice (Fig. 2–3) and under normoxic conditions (Fig. 5). These findings suggest that IRF5 needs to be activated by a pathogenic stimulus to impact cytokine production. The phosphorylation of IRF5 occurs in peripheral macrophages under pathological conditions [36, 39]. Injured tissues release damage-associated molecular patterns (DAMPs) that combine with Toll-like receptor 4 (TLR4) on the membranes of macrophages. This TLR outside-in signal induces formation of a phosphorylating complex including TNF receptor associated factor 6 (TRAF6), interlukin-1 receptor associated kinase 1 and 4 (IRAK 1 and 4), and the adaptor protein MyD88 [40–42]. This complex subsequently phosphorylates IRF5 (p-IRF5) and only the p-IRF5 can translocate from the cytoplasm to the nucleus to combine with IFN stimulated response element (ISRE) and promote transcription of pro-inflammatory cytokines [43]. The same mechanism of IRF5 translocation most likely also exists in ischemic microglia. These pro-inflammatory cytokines not only activate immune cells, but are also cytotoxic and kill neurons directly in the ischemic brain. For example, TNFα and IL-1β act to destroy myelin or oligodendrocytes, leading to demyelinating lesions and neuronal death [44, 45]. In the present study, we co-cultured neurons with IRF5-overexpressed microglia under OGD conditions, and observed neuronal apoptosis and process fragmentation, a pathological process mediated by the cytotoxic factors released from microglia (Fig. 6). Our result is consistent with a previous report showing that IRF5 promoted Fas-induced apoptosis in dendritic cells [46].
With IRF5 deletion in phagocytes, an enhanced anti-inflammatory profile was seen in microglia, suggesting the pro- and anti-inflammatory responses are tightly controlled in a coupled, oscillating manner. Our previous study tested the role of another IRF, IRF4, in post-HIE inflammation, and found IRF4 mediates gene expression of anti-inflammatory cytokines in microglia [2]. It was reported that IRF4 competes with IRF5 for binding to the adaptor MyD88, a critical component of phosphorylating complex for IRF5 downstream to TLR4 signaling [47, 11]. As a result, the pro- and anti-inflammatory cytokine production reaches a balance. However, the balance is broken and exhibits a see-saw pattern if the expression of IRF5 or IRF4 is changed. We have previously found that these two IRFs are expressed in a see-saw pattern at the acute and chronic stages of injury in microglia after ischemic stroke [11], suggesting the pro- and anti-inflammatory responses are dynamic and contribute to the pathology of cerebral ischemia. In the present study, an anti-inflammatory profile predominated after IRF5 deletion, further proving the existence of a dual and balanced IRF5-IRF4 regulatory axis in microglial activation.
The present study has limitations that should be kept in mind when interpreting the data. Firstly, FC sorted microglia were gated with low-to-intermediate expression levels of CD45 combined with high expression of CD11b (CD45low-intCD11b+) [48]. Recent findings revealed that microglial CD45 expression may increase to a high level in AD and with aging, so that some of the cells are gated up to CD45high by the conventional strategy in FC [49–51]. Although it is unknown whether this is also true in the neonatal HIE brains, the gating strategy for ischemic microglia may need to be further confirmed with more microglial markers in future studies. Secondly, we generated phagocytic IRF5 CKO mice by mating IRF5 floxed mice with LysMCre mice, in which Cre expression is under the control of the lysozyme M promoter (LysMCre mice). LysMCre leads to deletion of floxed genes in both microglia and monocytes [52, 53]. Thus, the improved HIE outcomes reflected in figure 4 might be a combined effect of IRF5 depletion from both phagocytes. Future studies are warranted to use bone marrow chimera animal models generated from IRF5 CKO and WT mice, so that the contribution of IRF5 signaling from microglia vs. brain-infiltrating monocytes to ischemic outcomes can be separated for cell specific effect studies.
