

Abstract
The type II transmembrane serine protease TMPRSS2 facilitates the entry of coronaviruses, such as SARS-CoV-2, into host cells by cleaving the S1/S2 interface of the viral spike protein. Based on structural data derived from X-ray crystallographic data of related trypsin-like proteases, a homology model of TMPRSS2 is described and validated using the broad spectrum COVID-19 drug candidate camostat as a probe. Both active site recognition and catalytic function are examined using quantum mechanics/molecular mechanics molecular dynamic (QM/MM MD) simulations of camostat and its active metabolite, 4-(4-guanidinobenzoyloxy) phenylacetate (GBPA). Substrate binding is shown to be primarily stabilized through salt bridge formation between the shared guanidino pharmacophore and D435 in pocket A (flanking the catalytic S441). Based on the binding mode of GBPA, residues K342 and W461 have been identified as potential contacts involved in TMPRSS2 selective binding and activity. Additional data is reported that indicates the transition state structure is stabilized through H-bonding interactions with the backbone N–H groups within an oxyanion hole following bottom-side attack of the carbonyl by S441. This is supported by prior work on related serine proteases suggesting further strategies to exploit in the design of more potent inhibitors. Taken overall, the proposed structure along with the key contact sites and mechanistic features identified should prove highly advantageous to the design and rational development of safe and effective therapeutics that target TMPRSS2 and avoid inhibition of other trypsin-dependent processes.
Keywords: SARS-CoV-2, Camostat, Structure-based design, TMPRSS2, Serine protease, Molecular dynamics
Introduction
Many viruses, including the novel SARS-CoV-2, require host mediated spike (S) protein processing to gain cell entry [1, 2]. The spike protein is comprised of two functional subunits responsible for binding to the host cell surface receptor (S1 subunit) and fusion of the viral and cellular membranes (S2 subunit) [3]. Recent work has shown that the SARS-CoV-2 virus uses the angiotensin-converting enzyme 2 (ACE2) as a receptor for the unprimed S protein [2]. Once bound to ACE2, the type II transmembrane serine protease (TTSP) subfamily member, TMPRSS2, primes S by cleaving the S1/S2 interface found in the polybasic sequence P681RRAR↓SVA688 [4]. This step promotes a sequence of conformational changes that induce the fusion between the virus envelope and the host cell membrane [4]. While both ACE2 and TMPRSS2 are intimately involved in S protein processing, the latter is essential for not only SARS-CoV-2, but most other clinically relevant influenza and coronavirus infections [5–7]. In vivo studies have shown that TMPRSS2-knockout mice exhibit milder influenza symptoms as well as reduced death rates when compared to their wild type counterparts [8]. Similarly, mice with under expressed TMPRSS2 in their bronchial path cells show less severe lung pathologies after being infected with the coronaviruses SARS-CoV and MERS-CoV [9].
The catalytic mechanism of TMPRSS2 is well documented. The enzyme belongs to the trypsin-like serine protease family, one of the largest protease families in humans and mammals [10]. Sequence analyses have shown the multi-domain structure consists of an N-terminal membrane-spanning domain, a low-density lipoprotein receptor class A domain, and a protease domain [10, 11] containing the catalytic triad H296/D345/S441 [5]. Proteases in this family cleave peptide bonds at the carboxy-side of positively charged amino acid sidechains, such as arginine and lysine [12]. Substrate recognition of the substrate is driven by the formation of a salt link between the charged sidechain and a highly conserved aspartate within a polar pocket of the active site [12]. They also share common mechanistic features including the formation of an acyl-enzyme intermediate that forms in a rapid burst phase following substrate recognition. This intermediate, however, is short lived and undergoes general base catalyzed hydrolysis mediated by the polarized histidine to complete the catalytic cycle [13]. Although no selective inhibitors of TMPRSS2 are known, recent work has shown SARS-CoV-2 entry is inhibited by two broad spectrum serpins, camostat [14] and nafamostat shown in Fig. 1 [15]. Both drugs display the generic guanidino pharmacophore recognized by all trypsin-like proteases and form stable acyl-enzyme intermediates to effectively block substrate binding [12]. However, the lack of selectivity is problematic in the development of safe and effective therapeutics that target TMPRSS2 and avoid wholesale inhibition of trypsin-dependent processes.
Fig. 1.
Clinically used suicide inhibitors of TMPRSS2 highlighting the scissile ester bond (bracketed) and key guanidino pharmacophore
Despite the significance of TMPRSS2 in fighting SARS, MERS, and influenza, the discovery of selective drugs has proven elusive. To date no X-ray crystallographic data has surfaced to structurally characterize substrate or inhibitor complexes with TMPRSS2 or other related TTSP’s. This is most likely due to the inherent difficulties associated with solving crystal structures of membrane bound proteins. The lack of structural data, however, has significantly hindered structure-based design efforts to identify selective TMPRSS2 agents. Fortunately, TMPRSS2 shows significant sequence identity with other trypsin-like serine proteases that have been structurally resolved including human plasma kallikrein, factor Xia, and hepsin [16–19]. In addition, crystallographic data are available to validate binding site models of camostat [20] and nafamostat [21]. In this study, we apply model building techniques and molecular dynamics (MDs) simulations to construct a 3-dimensional model of TMPRSS2. Enzyme complexes of camostat bound through all stages of catalysis (substrate binding, transition state formation, and acylation) are also presented and compared with X-ray crystallographic data from related proteases. A particular emphasis is placed on active site analysis (steric fit and polarity) and the identification of structural features that provide new insight to the design of selective compounds.
