

Abstract
Post-translational modification of proteins with ubiquitin (ubiquitination) provides a rapid and versatile mechanism for regulating cellular signalling systems. Met1-linked (or ‘linear’) ubiquitin chains have emerged as a key regulatory signal that controls cell death, immune signalling, and other vital cellular functions. The molecular machinery that assembles, senses, and disassembles Met1-linked ubiquitin chains is highly specific. In recent years, the thorough biochemical and genetic characterisation of the enzymes and proteins of the Met1-linked ubiquitin signalling machinery has paved the way for substantial advances in our understanding of how Met1-linked ubiquitin chains control cell signalling and biology. Here, we review current knowledge and recent insights into the role of Met1-linked ubiquitin chains in cell signalling with an emphasis on their role in disease biology. Met1-linked ubiquitin has potent regulatory functions in immune signalling, NF-κB transcription factor activation, and cell death. Importantly, mounting evidence shows that dysregulation of Met1-linked ubiquitin signalling is associated with multiple human diseases, including immune disorders, cancer, and neurodegeneration. We discuss the latest evidence on the cellular function of Met1-linked ubiquitin in the context of its associated diseases and highlight new emerging roles of Met1-linked ubiquitin chains in cell signalling, including regulation of protein quality control and metabolism.
Subject terms: Cancer, Cell death and immune response, Immune cell death, Immunological disorders, Signal transduction

Facts
Met1-linked ubiquitin chains are conjugated, sensed, and disassembled by a dedicated and highly specific molecular machinery
Met1-linked ubiquitin chains control immune signalling, NF-κB activation, and programmed cell death
Met1-linked ubiquitin signalling is critical for proper regulation of inflammation, immunity, and cellular homeostasis in animals and humans
Dysregulation of Met1-linked ubiquitin signalling causes different responses or reactions in different cell types
Genetic defects in Met1-linked ubiquitin signalling are associated with severe human pathologies, including autoinflammation, immunodeficiency, cancer, and neurodegeneration
Open Questions
What are the cellular functions of Met1-linked ubiquitin chains beyond regulation of immune signalling, NF-κB, and programmed cell death?
Which pathways and cellular mechanisms are responsible for disease development associated with dysfunctional Met1-linked ubiquitin signalling?
What are the mechanisms underlying the cell type-specific responses to dysregulation of Met1-linked ubiquitin signalling?
Is the Met1-linked ubiquitin machinery affected by external stresses or environmental agents, such as toxins, nutrition, or the microbiota?
To what extent is dysregulation of Met1-linked ubiquitin involved in development of sporadic disease not caused by mutations in the Met1-linked ubiquitin machinery?
Introduction
Cellular homeostasis depends on adaptation to the environment through numerous signalling cascades that translate extrinsic and intrinsic cues and stresses into appropriate responses. Signalling is most commonly propagated through rapid and specific post-translational modifications (PTMs) of signalling proteins, resulting in adaptive changes at the transcriptional and translational levels. Failure to adapt can cause cell damage or cell death and may lead to disease. Met1-linked ubiquitin (also known as linear ubiquitin) is a PTM that over the past decade has emerged as critically important for controlling vital signalling cascades and preventing cell death [1]. Consequently, dysregulation of Met1-linked ubiquitin modifications is associated with severe pathologies, including immune disorders, cancer, and neurodegeneration (Fig. 1; Table 1), emphasising that the balance of one single subtype of PTM can be essential for maintaining homeostasis and preventing life-threatening diseases.
Fig. 1. Pathologies associated with Met1-linked ubiquitin.
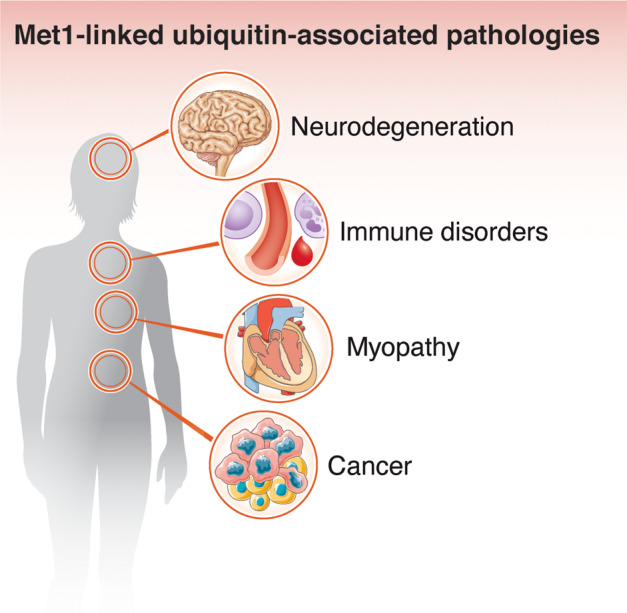
In recent years, the understanding of the (patho)physiological importance of Met1-linked ubiquitin signalling has increased substantially. Dysregulation or defects in Met1-linked ubiquitin signalling is now recognised to be associated with severe human disease, including neurodegeneration, myopathy, immune disorders, and cancer.
Table 1.
Genetic alterations and phenotypes in humans.
| Gene | DNA | Protein | Disease | Pathology | Penetrance | Ref. |
|---|---|---|---|---|---|---|
| RNF31 (HOIP) | c.215T > C | p.Leu72Pro | LUBAC deficiency [OMIM ID: 615895] | Common variable immunodeficiency, multiorgan autoinflammation, recurrent fevers, diarrhoea, with or without amylopectinosis and lymphangiectasia | 2/2 | [33, 126] |
| c.1197G > C | p.Gln399His | |||||
| c.1737 + 3A > G | N/A | |||||
| SNP ID rs184184005 | p.Gln584His | Activated B cell diffuse large B cell lymphoma (ABC-DLBCL) | Enhanced BCR-mediated NF-κB activity in ABC-DLBCL cells. Gain-of-function variants | N/A | [132] | |
| SNP ID rs149481717 | p.Gln622Leu | |||||
| RBCK1 (HOIL-1) | c.121_122delCT | p.Leu41fsX7 | LUBAC deficiency [OMIM ID: 615895] | Chronic autoinflammation, immunodeficiency, amylopectinosis and associated (cardio)myopathy, lymphadenopathy, hepatosplenomegaly | 3/17 | [32] |
| c.553C > T | p.Gln185X | |||||
| c.ex1_ex4del | N/A | |||||
| c.727G > T | p.Glu243X | Amylopectinosis, muscle weakness, cardiomyopathy | 14/17 | [129, 130] | ||
| c.1160A > G | p.Asn387Ser | |||||
| c.896_899delAGTG | p.Glu299ValfsX18 | |||||
| c.722delC | p.Ala241GlyfsX34 | |||||
| c.52G > C | p.Ala18Pro | |||||
| 727_728ins GGCG | p.Glu243GlyfsX114 | |||||
| c.ex1_ex4del | N/A | |||||
| c.1054C > T | p.Arg352X | |||||
| c.91713_91714insG | p.Arg298ArgfsX40 | |||||
| c.494delG | p.Arg165ArgfsX111 | |||||
| N/A | p.Gln222X; | |||||
| N/A | p.Glu190fs | |||||
| c.456 + 1G > C | N/A | |||||
| SHARPIN (SHARPIN) | SNP ID rs572750141 | p.Gly186Arg | Late onset Alzheimer’s disease | Dementia | N/A | [144] |
| OTULIN (OTULIN) | c.815T > C | p. Leu272Pro | OTULIN-Related Autoinflammatory Syndrome (ORAS) [OMIM ID: 617099] | Chronic autoinflammation and autoimmunity; recurrent fevers, diarrhoea, arthritis, panniculitis, neutrophilia, increased blood immunoglobulin levels, and autoantibodies | 7/7 | [58, 60, 127, 128] |
| c.731A > G | p. Tyr244Cys | |||||
| c.517delC | p.Gly174AspfsX2 | |||||
| c.841G > A | p.Gly281Arg | |||||
| c.864 + 2T > C | p.Leu157GlyfsX12 |
Penetrance reflects the number of patients that display the indicated symptoms out of the total number of patients reported with mutations in the given gene. BCR, B cell receptor; del, deletion; ex, exon; fs, frameshift; ins, insertion; N/A, not available; ref., reference; SNP, single-nucleotide polymorphism.
The post-translational modification of proteins with ubiquitin (Ub), a 76 amino acid residue protein, regulates a plethora of signalling cascades and controls virtually all aspects of eukaryotic biology. Dedicated ‘writers’ assemble the Ub modifications, ‘readers’ decipher the signals, and ‘erasers’ reverse them (Fig. 2A). Ub is attached to lysine residues in substrates by Ub (E3) ligases [2, 3]. Ubiquitinated proteins are recognised by receptors that contain ubiquitin-binding domains (UBDs) and transmit the ubiquitin signal [4]. Finally, ubiquitination is reversible and specialised proteases termed deubiquitinases (DUBs) cleave the Ub signals to terminate signalling [5].
Fig. 2. The Met1-Ub Machinery.

A The proteins involved in ‘writing’ (assembling), ‘reading’ (binding), and ‘erasing’ (disassembling) Met1-Ub signals. LUBAC, together with E1 and E2 enzymes, assembles Met1-Ub in an ATP-dependent process. The first Ub conjugated to a substrate (shown in grey) does not have a defined linkage as the lysine residue (K) is unique to the substrate. Several proteins can bind Met1-Ub signals, and these are involved in ‘interpreting’ the biochemical modification of the substrate and funnelling it into the correct cellular pathway or response. OTULIN and CYLD (in complex with SPATA2) are the best-described DUBs that disassemble Met1-Ub. OTULIN exclusively disassembles Met1-Ub whereas CYLD-SPATA2 is less specific and also disassembles Lys63-Ub and some other linkages in addition to Met1-Ub (this activity is not shown). B Domain architecture of LUBAC and associated Met1-Ub DUBs. Domain interactions are indicated by arrows. CAP-Gly, cytoskeleton-associated protein glycine-rich; IBR, in-between-RING; LDD, linear ubiquitin chain-determining domain; LTM, LUBAC-tethering motif; NZF, Npl4 zinc finger; OTU, ovarian tumour; PH, pleckstrin homology; PIM, peptide:N-glycanase/ubiquitin-associated UBA-containing or UBX-containing protein-interacting motif; PUB, peptide:N-glycanase/ubiquitin-associated UBA-containing or UBX-containing protein; RING, Really Interesting New Gene; UBA, ubiquitin-associated; UBL, ubiquitin-like; USP, Ubiquitin specific protease; Znf, Zinc finger.
The Ub system serves two main purposes: control of protein turnover by targeting substrates for proteasomal or lysosomal degradation and orchestration of cell signalling networks by regulation of protein-protein interactions, localisation, and activity, akin to phosphorylation. This functional versatility arises from the many different types of Ub signals that can be assembled [6, 7]. In addition to modification with a single Ub moiety (mono-ubiquitination), polyUb chains can be assembled through ubiquitination of Ub itself on one of the seven internal lysine residues (Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63) or the N-terminal methionine (Met1), resulting in the formation of eight structurally and functionally distinct Ub chains [6, 8]. All eight Ub chain types exist within cells and determine different fates of the modified proteins [9–13]. Lys48-linked Ub chains function as signals for proteasomal degradation, while non-degradative ubiquitin signals most commonly involve Lys63-linked Ub chains (Lys63-Ub) [6]. Cellular roles of the remaining ‘atypical’ chains (Met1, Lys6, Lys11, Lys27, Lys29, and Lys33 linkages) are less well understood, but they are emerging as important and specific signals in a range of processes [14, 15].
Writing, reading, and erasing Met1-linked Ub signals
For decades, Met1-linked Ub chains were thought only to exist as the translation product of the polyubiquitin genes UBB and UBC. But in 2006, Iwai and colleagues identified a Ub ligase complex that de novo assembles Ub chains in a head-to-tail or linear fashion through the N-terminal methionine (Met1-Ub) [16]. They aptly named this complex the linear Ub chain assembly complex (LUBAC). Since then, specific or highly selective UBDs and DUBs for Met1-Ub have been described, ensuring accurate control of Met1-Ub signalling (reviewed in [1, 17]). As discussed in this review, Met1-Ub is now recognised as a potent signalling modification involved in inflammation, immunity, and range of other functions important for maintaining normal cellular function and coping with various stresses.
