

Abstract
Pancreatic ductal adenocarcinoma (PDAC) has poor prognosis due to limited therapeutic options. This study examines the roles of genome-wide association study identified PDAC-associated genes as therapeutic targets. We have identified HNF4G gene whose silencing most effectively repressed PDAC cell invasiveness. HNF4G overexpression is induced by the deficiency of transcriptional factor and tumor suppressor SMAD4. Increased HNF4G are correlated with SMAD4 deficiency in PDAC tumor samples and associated with metastasis and poor survival time in xenograft animal model and in patients with PDAC (log-rank P = 0.036; HR = 1.60, 95% CI = 1.03–2.47). We have found that Metformin suppresses HNF4G activity via AMPK-mediated phosphorylation-coupled ubiquitination degradation and inhibits in vitro invasion and in vivo metastasis of PDAC cells with SMAD4 deficiency. Furthermore, Metformin treatment significantly improve clinical outcomes and survival in patients with SMAD4-deficient PDAC (log-rank P = 0.022; HR = 0.31, 95% CI = 0.14–0.68) but not in patients with SMAD4-normal PDAC. Pathway analysis shows that HNF4G may act in PDAC through the cell-cell junction pathway. These results indicate that SMAD4 deficiency-induced overexpression of HNF4G plays a critical oncogenic role in PDAC progression and metastasis but may form a druggable target for Metformin treatment.
Electronic supplementary material
The online version of this article (10.1007/s13238-020-00760-4) contains supplementary material, which is available to authorized users.
Keywords: pancreatic cancer, HNF4G, SMAD4 deficiency, SMAD4-deficient PDAC, Metformin
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the seventh leading cause of cancer deaths worldwide (Bray et al., 2018; Ferlay et al., 2019). Globally, the incidence of pancreatic cancer is nearly the same as mortality and the five-year survival rate is about 8% (Siegel et al., 2018). A formidable challenge remains for PDAC therapy. Although surgery followed by adjuvant chemotherapy is the first-line therapy (Neoptolemos et al., 2018), such treatment has not yet resulted in a desired outcome for PDAC patients. The high lethality of this disease has driven to develop new therapeutic strategies. However, numerous target agents under evaluation have so far failed to significantly improve patient survival although Erlotinib and Olaparib have been shown to be of statistically significant, yet clinically marginal, benefit for the prognosis of PDAC patients (Moore et al., 2007; Golan et al., 2019). The failure of target therapies to pancreatic cancer might be attributable to high molecular heterogeneity of the cancer (Neoptolemos et al., 2018). In recent years, whole-exome or whole-genome sequencing studies have discovered many genomic alternations in PDAC (Bailey et al., 2016) and the results may help to develop future new precision therapies; however, addressing other molecular mechanisms for PDAC development and progression such as genetic variants and epigenetic modifications of genes are also needed for precision care of this malignancy.
Genome-wide association study (GWAS) is a powerful tool to identify genetic variants associated with risk and phenotypes of diseases. In the past 10 years, 8 GWAS on different racial populations have been published and identified 79 genetic variants associated with PDAC (Amundadottir et al., 2009; Low et al., 2010; Petersen et al., 2010; Wu et al., 2011; Li et al., 2012; Wolpin et al., 2014; Childs et al., 2015; Klein et al., 2018). However, the functions and the action mechanisms of these associated variants remain largely unknown. It has been proposed that some GWAS-identified disease risk variants may also contribute to the disease progression or even outcomes through their molecular functions on disease phenotypes including response to therapies. Indeed, we have previously demonstrated that a GWAS-identified PDAC risk variant BACH1 is also associated with poor survival of patients, which may be mediated by resistance of the variant genotype to gemcitabine treatment (Huang et al., 2018). Another example is that we found a genetic variant in the 5′UTR of SLC39A6 associated with shorter survival in esophageal cancer (Wu et al., 2013), which may be attributed to alleviating SLC39A6 repression and promoting cancer cell invasion and metastasis (Cheng et al., 2017). Therefore, it is warranted to explore the potential roles of the genetic susceptibility variants in disease progression and in therapeutic targeting.
In this study, we have investigated the GWAS-identified 36 genes whose variants are associated with risk of PDAC by small interfering RNAs and found that the hepatic nuclear factor 4γ (HNF4G) was an important player promoting PDAC progression and invasiveness. HNF4G was previously identified as a transcription factor belonging to nuclear hormone receptor superfamily. Several studies have suggested that HNF4G might act as an oncogene modulating cell proliferation and invasion in cancer (Okegawa et al., 2013; Shukla et al., 2017; Wang et al., 2018). However, the function and action mechanism of HNF4G in pancreatic cancer remain unknown. Here, we demonstrate that SMAD4 deficiency upregulates HNF4G expression in PDAC, which can be suppressed by Metformin via AMPK-mediated phosphorylation-coupled ubiquitination degradation. Metformin treatment significantly represses progression and metastasis of transplanted PDAC in mice and improves the clinical outcomes in patients with SMAD4-deficient PDAC.
RESULTS
HNF4G is an important player in the progression and invasiveness of PDAC
By joint analysis and fine mapping of previously published 8 GWAS data (See Methods; Table S1), we discovered 36 genes significantly associated with PDAC (P < 1 × 10−4; Fig. 1A and Table S2). We then conducted high-throughput siRNA screening of these 36 genes to examine whether they have effects on PDAC cell phenotypes (Fig. 1B) and found that knocking down expression of 9 genes significantly repressed but 6 genes significantly promoted PDAC cells invasiveness indicated by cell roundness and ratio (Figs. 1C, S1A, S1B and Table S3). In this study, we focused on genes having potential oncogenic roles because they are more likely to be therapeutic targets. We then examined the effects of the 9 genes that have potential oncogenic role by using their individual siRNAs. Transwell assays showed that, among these genes, HNF4G was the most effective in repressing PDAC cell migration and invasion when its expression was silenced (Figs. 1D and S1C). IHC staining of tissue arrays showed that HNF4G expression levels were significantly higher in PDAC tumor than in adjacent normal tissues (P < 0.0001; Fig. 1E). Combined analysis of PDAC data in both The Cancer Genome Atlas (TCGA) and the genotype-tissue expression (GTEx) also showed significantly higher HNF4G RNA levels in tumor than in normal samples (P < 0.001; Fig. 1F). Furthermore, we found that the elevated HNF4G RNA levels were significantly correlated with advanced tumor stage (P = 0.008, Fig. 1G) and poor survival (HR = 1.60, 95% CI = 1.03–2.47; Fig. 1H). Collectively, these results strongly support that HNF4G is an important player in PDAC development and progression.
Figure 1.