Results and discussion
Homology model
The total sequence identity between the templates and TMPRSS2 ranged between 34.9 and 43.6%, with prostasin and plasma Kallikrein A having the lowest and highest sequence identity, respectively. The total sequence identity between the templates and TMPRSS2, as well as the amino acid sequence range corresponding to the trypsin-like region are presented in Table S1. The template with the highest overall sequence identity was that of the plasma Kallikrein A (PDB: 2ANY). A total of 61 residues were fully conserved in all seven sequences, including the catalytic triad: H296, D345, and S441 (based on TMPRSS2 sequence numbering), and the anchoring salting bridge D435. Figure 2 shows the multiple sequence alignment (MSA) and consensus of TMPRSS2, at the 50 and 100% level, with the template structure sequences. The MSA for all sequences used in this study is shown in Fig. S1.
Fig. 2.

Multiple sequence alignment of trypsin-like portion of TMPRSS2 showing the percent sequence identity with respect to the template structures and secondary structure-forming residues. Pockets A–E refer to the identified binding sites, see Fig. 3
The tertiary structure and solvent accessible surface of the consensus homology model (TMPRSS2hm) created by Prime is shown in Fig. 3A. The atomic coordinates of the homology model are provided as a PDB file in the electronic Supporting Information section. The extracellular structure of TMPRSS2hm is characterized by a solvent exposed cleft that harbors the catalytic triad and several polar pockets. The N-terminal region of TMPRSS2hm which connects to the transmembrane domain of the protein is labeled for clarity but is not included here. The catalytic triad is shown in Fig. 3B. A brief analysis of the active site geometry indicates H296 forms a hydrogen bond network with S441 and D345. This network allows a proton to be efficiently transferred from S441 to H296 which activates the serine for a nucleophilic attack of the bound substrate. In addition, D345 is in the correct hydrogen bonding position to stabilize the positive charge of H296 in the transition state. For this reason, the triad is often referred to as a charge relay that functions to polarize S441 while reducing the activation energy of T.S. complex formation. The highly conserved aspartate D435 lies in a partially buried polar pocket adjacent to S441. This residue is the primary recognition element of all trypsin-like proteases and represent the key anchor point to the positively charged sidechains of lysine and arginine residues of the substrate. The pKa values for the titratable residues in the active site, calculated using Epik, are 7.05 (H296) and 3.09 (D345), and 7.01 for the anchoring aspartate (D435).
Fig. 3.
TMPRSS2 homology model. A Tertiary structure of TMPRSS2hm showing its relative position to the extracellular matrix and transmembrane anchor. The gray surface corresponds to the van der Waals surface area, loops are shown in mauve, α-helices in aqua, and β-sheets in purple. B Catalytic triad of TMPRSS2hm, S441, H296 and D345, showing the distance between donor protons and acceptor heavy atoms
Apo TMPRSS2hm
The final TMPRSS2 homology model (TMPRSS2hm) passed the “Verify 3D” check after the serial refinement of the six loops outlined in the methods section. Also, all residues of TMPRSS2hm, except A69 and V179, are in favorable regions of the Ramachandran plot (Fig. S2). Next, the structural stability of the apo form of TMPRSS2hm was studied via MD simulations. Two independent simulations starting from the same initial pose were carried out using two force field models including the conventional ff14SB (with a TIP3P water box) and the implicitly polarized ff15ipq (with a SPCE/b water box). Two sets of geometric parameters were calculated to determine the relative stability and strength of the hydrogen bond network in the catalytic triad. The first set involved the distance between OγS441---NδH296 and the angle formed between OγS441---HγS441---NδH296. The second set involved the distance between NεH296---OδD345 and the angle formed between NεH296--- HεH296---OδD345, where Oδ is the geometric mean between Oδ1 and Oδ2 of the aspartic acid residue. An analysis of the ff14SB trajectory indicates the hydrogen bond formed between H296 and D345 ranges from almost ionic interactions (<3 Å and 120–180°) to weak dipolar electrostatic interactions (>5 Å). The variance from ideal H-bonding parameters is even more pronounced in the S441 to H296 geometry which adopts an OγS441---HγS441---NδH296 angle from 30 to 60°. A visual inspection of the frames containing this angle show this corresponds to a weak hydrogen bond between HγS441 and an adjacent backbone carbonyl oxygen of residue G449. On the other hand, the ff15ipq trajectories produce hydrogen bonding parameters that are much closer to published X-ray crystallographic data. The hydrogen bond interaction between H296 and D345 is tightened to a narrower range (2.5–3.0 Å and 150–180°) and the network involving S441 adopts a more linear orientation (135–160°) that forms over a shorter heteroatom distance (4.5–5.5 Å), the results are compared in Fig. S3. The results suggest that ff14SB is capable of keeping the stability of the active site geometry for shorter simulations (<2.5 ns) however, for longer time scales the terms of the ff14SB force field are not adequate to capture the dynamics of the active site geometry. On the other hand, the ff15ipq force field is capable of properly keeping the active site geometry for up to 10 ns simulation time scales. The data highlights the importance of including implicitly polarized terms in force field calculations involving strong H-bonding and ionic interactions and is the method of choice in all subsequent calculations reported here. The salt bridge stability results obtained from our simulations are in agreement with the data presented by Ahmed et al. [22]. This group has demonstrated in an extensive benchmarking study that the ff15ipq force field is the best at replicating the salt bridge interactions observed from NMR experiments.