Met1-Ub conjugation: writing the signal
LUBAC is the only known Ub ligase in vertebrates that assembles Met1-Ub. The complex is composed of three proteins, HOIL-1-interacting protein (HOIP; also known as RNF31), Heme-Oxidised IRP2 Ub Ligase-1 (HOIL-1; also known as RBCK1), and SHANK-Associated RH Domain-Interacting Protein (SHARPIN) [16, 18–20] (Fig. 2B). Both HOIP and HOIL-1 belong to the Really Interesting New Gene (RING)-in-between-RING (RBR) family of Ub ligases [21]. Even though both HOIP and HOIL-1 are bona fide Ub ligases, only HOIP’s RBR is required for Met1-Ub assembly in vitro [16]. SHARPIN has no Ub ligase activity (Fig. 2B). HOIP has remarkable specificity and exclusively assembles Met1-Ub (reviewed in [17]). This is achieved through a unique domain, the linear ubiquitin chain-determining domain (LDD), which orients the proximal ‘acceptor’ Ub moiety such that its N-terminus is the only amino group available for conjugation to the distal ‘donor’ ubiquitin moiety [22–24]. Because of this exquisitely specific mechanism, HOIP’s preferred (if not only) substrate is likely Ub itself. In support of this notion, LUBAC assembles Met1-Ub on existing Lys63-Ub (and possibly other Ub-linkages) during immune signalling, forming hybrid Lys63/Met1-Ub chains [25–28].
In isolation, HOIP is autoinhibited and has little Ub ligase activity [23]. HOIL-1 and SHARPIN function as co-factors in LUBAC and are critical for HOIP activation. Binding of the Ub-like (UBL) domains of HOIL-1 and SHARPIN to the Ub-Associated (UBA) domain of HOIP releases the autoinhibition and promotes Met1-Ub assembly [22, 23, 29]. In cells, HOIP, HOIL-1, and SHARPIN form a ~600 kDa complex [16, 20]. The structure and stoichiometry of LUBAC remains unclear, but the complex is maintained through multiple inter-molecular interactions between the three components [16, 18–20, 30] (Fig. 2B). These interactions are critical for the integrity of the complex and loss of any of the three LUBAC components destabilises the entire complex and impairs LUBAC function in cells [18–20, 31–34].
Met1-Ub binding proteins: reading the signal
Once an Met1-Ub signal has been assembled, UBD-containing proteins, often termed Ub receptors, ‘read’ the signal and translate it into specific cellular outcomes (reviewed in [35]) (Fig. 2A). Several proteins have been described to bind to Met1-Ub via specific UBDs. The best characterised Met1-Ub binding protein is the non-catalytic subunit of the IκB kinase (IKK) complex: NF-κB Essential Modifier (NEMO). NEMO binds Met1-Ub through its UBD in ABIN and NEMO (UBAN) domain [36] and facilitates activation of the IKK complex and Nuclear Factor-κB (NF-κB). A20-Binding Inhibitors of NF-κB (ABIN) proteins (ABIN 1–3) and Optineurin also contain UBAN domains that bind to Met1-Ub [36–39]. Other Met1-Ub binding proteins include HOIL-1 and the DUB A20 [40–42], the cellular Inhibitor of Apoptosis (IAP) 1 (cIAP1), cIAP2, X-linked IAP (XIAP) [39, 43], and the kinetochore protein KNL1 [44].
Met1-Ub disassembly: erasing the signal
Regulation of Ub signals is crucial to prevent excessive or aberrant signalling. This is achieved by finely balancing assembly of Ub modifications with disassembly by DUBs [5] (Fig. 2A). A unique characteristic of Met1-Ub is that the Ub moieties are linked via normal peptide bonds rather than isopeptide bonds as the seven Lys-linked chain types. Of the ~100 DUBs in humans [5], few are able to specifically cleave Met1-linkages. A range of Ubiquitin Specific Protease (USP) family DUBs, including USP2b, USP4, and USP15, and USP21, can cleave Met1-Ub, albeit with low specificity (i.e., they cleave a range of other chain types as well) and efficiency [45]. To date, only two DUBs that selectively and effectively disassemble Met1-Ub have been identified: OTU deubiquitinase with linear linkage specificity (OTULIN) [46, 47] and USP-DUB Cylindromatosis (CYLD) [39, 48] (Fig. 2A, B).
OTULIN (formerly known as FAM105B and Gumby) is remarkably specific for Met1-Ub [49] and it is the only known DUB in vertebrates that exclusively disassembles Met1-Ub and none of the Lys linkages [46, 47]. OTULIN binds directly to the PNGase/UBA or UBX-containing proteins (PUB) domain in HOIP via its PUB-interacting Motif (PIM), forming an OTULIN-LUBAC complex in cells [50, 51] (Fig. 2B). The USP domain of CYLD cleaves both Met1- and Lys63-linkages [39, 45], but also has activity towards other chain types [45]. CYLD also forms a complex with LUBAC by associating with HOIP’s PUB domain [28, 52, 53]. However, unlike OTULIN, this interaction is indirect and requires the PIM-containing Spermatogenesis-Associated 2 (SPATA2) to act as a bridging factor between HOIP and CYLD [54–57]. Consistent with the fact that OTULIN and CYLD-SPATA2 bind the same site on LUBAC, their binding is mutually exclusive [52, 54] (Fig. 2B). This indicates that at least two distinct LUBAC-DUB complexes – LUBAC-OTULIN and LUBAC-SPATA2-CYLD – exist. In cells, the absence of OTULIN results in a striking increase in the steady-state abundance of Met1-Ub [47, 52, 58, 59]. In contrast, CYLD deficiency does not lead to any increase in the steady-state level of Met1-Ub [52], suggesting that OTULIN and CYLD regulate Met1-Ub conjugation differently.
Studying the signal: from biochemistry to biology and disease
Functional studies of PTMs in biological systems are notoriously difficult as the modifications are commonly not genetically tractable. For Met1-Ub, its low abundance hampers detection and manipulation even further. Met1-Ub constitutes only ~0.5% of all Ub chains in cells [12, 13, 60]. The identification and detailed biochemical characterisation of specific or highly selective regulators have in recent years revolutionised the functional studies of Met1-Ub. Because of the remarkable Met1-Ub-specificity of LUBAC (HOIP) and OTULIN, we now have a set of genetic proxies to study Met1-Ub signalling in a range of biological systems; from cell lines to animal models to humans. Knock-out mice of LUBAC components or OTULIN – or knock-in mice expressing catalytically inactive proteins – have proven to be powerful tools to dissect Met-Ub signalling [25, 34, 47, 58, 61–65]. Moreover, Met1-Ub-specific antibodies [66] or affinity reagents based on NEMO-UBAN [27] or catalytically inactive OTULIN [52], as well as ubiquitin chain restriction (UbiCRest) analysis using active OTULIN [27, 67, 68], have allowed enrichment and detailed studies of Met1-Ub conjugation and disassembly in signalling complexes. These tools have paved the way for transformative discoveries on the molecular function and (patho)physiological importance of Met1-Ub signalling. Intriguingly, an ortholog of LUBAC, termed LUBEL, was recently discovered in Drosophila, showing that non-vertebrates also provide a model system for studying Met1-Ub signalling [69, 70].
Met1-Ub in immune signalling
The best-studied functions of Met1-Ub are in innate and adaptive immunity where it controls signalling from a multitude of receptors, including cytokine receptors, pattern recognition receptors (PRRs), and antigen receptors (Fig. 3). Met1-Ub regulates multiple aspects of these pathways, most notably NF-κB activation and cell death, which orchestrate inflammation and are vital for organism homeostasis [71, 72].
Fig. 3. Principles of immune signalling regulation by Met1-Ub.

Met1 (green) and Lys63 (blue) Ub linkages control signalling from a large range of innate and adaptive immune receptors. Generally, activation of immune receptors leads to the recruitment of adaptor proteins that become ubiquitinated with Met1-Ub and Lys63-Ub by different Ub ligases. Notably, Met1-Ub can be formed on pre-existing Lys63-Ub leading to the generation of hybrid Lys63/Met1-Ub chains. For canonical NF-κB activation triggered by TNF-R1, BCR or TCR, TLRs, IL-1R, or NOD1/2, engagement of these receptors leads to the formation of large, multi-protein receptor signalling complexes (RSCs) that contain adaptor proteins, such as TRADD and RIPK1 for TNF-R1, CARMA1-BCL10-MALT1 for BCR and TCR, MyD88 and IRAK1/4 for IL-1R and TLRs, and RIPK2 for NOD1/2, as well as Ub ligases, including cIAP1/2 for TNF-R1, XIAP for NOD2, and TRAF6 for IL-1R, TLRs, and BCR/TCR. LUBAC is recruited to these RSCs through binding to Lys63-Ub chains and conjugates Met1-Ub to existing Lys63-Ub to form hybrid Lys63/Met1-Ub chains or directly to adaptors in the RSCs. Lys63- and Met1-Ub modifications are recognised by the Ub-dependent kinase complexes TAK1-TAB2/3 and IKKα/β-NEMO, respectively. NEMO binding to Met1-Ub causes a conformational change in the IKK complex, leading to phosphorylation and activation NF-κB, which facilitates transcription of a plethora of genes encoding inflammatory mediators and pro-survival factors. For signalling triggered by TNF receptor-1 (TNF-R1), Met1-Ub also prevents the formation of cell death-inducing complexes that trigger either caspase-8-mediated apoptosis or RIPK3-MLKL-mediated necroptosis. Furthermore, LUBAC positively regulates the NLRP3 inflammasome where Met1-Ub conjugated to ASC promotes caspase-1 activation and IL-1β secretion. In contrast, LUBAC is a negative regulator of RIG-I signalling by conjugating Met1-Ub to TRIM25, which induces its proteasomal degradation and thereby inhibits IRF activation. OTULIN and CYLD disassembles Met1-Ub modifications in the TNF-R1- and NOD1/2-RSCs. It has not been reported which DUB(s) antagonises Met1-Ub in other immune signalling pathways. Lastly, OTULIN is required for LUBAC ‘maintenance’ by removing Met1-Ub autoubiquitination by HOIP. LUBAC autoubiquitination may contribute to signalling but can also lead to proteasomal degradation of HOIP, thereby destabilising LUBAC. ASC, apoptosis-associated speck-like protein containing a CARD; BCL10, B cell lymphoma 10; CARMA1, Caspase recruitment domain-containing membrane-associated guanylate kinase protein-1; Casp, caspase; FADD, Fas-associated protein with Death Domain; IRAK, interleukin-1 associated kinase; IRF, interferon regulatory factor; MALT1, Mucosa-associated lymphoid tissue lymphoma translocation protein 1; MLKL, Mixed Linkage Kinase Domain-Like; MyD88, myeloid differentiation primary response 88; PKC, protein kinase C; RIG-I, Retinoic acid-inducible gene I; RIPK, Receptor-interacting serine/threonine-protein kinase; TAB, TAK1- binding protein; TAK1, TGF-beta-activated kinase1; TANK, TRAF family member-associated NF-κB activator; TBK1, TANK-binding kinase 1; TRADD, Tumour necrosis factor receptor type 1-associated death domain protein; TRAF, TNF-receptor associated factor; TRIM25, Tripartite motif-containing protein 25.