In conclusion, our data provide evidence that IRF5 signaling in phagocytes is detrimental in neonatal HIE. While IRF5 signaling mainly mediates pro-inflammatory responses, it also affects anti-inflammatory responses in an oscillating manner. More importantly, phagocytic IRF5 signaling impacts HIE outcomes by shaping post-ischemic inflammation, potentially representing an effective therapeutic target for HIE. Cell-specific therapy has been very promising with the rapid development of lentiviral vectors and siRNA techniques [37, 38]. The present study highlights the rational that therapeutic strategies aimed at immune responses can focus on a specific component of the immune response to injury, instead of targeting the entire immune system, which may avoid the side effects of immune compromise.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by funding from National Institutes of Health: R01 NS093042/NS108779 (Fudong Liu) and R37 NS096493 (Louise McCullough).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Disclosures
All authors report no conflicts.
References:
- 1.Kaur H, Xu N, Doycheva DM, Malaguit J, Tang J, Zhang JH. Recombinant Slit2 attenuates neuronal apoptosis via the Robo1-srGAP1 pathway in a rat model of neonatal HIE. Neuropharmacology. 2019;158:107727. doi: 10.1016/j.neuropharm.2019.107727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Mamun A, Yu H, Mirza MA, Romana S, McCullough LD, Liu F. Myeloid cell IRF4 signaling protects neonatal brains from hypoxic ischemic encephalopathy. Neurochemistry international. 2019;127:148–57. doi: 10.1016/j.neuint.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al Mamun A, Yu H, Romana S, Liu F. Inflammatory Responses are Sex Specific in Chronic Hypoxic-Ischemic Encephalopathy. Cell transplantation. 2018:963689718766362. doi: 10.1177/0963689718766362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu X, Li P, Guo Y, Wang H, Leak RK, Chen S et al. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43(11):3063–70. doi: 10.1161/strokeaha.112.659656. [DOI] [PubMed] [Google Scholar]
- 5.Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. International journal of molecular sciences. 2017;18(10). doi: 10.3390/ijms18102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitt AB, Buss A, Breuer S, Brook GA, Pech K, Martin D et al. Major histocompatibility complex class II expression by activated microglia caudal to lesions of descending tracts in the human spinal cord is not associated with a T cell response. Acta neuropathologica. 2000;100(5):528–36. doi: 10.1007/s004010000221. [DOI] [PubMed] [Google Scholar]
- 7.Burke NN, Kerr DM, Moriarty O, Finn DP, Roche M. Minocycline modulates neuropathic pain behaviour and cortical M1-M2 microglial gene expression in a rat model of depression. Brain, behavior, and immunity. 2014;42:147–56. doi: 10.1016/j.bbi.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Butturini E, Boriero D, Carcereri de Prati A, Mariotto S. STAT1 drives M1 microglia activation and neuroinflammation under hypoxia. Archives of biochemistry and biophysics. 2019;669:22–30. doi: 10.1016/j.abb.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Crain JM, Nikodemova M, Watters JJ. Microglia express distinct M1 and M2 phenotypic markers in the postnatal and adult central nervous system in male and female mice. Journal of neuroscience research. 2013;91(9):1143–51. doi: 10.1002/jnr.23242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime reports. 2014;6:13. doi: 10.12703/p6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al Mamun A, Chauhan A, Yu H, Xu Y, Sharmeen R, Liu F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. The European journal of neuroscience. 2018;47(2):140–9. doi: 10.1111/ejn.13778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Almuttaqi H, Udalova IA. Advances and challenges in targeting IRF5, a key regulator of inflammation. The FEBS journal. 2019;286(9):1624–37. doi: 10.1111/febs.14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirza MA, Ritzel R, Xu Y, McCullough LD, Liu F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. Journal of neuroinflammation. 2015;12:32. doi: 10.1186/s12974-015-0251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swonger AK, Rech RH. Serotonergic and cholinergic involvement in habituation of activity and spontaneous alternation of rats in a Y maze. Journal of comparative and physiological psychology. 1972;81(3):509–22. [DOI] [PubMed] [Google Scholar]