Enzyme–ligand complexes
Docking
Static enzyme–ligand complexes were generated using Glide and the equilibrated apo form of TMPRSS2hm as a template. In all cases, the best scored poses located the benzoyl guanidino moiety in the primary recognition pocket A as shown in Fig. 4A. This pocket contains the highly conserved aspartate that is common to all trypsin-like proteases and the key anchor point to the positively charged guanidino group [10, 11]. The terminal dimethyl amide group was found to dock in four favorable poses in discrete sites referred to as Pockets B–E (highlighted in Fig. 4B). Three out the four pockets (B, C, and E) have highly polar, electronegative surfaces, whereas Pocket D shows a neutral surface charge. The cdock algorithm was applied to simulate nucleophilic attack by S441 to any of the three carbonyl carbons of the substrate. Two transition state poses were observed, and both were due to selective addition of the catalytic serine to the carbonyl carbon between the two benzene rings. In the first pose, S441 attacks the carbonyl from above which results in the oxyanion pointing towards H296, Fig. 4C. In the second pose, S441 attacks the carbonyl from below which results in the oxyanion pointing towards the oxyanion hole formed by G439 and S441, Fig. 4D. The cdock score for the first and second pose was −6.41 kcal/mol and −7.08 kcal/mol, respectively.
Fig. 4.

Standard and covalent docking. A Camostat docked into TMPRSS2hm showing the guanidino group in Pocket A relative to the position of the catalytic S441. Three residues, D435, S436, and G464, were identified to be forming a salt bridge and hydrogen bonds with the guanidino moiety in camostat. B Solvent accessible binding cleft formed by pockets A–E identified from standard docking. In sub-figures A and B, the red surfaces indicates electronegative regions, the white surface indicates electroneutral regions, and the blue surface indicates electropositive regions. C The transition state acyl intermediate after a top nucleophilic attack of S441 causing the negatively charged oxyanion to point down towards the positively charged catalytic H291; cdock score = −6.41 kcal/mol. D The transition state acyl intermediate after a bottom-side nucleophilic attack of S441 causing the negatively charged oxyanion to point up toward the oxyanion hole formed by G439 and S441; cdock score = −7.08 kcal/mol
In addition, literature reports have shown that camostat is hydrolyzed by carboxylesterase (CES) to form an active metabolite 4-(4-guanidinobenzoyloxy) phenylacetate (GBPA, see Methods) [23]. Similar to camostat, GBPA standard docking results located the benzoyl guanidino moiety in the primary recognition pocket A. The terminal carboxyl group, however, did not make any contacts with any of the residues that form the binding cleft. The cdock algorithm was applied to simulate nucleophilic attack by S441 to any of the carbonyl carbons in GBPA. A single transition state pose observed due to selective addition of the catalytic serine to the carbonyl carbon between the two benzene rings. The pose obtained is due to an attack by S441 from below which results in the oxyanion pointing towards the oxyanion hole formed by G439 and S441, akin to the camostat pose shown in Fig. 4D. The terminal carboxyl group of GBPA is <3 Å away from K342, suggesting the formation of an anchoring salt bridge.
Molecular dynamics
Substrate complexes
The enzyme–substrate and TS complexes were evaluated using quantum mechanics/molecular mechanics (QM/MM) MDs simulations. In all cases, the ff15ipq and SPCE/b implicitly polarized force fields were applied as the default MM parameters. An initial set of simulations were performed on the apo form of the enzyme using QM to model the residues of the catalytic triad. The resulting trajectories produced H-bonding distances and geometries in close agreement with those reported above, indicating the ff15ipq force field is comparable in capturing these critical interactions. Based on these results, a second system was constructed to simulate active site dynamics in which all atoms of the ligand (camostat) were assigned to the QM portion of the energy function. Following equilibration, an analysis of the trajectory showed substrate binding induced a significant strengthening of the S441 to H296 H-bonding interaction. The O–N distance tightens from ~5.0 to 2.9 and the O–H–N angle is less variable and closer to linearity as shown in Fig. 5A(left). This geometry, as well as the orientation of the H296-D435 H-bonding interaction, Fig. 5A(right), is in good agreement with published parameters derived from X-ray crystallographic data of related serine proteases (see Table S2) [16–19]. The average distance of between the oxygen from S441 to the scissile carbonyl bond of camostat is 3.7 Å, as shown in Fig. 5B. Over the course of the simulation, the substrate maintains a network of polar interactions within pocket A. An analysis of the trajectory indicates that four residues, D435, S436, S463, and G464, are involved in the formation of hydrogen bonds and salt bridge interactions with the guanidino group of the ligand. In 100% of the analyzed frames, at least one hydrogen bond is formed between the guanidino donor and the acceptor residues. The maximum number of identified hydrogen bonds was calculated to be 5. As shown in Fig. 6, the cumulative distribution function for the number of hydrogen bonds indicates that majority of the time there are at least three hydrogen bonds anchoring the ligand in place.