Innate immune signalling
LUBAC was first described to regulate signalling in response to activation of tumour necrosis factor (TNF) receptor 1 (TNF-R1) [31, 40]. Since then, important functions of LUBAC and Met1-Ub have been described in nucleotide oligomerisation domain (NOD)1 and NOD2 signalling [73, 74], interleukin-1 (IL-1) receptor (IL-1R) signalling [18, 19, 32, 33], and Toll-like receptor (TLR) signalling [19, 63, 75–77] (Fig. 3). As a general model, activation of these receptors leads to the formation of large, multi-protein receptor signalling complexes (RSCs) that contain adaptor proteins, such as TRADD and RIPK1 for TNF-R1, and Ub ligases, including cIAP1/2 and XIAP for TNF-R1 and NOD2, respectively (reviewed in [78, 79]) (Fig. 3). Formation of these RSCs leads to the assembly of Lys63-Ub on signalling proteins, which facilitate the recruitment of LUBAC to the RSCs [40, 73]. Recruitment of LUBAC results in the conjugation of Met1-Ub to RSC components and together the Lys63-Ub and the M1-Ub recruits and activates the Ub-dependent kinases TGFβ-activated kinase 1 (TAK1) and the IKK complex through their Ub receptors TAK1-binding protein (TAB)2 and TAB3 [80] and NEMO [36], respectively (Fig. 3). Binding of NEMO’s UBAN domain to Met1-Ub induces a conformational change in NEMO [36, 81], which—together with phosphorylation by TAK1—activates IKK, leading to phosphorylation and activation of NF-κB and transcription of target genes [82].
Upon ligand binding, TNF-R1 forms a membrane-bound RSC termed ‘complex-I’ [72] (Fig. 3). LUBAC is recruited to complex-I through Lys63-Ub conjugated by cIAP1/2 on RIPK1 and possibly other proteins [18, 40]. In turn, LUBAC assembles Met1-Ub on RIPK1, TRADD, NEMO, and TNF-R1 itself [18, 31, 40, 46, 52, 68]. Importantly, LUBAC is not essential for TNF-induced NF-κB activation per se but rather functions to stabilise the RSC and enhance signalling output [18, 40]. TNF-R1-RSC stability, however, is likely still important for proper cytokine responses and immune function in vivo as mice with reduced LUBAC function are immunodeficient [83]. NOD2 forms a signalling complex resembling the TNF-R1-RSC where the Ub ligase XIAP ubiquitinates RIPK2 to recruit LUBAC [73, 84] (Fig. 3). In turn, LUBAC assembles Met1-Ub on RIPK2 to facilitate signalling and NF-κB activation [27, 73]. In contrast to TNF-R1, Met1-Ub appears to be essential for NOD-mediated signalling, NF-κB activation, and cytokine production [73]. For IL-1R- and TLR-RSCs, IRAK1/4 and MyD88 are the targets for LUBAC-mediated Met1-ubiquitination (Fig. 3). However, the laboratory of Philip Cohen discovered that the actual substrate of LUBAC in the TLR1/2- and IL-1R-RSCs are existing Lys63-Ub on IRAK1/4 and MyD88, giving rise to hybrid Lys63/Met1-Ub chains [25]. Hybrid Lys63/Met1-Ub has also been found in the TNF-R1-, TLR3-, and NOD2-RSCs [26–28] and is emerging as a general signalling principle in immune signalling. The hybrid Lys63/Met1 chains are thought to ensure efficient activation of the TAB2/3-TAK1 and NEMO-IKK complexes by juxtapositioning them on the same Ub chain [85].
In the RSCs, Ub chains exist in a dynamic equilibrium of Ub ligase-mediated conjugation and DUB-mediated disassembly. The LUBAC-associated DUBs OTULIN and CYLD-SPATA2 regulate Met1-Ub in the TNF-R1- and NOD2-RSCs. CYLD-SPATA2 removes both Lys63-Ub [52, 86–89] and Met1-Ub from RIPK1 [52, 54, 55, 57, 90] and RIPK2 [28, 54], restricting NF-κB activation and promoting cell death (in the case of TNF-R1-RSC) [52, 55–57, 90] (Fig. 3). OTULIN also removes Met1-Ub from RIPK2 after NOD2 stimulation to limit signalling [27, 28], although it appears not to be stably associated with the NOD2-RSC [52]. The role of OTULIN in TNF-R1 signalling is a little less clear. Studies have found OTULIN in the TNF-R1-RSC [51, 56] and shown that it limits Met1-Ub accumulation on complex components, including RIPK1 and TNF-R1 [27, 46, 91]. However, others have reported that OTULIN is not recruited to TNF-R1 and does not regulate Met1-ubiquitination in the RSC, but rather controls LUBAC autoubiquitination and activity outside the RSC [52]. Consistent with this notion, increased autoubiquitination and subsequent degradation of LUBAC has been observed in some OTULIN-deficient cells, leading to reduced Met1-Ub conjugation and signalling in the TNF-R1-RSC [58, 60, 65]. This indicates that OTULIN is necessary for LUBAC ‘maintenance’ by deubiquitinating LUBAC components, primarily HOIP [27, 28, 50] (Fig. 3). Notably, these data also suggest that OTULIN might not stably associate with Met1-Ub-containing RSCs or that LUBAC-SPATA2-CYLD, but not LUBAC-OTULIN, is specifically recruited to the RSCs. Alternatively, LUBAC-OTULIN could also be recruited to the complexes, but OTULIN’s retention—or active displacement—depends on cellular context or other Met1-Ub-regulatory mechanisms, such as phosphorylation of Tyr56 in its PIM, which abrogates binding to HOIP [50]. Consistent with the idea of active displacement, Douglas et al. showed that OTULIN is phosphorylated on Tyr56 during TNF-induced necroptosis, likely displacing OTULIN from LUBAC, and that this increases Met1-ubiquitination of RIPK1 [91].
Other PRRs that Met1-Ub regulates include the NLRP3 inflammasome as well as the viral RNA sensor RIG-I, but the mechanisms are less well understood. LUBAC is required for assembly and activation of the NLRP3 inflammasome independent of NF-κB activation [92–94], whereas it – in contrast to its role in NF-κB signalling – appears to be a negative regulator of the RIG-I-dependent anti-viral interferon response by inducing TRIM25 degradation [95, 96] (Fig. 3). However, a recent study of Influenza A infection in mice showed a blunted RIG-I-dependent immune response in HOIL-1- and HOIP-deficient mice [97], thus contradicting the notion of LUBAC being a negative regulator of RIG-I signalling.
Regulation of cell death by Met1-Ub
In addition to its role in immune signalling and NF-κB activation, Met1-Ub also controls programmed cell death (reviewed in [98, 99]). If TNF-R1-RSC formation or signalling is perturbed, the outcome of receptor engagement is switched from NF-κB activation to induction of cell death, which is mediated by the membrane-detached RIPK1-containing ‘complex-II’ [100] (Fig. 3). LUBAC-mediated Met1-ubiquitnation of RIPK1 is necessary to prevent TNF-induced complex-II formation and cell death [18, 19]. HOIP and HOIL-1 knockout embryos [34, 62], or catalytically inactive knock-ins [25, 63, 64], die at midgestation from aberrant cell death. However, embryonic lethality is not rescued by the deletion of TNF-R1 or RIPK1 but only by co-deletion of caspase-8 and MLKL [62], which completely abrogates both apoptosis and necroptosis [101], indicating that TNF-R1 is not the only death-inducing receptor that LUBAC regulates. To this end, LUBAC has been shown to prevent cell death in response to engagement of both tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor [102, 103], CD95 [103], TLR3 [76], and TLR4 [104]. SHARPIN-deficient mice are viable but develop chronic proliferative dermatitis (cpdm) and multi-organ inflammation due to a spontaneous mutation in Sharpin [61], which results in SHARPIN loss and LUBAC destabilisation [18–20]. These Sharpincpdm mice have been studied extensively (reviewed in [1]). The dermatitis in Sharpincpdm mice is caused by TNF-R1-induced apoptosis of keratinocytes [18, 105, 106]. The Sharpincpdm phenotype is largely cell or tissue intrinsic and is not mediated by immune cells as bone marrow or splenocyte transplantations fail to transfer the disease to wild-type mice [61, 106].
In summary, defective Met1-Ub signalling causes impaired NF-κB activation and production of inflammatory mediators after engagement of PRRs, such as TLRs and NODs [19, 63, 73–77], and cytokine receptors, including TNF-R1 and IL-1R [18, 19, 31–33, 40], as well as excessive cell death [18, 19, 62, 103, 105, 106]. Together, these defects likely lead to an overall dysfunctional or uncoordinated innate immune response to pathogens [76, 83, 97, 107], which contributes to immunodeficiency and inflammation in LUBAC-deficient mice.
Met1-Ub in B and T cells
Met1-Ub also plays an important role in the adaptive immune system where NF-κB activation is critical for lymphocyte development and antigen receptor-mediated immune responses [108]. LUBAC regulates B-cell receptor (BCR)- and T-cell receptor (TCR)-induced NF-κB activation through Met1-ubiquitination of BLC10 in the CARMA1-BCL10-MALT1 complex [109, 110] (Fig. 3). In B cells, BCR-induced NF-κB activation depends on Met1-Ub in lymphoma cell lines [109], whereas LUBAC activity is dispensable for BCR signalling in primary murine B cells [63]. In contrast, B cells invariably depend on Met1-Ub for CD40-induced NF-κB activity [18, 20, 63, 111], and mice lacking HOIP activity in B cells have substantially impaired antibody responses and defective B1 cell development [63]. In T cells, LUBAC activity is crucial for differentiation, survival, and homeostasis of CD4+ and CD8+ T cells, natural killer T cells, and regulatory T cells (Tregs) [112–116]. Consistently, ablation of HOIP in Tregs in mice causes Treg deficiency, breakdown of peripheral tolerance, and lethal immune pathology [115, 116]. Intriguingly, LUBAC appears to maintain T cell differentiation and survival through induction of NF-κB-dependent and -independent transcriptional programmes and not by preventing TNF-induced cell death [115]. This suggests that LUBAC coordinates multiple signalling pathways required for T cell development and homeostasis.
Met1-Ub, infection, and host-pathogen interactions
A clear testament to the importance of Met1-Ub in immune responses is that bacteria and viruses have evolved an armoury of secreted effector proteins that target or hijack Met1-Ub-dependent signalling processes within host cells to evade or restrict the response to infection (Table 2; Fig. 4).
Table 2.
Pathogen effectors targeting Met1-Ub signalling.
| Pathogen | Effector | Intracellular activity | Host target | Ref. |
|---|---|---|---|---|
| Bacteria | ||||
| Legionella Pneumophilia | RavD | Limits Met1-Ub accumulation on Legionella-containing vacuoles, inhibits NF-κB activation | Met1-Ub | [117] |
| Salmonella enterica ser. Typhimurium | SopE | Activates LUBAC for downstream signalling post-infection | N/A | [158] |
| Shigella flexneri | IpaH1.4 | Inhibits NF-κB activation via Lys48-linked ubiquitination and proteasomal degradation of HOIP | HOIP | [120] |
| IpaH2.5 | ||||
| IpaH9.8 | Inhibits NF-κB activation via Lys48-linked ubiquitination and proteasomal degradation of NEMO | NEMO and ABIN-1 | [119] | |
| Mycobacterium tuberculosis | PPE60 | Manipulation of LUBAC, NF-κB signalling, and pyroptosis for Mycobacterium survival | N/A | [159] |
| Viruses | ||||
| Hepatitis B virus (HBV) | HBx | Inhibits IRF3 activation by recruiting Parkin-associated LUBAC to RIG-I/MAVS | Parkin | [123] |
| Hepatitis C virus (HCV) | NS3 | Inhibits Met1-Ub conjugation and NF-κB activity by blocking LUBAC-NEMO interaction | HOIP | [160] |
| Porcine Reproductive and Respiratory Syndrome virus (PRRSV) | NSP1 | Inhibits Met1-Ub conjugation and NF-κB activity by blocking the HOIP-SHARPIN interaction | HOIP and HOIL-1 | [161] |
| NSP11 | Inhibits NF-κB activation by recruiting OTULIN to NEMO | OTULIN | [121] | |
| Epstein-Barr virus (EBV) | LMP1 | Recruits LUBAC to ubiquitinate and inactivate IRF7, limits interferon production | HOIP | [122] |
| Activates oncogenic NF-κB and MAP kinase signalling by recruiting LUBAC to TRAF1 | LUBAC and TRAF1 | [124] | ||
| Human T Cell Leukemia virus type 1 (HTLV-1) | Tax | Activates leukaemogenic IKK signalling by recruiting LUBAC to NEMO | LUBAC and NEMO | [125] |
HBx, Hepatitis B virus X protein; IpaH1.4, Invasion plasmid antigen H1.4; IpaH2.5, Invasion plasmid antigen H2.5; IpaH9.8, Invasion plasmid antigen H9.8; IRF7, Interferon regulatory factor 7; LMP1, Latent membrane protein 1; MAVS, Mitochondrial antiviral signalling protein; NS3, Nonstructural protein 3; NSP1, Nonstructural protein 1; NSP11, Nonstructural protein 11; PPE60, ProProGlu family protein 60; RavD, region allowing vacuole colocalization D; SopE, Salmonella outer protein E; Tax, transactivator from the X-gene region.