- 15.Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15(5):1203–18. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 16.Tasca CI, Dal-Cim T, Cimarosti H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol Biol. 2015;1254:197–210. doi: 10.1007/978-1-4939-2152-2_15. [DOI] [PubMed] [Google Scholar]
- 17.Giordano G, Costa LG. Primary neurons in culture and neuronal cell lines for in vitro neurotoxicological studies. Methods in molecular biology (Clifton, NJ). 2011;758:13–27. doi: 10.1007/978-1-61779-170-3_2. [DOI] [PubMed] [Google Scholar]
- 18.Das G, Gupta V, Khan J, Mukherjee D, Ghosh S. Generation of Neurospheres from Mixed Primary Hippocampal and Cortical Neurons Isolated from E14-E16 Sprague Dawley Rat Embryo. Journal of visualized experiments : JoVE. 2019(150). doi: 10.3791/59800. [DOI] [PubMed] [Google Scholar]
- 19.Jiang XS, Ni YQ, Liu TJ, Zhang M, Ren H, Jiang R et al. CCR2 overexpression promotes the efficient recruitment of retinal microglia in vitro. Molecular vision. 2012;18:2982–92. [PMC free article] [PubMed] [Google Scholar]
- 20.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280(21):20493–502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 21.Zhao SC, Wang C, Xu H, Wu WQ, Chu ZH, Ma LS et al. Age-related differences in interferon regulatory factor-4 and -5 signaling in ischemic brains of mice. Acta pharmacologica Sinica. 2017;38(11):1425–34. doi: 10.1038/aps.2017.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bok E, Chung YC, Kim KS, Baik HH, Shin WH, Jin BK. Modulation of M1/M2 polarization by capsaicin contributes to the survival of dopaminergic neurons in the lipopolysaccharide-lesioned substantia nigra in vivo. Exp Mol Med. 2018;50(7):76. doi: 10.1038/s12276-018-0111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porro C, Cianciulli A, Trotta T, Lofrumento DD, Panaro MA. Curcumin Regulates Anti-Inflammatory Responses by JAK/STAT/SOCS Signaling Pathway in BV-2 Microglial Cells. Biology (Basel). 2019;8(3). doi: 10.3390/biology8030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monga S, Nagler R, Amara R, Weizman A, Gavish M. Inhibitory Effects of the Two Novel TSPO Ligands 2-Cl-MGV-1 and MGV-1 on LPS-induced Microglial Activation. Cells. 2019;8(5). doi: 10.3390/cells8050486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ormstad H, Aass HC, Lund-Sorensen N, Amthor KF, Sandvik L. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. Journal of neurology. 2011;258(4):677–85. doi: 10.1007/s00415-011-6006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta pharmacologica Sinica. 2013;34(9):1121–30. doi: 10.1038/aps.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverstein FS. Do seizures contribute to neonatal hypoxic-ischemic brain injury? The Journal of pediatrics. 2009;155(3):305–6. doi: 10.1016/j.jpeds.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kossoff E Neonatal seizures due to hypoxic-ischemic encephalopathy: should we care? Epilepsy currents. 2011;11(5):147–8. doi: 10.5698/1535-7511-11.5.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co-expression meta-analysis. Acta neuropathologica communications. 2015;3:31. doi: 10.1186/s40478-015-0203-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao XF, Alam MM, Liao Y, Huang T, Mathur R, Zhu X et al. Targeting Microglia Using Cx3cr1-Cre Lines: Revisiting the Specificity. eNeuro. 2019;6(4). doi: 10.1523/eneuro.0114-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Al Mamun A, Chauhan A, Qi S, Ngwa C, Xu Y, Sharmeen R et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proceedings of the National Academy of Sciences of the United States of America. 2020;117(3):1742–52. doi: 10.1073/pnas.1914742117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5(2):73–90. [PMC free article] [PubMed] [Google Scholar]
- 33.Chiba T, Umegaki K. Pivotal roles of monocytes/macrophages in stroke. Mediators of inflammation. 2013;2013:759103. doi: 10.1155/2013/759103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gliem M, Schwaninger M, Jander S. Protective features of peripheral monocytes/macrophages in stroke. Biochimica et biophysica acta. 2016;1862(3):329–38. doi: 10.1016/j.bbadis.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 35.Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nature immunology. 2011;12(3):231–8. doi: 10.1038/ni.1990. [DOI] [PubMed] [Google Scholar]