Fig. 5.

Camostat QM/MM MD simulation. TMPRSS2hm as parameterized using ff15ipq force field and camostat was treated as the QM region. A Hydrogen bond network in catalytic triad. (left) hydrogen bond between S441 and H296. (right) hydrogen bond between H296 and D345. B Histogram showing the distribution of the distance between the oxygen atom of the catalytic S441 and the carbonyl carbon in the scissile bond of camostat. C Distance and direction of carbonyl oxygen in relation to oxyanion hole formed by G439 and S441. In sub-figures A and C, the yellow regions indicate the highest conformational probability, whereas the blue regions indicate accessible conformations albeit with a lower probability of being observed
Fig. 6.
Cumulative distribution function showing the number of hydrogen bonds formed between the guanidino group and the four anchor residues D435, S436, S463, and G464
The last geometric parameter studied was the direction that the carbonyl oxygen points during the course of the 5 ns simulated time. A dummy point (DUG439,S441) was defined as the geometric mean of the oxyanion-hole-forming backbone nitrogen atoms of G439 and S441. The distance between the carbonyl oxygen in camostat and the dummy point (C=O---DUG439,S441), and the angle formed between the carbonyl and dummy point are shown in Fig. 5C. The average distance of 3.6 Å and angle of 130° positions the carbonyl for a bottom-side attack by the catalytic serine. Angles of <90° were not observed, suggesting that top side addition to the carbonyl is highly unlikely. This is consistent with the mode of attack of other serine proteases, in which the oxyanion formed during the transition state is stabilized by the oxyanion hole formed by N–H groups of the conserved residues G439 and S441.
The QM/MM MD simulation for GBPA showed that the benzoyl guanidino portion of the molecule behaves identically to camostat. The average number of anchoring hydrogen bonds of the guanidino group to pocket A is 3 and the average distance of between the oxygen from S441 to the scissile carbonyl bond is 3.6 Å. The opposite end of the GBPA molecule has a negatively charged carboxy group that initially floats pointing towards the solvent (i.e., the initial pose from Glide docking). The position of this carboxy group rapidly changes and within 100 ps of start of the simulation it moves to make a salt bridge with K342 that lies at the intersection of pockets D and E as shown in Fig. 7A. The salt bridge contact is conserved throughout the rest of the simulation time (1.9 ps) and its average length (DU--NηK342) is 2.1 Å. In contrast to camostat, the distance and direction of the carbonyl oxygen of GBPA in relation to the N–H groups of oxyanion hole is closer. The distance ranges between 1.8 and 3.2 Å and the angle is between 105 and 175° (compare Fig. 5C vs. Fig. S4). Within the first 100 ps of the simulation, a π–π contact is also observed between the aromatic phenyl ring of GPBA and the aryl ring of W461. The center-of-mass distance between the two ring is 3.8 Å over a short window of 20 ps. Despite the brevity of the π–π interaction, it is another possible contact point that can be exploited to enhance the binding affinity of TMPRSS2 inhibitors.
Fig. 7.
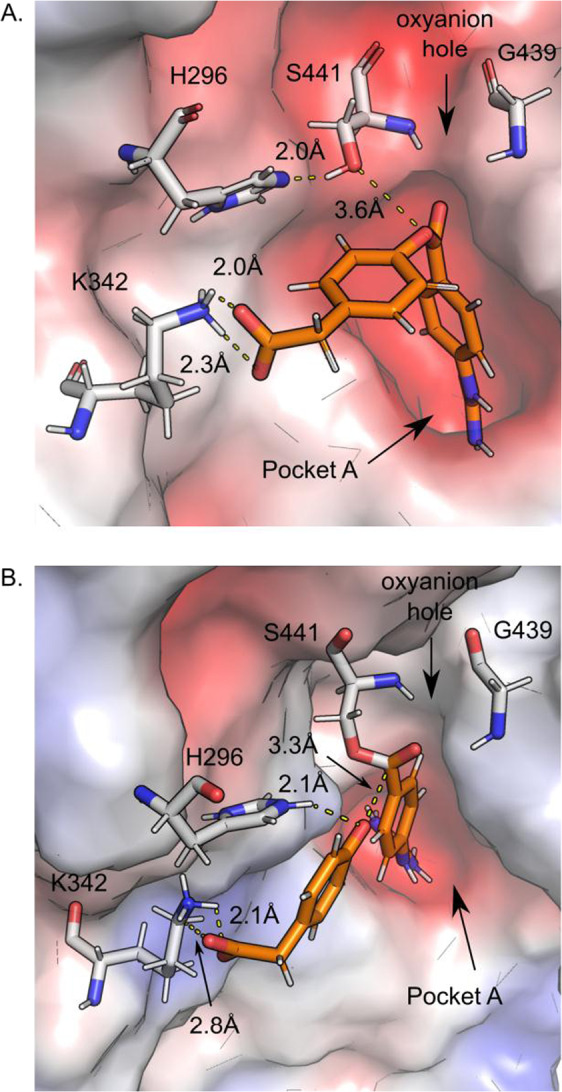
GBPA active site interactions. A Substrate binding is conferred through salt-link interactions between the guanidino group and D435 in pocket A and the phenylacetate group and K342. The catalytic S441 is 3.6 Å away from the reactive carbonyl carbon, and the oxygen points toward the oxyanion hole formed by G439 and S441. B Acyl enzyme intermediate and phenolate products formed by collapse of the TS structure during equilibration. The two salt bridges at the terminal ends of the molecule (D435 and K342) are conserved. In sub-figures A and B the red surfaces indicates electronegative regions, the white surface indicates electroneutral regions, and the blue surface indicates electropositive regions
Transition state complexes
To adequately model the covalently bound state of the substrate, the catalytic serine residue (S441) was included in the QM grouping of substrate atoms. The starting points for the MD simulations were taken from the docked structures as described above. In all cases, the primary pharmacophore maintained strong salt-link and H-bonding interactions between the guanidino group and polar sidechains within pocket A. The degree of flexibility of the tetrahedral intermediate was assessed by measuring the distance and angle between the oxyanion and center-point between the N–H groups of the oxyanion hole (as previously described). An analysis of the trajectories derived from bottom-side addition S441 to the carbonyl show