Fig. 4. Pathogen interference with Met1-Ub signalling.

In response to bacterial and viral infections, Met1-Ub is assembled in activated immune receptor signalling complexes. In the receptor complexes, Met1-Ub stimulate the IKK complex and downstream NF-κB transcriptional activation. Met1-Ub can also be assembled on intracellular bacteria or their vacuoles (not shown), which promotes NF-κB activation and autophagosome-mediated killing of the bacteria (xenophagy). Some pathogens have evolved secreted effector proteins to specifically interfere with Met1-Ub signalling. Legionella pneumophila, the causative agent of Legionnaires’ disease, encodes the DUB RavD that specifically removes Met1-Ub signals to inhibit IKK activation and the NF-κB response and to possibly interfere with xenophagy of the invading Legionella. Shigella flexneri secretes the ubiquitin ligases IpaH1.4, IpaH2.5 and IpaH9.8 that promote the proteasomal degradation of LUBAC components or the Met1-Ub receptor NEMO, which prevents assembly or sensing of Met1-Ub for activation of the IKK complex and NF-κB. Porcine reproductive and respiratory syndrome virus (PRRSV) secretes NSP11 that hijacks endogenous host OTULIN to remove Met1-Ub from NEMO-containing signalling complexes and inhibit NF-κB activation. Epstein-Barr virus (EBV) encodes the membrane protein LMP1, which recruits LUBAC to ubiquitinate IRF7 and inhibit its transcriptional activity to limit the anti-viral interferon (IFN) response.
Perhaps the most obvious strategy for pathogens to restrict the Met1-Ub signalling is to directly inhibit it. A striking example of how specifically pathogens can target Met1-Ub is the Legionella pneumophila-encoded DUB RavD. RavD is, similar to OTULIN, exquisitely specific for Met1-linkages [117]. Upon infection, RavD is secreted and removes Met1-Ub from invading Legionella and their vacuoles to impair NF-κB signalling [117] and possibly xenophagy of invading bacteria, as shown for Salmonella Typhimurium [59, 118] (Fig. 4). Shigella flexneri employs a similar but distinct strategy to inhibit Met1-Ub signalling where multiple effectors target LUBAC signalling. Upon infection, the Shigella Ub ligases IpaH1.4 and IpaH2.5 modify HOIP with Lys48-Ub and IpaH9.8 modifies NEMO with Lys48-Ub, targeting them for proteasomal degradation, thereby impairing assembly and sensing of Met1-Ub and inhibiting NF-κB activation [119, 120] (Fig. 4).
Another strategy employed by pathogens to manipulate the host response is to hijack the host’s own Met1-Ub machinery and use it against the host. To promote viral replication, porcine reproductive and respiratory syndrome virus (PRRSV)-encoded NSP11 recruits host cell OTULIN to disassemble Met1-Ub in host immune signalling complexes and limit NF-κB activation [121] (Fig. 4). Epstein-Barr virus (EBV), on the other hand, uses the effector LMP1 to recruit LUBAC to ubiquitinate and inactivate the transcription factor IRF7, thereby impairing anti-viral interferon production [122] (Fig. 4). A similar mechanism is employed by hepatitis B virus (HBV), which recruits LUBAC via its HBx effector to impair RIG-I-MAVS-dependent IRF3 activation and interferon production [123].
In contrast to pathogens that restrict Met1-Ub signalling, some oncogenic viruses exploit LUBAC for NF-κB activation, cell survival, and cellular transformation. In addition to IRF7 inhibition, EBV-encoded LMP1 recruits LUBAC and TRAF1/2, leading to Met1-Ub conjugation to NEMO and TRAF1 to promote NF-κB activation and cell proliferation [122, 124]. Similarly, the Tax protein of human T cell leukemia virus type-1 (HTLV-1) interacts with LUBAC to promote Met1-Ub assembly and IKK-mediated NF-κB activation [125].
Dysregulation of Met1-Ub in human disease
Defects in Met1-Ub signalling are associated with a range of severe and potentially fatal human diseases, including immune disorders, cancer, and neurodegenerative diseases, underscoring its physiological importance (Fig. 1; Table 1). Dysregulation of NF-κB and cell death are prominent components of these diseases, consistent with the models and molecular functions of Met1-Ub as discussed above. Notably, these disease associations also hint at novel and previously unappreciated functions or regulatory aspects of Met1-Ub signalling.
LUBAC deficiency and ORAS
The hallmark disorders associated with Met1-Ub are rare, monogenic, autosomal recessive diseases caused by loss-of-function (LOF) mutations in HOIP, HOIL-1, or OTULIN [32, 33, 58, 60, 126–128]. Mutations in HOIP or HOIL-1 cause LUBAC deficiency, a combined autoinflammatory, immunodeficiency, and glycogen storage disease associated with destabilisation of LUBAC and impaired Met1-Ub signalling [32, 33, 126, 129, 130] (Table 1; Fig. 5A). Intriguingly, excessive Met1-Ub formation causes a similar critical illness. Mutations in OTULIN causes OTULIN-Related Autoinflammatory Syndrome (ORAS) (also known as otulipenia), a TNF-driven autoinflammatory and autoimmune syndrome associated with LUBAC deregulation and Met1-Ub accumulation [58, 60, 127, 128] (Table 1).
Fig. 5. Clinical manifestations and cell type-specific reactions in LUBAC deficiency and ORAS.

(A) Comparison of clinical manifestations of LUBAC deficiency and ORAS. Black text indicates overlapping manifestations, the green text indicates manifestations specific to LUBAC deficiency, and blue text indicates manifestations specific to ORAS. (B) Cell type-specific reactions to IL-1β or TNF stimulation. In LUBAC deficiency, patient-derived B cells and fibroblasts from HOIP- or HOIL-1-mutated patients show blunted NF-κB responses and decreased cytokine production. In contrast, patient monocytes have a hyperinflammatory transcriptional profile and hyper-secrete IL-6 in response to IL-1β stimulation. Decreased immune signalling in fibroblasts and B cells likely contribute to immunodeficiency in LUBAC deficiency patients, while the inflammatory profile of monocytes likely contributes to the paradoxical hyperinflammation. OTULIN-deficient lymphocytes (from mice) or patient-derived fibroblasts degrade and downregulate LUBAC. In these cells, TNF-induced NF-κB activity and cytokine production is decreased, and cells are sensitised to cell death. In contrast, OTULIN-deficient myeloid cells (primary murine or human cell lines) retain LUBAC expression and show increased basal and TNF-induced NF-κB activity, but the cells are still sensitive to TNF-induced cell death. TNF hyper-signalling and cytokine secretion from OTULIN-deficient myeloid cells combined with TNF-induced cell death of multiple cell types may in part explain the TNF-dependent inflammation and pathogenesis of ORAS. The molecular mechanism underlying these cell type-specific differences are unknown.
Most mutations in HOIP, HOIL-1, and OTULIN are reported or predicted to severely destabilise or truncate the affected protein, while a few are hypomorphic (and partially destabilised) missense variants with reduced function, such as OTULINL272P that exhibits impaired substrate binding and activity towards Met1-Ub [58]. Not all reported mutants have been characterised biochemically, and it is possible that missense mutations may affect cellular localisation (see below for SHARPIN) or protein-protein interactions in addition to stability and enzymatic function. No clinical manifestations have been reported in heterozygous carriers of HOIP, HOIL-1, or OTULIN mutations [32, 33, 58, 60, 126–130].
Even though LUBAC deficiency and ORAS are aetiologically distinct and molecularly have opposite effects on Met1-Ub, there are several seemingly paradoxical similarities in the clinical manifestations of the two diseases (Fig. 5A). However, significant differences in the disease presentations and pathogeneses exist. LUBAC deficiency and ORAS patients commonly present with early-onset (often neonatal), systemic autoinflammatory manifestations, including fevers, elevated cytokines and acute-phase proteins in the blood, increased white blood cell counts (leukocytosis), as well as diarrhoea [32, 33, 58, 60, 126–128] (Fig. 5A). ORAS patients also present with inflammation of the skin (panniculitis) and joints (arthritis) [58, 60, 127, 128], autoantibodies [58], as well as lipodystrophy [127]. In addition to the inflammatory manifestations, patients with HOIP or HOIL-1 mutations generally display muscular accumulation of glycogen (amylopectinosis) and amylopectinosis-associated muscle weakness, which may cause cardiomyopathy and heart failure [32, 33, 129, 130] (Fig. 5A). Notably, the clinical presentation of patients with HOIL-1 deficiency is variable and some present with muscular amylopectinosis only and no obvious immunological manifestations [62, 63] (Table 1). The reason for this variability is unclear but it may be related to the impact of other genetic variants in these patients. Of note, mutations in SHARPIN have not been found in patients with LUBAC deficiency symptoms but are instead associated with Alzheimer’s disease (Table 1) (discussed below).
Interestingly, LUBAC deficiency is associated with primary immunodeficiency in addition to the inflammatory manifestations, and patients with HOIL-1 and HOIP mutations suffer from severe and repeated bacterial and viral infections [32, 33, 126]. In contrast, there is no clear evidence for immunodeficiency in ORAS patients [58, 60, 127]. This difference is likely due to the fact that there is still functional LUBAC left in ORAS patient cells [58, 60, 127], even in cells that downregulate and degrade LUBAC in response to OTULIN loss [60, 65], but likely very little (if any) functional LUBAC left in patients with HOIP and HOIL-1 mutations [32, 33, 126]. Consequently, the lack of functional Met1-Ub signalling in LUBAC deficiency patients causes defects in both innate and adaptive immune function, including cytokine production and B cell activation [32, 33], which likely cause susceptibility to infection.
LUBAC-deficient patients present with the apparently enigmatic co-occurence of hyperinflammation and immunodeficiency (Fig. 5A). This paradox may be explained in part by cell type-specific responses to Met1-Ub dysregulation. Fibroblasts and B cells from patients with HOIP or HOIL-1 mutations have blunted NF-κB responses after stimulation with TNF or IL-1β [32, 33], consistent with the model of LUBAC’s role in these pathways (Fig. 5B). Strikingly, monocytes from the same patients have a clear hyperinflammatory profile with increased transcription of pro-inflammatory genes and hypersecretion of IL-6 [32, 33]. Cell type-specific differences have also been reported for OTULIN-deficient cells (Fig. 5B). OTULIN-deficient myeloid cells retain LUBAC expression and have both increased basal NF-κB activation and increased responses to TNF stimulation [58, 60]. In contrast, OTULIN-deficient lymphocytes and fibroblasts lose LUBAC due to autoubiquitination and proteasomal degradation, which impairs TNF-induced NF-κB activation and sensitises to TNF-induced cell death [58, 60, 65]. The mechanisms underpinning these differences are unknown, but the findings suggest that the ‘demographics’ of the Met1-Ub machinery – i.e., the expression levels and activities of LUBAC, Met1-Ub-DUBs, Met1-Ub-receptors, as well as possible unknown regulators – could be different in different cell types and that this might determine how the perturbed system reacts. As in the case of OTULIN deficiency, do myeloid cells for example express a DUB that at least to some extent can take over the function of OTULIN and prevent LUBAC autoubiquitination and degradation? Another possibility is that Met1-Ub signals could serve different functions in different cell types. In the case of LUBAC deficiency, it is possible that impairment of Met1-Ub signalling in monocytes leads to inhibition of a negative regulatory mechanism for IL-1β and immune signalling.