HNF4G is an important player in PDAC progression and invasiveness. (A) Scheme of reanalyzing PDAC susceptibility genes using combined GWAS data. (B) High content screening strategy for 36 candidate genes in PDAC cells. Scale bar: 100 μm. (C) Heatmap showing the results of high content screening with a siRNAs library in PDAC cells. (D) The effect of siRNA knockdown of indicated genes on migration ability of PANC-1 cells. Data represent mean ± SEM from 3 experiments. (E) Immunohistochemical (IHC) staining of HNF4G in tissue array consisting of 65 PDAC samples. Left panel, representative IHC images, Scale bar: 700 μm (left images) and 60 μm (right images); right panel, quantification statistic. (F) Scatter dot plots showing HNF4G expression levels in PDAC tumor and normal samples. Data are derived from the Gene Expression Profiling Interactive Analysis (GEPIA). (G) Scatter dot plots showing HNF4G expression levels in early and latter stage of PDAC. Data were derived from the TCGA PDAC dataset. (H) Kaplan-Meier plots of overall survival of patients derived from the TCGA PDAC dataset stratified by HNF4G expression. The best performing threshold is used as a cutoff. HR, hazard ratio; CI, confidence interval. Statistical significance: *, P < 0.05, **, P < 0.01 and ****, P < 0.0001 of Student’s t-test or Wilcoxon rank-sum test. ns, not significant
HNF4G overexpression is associated with SMAD4 deficiency in PDAC
To seek for why HNF4G is overexpressed in PDAC, we looked at genomic alterations including SNP, CNV and methylation status of HNF4G in PDAC tissues and cell lines derived from different datasets but the results were negative (Fig. S2A and S2B). Nevertheless, analysis of HNF4G promoter and enhancer sequences using the GIAGEN software suggested 115 transcription factor binding sites (Table S4). Further analysis of TCGA data showed that PDAC tumors with deletion of SMAD4, a driver gene frequent loss-of-function in PDAC and a gene previously reported to regulate HNF4G expression in enterocyte (Chen et al., 2019), had higher HNF4G RNA levels than those without SMAD4 deletion (P = 0.004; Fig. 2A). Furthermore, PDAC tumors with SMAD4 truncation mutations also had higher HNF4G RNA levels compared with those without such mutations or with other mutation types (Fig. S2C). These results were confirmed in our PDAC samples analyzed by Immunohistochemical (IHC) staining showing that SMAD4 expression levels were inversely correlated with HNF4G levels (Fig. 2B). The inverse correlation between SMAD4 and HNF4G expression levels were also seen in PDAC cell lines where the SMAD4 gene deletion status was known (Fig. 2C). Additionally, we found that knockdown of SMAD4 expression in PDAC cells, where SMAD4 is normal, significantly increased HNF4G expression at both RNA and protein levels, while ectopic expression of SMAD4 in PDAC cells, where SMAD4 is deficient, significantly repressed HNF4G expression (Fig. 2D and 2E).
Figure 2.

HNF4G upregulation is caused by SMAD4 deficiency in PDAC. (A) HNF4G mRNA levels in PDAC as function of SMAD4 copy-number variation. Data were derived from the TCGA database. (B) The relationship between HNF4G and SMAD4 protein levels in PDAC determined by IHC staining. Left panel shows representative IHC images of HNF4G and SMAD4 in serial sections of PDAC tissue array (n = 185). Scale bar in left images = 600 μm. Scale bar in right images = 200 μm. Right panel shows HNF4G levels as function of SMAD4 levels both expressed as IHC scores: low, 0; medium, 1–4; and high, 6–12. (C) The expression levels of HNF4G RNA (left) and protein (right) in 4 PDAC cell lines with SMAD4 deficiency and 3 cell lines without SMAD4 deficiency. Data represent mean ± SEM from 3 independent determinations and each had triplicates. (D and E) The effects of SMAD4 knockdown (D) or overexpression (E) on HNF4G RNA (upper panel) and protein levels (lower panel) in PDAC cells. Data are mean ± SEM from 3 independent determinations and each had triplicates. (F and G) Relative expression levels of reporter gene bearing the HNF4G promoter region in T3M4 cells with or without SMAD4 overexpression (F) and in MIAPaCa-2 cells with or without SMAD4 knockdown (G). (H) Relative expression levels of reporter gene bearing the mutated HNF4G promoter region in PDAC cells. Each promoter harbors a mutated SBE. Mutation in SBE 1 had the most significant impact on reporter gene expression compared with vector control and the mutation in other SBEs. Results are mean ± SEM from 3 experiments and each had 6 replicates. (I and J) Chromatin immunoprecipitation assays showing binding of SMAD4 to HNF4G promoter region SBE 1 in PDAC cells (I) and knockdown of SMAD4 expression in these cells substantially decreased the binding (J). Fold enrichment represents DNA levels associated with HNF4G or IgG (as control) relative to an input control from 3 independent experiments. Data are mean ± SEM of 3 experiments. Statistical significance: *, P < 0.05, **, P < 0.01, ***, P < 0.001 and ****, P < 0.0001 of Student’s t-test, χ2 test or Wilcoxon rank-sum test. ns, not significant
Next, we examined how SMAD4 regulates HNF4G expression. Bioinformatics indicated 5 SMAD binding elements (SBEs; GTCTG) and one stretch of transforming growth factor-β (TGF-β) inhibitory elements (TIE; GCCAAGC) within the HNF4G promoter region (Fig. S2D and S2G). These findings suggested that the canonical TGF-β signaling inhibits transcription of HNF4G gene through SMAD4. We thus conducted reporter gene assays with a plasmid construct carrying the −1,200 base pairs to +1,400 base pairs of HNF4G promoter sequences centered by transcriptional start site (TSS) predicted by Promoter 2.0 Prediction Server. The reporter expression levels were significantly decreased when SMAD4 was overexpressed in T3M4 and BxPC-3 cells (Figs. 2F and S2E); however, the levels were significantly increased in MIAPaCa-2 and PANC-1 cells when SMAD4 expression was knocked down (Figs. 2G and S2F). In addition, the reporter assays with mutated HNF4G promoter at each SBE (Fig. S2G) showed that only mutation at the 1st SBE dramatically increased reporter expression level compared with wild-type HNF4G promoter (Fig. 2H). ChIP-qPCR analysis showed a significant enrichment of HNF4G in cells with normal SMAD4 but not in cells with deficient SMAD4 (Fig. 2I). Knockdown of SMAD4 expression in cells with normal SMAD4 also significantly reduced the interaction between SMAD4 and HNF4G promoter (Figs. 2J and S2H). Together, these results demonstrate an inhibitory role of SMAD4 in HNF4G transcription.
HNF4G activates the cell-cell junction pathway to promote PDAC metastasis
We then explore the effect of HNF4G on the capabilities of in vitro migration and invasion and in vivo metastasis in xenograft tumor models by stable overexpression or knockout of this gene in PDAC cells (Fig. S3A). We found that HNF4G knockout significantly suppressed (Fig. S3B) but HNF4G overexpression substantially enhanced the activities of cell migration and invasion in vitro (Fig. 3A). Similar results were obtained in mouse pancreatic orthotopic implantation of T3M4 cells, showing that HNF4G overexpression significantly promote metastasis of the xenografts to other organs such as liver, spleen and lung as measured by luminescence imaging (Fig. 3B) and confirmed by histopathology (Fig. 3C). These results suggest that HNF4G plays a pivotal role in PDAC metastasis.
Figure 3.