the distance between the oxyanion of the tetrahedral carbon and DU atom remains between 2.8 and 3.1 Å, while the range of the angle is 175–180° over the entire simulation time for both camostat and GBPA. A third simulation was performed on the TS structure of camostat generated from top side attack that directs the oxyanion towards the protonated histidine. In this case, the oxyanion is stabilized by the protonated histidine in <20% of the simulation time. The rest of the simulation time the oxyanion is stabilized by water molecules in the solvent box. This highly unstructured transition state provides additional evidence to conclude that a top side attack by the nucleophilic serine is highly unlikely. Significant differences also were noted in the position of the orientation of the diethylamide of camostat and the carboxy group of GBPA. In the former, the terminal end (that is ultimately cleaved through catalysis) was free floating away from the surface of the cleft into the solvent. In contrast, the carboxy group of GBPA remained tightly anchored to K342 which in turn may aid in the induction of ester bond cleavage. Within 2 ps of equilibration, the O⇨C=O bond was cleaved, generating the desired acyl-enzyme intermediate and an ion-pair interaction between the protonated NδH296 and the negatively charged phenolate oxygen. The structural features are highlighted in Fig. 7B. Since the histidine is not treated within the QM designated set of atoms, proton transfer is not observed but the results are quite remarkable and shows the model is capable of capturing the key mechanistic features of catalysis.
Conclusion
The significance of TMPRSS2 as a potential therapeutic target in the design of strategies to inhibit SARS-CoV-2 cell entry and infection cannot be overstated. Here, we reported a structural model of the catalytic domain of TMPRSS2 using a combination of homology modeling and MDs techniques. The fidelity of the model was vetted using camostat as a model substrate. As expected from prior work on related trypsin-like proteases, substrate recognition is conferred through a salt-link interaction between the positively charge gunidino group and a conserved aspartate (D345) in a buried pocket adjacent to the catalytic triad. The alignment of the scissile bond of the ester was shown to be ideally positioned for nucleophilic attack of the carbonyl group by S441. The equilibrated substrate-bound structures also indicate S441 adds to the bottom-side of the carbonyl group, leading to a TS structure in which the oxygen anion is stabilized through backbone N–H hydrogen bonds with S441 and G439. This pocket was not evident in the initial apo enzyme model built structure but adopted a more recognizable motif in response to substrate binding following equilibration. While we cannot rule out the possibility of a topside attack of the carbonyl leading to stabilization through salt-link formation with the protonated catalytic histidine, the resulting structure is less ordered and unlikely to form based on the data presented here. Significant differences in the binding modes observed for camostat and its active metabolite GBPA were also noted. Although there is evidence that both camostat and GBPA have similar potencies, our data suggests that GBPA may recognize additional epitopes in the cleft of the active site of TMPRSS2. In particular, the equilibrated structures of GBPA bound show the phenyl carboxy terminal group forms a strong salt-link with K342 and a pi stacking interaction with W461. Given the solvent exposed surface of the active site cleft is primarily electronegative in character (due to the presence of backbone C=O groups), the result is quite striking and points to a potential element that could be further exploited in the design of new analogs. Further support of the significance of these interactions can be found in the results of GBPA-TS simulation that resulted in ester bond breakage and product formation. This was enabled by treating the substrate and catalytic serine quantum mechanically with the QM/MM hybrid model. It is reasonable to conclude that the secondary anchor points of GBPA facilitated the release of product and formation of the resulting ion-pair interaction with the protonated H296. In contrast, the terminal dimethyl amide of camostat was found to adopt multiple positions on the surface of the active site cleft in the substrate-bound complex and remained solvent exposed in the TS complex. In terms of inhibitory activity, however, the significance of these results is unclear since the first step of the enzymatic reaction of serine proteases is known to be kinetically facile and both camostat and GBPA display the same primary pharmacophore.
