

Abstract
Japan has had a substantial increase in syphilis cases since 2013. However, research on the genomic features of the Treponema pallidum subspecies pallidum (TPA) strains from these cases has been limited. Here, we elucidated the genetic variations and relationships between TPA strains in Japan (detected between 2014 and 2018) and other countries by whole-genome sequencing and phylogenetic analyses, including syphilis epidemiological surveillance data and information on patient sexual orientation. Seventeen of the 20 strains in Japan were SS14- and the remaining 3 were Nichols-lineage. Sixteen of the 17 SS14-lineage strains were classified into previously reported Sub-lineage 1B. Sub-lineage 1B strains in Japan have formed distinct sub-clusters of strains from heterosexuals and strains from men who have sex with men. These strains were closely related to reported TPA strains in China, forming an East-Asian cluster. However, those strains in these countries evolved independently after diverging from their most recent common ancestor and expanded their genetic diversity during the time of syphilis outbreak in each country. The genetic difference between the TPA strains in these countries was characterized by single-nucleotide-polymorphism analyses of their penicillin binding protein genes. Taken together, our results elucidated the detailed phylogenetic features and transmission networks of syphilis.
Subject terms: Genome assembly algorithms, Computational biology and bioinformatics, Evolution, Molecular evolution, Microbiology, Bacteria, Bacterial genomics
Introduction
Treponema pallidum subspecies pallidum (TPA) is the etiologic agent of syphilis. An estimated 6 million new cases are diagnosed globally every year, with the majority in low- and middle-income countries1. However, a resurgence of syphilis has been reported in developed countries worldwide beginning in the twenty-first century2, including a substantial increase in syphilis cases in Japan since 20133.
To date, most molecular studies of TPA have been carried out by PCR amplification of several TPA genetic loci4,5. These have identified some epidemiological features related to the subtype and to the prevalence of macrolide resistance in strains in different countries6. Although whole-genome sequence (WGS) analyses have provided phylogenetic information on TPA strains in several countries7–14, those data have not necessarily been accumulated enough for comprehension of the overall aspects of the TPA circulations in the world.
The annual number of syphilis cases in Japan exceeded 7000 in 2018, which was the highest annual number of syphilis cases in 50 years15. However, research on the genomic features of the TPA strains from these cases has not been reported. Therefore, we conducted molecular typing and WGS analyses of TPA strains detected between 2014 and 2018 in Tokyo and Osaka prefectures which accounted for about 40% of all the cases in Japan. Then, we reconstructed the phylogenetic relationships between these TPA genomes and the publicly available genomes in other countries. Moreover, the genetic variations and global relationships of TPA strains, and the transmission networks of syphilis have been discussed using syphilis epidemiological surveillance data and patient gender and sexual orientation data.
Results
Molecular typing
For this study, 139 Treponema pallidum genomes that satisfied the criteria described in “Methods” were used for phylogenetic analysis: 20 genomes of TPA strains detected in Japan and, in GenBank, 117 genomes of TPA strains and 2 genomes of Treponema pallidum subspecies pertenue (TPE) strains. The patient and clinical information, and molecular typing results of the 20 TPA strains detected in Japan are shown in Table 1. These 20 TPA strains were from 9 heterosexual males, 6 men who have sex with men (MSM), 4 heterosexual females, and 1 case with missing data on gender and sexual orientation. These 20 were selected as follows. We identified 49 TPA strains among the samples collected from patients in Japan with suspected syphilis between 2014 and 2018, that were Treponema pallidum DNA-positive, and had been fully typed by the Enhanced Centers for Disease Control and Prevention (ECDCT) typing4. These 49 strains were analyzed by WGS analysis, and 20 strains satisfied the criteria of at least 90% coverage of the Nichols genome and a minimum of 10 reads in the WGS analysis (see “Methods” for details). 117 previously published treponemal genomes met the same criteria and were included in our study. According to the ECDCT system, all the strains from heterosexual male and heterosexual female cases were the 14d/f subtype, and the strains from MSM cases included the predominant 14d/f subtype and several other subtypes (Table 1).
Table 1.