The inflammatory symptoms in ORAS are controlled effectively by TNF blockade (infliximab or etanercept) but generally not by corticosteroid treatment [58, 60, 127]. In contrast, patients with HOIL-1 deficiency generally respond well to corticosteroids while TNF blockade only has moderate effect and has even been reported to exacerbate the inflammatory condition in one case [32]. Importantly, preliminary results indicate that bone marrow transplantations ameliorate the inflammatory symptoms in both LUBAC deficiency and ORAS patients [32, 60], emphasising that immune cells are critical mediators of these diseases [58, 60].
Cancer
Chronic inflammation and NF-κB activation is associated with cancer development, progression, and resistance to therapy [131]. Mounting evidence indicates that Met1-Ub signalling plays a part in multiple aspects of cancer through NF-κB regulation. Enhanced LUBAC activity and expression has been implicated in the development of activated B cell-like diffuse large B cell lymphoma (ABC-DLBCL). Rare gain-of-function variants in human HOIP are enriched 8-fold in patients with ABC-DLBCL relative to healthy individuals [132] (Table 1). These variants increase the HOIP–HOIL-1 interaction and enhances LUBAC activity [132]. Moreover, HOIP expression is elevated overall in ABC-DLBCL cells [133]. Increased LUBAC expression and activity enhances BCR-mediated NF-κB activation and protects against genotoxic stress-induced cell death, thereby promoting accumulation of somatic mutations and lymphomagenesis [132–134] (Fig. 6). LUBAC has also been shown to promote NF-κB activation and tumourigenesis in breast cancer in mice [135], suggesting that LUBAC-mediated NF-κB activation could be a means of tumour cell survival in multiple cancer types.
Fig. 6. Roles of the Met1-Ub machinery in cancer.

Examples of the involvement of the Met1-Ub machinery in cancer. Enhanced LUBAC activity, either as a consequence of HOIP mutations or overexpression, leads to constitutive NF-κB activation in ABC-DLBCL, which promotes B cell survival and proliferation as well as protects against DNA damage-induced cell death during B cell activation. In combination, this leads to sustained B cell survival and accumulation of somatic mutations, which contributes to lymphoma development. LUBAC activity and lymphoma cell survival can be inhibited by the N-Q622L peptide, which targets the HOIP-HOIL-1 interaction [132]. In LSCC, LUBAC overexpression leads to enhanced NF-κB activation, which confers tumour resistance to the chemotherapeutic agent cisplatin. Inhibition of LUBAC using the small-molecule LUBAC inhibitor gliotoxin reduces NF-κB activation and LSCC cell survival and sensitises tumours to cisplatin treatment [136]. OTULIN deficiency causes HCC in mice. The associated Met1-Ub accumulation causes aberrant mTOR activation, dysmetabolism, and increased RIPK1-mediated apoptosis and compensatory proliferation. Inhibition of mTOR by rapamycin leads to reduced tumour formation in mice [140]. In breast cancer patients, high OTULIN expression is associated with aggressive tumour subtypes and correlates with poor survival. Increased expression of OTULIN leads to removal of Met1-Ub from β-catenin, which stabilises the protein and allows Wnt/β-catenin-driven oncogenic signalling that promotes breast cancer cell survival, tumour growth, chemotherapy resistance, and metastasis in mice [142]. ABC-DLBCL, activated B cell diffuse large B cell lymphoma; HCC, hepatocellular carcinoma; LSCC, lung squamous cell carcinoma; mTOR, mammalian target, or rapamycin.
LUBAC-dependent NF-κB activation has also been found to promote therapy resistance. In a recent study, Ruiz et al. demonstrated that human and mouse lung squamous cell carcinoma (LSCC) cells have increased LUBAC expression, which leads to Met1-Ub accumulation and increased NF-κB activation in tumours, resulting in resistance to cisplatin [136] (Fig. 6). Strikingly, inhibition of LUBAC in LSCC cells using the small-molecule inhibitor gliotoxin decreased NF-κB activity, sensitised LSCC cells to cisplatin, and led to reduced tumour growth in mice [136]. Consistently, HOIP has been shown to confer resistance to cisplatin in a range of cancer cell types, albeit in an ATM-dependent and NF-κB-independent manner [137]. Regardless, these observations suggest that LUBAC inhibition might be a valid therapeutic strategy in certain cancers.
The Met1-Ub-targeting DUBs are also implicated in cancer (Fig. 6). CYLD is a well-established tumour suppressor originally discovered in patients with familial cylindromatosis, a genetic condition that leads to tumours of skin appendages [86–88]. CYLD is involved in the development of many different cancers through NF-κB, cell death, and other cellular functions (reviewed in [138, 139]), although it has not been shown experimentally that this is due to dysregulated Met1-Ub. Intriguingly, OTULIN was recently reported to be associated with cancer in humans and mice. In mice, OTULIN acts as a tumour suppressor in the liver [140, 141]. OTULIN deficiency in hepatocytes causes substantial accumulation of Met1-Ub and leads to steatohepatitis, fibrosis, and the development of hepatocellular carcinoma (HCC). This results from the aberrant mammalian target of rapamycin (mTOR)-dependent signalling [140] and compensatory proliferation due to increased FADD- and RIPK1-dependent cell death [141]. In humans, high OTULIN expression is associated with aggressive breast cancer subtypes and correlates with poor survival, likely by promoting Wnt/β-catenin-mediated tumour cell survival and growth [142] (Fig. 6).
Neurodegenerative disease
Neurodegenerative diseases, such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease are characterised by progressive neuronal degeneration associated with the formation of protein aggregates in neurons [143]. Recently, a rare functional SHARPIN variant was identified as a genetic risk factor associated with the most common form of dementia, late-onset AD [144]. The identified SHARPING186R variant (Table 1) causes abnormal, granular/speckled cellular localisation and impaired NF-κB activity [144]. In addition to SHARPIN, the Met1-Ub binding protein Optineurin is also genetically linked to neurodegeneration. Mutations in Optineurin have been identified in familial ALS [145], and these mutations impair Optineurin’s Met1-Ub-binding and NF-κB-inhibitory capacity [145, 146]. This suggests that dysregulated NF-κB due to imbalanced Met1-Ub signalling could be involved in the pathogeneses of AD and ALS. Intriguingly, neuronal protein aggregates in both AD and ALS have been found to be decorated with Met1-Ub [146–148]. In AD patients, tau aggregates are Met1-Ub positive [147], and in ALS, TDP-43 aggregates in brains from patients with both sporadic and familial disease contain Met1-Ub [146, 148]. This indicates that Met1-Ub may be part of the development of—or response to—protein aggregation. Strikingly, LUBAC was recently reported to be involved in protein quality control by Met1-ubiquitinating protein aggregates and facilitating their degradation [149] (discussed below). Currently, there are no reports of neurological symptoms in patients with HOIP, HOIL-1, or OTULIN mutations, but this could be related to the age of the patients. However, a preprint article has indicated that HOIL-1-deficient mice develop neurodegenerative disease and neurobehavioral deficits due to the accumulation of aberrant forms of glycogen (polyglucosan) in the brain and spinal cord [150], similar to the muscular phenotype of patients with HOIL-1 mutations [32, 129, 130].
Expanding the cellular functions of Met1-Ub
The role of Met1-Ub in NF-κB signalling and cell death is well-established. A range of recent studies, however, have expanded the portfolio of pathways and cellular functions controlled by Met1-Ub (Fig. 7). Although these emerging functions are not all well-understood mechanistically, they provide new avenues of research and may offer novel insights into the role of Met1-Ub signalling in disease development.
Fig. 7. Cellular functions of Met1-Ub.

In addition to the established functions of Met1-Ub, on which we have witnessed remarkable progress towards understanding their mechanistic underpinnings, recent studies have indicated new and emerging functions of Met1-Ub in the regulation of cellular signalling, function, and homeostasis.
Wnt signalling
The first pathway genetically associated with OTULIN upon its discovery was Wnt signalling [47]. Mice homozygous for missense LOF mutations in OTULIN die at midgestation due to defective Wnt signalling and abnormal vascularisation [47]. OTULIN interacts with DVL2, a key mediator of the Wnt pathway, and promotes Wnt signalling by antagonising the inhibitory effect of LUBAC and Met1-Ub on Wnt-induced transcriptional activity [47, 53]. The mechanism by which Met1-Ub controls Wnt signalling, however, remained enigmatic. Recently, Wang et al. shed new mechanistic light on this and showed that LUBAC assembles Met1-Ub on β-catenin at steady-state to facilitate its proteasomal degradation (likely via additional Lys48-Ub modifications), thereby keeping Wnt signalling in check [142]. Phosphorylation of OTULIN at Tyr56 by the tyrosine protein kinase ABL1 (c-Abl) recruits OTULIN to β-catenin and displaces LUBAC, leading to removal of Met1-Ub from β-catenin, which promotes its stabilisation and activation of the Wnt/β-catenin pathway [142].
Protein quality control
Accumulation of misfolded and aggregated proteins can be disastrous for cells and organisms. This is highlighted by the large number of neurodegenerative diseases that are associated with cytotoxic protein aggregates [143]. Although generally considered a non-proteolytic modification [6], Met1-Ub was recently found to promote proteasomal degradation of toxic, disease-associated protein aggregates, including Huntingtin (Htt; Huntington’s disease), Ataxin-3, SOD1, and TDP-43 [149]. LUBAC is recruited to misfolded protein aggregates, such as pathogenic polyglutamine expansions of Htt (Htt-polyQ), through the AAA-type ATPase p97/VCP [149], which contains a PIM that interacts with HOIP in a manner similar to OTULIN and SPATA2 [50, 51, 53]. Intriguingly, LUBAC-mediated Met1-Ub conjugation to protein aggregates facilitates their proteasomal degradation in a process independent of NF-κB activation [149]. Interestingly, the Drosophila LUBAC ortholog, LUBEL, protects flies against heat stress [69], and sustained Met1-ubiquitination of substrates such as PCNA and HOIP itself has been shown to induce proteasomal degradation [60, 65, 151], supporting a role of Met1-Ub in protein degradation and quality control. Collectively, this indicates that Met1-Ub could be a putative degradation signal in specific contexts and suggests that LUBAC activity may be crucial for prevention of neurodegenerative disease.
Autophagy and xenophagy
Autophagy is a membrane-dependent intracellular degradation pathway, through which cytoplasmic content is degraded in lysosomes [152]. Met1-Ub has recently been found to control autophagy-related processes. In a process known as xenophagy, infection by cytosol-invading bacteria, e.g., Salmonella Typhimurium, leads to ubiquitination of the bacterial surface [153] and recruitment of the autophagy machinery to target the bacterium for degradation. LUBAC is recruited to the surface of cytosol-invading Salmonella and modifies the bacterial surface with Met1-Ub, which simultaneously turns the bacterial surface into an NF-κB-activating signalling platform and recruits the autophagy machinery via the Met1-Ub-receptor Optineurin for bacterial destruction [59, 118]. Recently, Met1-Ub was also found to regulate autophagy induction in response to nutrient starvation [154]. Chu et al. showed that LUBAC promotes autophagy by stabilising the ULK1 autophagy initiation complex through Met1-ubiquitination of ATG13, which is reversed by OTULIN [154]. These reports indicate that Met1-Ub is important both for defending the cytosol against pathogens as well as maintaining cellular homeostasis via autophagy regulation.