Finally, significant effort was devoted here to the evaluation and selection of computational methods to best represent the active site geometry of the catalytic triad and enzyme–substrate interactions. This was accomplished through a trial and error process that compared computed geometries and key contacts with those derived from X-ray crystallographic data. One of the primary conclusions reached here pertains to the inherent limitation of conventional pairwise force field terms in capturing induce dipole many body effects in polar systems. As reported above, the standard ff14SB force field trajectories failed to capture the close-knit H-bonding geometries of the catalytic triad in the apo form of the enzyme. Fortunately, this issue was addressed by inclusion of the implicitly polarized terms using the ff15ipq force field. The resulting H-bonding geometries mimicked those derived from published data on related serine proteases. The application of QM/MM hybrid models were also evaluated here. In addition to alleviating complex protocols to parameterize the substrate, the use of QM to treat the substrate was found to produce active site models that better account for the geometry of attack of the catalytic serine to the carbonyl carbon of the cleavable ester (in both camostat and GBPA). Furthermore, the approach allowed bond breaking to occur providing significant advantages in in modeling the TS structure and product formation. These protocols should prove useful in facilitating future work on TMPRSS2 and related proteases using computational techniques. In particular, the structures and methods described here may provide much needed tools in the design of more potent and selective inhibitors that block SARS-CoV-2 spike protein by TMPRSS2. The coordinates of the refined homology model are provided as a PDB file in the Supporting Information section.
Methods
Homology modeling
The amino acid sequence of TMPRSS2 was retrieved from the UniProt database (Gene ID: O15393) and crystal structures with high sequence identity were identified using the BLASTp algorithm. A total of six crystal structures were chosen to serve as templates for the homology model—PDB IDs: 2ANY, 1XX9, 2OQ5, 3P8F, 5CE1, and 3FVF, the latter one has camostat co-crystallized. All crystal structure templates were truncated to contain only the trypsin domain as identified by the Pfam database [24]. The Prime homology model suite (Schrodinger, Inc) was used to align the sequences and identify conserved secondary structure assignments. Six homology models were constructed based on each of the six crystal structure templates. The camostat ligand was included in the model constructed from the 3FVF template. This was followed by the construction of a single final consensus model using the six homology models previously built. Similar techniques were applied in previous work on TMPRSS2 in which models were built based on a single sequence homology [25–27]. By using multiple templates we have ensured 100% coverage of the TMPRSS2 amino acid sequence as shown in the Supplementary Fig. S1. The final homology model obtained (with camostat bound to the active site) is referred to as TMPRSS2hm. A set of six loop sections near the active site triad were refined using the loop refinement functionality of the Prime program. The refined looped sections were: 39–53, 58–75, 79–91, 172–177, 181–190, and 193–198. The loop structures were verified using “Verify 3D” and checking that all residues fall in favorable regions of the Ramachandran plot. The final structure was further refined through a series of short MD simulations as described below.
Docking
The ligands were constructed using the 2D builder functionality in the Maestro suite (Schrodinger Inc) and assigned parameters using the OPLS3 force field [28]. Epik was used to generate all possible protonation states at pH 7. The receptor docking grid was prepared using the structure with the lowest total energy from the production MD simulation run. A cubic sampling box with 15 Å sides was defined at the center-of-mass of the camostat molecule. Glide’s extra precision (XP) algorithm was applied to sample ligand poses while keeping the receptor fixed, except for the following hydroxyl groups which were allowed to rotate: S181, C182, S186, T204, C210, and Y219. The transition state intermediate of camostat was generated using the Glide covalent docking (cdock) package [29] and using the XP docking pose as the input structure. In brief, the cdock algorithm follows the steps: (i) if necessary, remove the proton from the nucleophilic serine; (ii) increase the bond order between the nucleophilic oxygen (from serine) and carbonyl carbon (from ligand) from 0 to 1, i.e., form the covalent bond; (iii) reduce the formal charge of the carbonyl
oxygen (from ligand) to −1; finally, (iv) reduce the bond order between the carbonyl carbon and oxygen from a double to a single bond. In order to properly account for the proton transfer that occurs during the formation of the transition state structure, the proton was shifted from S441 to H296. The final step of the reaction was constructed by deprotonating H296 to its original state following acylation of S441 and elimination of the phenolic leaving group and product. The structures considered are shown in Fig. 8.
Fig. 8.