HNF4G overexpression promotes PDAC cell invasiveness and activates the cell-cell junction pathway. (A) HNF4G overexpression promoted migration and invasion of PDAC cells in vitro. Left panel shows representative images of transwell assays and right panel shows quantification statistic. Data are mean ± SEM from 3 independent experiments and each had duplicate. (B) HNF4G overexpression promoted migration and invasion of PDAC cells transplanted in the pancreas of mice (n = 3). Left panel shows representative bioluminescence images taken at 7 and 40 days of implantation; Right panel shows quantitative fluorescent intensity of the transplanted PDAC. (C) Representative H&E staining pictures of the pancreas, liver and lung from mice implanted orthotopically with PDAC cells with or without HNF4G overexpression. Scale bars: 100 μm. (D) The work flow schematic for analyzing the candidate genes targeted by HNF4G. (E) Functional enrichment of the 293 HNF4G-targeted genes by Metascape. (F) The expression levels of some downstream genes of HNF4G in PDAC cells with or without HNF4G overexpression. Results are mean ± SEM from 3 independent determinations and each had triplicate. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001 and ****, P < 0.0001 of Student’s t-test or Wilcoxon rank-sum test. ns, not significant
Based on analyzing the chromatin immunoprecipitation sequencing (ChIP-seq) data in the Gene Expression Omnibus (GEO) database and the RNA-sequencing data in the TCGA PDAC database, we found 6,783 genes that might be regulated by HNF4G and among them, 293 genes were significantly co-expressed with HNF4G (Fig. 3D and Table S5). Of the 293 genes, 32 were enriched in the cell-cell junction pathway (Fig. 3E and Table S6), an important expression program relative to cell junction integrity and cancer metastasis (Runkle and Mu, 2013; Martin, 2014; De Pascalis and Etienne-Manneville, 2017). We found that in PDAC cells, the expressions of several genes in the cell-cell junction pathway were significantly altered by ectopic overexpression or knockdown of HNF4G (Figs. 3F and S3C), suggesting that the role of HNF4G overexpression in promoting PDAC metastasis may be through regulating the cell-cell junction pathway.
Metformin activates AMPK that induces HNF4G phosphorylation-ubiquitination degradation
Since HNF4G overexpression promotes PDAC metastasis, inhibiting its activity might be an option for PDAC therapy. By analyzing HNF4G protein sequences using the Scansite4, we found 29 motifs that could be phosphorylated by several kinases including PK3R1 and PRKAA1 (Table S7) for that the agonists are currently available. We treated PDAC cells with Isoprenaline (PIK3R1 agonist) or Metformin (PRKAA1 agonist) and found that Metformin but not Isoprenaline substantially enhanced HNF4G phosphorylation and consequent ubiquitination degradation (Figs. 4A–D and S4A–D). This effect was abolished when adenosine 5′-monophosphate (AMP)-activated protein kinase α (AMPKα) expression was knocked down in cells (Fig. 4E), indicating that Metformin-induced HNF4G phosphorylation and degradation was likely mediated by AMPKα. By analyzing HNF4G sequence, we identified 3 potential AMPK-phosphorylation sites including threonine 143 (T143), serine 161 (S161) and serine 382 (S382). Indeed, in vitro assays showed that the HNF4G-alanine 382 mutant (A382) had significantly reduced phosphorylation by AMPK compared with the HNF4G-serine 382 wild-type (S382), while the HNF4G-arginine 143 (R143) or HNF4G-alanine 161 (A161) mutant had little effect (Figs. 4F, 4G, S4E and S4F). Consistent with these results, motif analysis also showed a great similarity between the canonical AMPK substrate motif and the HNF4G S382 site that is highly conserved in different species (Fig. S4G). In addition, the expressions of HNF4G target genes were abrogated in HNF4G-overexpressing cells treated with Metformin (Fig. 4H). Collectively, these results indicate that the effect of Metformin on promoting phosphorylation/ubiquitination-coupled degradation of HNF4G is mediated by AMPK activation.
Figure 4.

Metformin activates AMPK that induces HNF4G phosphorylation-ubiquitination coupled degradation. (A) Effect of Metformin (10 μmol/L) on HNF4G and AMPKα phosphorylation in T3M4 cells. (B) Metformin (10 μmol/L) promoted AMPKα phosphorylation and HNF4G degradation in T3M4 cells. (C) Metformin promoted HNF4G degradation but not inhibited its synthesis in T3M4 cells. Left panel, Metformin treatment substantially decreased the HNF4G levels with time in cells exposed to protein synthesis inhibitor cycloheximide (CHX; 20 μg/mL) compared with cells exposed to vehicle; right panel, Metformin treatment no longer substantially decreased HNF4G level in cells exposed to proteasome inhibitor MG132 (5 μmol/L). (D) Metformin promotes HNF4G ubiquitination. T3M4 cells were treated with or without Metformin (10 μmol/L). Cell lysates were immunoprecipitated (IP) with either control IgG or antibody against HNF4G and analyzed by immunoblotting with a ubiquitin (Ub)-specific antibody. Bottom panels, input from cell lysates. (E) Immunoblot analysis of HNF4G phosphorylation status in T3M4 cells with or without AMPKα knockdown treated with Metformin (10 μmol/L). (F and G) Immunoblot analysis of phosphorylated HNF4G and AMPKα in T3M4 cells, cells with HNF4G knockout or cells with ectopic expression of S382A-mutated HNF4G exposed to Metformin (10 μmol/L) for different times. (H) Metformin treatment significantly decreased the expression levels of some oncogenes in PDAC cells compared with vehicle controls. Results are mean ± SEM from 3 independent determinations and each had triplicate. Statistical significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 of Student’s t-test
Metformin suppress HNF4G-induced PDAC metastasis depending on SMAD4 status
We next examined whether Metformin suppresses cell invasiveness and metastasis induced by HNF4G overexpression. In vitro assays showed that Metformin significantly inhibited PDAC cell migration and invasion in a concentration-dependent manner; however, this effect was only seen in T3M4 and BxPC-3 cell lines that are known SMAD4 loss-of-function mutants but not in PANC-1 cell line that is SMAD4 wild-type (Figs. 5A and S5A). The dependency of Metformin action upon the SMAD4 status was further verified in SMAD4-wild type cells with SMAD4 knockdown by its shRNA (Figs. 5B and S5B). In vivo experiments showed that treatment with Metformin of mice carrying PDAC xenograft derived from HNF4G-overexpressing T3M4 cells in the pancreas significantly reduced the tumor burden and prolonged survival time compared with treatment with vehicle (Fig. 5C and 5D). Furthermore, histopathological analysis demonstrated that Metformin treatment significantly decreased metastatic tumor number in the liver (Fig. 5E). However, we did not observe significant differences in the body weight of mice treated with Metformin or vehicle (Fig. S5C), indicating that Metformin is not toxic at this effective dose. We then performed immunohistochemical analysis in serial sections of xenograft tumors and liver metastases and the results showed that treatment with Metformin substantially increased the p-AMPK levels but reduced the HNF4G levels as compared with treatment with vehicle (Fig. 5F). Collectively, both in vitro and in vivo results indicated that Metformin is an effective therapeutic agent targeting HNF4G for treatment of SMAD4 deficient PDAC.
Figure 5.