Metabolic control
The first indication that Met1-Ub could be involved in metabolic regulation came from patients with LUBAC deficiency. In most patients, HOIP and HOIL-1 mutations cause amylopectinosis, a form of glycogen storage disease where aberrant forms of glycogen accumulate, primarily in muscle tissue [32, 33, 129, 130] (Table 1). In contrast, mice with hepatocyte-specific OTULIN deletion show an almost complete lack of glycogen in their livers [140], lending support to a role of Met1-Ub in glycogen metabolism. Transcriptional analysis of blood cells from a patient with HOIL-1 deficiency showed that, in addition to inflammatory pathways, genes associated with carbohydrate metabolism are dysregulated [32]. Although, LUBAC has been shown to form an immuno-regulatory complex with the glucose metabolism modulator TIGAR [155], the mechanism by which Met1-Ub may control carbohydrate/glycogen metabolism remains unknown.
Findings also indicate that Met1-Ub controls lipid metabolism. Some ORAS patients present with lipodystrophy [127], a disorder characterised by varying degrees of body fat loss [156]. Moreover, mice with hepatocyte-specific OTULIN deletion show severe neonatal steatosis and increased cholesterol in serum, which is associated with apoptosis and aberrant activation of mTOR complex 1 (mTORC1) in hepatocytes [140]. However, like for carbohydrates, the role of Met1-Ub in control of lipid metabolism is unclear. Intriguingly, diet-induced steatosis in adult mice impairs LUBAC formation [157]. This shows that environmental factors, such as diet, may affect Met1-Ub signalling in disease states and suggests that reduced LUBAC function and Met1-Ub signalling could be an underlying molecular mechanism in the pathogenesis of non-alcoholic steatohepatitis (NASH). Notably, one ORAS patient was found to have progressive steatotic liver disease [140], further strengthening the association between Met1-Ub dysregulation and NASH.
Chromosome alignment
In a recent study, Met1-Ub was shown to be important for chromosome alignment and mitotic progression [44]. Failure of chromosome alignment can lead to chromosome missegregation, which can cause aneuploidy and cancer. All three LUBAC components were identified in an RNAi-based screen for regulators of mitosis [44]. In cells, LUBAC Met1-ubiquitinated the kinesin motor CENP-E and facilitated CENP-E’s localisation to the kinetochore through recruitment to kinetochore-associated KNL1, a newly identified Met1-Ub-binding protein [44]. LUBAC deficiency led to unstable microtubule attachment to the kinetochore and caused chromosome misalignment during mitosis [44]. This suggests that LUBAC deficiency could cause aneuploidy and increased cancer risk, but further studies are needed to clarify this potential association.
Concluding remarks
It is clear that Met1-Ub is a central and potent signalling modification for regulation of NF-κB activity and cell death. Both NF-κB and cell death play key roles in maintaining immune homeostasis, and the crucial physiological role of Met1-Ub in these pathways is explicitly underscored by the monogenic autoinflammatory syndromes associated with dysregulation of either conjugation or disassembly of Met1-Ub. Intriguingly, Met1-Ub is also emerging to be involved in the development of more genetically complex and sporadic diseases, such as cancer and neurodegeneration, seemingly by mechanisms that also go beyond the regulation of NF-κB and cell death. Furthermore, an important and developing concept is cell- and context-dependent functions of the Met1-Ub signals, LUBAC, and the Met1-Ub-DUBs. Careful analysis of cell type-specific knock-out or knock-in mice, as well as patient-derived cells, will likely reveal novel context-specific functions and mechanisms of Met1-Ub regulation. Importantly, these mechanisms and functions may go far beyond the control of NF-κB and cell death, as indicated by recent findings that are currently expanding the repertoire of pathways and cellular functions regulated by Met1-Ub. It will be important to elucidate the basis of these context-dependent roles and the mechanisms of the emerging functions to understand the complex phenotypes and diseases associated with dysfunction of the Met1-Ub machinery.
Acknowledgements
We apologise to those whose work could not be cited here due to space restrictions. R.B.D. is supported by the Technical University of Denmark and the Novo Nordisk Foundation [NNF19OC0054248]. A.S.J. is supported by The Croucher Foundation, Hong Kong. The figures were prepared using Servier Medical Art (https://smart.servier.com).
Author contributions
A.S.J. and C.R.E. drafted and edited the manuscript text and prepared the figures. R.B.D. conceptualised, edited, and revised the manuscript and figures.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Akhee Sabiha Jahan, Camilla Reiter Elbæk
References
- 1.Hrdinka M, Gyrd-Hansen M. The Met1-linked ubiquitin machinery: emerging themes of (De)regulation. Mol Cell. 2017;68:265–80. doi: 10.1016/j.molcel.2017.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 3.Kerscher O, Felberbaum R, Hochstrasser M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Bi. 2006;22:159–80. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 4.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. doi: 10.1146/annurev-biochem-051810-094654. [DOI] [PubMed] [Google Scholar]
- 5.Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Bio. 2019;20:338–52. doi: 10.1038/s41580-019-0099-1. [DOI] [PubMed] [Google Scholar]
- 6.Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–29. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 7.Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:579–86. doi: 10.1038/ncb3358. [DOI] [PubMed] [Google Scholar]
- 8.Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26:399–422. doi: 10.1038/cr.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–45. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteom. 2011;10:M111.013284. doi: 10.1074/mcp.M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–40. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dammer EB, Na CH, Xu P, Seyfried NT, Duong DM, Cheng D, et al. Polyubiquitin linkage profiles in three models of proteolytic stress suggest the etiology of alzheimer disease. J Biol Chem. 2011;286:10457–65. doi: 10.1074/jbc.M110.149633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ziv I, Matiuhin Y, Kirkpatrick DS, Erpapazoglou Z, Leon S, Pantazopoulou M, et al. A perturbed ubiquitin landscape distinguishes between ubiquitin in trafficking and in proteolysis. Mol Cell Proteom. 2011;10:M111.009753. doi: 10.1074/mcp.M111.009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulathu Y, Komander D. Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Bio. 2012;13:508–23. doi: 10.1038/nrm3394. [DOI] [PubMed] [Google Scholar]
- 15.Huizen M, van, Kikkert M. The role of atypical ubiquitin chains in the regulation of the antiviral innate immune response. Front Cell Dev Biol. 2020;7:392. doi: 10.3389/fcell.2019.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. Embo J. 2006;25:4877–87. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elliott PR. Molecular basis for specificity of the Met1-linked polyubiquitin signal. Biochem Soc T. 2016;44:1581–602. doi: 10.1042/BST20160227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–6. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda F, Deribe YL, Skånland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature. 2011;471:637–41. doi: 10.1038/nature09814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–6. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 21.Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J. 2014;458:421–37. doi: 10.1042/BJ20140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smit JJ, Monteferrario D, Noordermeer SM, Dijk WJ, van, Reijden BA, van der, Sixma TK. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012;31:3833–44. doi: 10.1038/emboj.2012.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012;13:840–6. doi: 10.1038/embor.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stieglitz B, Rana RR, Koliopoulos MG, Morris-Davies AC, Schaeffer V, Christodoulou E, et al. Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature. 2013;503:422–6. doi: 10.1038/nature12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emmerich CH, Ordureau A, Strickson S, Arthur JSC, Pedrioli PGA, Komander D, et al. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA. 2013;110:15247–52. doi: 10.1073/pnas.1314715110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emmerich CH, Bakshi S, Kelsall IR, Ortiz-Guerrero J, Shpiro N, Cohen P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem Bioph Res Co. 2016;474:452–61. doi: 10.1016/j.bbrc.2016.04.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker-Jensen S, et al. OTULIN restricts Met1-linked ubiquitination to control innate immune signaling. Mol Cell. 2013;50:818–30. doi: 10.1016/j.molcel.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hrdinka M, Fiil BK, Zucca M, Leske D, Bagola K, Yabal M, et al. CYLD limits Lys63- and Met1-linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep. 2016;14:2846–58. doi: 10.1016/j.celrep.2016.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Wang Y, Gong Y, Fu T, Hu S, Zhou Z, et al. Structural insights into SHARPIN-mediated activation of HOIP for the linear ubiquitin chain assembly. Cell Rep. 2017;21:27–36. doi: 10.1016/j.celrep.2017.09.031. [DOI] [PubMed] [Google Scholar]
- 30.Fujita H, Tokunaga A, Shimizu S, Whiting AL, Aguilar-Alonso F, Takagi K, et al. Cooperative domain formation by homologous motifs in HOIL-1L and SHARPIN plays a crucial role in LUBAC stabilization. Cell Rep. 2018;23:1192–204. doi: 10.1016/j.celrep.2018.03.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol. 2009;11:123–32. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 32.Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol. 2012;13:1178–86. doi: 10.1038/ni.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med. 2015;212:939–51. doi: 10.1084/jem.20141130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peltzer N, Rieser E, Taraborrelli L, Draber P, Darding M, Pernaute B, et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 2014;9:153–65. doi: 10.1016/j.celrep.2014.08.066. [DOI] [PubMed] [Google Scholar]
- 35.Fennell LM, Rahighi S, Ikeda F. Linear ubiquitin chain-binding domains. FEBS J. 2018;285:2746–61. doi: 10.1111/febs.14478. [DOI] [PubMed] [Google Scholar]
- 36.Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell. 2009;136:1098–109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Oshima S, Turer EE, Callahan JA, Chai S, Advincula R, Barrera J, et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature. 2009;457:906–9. doi: 10.1038/nature07575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Löhr F, et al. Ubiquitin binding mediates the NF-κB inhibitory potential of ABIN proteins. Oncogene. 2008;27:3739–45. doi: 10.1038/sj.onc.1211042. [DOI] [PubMed] [Google Scholar]
- 39.Komander D, Reyes-Turcu F, Licchesi JDF, Odenwaelder P, Wilkinson KD, Barford D. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. Embo Rep. 2009;10:466–73. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–44. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 41.Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, et al. A20 inhibits LUBAC-mediated NF-κB activation by binding linear polyubiquitin chains via its zinc finger 7. Embo J. 2012;31:3845–55. doi: 10.1038/emboj.2012.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-κB regulation. Embo J. 2012;31:3856–70. doi: 10.1038/emboj.2012.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gyrd-Hansen M, Darding M, Miasari M, Santoro MM, Zender L, Xue W, et al. IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-κB as well as cell survival and oncogenesis. Nat Cell Biol. 2008;10:1309–17. doi: 10.1038/ncb1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu M, Chang Y, Hu H, Mu R, Zhang Y, Qin X, et al. LUBAC controls chromosome alignment by targeting CENP-E to attached kinetochores. Nat Commun. 2019;10:273. doi: 10.1038/s41467-018-08043-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ritorto MS, Ewan R, Perez-Oliva AB, Knebel A, Buhrlage SJ, Wightman M, et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat Commun. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell. 2013;153:1312–26. doi: 10.1016/j.cell.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivkin E, Almeida SM, Ceccarelli DF, Juang Y-C, MacLean TA, Srikumar T, et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature. 2013;498:318–24. doi: 10.1038/nature12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato Y, Goto E, Shibata Y, Kubota Y, Yamagata A, Goto-Ito S, et al. Structures of CYLD USP with Met1- or Lys63-linked diubiquitin reveal mechanisms for dual specificity. Nat Struct Mol Biol. 2015;22:222–9. doi: 10.1038/nsmb.2970. [DOI] [PubMed] [Google Scholar]