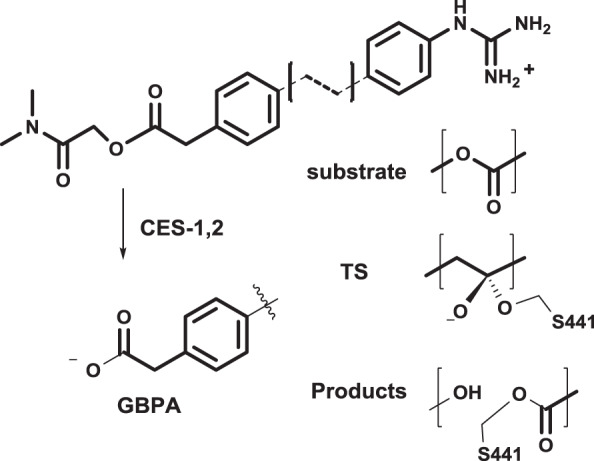
Substrate, transition state (TS), and product structures and metabolic function of carboxylesterase (CES)
Molecular dynamics
All of the MD simulation stages, unless noted otherwise, were carried out using the SANDER.MPI function of the AMBER 18 software package [30]. Ligand structures were preprocessed using the Antechamber package to assign AM1-BCC partial charges. The ligand and enzyme structures were then processed using LEaP to assign gaff [31] and ff14SB [32] or ff15ipq [33] force field parameters for the ligand and enzyme, respectively. The transition state and acyl-enzyme models involving a covalent bond to S441 were defined by unique residues. The covalently bound serine residue we first assigned Amber atom types and AM1-BCC partial charges using the antechamber program. Bonded terms (lengths, angles, and dihedrals) were derived from the existing Amber parameter databases in a two-step process. Firstly, the serine atom types were assigned bonded terms from the standard amino acid parameters in the ff19SB force field. Secondly, all ligand atom types were assigned bonded terms from the gaff force field, including the parameters that describe the linking OS441–Cligand bond. All complexes were submerged into a periodic box of TIP3P water with a 10 Å buffer region. Each system was initially minimized using the steepest descent method for 100 steps followed by 9900 steps of conjugate gradient minimization. This was followed by a step-wise heating procedure in which the system temperature was gradually ramped from 0 to 300 K over 15,000 steps and subsequently relaxed over 5000 steps in which the average temperature was kept constant at 300 K using the weak-coupling algorithm. During both heating stages the position of all non-solvent atoms was restrained with a harmonic potential with a force constant of 25 kcal/mol Å. This was followed by a two-step procedure in which the average pressure was maintained at 1 bar for 20,000 steps (using the Berendsen barostat and pressure relaxation time of 0.2 ps), the position of all non-solvent atoms was restrained with a harmonic potential with a force constant of 5 kcal/mol Å. This was followed a relaxation stage of 20,000 steps in which the pressure relaxation time was raised to 2 ps, and only the position of the receptor Cα were restrained with a harmonic potential with a force constant of 0.5 kcal/mol Å. Finally, all production runs were carried out without any restraints and kept at constant temperature of 300 K and a pressure of 1 bar. The total simulation time for all production runs was 10 ns. In addition, we used the refined homology model structure, obtained using the ff15ipq force field, to start QM/MM MD simulations. The QM/MM simulations defined the catalytic triad and the scissile bond region of the ligand as the quantum region and the rest of the system as the molecular mechanics region. The semiempirical Hamiltonian PM3 level of theory was used to describe the quantum region. At each simulation time step, Amber calculates the effective total energy by partitioning the system into the QM, and MM regions, thus quantum energies are calculated at each time step. Equilibration of all systems, in both MD and QM/MM MD simulations, was ensured by checking the root-mean-squared-deviation of the Cα of TMPRSS2hm and the heavy atoms of any bound ligand, shown in Supplementary Figs S6–S9.
Supplementary information
Acknowledgements
This work was supported by the National Institutes of Health AI14378. Computing time was provided in part by the Minnesota Supercomputer Institute.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1007/s00044-021-02708-7.
References
- 1.Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R, Nunneley JW, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020. 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed]
- 3.Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–47. doi: 10.1016/s0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. 2019;73:529–57. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 5.Meyer D, Sielaff F, Hammami M, Böttcher-Friebertshäuser E, Garten W, Steinmetzer T. Identification of the first synthetic inhibitors of the type II transmembrane serine protease TMPRSS2 suitable for inhibition of influenza virus activation. Biochem J. 2013;452:331–43. doi: 10.1042/bj20130101. [DOI] [PubMed] [Google Scholar]
- 6.Bertram S, Dijkman R, Habjan M, Heurich A, Gierer S, Glowacka I, et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J Virol. 2013;87:6150–60. doi: 10.1128/jvi.03372-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci USA. 2020;117:7001–3. doi: 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatesuer B, Bertram S, Mehnert N, Bahgat MM, Nelson PS, Pöhlman S, et al. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathol. 2013;9:e1003774.. doi: 10.1371/journal.ppat.1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J Virol. 2019;93. 10.1128/jvi.01815-18. [DOI] [PMC free article] [PubMed]