- 36.Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434(7030):243–9. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- 37.Courties G, Heidt T, Sebas M, Iwamoto Y, Jeon D, Truelove J et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J Am Coll Cardiol. 2014;63(15):1556–66. doi: 10.1016/j.jacc.2013.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Liu Y, Xu H, Fu Q. Nanoparticle-Delivered IRF5 siRNA Facilitates M1 to M2 Transition, Reduces Demyelination and Neurofilament Loss, and Promotes Functional Recovery After Spinal Cord Injury in Mice. Inflammation. 2016;39(5):1704–17. doi: 10.1007/s10753-016-0405-4. [DOI] [PubMed] [Google Scholar]
- 39.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6(9):644–58. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 40.Balkhi MY, Fitzgerald KA, Pitha PM. Functional regulation of MyD88-activated interferon regulatory factor 5 by K63-linked polyubiquitination. Molecular and cellular biology. 2008;28(24):7296–308. doi: 10.1128/mcb.00662-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Balkhi MY, Fitzgerald KA, Pitha PM. IKKalpha negatively regulates IRF-5 function in a MyD88-TRAF6 pathway. Cellular signalling. 2010;22(1):117–27. doi: 10.1016/j.cellsig.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 42.Paun A, Reinert JT, Jiang Z, Medin C, Balkhi MY, Fitzgerald KA et al. Functional characterization of murine interferon regulatory factor 5 (IRF-5) and its role in the innate antiviral response. The Journal of biological chemistry. 2008;283(21):14295–308. doi: 10.1074/jbc.M800501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cushing L, Winkler A, Jelinsky SA, Lee K, Korver W, Hawtin R et al. IRAK4 kinase activity controls Toll-like receptor-induced inflammation through the transcription factor IRF5 in primary human monocytes. The Journal of biological chemistry. 2017;292(45):18689–98. doi: 10.1074/jbc.M117.796912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lock C, Oksenberg J, Steinman L. The role of TNFalpha and lymphotoxin in demyelinating disease. Annals of the rheumatic diseases. 1999;58 Suppl 1:1121–8. doi: 10.1136/ard.58.2008.i121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vela JM, Molina-Holgado E, Arevalo-Martin A, Almazan G, Guaza C. Interleukin-1 regulates proliferation and differentiation of oligodendrocyte progenitor cells. Molecular and cellular neurosciences. 2002;20(3):489–502. doi: 10.1006/mcne.2002.1127. [DOI] [PubMed] [Google Scholar]
- 46.Couzinet A, Tamura K, Chen HM, Nishimura K, Wang Z, Morishita Y et al. A cell-type-specific requirement for IFN regulatory factor 5 (IRF5) in Fas-induced apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2556–61. doi: 10.1073/pnas.0712295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eguchi J, Kong X, Tenta M, Wang X, Kang S, Rosen ED. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. 2013;62(10):3394–403. doi: 10.2337/db12-1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. Journal of immunology (Baltimore, Md : 1950). 1995;154(9):4309–21. [PubMed] [Google Scholar]
- 49.Masliah E, Mallory M, Hansen L, Alford M, Albright T, Terry R et al. Immunoreactivity of CD45, a protein phosphotyrosine phosphatase, in Alzheimer’s disease. Acta neuropathologica. 1991;83(1):12–20. doi: 10.1007/bf00294425. [DOI] [PubMed] [Google Scholar]
- 50.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB et al. New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(12):E1738–46. doi: 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritzel RM, Patel AR, Pan S, Crapser J, Hammond M, Jellison E et al. Age- and location-related changes in microglial function. Neurobiology of aging. 2015;36(6):2153–63. doi: 10.1016/j.neurobiolaging.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 52.Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. The American journal of pathology. 2009;175(1):132–47. doi: 10.2353/ajpath.2009.081002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S et al. A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nature neuroscience. 2013;16(11):1618–26. doi: 10.1038/nn.3531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.