- 49.Elliott PR, Komander D. Regulation of Met1-linked polyubiquitin signalling by the deubiquitinase OTULIN. FEBS J. 2016;283:39–53. doi: 10.1111/febs.13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elliott PR, Nielsen SV, Marco-Casanova P, Fiil BK, Keusekotten K, Mailand N, et al. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol Cell. 2014;54:335–48. doi: 10.1016/j.molcel.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schaeffer V, Akutsu M, Olma MH, Gomes LC, Kawasaki M, Dikic I. Binding of OTULIN to the PUB domain of HOIP controls NF-κB signaling. Mol Cell. 2014;54:349–61. doi: 10.1016/j.molcel.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 52.Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 2015;13:2258–72. doi: 10.1016/j.celrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takiuchi T, Nakagawa T, Tamiya H, Fujita H, Sasaki Y, Saeki Y, et al. Suppression of LUBAC-mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes Cells. 2014;19:254–72. doi: 10.1111/gtc.12128. [DOI] [PubMed] [Google Scholar]
- 54.Elliott PR, Leske D, Hrdinka M, Bagola K, Fiil BK, McLaughlin SH, et al. SPATA2 Links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol Cell. 2016;63:990–1005. doi: 10.1016/j.molcel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schlicher L, Wissler M, Preiss F, Brauns-Schubert P, Jakob C, Dumit V, et al. SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. Embo Rep. 2016;17:1485–97. doi: 10.15252/embr.201642592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wagner SA, Satpathy S, Beli P, Choudhary C. SPATA2 links CYLD to the TNF-α receptor signaling complex and modulates the receptor signaling outcomes. Embo J. 2016;35:1868–84. doi: 10.15252/embj.201694300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kupka S, De Miguel D, Draber P, Martino L, Surinova S, Rittinger K, et al. SPATA2-mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep. 2016;16:2271–80. doi: 10.1016/j.celrep.2016.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell. 2016;166:1215–.e20. doi: 10.1016/j.cell.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wijk SJL, van, Fricke F, Herhaus L, Gupta J, Hötte K, Pampaloni F, et al. Linear ubiquitination of cytosolic Salmonella typhimurium activates NF-κB and restricts bacterial proliferation. Nat Microbiol. 2017;2:17066. doi: 10.1038/nmicrobiol.2017.66. [DOI] [PubMed] [Google Scholar]
- 60.Damgaard RB, Elliott PR, Swatek KN, Maher ER, Stepensky P, Elpeleg O, et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. Embo Mol Med. 2019;11:e9324. doi: 10.15252/emmm.201809324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.HogenEsch H, Gijbels MJ, Offerman E, Hooft J, van, Bekkum DW, van, Zurcher C. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am J Pathol. 1993;143:972–82. [PMC free article] [PubMed] [Google Scholar]
- 62.Peltzer N, Darding M, Montinaro A, Draber P, Draberova H, Kupka S, et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature. 2018;557:112–7. doi: 10.1038/s41586-018-0064-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sasaki Y, Sano S, Nakahara M, Murata S, Kometani K, Aiba Y, et al. Defective immune responses in mice lacking LUBAC-mediated linear ubiquitination in B cells. Embo J. 2013;32:2463–76. doi: 10.1038/emboj.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kelsall IR, Zhang J, Knebel A, Arthur JSC, Cohen P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc Natl Acad Sci USA. 2019;116:13293–8. doi: 10.1073/pnas.1905873116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature. 2018;559:120–4. doi: 10.1038/s41586-018-0256-2. [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto ML, Dong KC, Yu C, Phu L, Gao X, Hannoush RN, et al. Engineering and structural characterization of a linear polyubiquitin-specific antibody. J Mol Biol. 2012;418:134–44. doi: 10.1016/j.jmb.2011.12.053. [DOI] [PubMed] [Google Scholar]
- 67.Hospenthal MK, Mevissen TET, Komander D. Deubiquitinase-based analysis of ubiquitin chain architecture using Ubiquitin Chain Restriction (UbiCRest) Nat Protoc. 2015;10:349–61. doi: 10.1038/nprot.2015.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mevissen TET, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, et al. OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell. 2013;154:169–84. doi: 10.1016/j.cell.2013.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asaoka T, Almagro J, Ehrhardt C, Tsai I, Schleiffer A, Deszcz L, et al. Linear ubiquitination by LUBEL has a role in Drosophila heat stress response. EMBO Rep. 2016;17:1624–40. doi: 10.15252/embr.201642378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aalto AL, Mohan AK, Schwintzer L, Kupka S, Kietz C, Walczak H, et al. M1-linked ubiquitination by LUBEL is required for inflammatory responses to oral infection in Drosophila. Cell Death Differ. 2019;26:860–76. doi: 10.1038/s41418-018-0164-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–8. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 72.Peltzer N, Walczak H. Cell death and inflammation – a vital but dangerous liaison. Trends Immunol. 2019;40:387–402. doi: 10.1016/j.it.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 73.Damgaard RB, Nachbur U, Yabal M, Wong WW-L, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46:746–58. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 74.Warner N, Burberry A, Franchi L, Kim Y-G, McDonald C, Sartor MA, et al. A genome-wide siRNA screen reveals positive and negative regulators of the NOD2 and NF-κB signaling pathways. Sci Signal. 2013;6:rs3–rs3. doi: 10.1126/scisignal.2003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zak DE, Schmitz F, Gold ES, Diercks AH, Peschon JJ, Valvo JS, et al. Systems analysis identifies an essential role for SHANK-associated RH domain-interacting protein (SHARPIN) in macrophage Toll-like receptor 2 (TLR2) responses. Proc Natl Acad Sci USA. 2011;108:11536–41. doi: 10.1073/pnas.1107577108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zinngrebe J, Rieser E, Taraborrelli L, Peltzer N, Hartwig T, Ren H, et al. LUBAC deficiency perturbs TLR3 signaling to cause immunodeficiency and autoinflammationLUBAC regulates TLR3 signaling. J Exp Med. 2016;213:2671–89. doi: 10.1084/jem.20160041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Z, Sokolovska A, Seymour R, Sundberg JP, HogenEsch H. SHARPIN is essential for cytokine production, NF-κB signaling, and induction of Th1 differentiation by dendritic cells. PLoS ONE. 2012;7:e31809. doi: 10.1371/journal.pone.0031809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fiil BK, Gyrd-Hansen M. Met1-linked ubiquitination in immune signalling. FEBS J. 2014;281:4337–50. doi: 10.1111/febs.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shimizu Y, Taraborrelli L, Walczak H. Linear ubiquitination in immunity. Immunol Rev. 2015;266:190–207. doi: 10.1111/imr.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kanayama A, Seth RB, Sun L, Ea C-K, Hong M, Shaito A, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell. 2004;15:535–48. doi: 10.1016/j.molcel.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 81.Hauenstein AV, Xu G, Kabaleeswaran V, Wu H. Evidence for M1-linked polyubiquitin-mediated conformational change in NEMO. J Mol Biol. 2017;429:3793–800. doi: 10.1016/j.jmb.2017.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hinz M, Scheidereit C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014;15:46–61. doi: 10.1002/embr.201337983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.MacDuff DA, Reese TA, Kimmey JM, Weiss LA, Song C, Zhang X, et al. Phenotypic complementation of genetic immunodeficiency by chronic herpesvirus infection. Elife. 2015;4:e04494. doi: 10.7554/eLife.04494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Damgaard RB, Fiil BK, Speckmann C, Yabal M, Stadt zur U, Bekker-Jensen S, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. Embo Mol Med. 2013;5:1278–95. doi: 10.1002/emmm.201303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cohen P, Strickson S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017;24:1153–9. doi: 10.1038/cdd.2017.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brummelkamp TR, Nijman SMB, Dirac AMG, Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-κB. Nature. 2003;424:797–801. doi: 10.1038/nature01811. [DOI] [PubMed] [Google Scholar]
- 87.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424:793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 88.Kovalenko A, Chable-Bessia C, Cantarella G, Israël A, Wallach D, Courtois G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature. 2003;424:801–5. doi: 10.1038/nature01802. [DOI] [PubMed] [Google Scholar]
- 89.Wright A, Reiley WW, Chang M, Jin W, Lee AJ, Zhang M, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–16. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 90.Wei R, Xu LW, Liu J, Li Y, Zhang P, Shan B, et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Gene Dev. 2017;31:1162–76. doi: 10.1101/gad.299776.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Douglas T, Saleh M. Post-translational modification of OTULIN regulates ubiquitin dynamics and cell death. Cell Rep. 2019;29:3652–.e5. doi: 10.1016/j.celrep.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 92.Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation linear ubiquitination regulates NLRP3/ASC assembly. J Exp Med. 2014;211:1333–47. doi: 10.1084/jem.20132486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gurung P, Lamkanfi M, Kanneganti T-D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J Immunol. 2015;194:2064–7. doi: 10.4049/jimmunol.1402951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Douglas T, Champagne C, Morizot A, Lapointe J-M, Saleh M. The inflammatory caspases-1 and -11 mediate the pathogenesis of dermatitis in sharpin-deficient mice. J Immunol. 2015;195:2365–73. doi: 10.4049/jimmunol.1500542. [DOI] [PubMed] [Google Scholar]
- 95.Inn K-S, Gack MU, Tokunaga F, Shi M, Wong L-Y, Iwai K, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. 2011;41:354–65. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Belgnaoui SM, Paz S, Samuel S, Goulet M-L, Sun Q, Kikkert M, et al. Linear Ubiquitination of NEMO Negatively Regulates the Interferon Antiviral Response through Disruption of the MAVS-TRAF3 Complex. Cell Host Microbe. 2012;12:211–22. doi: 10.1016/j.chom.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 97.Brazee PL, Morales-Nebreda L, Magnani ND, Garcia JGN, Misharin AV, Ridge KM, et al. Linear ubiquitin assembly complex regulates lung epithelial–driven responses during influenza infection. J Clin Invest. 2020;130:1301–14. doi: 10.1172/JCI128368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kupka S, Reichert M, Draber P, Walczak H. Formation and removal of poly-ubiquitin chains in the regulation of tumor necrosis factor-induced gene activation and cell death. FEBS J. 2016;283:2626–39. doi: 10.1111/febs.13644. [DOI] [PubMed] [Google Scholar]
- 99.Sasaki K, Iwai K. Roles of linear ubiquitinylation, a crucial regulator of NF-κB and cell death, in the immune system. Immunol Rev. 2015;266:175–89. doi: 10.1111/imr.12308. [DOI] [PubMed] [Google Scholar]
- 100.Annibaldi A, Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med. 2017;24:49–65. doi: 10.1016/j.molmed.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 101.Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64. doi: 10.1038/s41422-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lafont E, Kantari-Mimoun C, Draber P, Miguel DD, Hartwig T, Reichert M, et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. Embo J. 2017;36:1147–66. doi: 10.15252/embj.201695699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Taraborrelli L, Peltzer N, Montinaro A, Kupka S, Rieser E, Hartwig T, et al. LUBAC prevents lethal dermatitis by inhibiting cell death induced by TNF, TRAIL and CD95L. Nat Commun. 2018;9:3910. doi: 10.1038/s41467-018-06155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sasaki Y, Iwai K. Crucial role of linear ubiquitin chain assembly complex–mediated inhibition of programmed cell death in TLR4-mediated B cell responses and B1b cell development. J Immunol. 2018;200:3438–49. doi: 10.4049/jimmunol.1701526. [DOI] [PubMed] [Google Scholar]