Metformin suppress HNF4G-induced PDAC metastasis depending on SMAD4 status. (A) Metformin treatment significantly repressed in vitro migration and invasion of SMAD4-deficient T3M4 cells but not SMAD4-efficient PANC-1. Left panels show representative images of transwell assays and right panels represent quantitative statistic. Data are mean ± SEM from 3 independent experiments and each had triplicate. Shown are the results in cells treated with or without Metformin (10 μmol/L); see also Fig. S5 for the entire and detailed dose-dependent results. (B) Knockdown of SMAD4 expression significantly promoted Metformin to suppress the migration and invasion of PDAC cells. Upper panel are representative transwell images and lower panel are quantitative data (mean ± SEM from 3 independent experiments and each had triplicate). (C) Metformin treatment significantly repressed the spread of HNF4G-overexpressing T3M4 cells implanted in mouse pancreas (n = 4). Left panel shows bioluminescence images of mice and the right panel shows quantitative fluorescent intensities. (D) Metformin treatment significantly prolonged survival time of mice implanted with PDAC in the pancreas as compared with vehicle control. (E) Metformin treatment significantly reduced PDAC metastases in the liver compared with vehicle control. Left panel shows representative tumor nodes staining by H&E and right panel shows quantitative statistic. Scale bars: 100 μm (40×) and 10 μm (400×). (F) Representative IHC pictures showing that Metformin treatment substantially reduced HNF4G but increased p-AMPKα expression levels in serial sections of both pancreas and liver from mice with PDAC implantation as compared with vehicle control. Scale bars: 100 μm (100×) and 25 μm (400×). Statistical significance: **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 of Student’s t-test or Wilcoxon rank-sum test. ns, not significant
Metformin treatment improves clinical outcomes in patients with SMAD4-deficient PDAC
To examine the curative effect of Metformin on PDAC, we recruited 139 individuals who concurrently suffered from type 2 diabetes mellitus and therefore were administrated with Metformin or other drugs prior to PDAC diagnosis (Table S8). IHC analysis of SMAD4 and HNF4G expression in this PDAC sample set (Fig. 6A) showed 74.1% (103/139) of SMAD4 deficiency. The significant inverse correlation between SMAD4 levels and HNF4G levels were confirmed in non-Metformin treatment group (P = 0.0012) but not in Metformin group (Fig. 6B) because HNF4G was substantially degraded in Metformin group. We analyzed the effect of Metformin treatment on clinical outcomes in patients who had complete tumor staging data (n = 113) or survival data (n = 118). We found that among patients with SMAD4-deficient PDAC, the proportion of low-staged tumors (stages IA, IB and IIA), classified upon diagnosis, was significantly higher in those with Metformin treatment than that in those without Metformin treatment (60.9% versus 31.7%, P = 0.015); however, this significant difference was not seen in patients with SMAD4-normal PDAC (Fig. 6C). More importantly, we found that patients with SMAD4-defficient PDAC treated with Metformin had significantly longer survival time than those without Metformin treatment (log-rank P = 0.022), with HR of death for Metformin treatment being 0.31 (95% CI = 0.14–0.68). In contrast, no significant effect of Metformin treatment on survival time was observed in patients with SMAD4-normal PDAC (Fig. 6D). These results suggest that Metformin treatment may inhibit PDAC progression and thus improve survival in patients with SMAD4-deficient PDAC.
Figure 6.

Metformin treatment improves clinical outcomes of patients with SMAD4-deficient PDAC. (A) Representative images of IHC staining of SMAD4 and HNF4G proteins in PDAC. Scale bar = 100 μm. (B) The correlation between HNF4G and SMAD4 protein levels in PDAC determined by IHC score. SMAD4−, IHC score = 0; SMAD4+, IHC score > 0. HNF4G Low level, scores 0–4; HNF4G high level, scores 6–12. (C) The distribution of patients by PDAC tumor stage and SMAD4 status as function of Metformin treatment. (D) Kaplan-Meier estimate of survival time in 118 patients with PDAC by SMAD4 status and Metformin treatment. Hazard ratio (HR) and 95% confidence interval (CI) were calculated with age, sex, tumor stage as covariates. SMAD4−, IHC score = 0; SMAD4+, IHC score > 0. Statistical significance: *, P < 0.05 and**, P < 0.01 of χ2 test. ns, not significant
DISCUSSION
In the present study, we have demonstrated that GWAS identified PDAC-associated HNF4G functions as an oncogene that plays a pivotal role in PDAC invasiveness and metastasis. Overexpression of HNF4G activates the cell-cell junction pathway, which is likely the underlying molecular mechanism for promoting PDAC invasiveness and metastasis. We have verified for the first time that HNF4G overexpression can be caused by the deficiency of SMAD4, a tumor suppressor gene that is frequently deleted or mutated in PDAC. Furthermore, we have found that panacea Metformin can suppress HNF4G activity via activating AMPK that leads to phosphorylation coupled ubiquitination degradation of HNF4G protein. Treatment of mouse PDAC xenograft and clinical PDAC patients with Metformin have revealed a curative effect in SMAD4-deficient PDAC. These findings hold the potential for clinical trials to test HNF4G pathway inhibitors including Metformin in SMAD4-deficient/HNF4G-overexpressing PDAC (Fig. 7).
Figure 7.
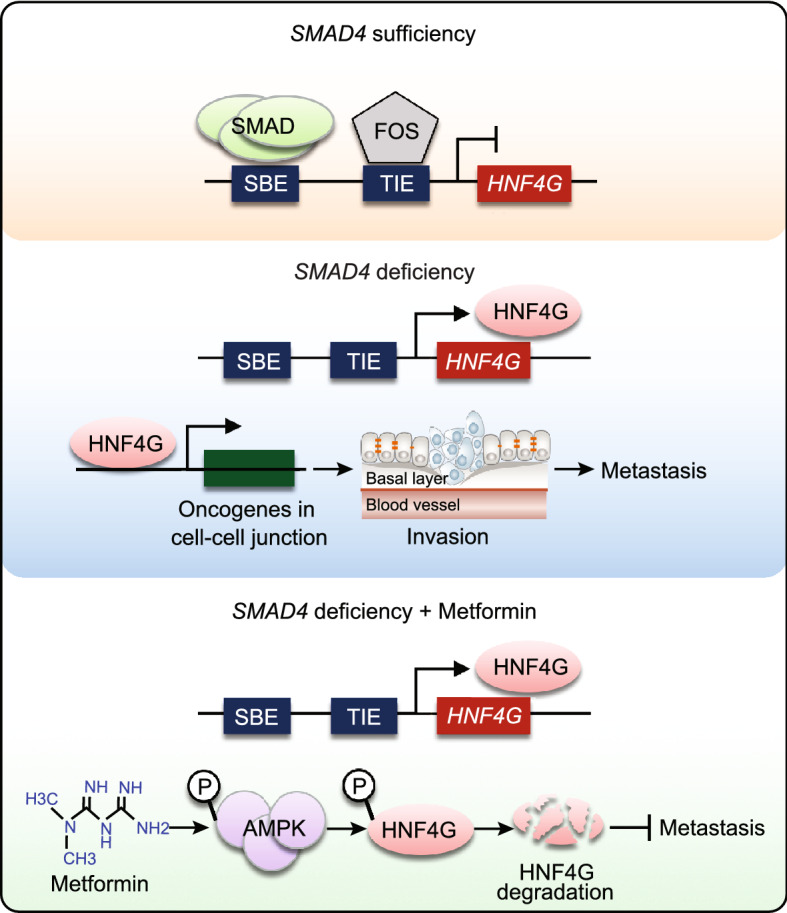
A proposed working model for aberrant SMAD4-HNF4G in PDAC invasiveness and the effect of Metformin. In PDAC cells where SMAD4 is sufficient, the expression of the downstream oncogene HNF4G that promotes PDAC invasiveness is physiologically inhibited by the SMAD complex. In PDAC cells where SMAD4 is deficient, the expression of HNF4G is over-activated, which evokes cancer cell invasion and metastasis. Metformin may act as a target drug repressing PDAC invasion and metastasis by activating AMPK that induces phosphorylation-ubiquitination coupled HNF4G degradation
Previous studies have suggested that HNF4G may act in promoting cell proliferation and invasion in several types of human cancer, such as bladder cancer (Okegawa et al., 2013), lung cancer (Wang et al., 2018) and prostate cancer (Shukla et al., 2017). Our results in the present study are consistent with these findings and extend the oncogenic role of HNF4G to pancreatic cancer. Furthermore, we have achieved several important novel findings about HNF4G. First, by combined analysis of the publically available ChIP-sequencing and RNA-sequencing data, we have identified cell-cell junction pathway may be the downstream of HNF4G that may promote PDAC cell invasion and metastasis although the ChIP-sequencing data were from hepatocellular carcinoma cells and therefore need confirmation in PDAC. Second, we have demonstrated for the first time that Metformin cause HNF4G degradation via activating AMPK and consequently suppress PDAC progression and metastasis. These findings provide the rationale for clinical trials testing HNF4G pathway inhibitors such as Metformin and others in patients with HNF4G-upregulated PDAC. In addition, because HNF4G is a transcription factor with broad regulatory functions (Lau et al., 2018), our findings might also be implicated in other types of gastrointestinal cancer with aberrant HNF4G.