- 10.Goettig P, Brandstetter H, Magdolen V. Surface loops of trypsin-like serine proteases as determinants of function. Biochimie. 2019;166:52–76. doi: 10.1016/j.biochi.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bugge TH, Antalis TM, Wu Q. Type II transmembrane serine proteases. J Biol Chem. 2009;284:23177–81. doi: 10.1074/jbc.r109.021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102:4501–24. doi: 10.1021/cr000033x. [DOI] [PubMed] [Google Scholar]
- 13.Fuhrmann CN, Daugherty MD, Agard DA. Subangstrom crystallography reveals that short ionic hydrogen bonds, and not a His-Asp low-barrier hydrogen bond, stabilize the transition state in serine protease catalysis. J Am Chem Soc. 2006;128:9086–102. doi: 10.1021/ja057721o. [DOI] [PubMed] [Google Scholar]
- 14.Shirato K, Kawase M, Matsuyama S. Middle east respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. 2013;87:12552–61. doi: 10.1128/jvi.01890-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto M, Matsuyama S, Li X, Takeda M, Kawaguchi Y, Inoue J-I, et al. Identification of Nafamostat as a potent inhibitor of middle east respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay. Antimicrob Agents Chemother. 2016;60:6532–9. doi: 10.1128/aac.01043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang J, Yu CL, Williams SR, Springman E, Jeffery D, Sprengeler PA, et al. Expression, crystallization, and three-dimensional structure of the catalytic domain of human plasma kallikrein. J Biol Chem. 2005;280:41077–89. doi: 10.1074/jbc.m506766200. [DOI] [PubMed] [Google Scholar]
- 17.Jin L, Pandey P, Babine RE, Gorga JC, Seidl KJ, Gelfand E, et al. Crystal structures of the FXIa catalytic domain in complex with ecotin mutants reveal substrate-like interactions. J Biol Chem. 2005;280:4704–12. doi: 10.1074/jbc.m411309200. [DOI] [PubMed] [Google Scholar]
- 18.Kyrieleis OJP, Huber R, Ong E, Oehler R, Hunter M, Madison EL, et al. Crystal structure of the catalytic domain of DESC1, a new member of the type II transmembrane serine proteinase family. FEBS J. 2007;274:2148–60. doi: 10.1111/j.1742-4658.2007.05756.x. [DOI] [PubMed] [Google Scholar]
- 19.Yuan C, Chen L, Meehan EJ, Daly N, Craik DJ, Huang M, et al. Structure of catalytic domain of Matriptase in complex with Sunflower trypsin inhibitor-1. BMC Struct Biol. 2011;11:30. doi: 10.1186/1472-6807-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spraggon G, Hornsby M, Shipway A, Tully DC, Bursulaya B, Danahay H, et al. Active site conformational changes of prostasin provide a new mechanism of protease regulation by divalent cations. Protein Sci. 2009;18:1081–94. doi: 10.1002/pro.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rickert KW, Kelley P, Byrne NJ, Diehl RE, Hall DL, Montalvo AM, et al. Structure of human prostasin, a target for the regulation of hypertension. J Biol Chem. 2008;283:34864–72. doi: 10.1074/jbc.m805262200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahmed MC, Papaleo E, Lindorff-Larsen K. How well do force fields capture the strength of salt bridges in proteins? PeerJ. 2018;6:e4967.. doi: 10.7717/peerj.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Midgley I, Hood AJ, Proctor P, Chasseaud LF, Irons SR, Cheng KN, et al. Metabolic-fate of C-14 Camostat mesylate in man, rat and dog after intravenous administration. Xenobiotica. 1994;24:79–92. doi: 10.3109/00498259409043223. [DOI] [PubMed] [Google Scholar]
- 24.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–32. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rensi S, Keys A, Lo Y-C, Derry A, McInnes G, Liu T, et al. Homology modeling of TMPRSS2 yields candidate drugs that may inhibit entry of SARS-CoV-2 into human cells. ChemRxiv. 2020. 10.26434/chemrxiv.12009582.v1.
- 26.Elmezayen AD, Al-Obaidi A, Şahin AT, Yelekçi K. Drug repurposing for coronavirus (COVID-19): in silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J Biomol Struct Dyn. 2020:1–13. 10.1080/07391102.2020.1758791. [DOI] [PMC free article] [PubMed]
- 27.Rahman N, Basharat Z, Yousuf M, Castaldo G, Rastrelli L, Khan H. Virtual screening of natural products against Type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2). Molecules. 2020;25. 10.3390/molecules25102271. [DOI] [PMC free article] [PubMed]
- 28.Roos K, Wu C, Damm W, Reboul M, Stevenson JM, Lu C, et al. OPLS3e: extending force field coverage for drug-like small molecules. J Chem Theory Comput. 2019;15:1863–74. doi: 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- 29.Toledo Warshaviak D, Golan G, Borrelli KW, Zhu K, Kalid O. Structure-based virtual screening approach for discovery of covalently bound ligands. J Chem Inf Model. 2014;54:1941–50. doi: 10.1021/ci500175r. [DOI] [PubMed] [Google Scholar]
- 30.Case D, Ben-Shalom I, Brozell S, Cerutti D, Cheatham III T, Cruzeiro V, et al. AMBER. San Francisco: University of California; 2018.
- 31.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Gr Model. 2006;25:247–60. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debiec KT, Cerutti DS, Baker LR, Gronenborn AM, Case DA, Chong LT. Further along the road less traveled: AMBER ff15ipq, an original protein force field built on a self-consistent physical model. J Chem Theory Comput. 2016;12:3926–47. doi: 10.1021/acs.jctc.6b00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.