- 105.Kumari S, Redouane Y, López-Mosqueda J, Shiraishi R, Romanowska M, Lutzmayer S, et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife. 2014;3:e03422. doi: 10.7554/eLife.03422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rickard JA, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. Elife. 2014;3:e03464. doi: 10.7554/eLife.03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.MacDuff DA, Baldridge MT, Qaqish AM, Nice TJ, Darbandi AD, Hartley VL, et al. HOIL1 is essential for the induction of type I and III interferons by MDA5 and regulates persistent murine norovirus infection. J Virol. 2018;92:JVI.01368–18. doi: 10.1128/JVI.01368-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gerondakis S, Siebenlist U. Roles of the NF-κB pathway in lymphocyte development and function. Csh Perspect Biol. 2010;2:a000182. doi: 10.1101/cshperspect.a000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Satpathy S, Wagner SA, Beli P, Gupta R, Kristiansen TA, Malinova D, et al. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol Syst Biol. 2015;11:810. doi: 10.15252/msb.20145880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yang Y-K, Yang C, Chan W, Wang Z, Deibel KE, Pomerantz JL. Molecular determinants of scaffold-induced linear ubiquitinylation of B cell lymphoma/leukemia 10 (Bcl10) during T cell receptor and oncogenic caspase recruitment domain-containing Protein 11 (CARD11) signaling. J Biol Chem. 2016;291:25921–36. doi: 10.1074/jbc.M116.754028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hostager BS, Kashiwada M, Colgan JD, Rothman PB. HOIL-1L interacting protein (HOIP) is essential for CD40 signaling. PLoS ONE. 2011;6:e23061. doi: 10.1371/journal.pone.0023061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Okamura K, Kitamura A, Sasaki Y, Chung DH, Kagami S, Iwai K, et al. Survival of mature T cells depends on signaling through HOIP. Sci Rep. 2016;6:36135. doi: 10.1038/srep36135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Redecke V, Chaturvedi V, Kuriakose J, Häcker H. SHARPIN controls the development of regulatory T cells. Immunology. 2016;148:216–26. doi: 10.1111/imm.12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park Y, Jin H, Lopez J, Lee J, Liao L, Elly C, et al. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat Immunol. 2016;17:286–96. doi: 10.1038/ni.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Teh CE, Lalaoui N, Jain R, Policheni AN, Heinlein M, Alvarez-Diaz S, et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat Commun. 2016;7:13353. doi: 10.1038/ncomms13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sasaki K, Himeno A, Nakagawa T, Sasaki Y, Kiyonari H, Iwai K. Modulation of autoimmune pathogenesis by T cell-triggered inflammatory cell death. Nat Commun. 2019;10:3878. doi: 10.1038/s41467-019-11858-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wan M, Wang X, Huang C, Xu D, Wang Z, Zhou Y, et al. A bacterial effector deubiquitinase specifically hydrolyses linear ubiquitin chains to inhibit host inflammatory signalling. Nat Microbiol. 2019;4:1282–93. doi: 10.1038/s41564-019-0454-1. [DOI] [PubMed] [Google Scholar]
- 118.Noad J, Malsburg A, von der, Pathe C, Michel MA, Komander D, Randow F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat Microbiol. 2017;2:17063. doi: 10.1038/nmicrobiol.2017.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ashida H, Kim M, Schmidt-Supprian M, Ma A, Ogawa M, Sasakawa C. A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat Cell Biol. 2009;12:66–73. doi: 10.1038/ncb2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jong MF, de, Liu Z, Chen D, Alto NM. Shigella flexneri suppresses NF-κB activation by inhibiting linear ubiquitin chain ligation. Nat Microbiol. 2016;1:16084. doi: 10.1038/nmicrobiol.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Su Y, Shi P, Zhang L, Lu D, Zhao C, Li R, et al. The superimposed deubiquitination effect of OTULIN and porcine reproductive and respiratory syndrome virus (PRRSV) Nsp11 promotes multiplication of PRRSV. J Virol. 2018;92:e00175–18. doi: 10.1128/JVI.00175-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wang L, Wang Y, Zhao J, Ren J, Hall KH, Moorman JP, et al. The linear ubiquitin assembly complex modulates latent membrane protein 1 activation of NF-κB and interferon regulatory factor 7. J Virol. 2016;91:e01138–16. doi: 10.1128/JVI.01138-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Khan M, Syed GH, Kim S-J, Siddiqui A. Hepatitis B virus-induced parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 2016;12:e1005693. doi: 10.1371/journal.ppat.1005693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Greenfeld H, Takasaki K, Walsh MJ, Ersing I, Bernhardt K, Ma Y, et al. TRAF1 coordinates polyubiquitin signaling to enhance Epstein-Barr virus LMP1-mediated growth and survival pathway activation. PLoS Pathog. 2015;11:e1004890. doi: 10.1371/journal.ppat.1004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shibata Y, Tokunaga F, Goto E, Komatsu G, Gohda J, Saeki Y, et al. HTLV-1 tax induces formation of the active macromolecular IKK complex by generating Lys63- and Met1-linked hybrid polyubiquitin chains. PLoS Pathog. 2017;13:e1006162. doi: 10.1371/journal.ppat.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oda H, Beck DB, Kuehn HS, Moura NS, Hoffmann P, Ibarra M, et al. Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front Immunol. 2019;10:479. doi: 10.3389/fimmu.2019.00479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. 2016;113:10127–32. doi: 10.1073/pnas.1612594113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nabavi M, Shahrooei M, Rokni-Zadeh H, Vrancken J, Changi-Ashtiani M, Darabi K, et al. Auto-inflammation in a patient with a novel homozygous OTULIN mutation. J Clin Immunol. 2019;39:138–41. doi: 10.1007/s10875-019-00599-3. [DOI] [PubMed] [Google Scholar]
- 129.Nilsson J, Schoser B, Laforet P, Kalev O, Lindberg C, Romero NB, et al. Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann Neurol. 2013;74:914–9. doi: 10.1002/ana.23963. [DOI] [PubMed] [Google Scholar]
- 130.Wang K, Kim C, Bradfield J, Guo Y, Toskala E, Otieno FG, et al. Whole-genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med. 2013;5:67. doi: 10.1186/gm471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18:309–24. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 132.Yang Y, Schmitz R, Mitala J, Whiting A, Xiao W, Ceribelli M, et al. Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer Disco. 2014;4:480–93. doi: 10.1158/2159-8290.CD-13-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jo T, Nishikori M, Kogure Y, Arima H, Sasaki K, Sasaki Y, et al. LUBAC accelerates B-cell lymphomagenesis by conferring resistance to genotoxic stress on B cells. Blood. 2020;136:684–97. doi: 10.1182/blood.2019002654. [DOI] [PubMed] [Google Scholar]
- 134.Niu J, Shi Y, Iwai K, Wu Z. LUBAC regulates NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. Embo J. 2011;30:3741–53. doi: 10.1038/emboj.2011.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Song K, Cai X, Dong Y, Wu H, Wei Y, Shankavaram U, et al. Epsins 1 and 2 promote NEMO linear ubiquitination via LUBAC to drive breast cancer development. J Clin Invest. 2020. 10.1172/JCI129374. [DOI] [PMC free article] [PubMed]
- 136.Ruiz EJ, Diefenbacher ME, Nelson JK, Sancho R, Pucci F, Chakraborty A, et al. LUBAC determines chemotherapy resistance in squamous cell lung cancerLUBAC determines chemoresistance in squamous lung cancer. J Exp Med. 2019;216:450–65. doi: 10.1084/jem.20180742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.MacKay C, Carroll E, Ibrahim AFM, Garg A, Inman GJ, Hay RT, et al. E3 ubiquitin ligase HOIP attenuates apoptotic cell death induced by cisplatin. Cancer Res. 2014;74:2246–57. doi: 10.1158/0008-5472.CAN-13-2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun S-C. CYLD: a tumor suppressor deubiquitinase regulating NF-κB activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. doi: 10.1038/cdd.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mathis B, Lai Y, Qu C, Janicki J, Cui T. CYLD-mediated signaling and diseases. Curr Drug Targets. 2015;16:284–94. doi: 10.2174/1389450115666141024152421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Damgaard RB, Jolin HE, Allison MED, Davies SE, Titheradge HL, McKenzie ANJ, et al. OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ. 2020;27:1457–74. doi: 10.1038/s41418-020-0532-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Verboom L, Martens A, Priem D, Hoste E, Sze M, Vikkula H, et al. OTULIN prevents liver inflammation and hepatocellular carcinoma by inhibiting FADD- and RIPK1 kinase-mediated hepatocyte apoptosis. Cell Rep. 2020;30:2237–.e6. doi: 10.1016/j.celrep.2020.01.028. [DOI] [PubMed] [Google Scholar]
- 142.Wang W, Li M, Ponnusamy S, Chi Y, Xue J, Fahmy B, et al. ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nat Commun. 2020;11:3965. doi: 10.1038/s41467-020-17770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 144.Asanomi Y, Shigemizu D, Miyashita A, Mitsumori R, Mori T, Hara N, et al. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol Med. 2019;25:20. doi: 10.1186/s10020-019-0090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–6. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 146.Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun. 2016;7:12547. doi: 10.1038/ncomms12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nakayama Y, Sakamoto S, Tsuji K, Ayaki T, Tokunaga F, Ito H. Identification of linear polyubiquitin chain immunoreactivity in tau pathology of Alzheimer’s disease. Neurosci Lett. 2019;703:53–7. doi: 10.1016/j.neulet.2019.03.017. [DOI] [PubMed] [Google Scholar]
- 148.Nakayama Y, Tsuji K, Ayaki T, Mori M, Tokunaga F, Ito H. Linear polyubiquitin chain modification of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2019;79:256–65. doi: 10.1093/jnen/nlz135. [DOI] [PubMed] [Google Scholar]
- 149.Well EM, Bader V, Patra M, Sánchez-Vicente A, Meschede J, Furthmann N, et al. A protein quality control pathway regulated by linear ubiquitination. Embo J. 2019;38:e100730. doi: 10.15252/embj.2018100730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sullivan MA, Nitschke F, Chown EE, DiGiovanni LF, Chown M, Perri AM, et al. Deficiency of the E3 Ubiquitin Ligase RBCK1 Causes Diffuse Brain Polyglucosan Accumulation and Neurodegeneration. Biorxiv. 2018;277392.
- 151.Zhao S, Ulrich HD. Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc Natl Acad Sci. 2010;107:7704–9. doi: 10.1073/pnas.0908764107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Bio. 2018;19:349–64. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 153.Perrin AJ, Jiang X, Birmingham CL, So NSY, Brumell JH. Recognition of bacteria in the cytosol of mammalian cells by the ubiquitin system. Curr Biol. 2004;14:806–11. doi: 10.1016/j.cub.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 154.Chu Y., Kang Y., Yan C., Yang C., Zhang T., Huo H. et al. LUBAC and OTULIN regulate autophagy initiation and maturation by mediating the linear ubiquitination and the stabilization of ATG13. Autophagy 2020; 1–16. 10.1080/15548627.2020.1781393. [DOI] [PMC free article] [PubMed]
- 155.Tang Y, Kwon H, Neel BA, Kasher-Meron M, Pessin JB, Yamada E, et al. The fructose-2,6-bisphosphatase TIGAR suppresses NF-κB signaling by directly inhibiting the linear ubiquitin assembly complex LUBAC. J Biol Chem. 2018;293:7578–91. doi: 10.1074/jbc.RA118.002727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207:245–55. doi: 10.1677/JOE-10-0272. [DOI] [PubMed] [Google Scholar]
- 157.Matsunaga Y, Nakatsu Y, Fukushima T, Okubo H, Iwashita M, Sakoda H, et al. LUBAC formation is impaired in the livers of mice with MCD-dependent nonalcoholic steatohepatitis. Mediat Inflamm. 2015;2015:1–10. doi: 10.1155/2015/125380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fiskin E, Bionda T, Dikic I, Behrends C. Global analysis of host and bacterial ubiquitinome in response to Salmonella typhimurium infection. Mol Cell. 2016;62:967–81. doi: 10.1016/j.molcel.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 159.Gong Z, Li H, Kuang Z, Li C, Ali MK, Huang F, et al. Regulation of host cell pyroptosis and cytokines production by Mycobacterium tuberculosis effector PPE60 requires LUBAC mediated NF-κB signaling. Cell Immunol. 2018;335:41–50. doi: 10.1016/j.cellimm.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 160.Chen Y, He L, Peng Y, Shi X, Chen J, Zhong J, et al. The hepatitis C virus protein NS3 suppresses TNF-α–stimulated activation of NF-κB by targeting LUBAC. Sci Signal. 2015;8:ra118–ra118. doi: 10.1126/scisignal.aab2159. [DOI] [PubMed] [Google Scholar]
- 161.Jing H, Fang L, Ding Z, Wang D, Hao W, Gao L, et al. Porcine reproductive and respiratory syndrome virus nsp1α inhibits NF-κB activation by targeting the linear ubiquitin chain assembly complex. J Virol. 2016;91:e01911–16. doi: 10.1128/JVI.01911-16. [DOI] [PMC free article] [PubMed] [Google Scholar]