Another interesting result is the identification of SMAD4 as a HNF4G transcriptional inhibitor. SMAD4 also known as DPC4 (deleted in pancreatic cancer) is a well-known tumor suppressor and inactivated in more than 50% of PDAC (Iacobuzio-Donahue et al., 2009). It has been shown that SMAD4 deficiency is associated with poor prognosis of PDAC, likely due to loss of SMAD4 control of some oncogenes (Bardeesy et al., 2006). We have demonstrated for the first time that SMAD4 can bind to the SBE site located in the HNF4G promoter region, repressing HNF4G transcription. This result is consistent with previous study showing that SMAD4 binds SBE site and recruits FOS-complex to bind TIE site, thus repressing gene expression (Kerr et al., 1990). SMAD4 deficiency due to genomic changes such as deletion or truncated mutations may abolish its ability to repress HNF4G expression. Moreover, we have detected an inverse correlation between SMAD4 and HNF4G expression in both PDAC cell lines and clinical specimens, further supporting a negative regulation of HNF4G expression by SMAD4. A previous study reported that SMAD4 upregulates HNF4 (HNF4A/HNF4G) expression and functions as a feed-forward regulatory module to promote and stabilize the enterocyte identity (Chen et al., 2019), which is inconsistent with our result. The reason for this discrepancy is not immediately evident; however, it might be due to the differences in organ- or tumor-specific transcriptional networks (Heinz et al., 2015; Assi et al., 2019).
The panacea Metformin has been used for treating type 2 diabetes mellitus and many diseases including cancer (Libby et al., 2009); however, several problems exist in its use as anticancer agent. First, the underlying mechanism for the anticancer action remains unclear, although several hypotheses have been proposed. It has been shown that Metformin may act, in an AMPK-independent manner, through decreasing blood glucose and insulin levels, reactive oxygen species (ROS) production and DNA damage (Foretz et al., 2010; Ros and Schulze, 2013; Pernicova and Korbonits, 2014). On the other hand, Metformin may also act, in an AMPK-dependent manner, through directly inhibiting ATP synthesis which in turn activates AMPK (Miller et al., 2013). Activated AMPK may activate or inactivate some important proteins by phosphorylation (Lee et al., 2012). In the present study, we have demonstrated that Metformin can inhibit HNF4G activity in an AMPK-dependent manner: Metformin activates AMPK and the latter degrades HNF4G through phosphorylation-couples ubiquitination. Importantly, we have found that upon inhibition of HNF4G, Metformin treatment significantly represses PDAC progression and metastasis in vitro and in vivo in mice. We have also found that long-term administration of Metformin significantly reduced tumor stage and improved survival in patients with SMAD4-deficient PDAC. These results with clear acting mechanism provide additional evidence for clinical trials testing Metformin in suppressing invasion and metastasis of SMAD4-deficient PDAC. Second, some preclinical trials and in vitro studies have used Metformin at doses many times higher than those used in clinic (Dowling et al., 2012; Wu et al., 2016), which may limit its real application. However, in the present study, we have found that 10 μmol/L of Metformin, equivalent to the plasma level of taking 1.0 g of this panacea, is able to significantly degrade HNF4G and repress PDAC cell migration and invasion, suggesting that standard clinical Metformin dose may be effective to suppress metastasis of SMAD4-deficient PDAC. Finally, our findings suggest that the effectiveness of Metformin in suppressing PDAC progression and invasiveness depends on SMAD4 status. Over the past decade, a number of preclinical studies have reported that Metformin used solely or in combination with other remedies could inhibit PDAC progression in vitro or in vivo in some animal models (Wu et al., 2016; Chen et al., 2017). However, previous clinical trials did not show any promising results for it as an anticancer agent in PDAC (Kordes et al., 2015; Chaiteerakij et al., 2016; Reni et al., 2016). We speculate that the inconsistent results between in vitro assays and in vivo in clinical trials might result from the heterogeneity of SMAD4 status in different PDAC tumors because we have demonstrated that only SMAD4-deficient subtype of PDAC is sensitive to Metformin.
It is worth noting that we found a higher proportion of SMAD4-deficient PDAC (74.1%) in our patient set compared with that (30%–50%) reported in literature (Iacobuzio-Donahue et al., 2009). The most likely reason for this disparity might be attributable to the antibody used in the present study. This antibody targets the C-terminal of SMAD4 and therefore can detect its deficiency caused by all silencing genetic and epigenetic events rather than the deleterious mutations. Another potential reason is patient selection bias, because PDAC patients recruited in the present study all suffered from diabetes. On the other hand, one previous report has shown that the percentage of overall inactivation or loss of SMAD4 in pancreatic cancer is 60%–90% (Moustakas and Heldin, 2005) and our result (74.1%) is within the range. However, despite some disparity, the high proportion of SMAD4-deficient PDAC in our patient set may not impact our conclusion that patients with SMAD4-deficient PDAC benefit from Metformin treatment.
In summary, by using genome-wide association analysis and functional characterization, we have identified HNF4G as an oncogene in PDAC, whose overexpression may be caused by SMAD4 deficiency. Overexpression of HNF4G evokes PDAC progression and invasiveness, but can be suppressed by the panacea Metformin. These findings shed light on an important mechanism underlying the acting effect of SMAD4 deficiency in PDAC development and progression and suggest that the SMAD4-HNF4G pathway may be the target for precision treatment of PDAC.
MATERIALS AND METHODS
Patients, biospecimens and tissue microarrays
We recruited patients from Peking Union Medical College Hospital (PUMCH, n = 103) and Chinese Academy of Medical Sciences Cancer Hospital (CAMSCH, n = 36) (Table S8). All patients were diagnosed as type 2 diabetes mellitus and PDAC. Patient survival time was measured from the date of diagnosis to the date of last follow-up or death. Whether and when a subject had died was obtained from inpatient and outpatient records, subject’s family, or through follow-up telephone calls. Informed consents were solicited from all individuals and this study was approved by PUMCH and CAMSCH. Tissue microarrays were purchased from Shanghai Outdo Biotech (HPan-Ade170Sur-01-M-138) or made using surgically removed PDAC samples at PUMCH.
Cell lines and cell culture
PDAC cell lines BxPC-3, CFPAC-1, PANC-1, Capan-2 and MIAPaCa-2 were obtained from the China Center for Type Culture Collection while AsPC-1 and T3M4 were kind gifts from Dr. G. Yang (PUMCH). BxPC-3, AsPC-1 and Capan-2 cells were maintained in RPMI 1640, PANC-1, T3M4 and MIAPaCa-2 Cells were maintained in DMEM and CFPAC-1 cells were maintained in IMDM. All the medium were supplemented with 10% fetal bovine serum (FBS, Hyclone). These cell lines were authenticated by RNA-seq analysis of SNPs compared to exome data from Cancer Cell Line Encyclopedia. Cell lines were mycoplasma free as determined by PCR.
Genome-wide association analysis and candidate gene selection
We combined previously published 8 PDAC GWAS (Amundadottir et al., 2009; Low et al., 2010; Petersen et al., 2010; Wu et al., 2011; Li et al., 2012; Wolpin et al., 2014; Childs et al., 2015; Klein et al., 2018) and identified 79 PDAC-associated SNPs. We then performed fine-mapping of a 2-mega base (Mb) region centered on each of these SNPs and reanalyzed our previous PDAC GWAS data (Wu et al., 2011) and found 85 SNPs in 47 genes significantly associated with PDAC (P < 1.0 × 10−4; Table S2). After excluding genes that encode anti-sense RNAs, microRNAs and genes that have been reported in our previous studies (Zheng et al., 2016; Huang et al., 2018), we selected 36 genes for further RNA interfering screen (Table S3).
High content screening assays
The small interfering RNA (siRNA) library provided by GenePharma (Shanghai GenePharma Co., Ltd, Shanghai, China) comprised 3 individual non-overlapping siRNA designs for each gene and the repression efficiency was guaranteed by the provider. The high content screening assays were performed as previously described (Laufer et al., 2013; Laufer et al., 2014). Baseline and main effects were computed from non-targeting controls and single-gene knockdowns for each siRNA design. siRNA targeting KRAS, a known driver gene in PDAC, was used as a positive control for the screening assays.
Immunohistochemical analysis
The sections were incubated with antibody against HNF4G (#25801-1-AP, Proteintech), SMAD4 (#46535, Cell Signaling), or p-AMPKα (#2535, Cell Signaling) at 4 °C overnight and then detected with the ABC Kit (Pierce). The labeling score of intensity was estimated as negative (0), weak (1), moderate (2) and strong (3). The extent of staining, defined as the percentage of positive stained cells, was scored as 1 (≤10%), 2 (11%–50%), 3 (51%–80%) and 4 (>80%). The total IHC score was obtained by multiplying the score of intensity and that of extent, ranking from 0 to 12.
RNA extraction and quantitative real-time PCR analysis
RNA extraction and reverse transcription were performed using Trizol reagent (Invitrogen) and PrimeScriptTM RT reagent kit (TaKaRa), respectively. Quantitative real-time PCR (qRT-PCR) was performed in triplicate using SYBR green (Life Technologies). The primer sequences used for qRT-PCR are shown in Table S9.
Analysis of invasion and migration
Invasion assays were performed in Millicell chambers which coated with 30 μg of Matrigel (BD Biosciences). Cells (5 × 104) were added to the coated filters in serum-free medium and incubated 16–24 h; migrated cells were then fixed with methanol and stained. The migration assay was conducted in a similar fashion without coating the filters with Matrigel.
Western blot assays
Protein extracts from cells were prepared using detergent-containing lysis buffer. Total protein (20 µg) was subjected to SDS-PAGE and transferred to PVDF membrane (Millipore). Antibody against SMAD4 (#46535), AMPK (#5831), p-AMPK (Thr172, #2535), or ubiquitin (#3933) was from Cell Signaling. Antibody against HNF4G (#25801-1-AP) or β-ACTIN (#TA-09) was from Proteintech and ZSGB-BIO. Membranes were incubated overnight at 4 °C with primary antibody and visualized with a Phototope Horseradish Peroxidase Western Blot Detection kit (Thermo Fisher). HNF4G phosphorylation assay was performed using Phos-tag Acrylamide (#F4002, APExBIO) according to the instructions.
Chromatin immunoprecipitation coupled quantitative PCR analysis
PDAC cells were treated with formaldehyde for cross linking, followed by chromatin Immunoprecipitation (ChIP) with antibody against SMAD4 (1:100; Cell Signaling, #38454) or mouse IgG. The level of HNF4G DNA in the immunoprecipitation was determined by qPCR using SYBR Green and the primers for HNF4G 1st SBE was showed in Table S9.
Construction of reporter plasmids and luciferase reporter gene assays
A reporter vector in the pGL4.10 backbone was generated containing the HNF4G promoter region using the restriction enzymes XhoI and HindIII. The authenticity of all the constructs was verified by sequencing. All primers used in plasmid construction are shown in Table S9. Luciferase reporter assays was performed according to the manufacturer’s instructions (Promega, Cat# E1960).
RNA interference of gene expression
Small interfering RNA (siRNA; Table S9) targeting HNF4G, SMAD4 or PRKAA1 was purchased from GenePharma. Transfections of each siRNA (75 nmol/L) were performed with Lipofectamine 2000 (Invitrogen).
Establishment of HNF4G-knockout cell lines by CRISPR editing
The CRISPR/Cas9 system was used to generate genomic deletion of HNF4G in PDAC cells. Single guide RNA (sgRNA) sequence (Table S9) targeting the genomic sequence of HNF4G designed using CRISPR design tool were cloned into plasmid pUC19-U6-sgRNA. The pCAG-Cas9-EGFP and pUC19-U6-sgRNA plasmids were co-transfected into PDAC cells and the fluorescent cells were sorted by flow cytometry into a 96-well plate for culture.
Lentiviral production and infection
Lentiviral for HNF4G stably overexpression (#41208-1), SMAD4 stably overexpression (#LV-27250-1) and RNAi targeted SMAD4 (#67236-1, #67236-2) were purchased from GENECHEM as viral particles. Lentiviral for luciferase stably overexpression (pHB-LV056) was purchased from Hanbio as viral particles. Virus infection of various cell lines was performed according to the manufacturer’s instructions.
Transient overexpression and site-directed mutagenesis
HNF4G (#23850-1) and SMAD4 (#27250-1) transient overexpressing plasmids were purchased from GENECHEM. Plasmid transfections were done using Lipofectamine 2000 (Invitrogen). Plasmids with mutant SBE or phosphorylation motifs in HNF4G were generated using the Muta-direct Kit (SBS) or gene synthesis by TSINGKE. The primers sequence and detail information are showed in Table S9.
Animal experiments
Pancreatic PDAC implantation was performed on female BALB/C nude mice aged 4–6 weeks as described previously (Qiu and Su, 2013; Aiello et al., 2016). One week after PDAC transplantation, mice were randomly divided into two groups and treated with Metformin in drinking water (0.93 mg/mL) or vehicle. The body weight of animals was measured every other day. The animal experiments and procedures were approved by PUMC Animal Care and Use Committees.
Analysis of relevant data from the public databases
Peaks annotation of ChIP-seq data (GEO accession: GSM803404) was performed as described (Yu et al., 2015). Integrative analysis was performed using ChIP-seq data and gene expression data from TCGA (Table S5). Gene Ontology analysis was performed by METASCAPE (Zhou et al., 2019) (http://metascape.org). HNF4G expression data in normal and PDAC tumor tissues were derived from the Gene Expression Profiling Interactive Analysis (Tang et al., 2017) (GEPIA, http://gepia.cancer-pku.cn).
Statistical analysis
The associations between SNPs and risk of PDAC were analyzed by an additive model in a logistic regression framework with age and sex as covariates. Odds ratios (ORs) and their 95% confidence intervals (CIs) were calculated in logistic regression models adjusting for age and sex. Pearson’s and Spearman’s correlations were used to measure the correlations between groups. The correlation was considered significant when P < 0.05 and |r| > 0.30. Kaplan-Meier analysis and log-rank test were used to evaluate associations with survival time. Hazard ratios (HRs) and 95% CIs were calculated using Cox proportional hazards models with age, sex, tumor stage as covariates. Chi-square and Fisher’s exact tests were used to examine the difference of IHC score between groups. Student’s t-test were used to evaluate the difference between two groups. P < 0.05 was considered significant for all statistical analyses. All statistical analyses were performed using Prism 6.0 (Graphpad Software Inc.).
Electronic supplementary material
Below is the link to the electronic supplementary material.
ACKNOWLEDGMENTS
We gratefully appreciate Y. L., Q. C. and L. P. for their assistances in data analysis and T. L., Y. C. and W. F. for their assistances in preparation of figures. We also thank G. Y. and Y. D. for their assistances in cell lines and animal experiments. This work was supported by National Science Fund for Distinguished Young Scholars (Grant No. 81725015 to C. W.), Beijing Outstanding Young Scientist Program (Grant No. BJJWZYJH01201910023027 to C. W.) and CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2016-I2M-3-019 to D.L., Grant No. 2016-I2M-4-002 to C.W. and Grant No. 2016-I2M-1-001 to W. T.).
AUTHOR CONTRIBUTIONS
Y.Z., C. W. and D.L. conceptualized and supervised this study. C. Wang contributed to the design and conducted most experiments. T. Z., Q. L., M. D. and J. G. responded to sample collection and preparation. X.Y. performed figure preparation. C. W., C. W. and D.L. prepared manuscript.
ABBREVIATIONS
AMPK, adenosine 5′-monophosphate (AMP)-activated protein kinase; bp, base pair; ChIP, chromatin immunoprecipitation; CHX, cycloheximide; CI, confidence interval; FBS, fetal bovine serum; GEO, Gene Expression Omnibus; GEPIA, the Gene Expression Profiling Interactive Analysis; GTEx, the Genotype-Tissue Expression; GWAS, genome-wide association study; HCS, high content screening; H&E, hematoxylin-eosin staining; HR, hazard ratio; IgG, immunoglobulin G; IHC, immunohistochemistry; Mb, mega base; mRNA, messenger RNA; OR, odds ratio; PCR, polymerase chain reaction; PDAC, pancreatic ductal adenocarcinoma; PVDF, polyvinylidene fluoride; qRT-PCR, quantitative real-time PCR; ROS, reactive oxygen species; SBE, smad binding element; siRNA, small interference RNA; SNP, single nucleotide polymorphism; TCGA, The Cancer Genome Atlas; TGF-β, transforming growth factor-β; TIE, TGF-β inhibitory element; TSS, transcriptional start site; Ub, ubiquitin.
COMPLIANCE WITH ETHICS GUIDELINES
Chengcheng Wang, Taiping Zhang, Quan Liao, Menghua Dai, Junchao Guo, Xinyu Yang, Wen Tan, Dongxin Lin, Chen Wu, Yupei Zhao declare that they have no conflict of interest. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study. All institutional and national guidelines for the care and use of laboratory animals were followed.
Footnotes
Dongxin Lin, Chen Wu, and Yupei Zhao have contributed equally.
Contributor Information
Dongxin Lin, Email: lindx@cicams.ac.cn.
Chen Wu, Email: chenwu@cicams.ac.cn.
Yupei Zhao, Email: zhao8028@263.net.
References
- Aiello, N.M., Rhim, A.D., and Stanger, B.Z. (2016). Orthotopic Injection of Pancreatic Cancer Cells. Cold Spring Harb Protoc 2016, pdb prot078360. [DOI] [PubMed]
- Amundadottir L, Kraft P, Stolzenberg-Solomon RZ, Fuchs CS, Petersen GM, Arslan AA, Bueno-de-Mesquita HB, Gross M, Helzlsouer K, Jacobs EJ, et al. Genome-wide association study identifies variants in the ABO locus associated with susceptibility to pancreatic cancer. Nat Genet. 2009;41:986–990. doi: 10.1038/ng.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assi SA, Imperato MR, Coleman DJL, Pickin A, Potluri S, Ptasinska A, Chin PS, Blair H, Cauchy P, James SR, et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat Genet. 2019;51:151–162. doi: 10.1038/s41588-018-0270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, Hezel AF, Horner J, Lauwers GY, Hanahan D, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–3146. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Chaiteerakij R, Petersen GM, Bamlet WR, Chaffee KG, Zhen DB, Burch PA, Leof ER, Roberts LR, Oberg AL. Metformin use and survival of patients with pancreatic cancer: a cautionary lesson. J Clin Oncol. 2016;34:1898–1904. doi: 10.1200/JCO.2015.63.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Qian W, Jiang Z, Cheng L, Li J, Sun L, Zhou C, Gao L, Lei M, Yan B, et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol Cancer. 2017;16:131. doi: 10.1186/s12943-017-0701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Toke NH, Luo S, Vasoya RP, Fullem RL, Parthasarathy A, Perekatt AO, Verzi MP. A reinforcing HNF4-SMAD4 feed-forward module stabilizes enterocyte identity. Nat Genet. 2019;51:777–785. doi: 10.1038/s41588-019-0384-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Wei L, Huang X, Zheng J, Shao M, Feng T, Li J, Han Y, Tan W, Tan W, et al. Solute carrier family 39 member 6 gene promotes aggressiveness of esophageal carcinoma cells by increasing intracellular levels of zinc, activating phosphatidylinositol 3-kinase signaling, and up-regulating genes that regulate metastasis. Gastroenterology. 2017;152(1985–1997):e1912. doi: 10.1053/j.gastro.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Childs EJ, Mocci E, Campa D, Bracci PM, Gallinger S, Goggins M, Li D, Neale RE, Olson SH, Scelo G, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nat Genet. 2015;47:911–916. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pascalis C, Etienne-Manneville S. Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell. 2017;28:1833–1846. doi: 10.1091/mbc.e17-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48:R31–43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M, Znaor A, Bray F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941–1953. doi: 10.1002/ijc.31937. [DOI] [PubMed] [Google Scholar]
- Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–327. doi: 10.1056/NEJMoa1903387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, Glass CK. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16:144–154. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zheng J, Li J, Che X, Tan W, Tan W, Shao M, Cheng X, Du Z, Zhao Y, et al. Functional role of BTB and CNC Homology 1 gene in pancreatic cancer and its association with survival in patients treated with gemcitabine. Theranostics. 2018;8:3366–3379. doi: 10.7150/thno.23978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, Vilardell F, Wang Z, Keller JW, Banerjee P, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–1813. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr LD, Miller DB, Matrisian LM. TGF-beta 1 inhibition of transin/stromelysin gene expression is mediated through a Fos binding sequence. Cell. 1990;61:267–278. doi: 10.1016/0092-8674(90)90807-Q. [DOI] [PubMed] [Google Scholar]
- Klein AP, Wolpin BM, Risch HA, Stolzenberg-Solomon RZ, Mocci E, Zhang M, Canzian F, Childs EJ, Hoskins JW, Jermusyk A, et al. Genome-wide meta-analysis identifies five new susceptibility loci for pancreatic cancer. Nat Commun. 2018;9:556. doi: 10.1038/s41467-018-02942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordes S, Pollak MN, Zwinderman AH, Mathot RA, Weterman MJ, Beeker A, Punt CJ, Richel DJ, Wilmink JW. Metformin in patients with advanced pancreatic cancer: a double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015;16:839–847. doi: 10.1016/S1470-2045(15)00027-3. [DOI] [PubMed] [Google Scholar]
- Lau HH, Ng NHJ, Loo LSW, Jasmen JB, Teo AKK. The molecular functions of hepatocyte nuclear factors - in and beyond the liver. J Hepatol. 2018;68:1033–1048. doi: 10.1016/j.jhep.2017.11.026. [DOI] [PubMed] [Google Scholar]
- Laufer C, Fischer B, Billmann M, Huber W, Boutros M. Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods. 2013;10:427–431. doi: 10.1038/nmeth.2436. [DOI] [PubMed] [Google Scholar]
- Laufer C, Fischer B, Huber W, Boutros M. Measuring genetic interactions in human cells by RNAi and imaging. Nat Protoc. 2014;9:2341–2353. doi: 10.1038/nprot.2014.160. [DOI] [PubMed] [Google Scholar]
- Lee CW, Wong LL, Tse EY, Liu HF, Leong VY, Lee JM, Hardie DG, Ng IO, Ching YP. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012;72:4394–4404. doi: 10.1158/0008-5472.CAN-12-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Duell EJ, Yu K, Risch HA, Olson SH, Kooperberg C, Wolpin BM, Jiao L, Dong X, Wheeler B, et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis. 2012;33:1384–1390. doi: 10.1093/carcin/bgs151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diab Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SK, Kuchiba A, Zembutsu H, Saito A, Takahashi A, Kubo M, Daigo Y, Kamatani N, Chiku S, Totsuka H, et al. Genome-wide association study of pancreatic cancer in Japanese population. PLoS ONE. 2010;5:e11824. doi: 10.1371/journal.pone.0011824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TA. The role of tight junctions in cancer metastasis. Semin Cell Dev Biol. 2014;36:224–231. doi: 10.1016/j.semcdb.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. doi: 10.1038/nature11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- Neoptolemos JP, Kleeff J, Michl P, Costello E, Greenhalf W, Palmer DH. Therapeutic developments in pancreatic cancer: current and future perspectives. Nat Rev Gastroenterol Hepatol. 2018;15:333–348. doi: 10.1038/s41575-018-0005-x. [DOI] [PubMed] [Google Scholar]
- Okegawa T, Ushio K, Imai M, Morimoto M, Hara T. Orphan nuclear receptor HNF4G promotes bladder cancer growth and invasion through the regulation of the hyaluronan synthase 2 gene. Oncogenesis. 2013;2:e58. doi: 10.1038/oncsis.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. doi: 10.1038/nrendo.2013.256. [DOI] [PubMed] [Google Scholar]
- Petersen GM, Amundadottir L, Fuchs CS, Kraft P, Stolzenberg-Solomon RZ, Jacobs KB, Arslan AA, Bueno-de-Mesquita HB, Gallinger S, Gross M, et al. A genome-wide association study identifies pancreatic cancer susceptibility loci on chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat Genet. 2010;42:224–228. doi: 10.1038/ng.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Su GH. Development of orthotopic pancreatic tumor mouse models. Methods Mol Biol. 2013;980:215–223. doi: 10.1007/978-1-62703-287-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reni M, Dugnani E, Cereda S, Belli C, Balzano G, Nicoletti R, Liberati D, Pasquale V, Scavini M, Maggiora P, et al. (Ir)relevance of metformin treatment in Patients with metastatic pancreatic cancer: an open-label, randomized phase II trial. Clin Cancer Res. 2016;22:1076–1085. doi: 10.1158/1078-0432.CCR-15-1722. [DOI] [PubMed] [Google Scholar]
- Ros S, Schulze A. Balancing glycolytic flux: the role of 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatases in cancer metabolism. Cancer Metab. 2013;1:8. doi: 10.1186/2049-3002-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkle EA, Mu D. Tight junction proteins: from barrier to tumorigenesis. Cancer Lett. 2013;337:41–48. doi: 10.1016/j.canlet.2013.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Cyrta J, Murphy DA, Walczak EG, Ran L, Agrawal P, Xie Y, Chen Y, Wang S, Zhan Y, et al. Aberrant activation of a gastrointestinal transcriptional circuit in prostate cancer mediates castration resistance. Cancer Cell. 2017;32(792–806):e797. doi: 10.1016/j.ccell.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang J, Xu L, Zheng Y, Ling D, Yang Z. Expression of HNF4G and its potential functions in lung cancer. Oncotarget. 2018;9:18018–18028. doi: 10.18632/oncotarget.22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpin BM, Rizzato C, Kraft P, Kooperberg C, Petersen GM, Wang Z, Arslan AA, Beane-Freeman L, Bracci PM, Buring J, et al. Genome-wide association study identifies multiple susceptibility loci for pancreatic cancer. Nat Genet. 2014;46:994–1000. doi: 10.1038/ng.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Li D, Jia W, Hu Z, Zhou Y, Yu D, Tong T, Wang M, Lin D, Qiao Y, et al. Genome-wide association study identifies common variants in SLC39A6 associated with length of survival in esophageal squamous-cell carcinoma. Nat Genet. 2013;45:632–638. doi: 10.1038/ng.2638. [DOI] [PubMed] [Google Scholar]
- Wu C, Miao X, Huang L, Che X, Jiang G, Yu D, Yang X, Cao G, Hu Z, Zhou Y, et al. Genome-wide association study identifies five loci associated with susceptibility to pancreatic cancer in Chinese populations. Nat Genet. 2011;44:62–66. doi: 10.1038/ng.1020. [DOI] [PubMed] [Google Scholar]
- Wu L, Zhou B, Oshiro-Rapley N, Li M, Paulo JA, Webster CM, Mou F, Kacergis MC, Talkowski ME, Carr CE, et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and Cancer. Cell. 2016;167:1705–1718. doi: 10.1016/j.cell.2016.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–2383. doi: 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- Zheng J, Huang X, Tan W, Yu D, Du Z, Chang J, Wei L, Han Y, Wang C, Che X, et al. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat Genet. 2016;48:747–757. doi: 10.1038/ng.3568. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.