Clinical information and molecular typing results of 20 Japanese Treponema pallidum subspecies pallidum strains.
| Sample ID | Sample type | Year detected | Detection location | Patient gender | Patient sexual orientation | Genome coverage (%) | Lineage | Sub-lineage | arp type | tpr type | tp0548 type | Macrolide sensitivity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 14B001UK | Swab | 2014 | Tokyo | NA | NA | 93.7 | Nichols | – | 14 | d | c | S |
| 15A011MM | Swab | 2015 | Tokyo | Male | MSM | 98.4 | SS14 | 1B | 14 | e | f | S |
| 15A019HM | Swab | 2015 | Tokyo | Male | HM | 98.4 | SS14 | 8 | 14 | d | f | R |
| 16A013MM | Swab | 2016 | Tokyo | Male | MSM | 91.1 | SS14 | 1B | 14 | d | f | R |
| 17A003MM | Swab | 2017 | Tokyo | Male | MSM | 98.3 | Nichols | – | 14 | b | c | S |
| 17A014MM | Swab | 2017 | Tokyo | Male | MSM | 97.3 | SS14 | 1B | 14 | d | f | S |
| 17A017MM | Swab | 2017 | Tokyo | Male | MSM | 98.3 | SS14 | 1B | 14 | d | f | R |
| 17A021MM | Swab | 2017 | Tokyo | Male | MSM | 98.3 | Nichols | – | 12 | a | c | R |
| 17C005HM | Swab | 2017 | Tokyo | Male | HM | 90.8 | SS14 | 1B | 14 | d | f | R |
| 17C009HM | Swab | 2017 | Tokyo | Male | HM | 96.0 | SS14 | 1B | 14 | d | f | R |
| 17D002HM | Swab | 2017 | Osaka | Male | HM | 98.3 | SS14 | 1B | 14 | d | f | R |
| 17D006HM | Swab | 2017 | Osaka | Male | HM | 92.7 | SS14 | 1B | 14 | d | f | R |
| 17D009HM | Swab | 2017 | Osaka | Male | HM | 96.0 | SS14 | 1B | 14 | d | f | R |
| 17E002HF | Swab | 2017 | Tokyo | Female | HF | 95.2 | SS14 | 1B | 14 | d | f | R |
| 18C004HM | Swab | 2018 | Tokyo | Male | HM | 94.5 | SS14 | 1B | 14 | d | f | R |
| 18D001HM | Swab | 2018 | Osaka | Male | HM | 95.2 | SS14 | 1B | 14 | d | f | R |
| 18D002HM | Swab | 2018 | Osaka | Male | HM | 96.6 | SS14 | 1B | 14 | d | f | R |
| 18E001HF | Swab | 2018 | Tokyo | Female | HF | 97.8 | SS14 | 1B | 14 | d | f | R |
| 18E002HF | Swab | 2018 | Tokyo | Female | HF | 98.3 | SS14 | 1B | 14 | d | f | R |
| 18E016HF | Swab | 2018 | Tokyo | Female | HF | 98.2 | SS14 | 1B | 14 | d | f | R |
NA, data not available; MSM, men who have sex with men; HF, heterosexual female; HM, heterosexual male; S, sensitive; R, resistant.
Phylogenetic and temporal analyses
A maximum likelihood phylogenetic tree was reconstructed for the 135 TPA and 2 TPE genomes (Fig. 1): these strains were from 17 countries. Of the TPA genomes, 112 (including 83 macrolide-resistant genomes) were classified as SS14-lineage and 23 (including 6 macrolide-resistant genomes) as Nichols-lineage. For the strains in Japan, 17 strains (including 15 macrolide-resistant genomes) were SS14-lineage and the other 3 strains (including 1 macrolide-resistant genome) were Nichols-lineage. Subsequently, temporal analyses were performed with clinical and low-rabbit passaged strains (Table S1). Root-to-tip regression analysis of the116 genomes did not indicate a clock-like signal (slope rate = − 0.0001, R2 = 0.0017). Meanwhile, clock-like signals were indicated when the root-to-tip analysis was performed separately in SS14 and Nichols. To further confirm the temporal signal in our tree, we performed date-randomization test in each lineage. The test indicated that the temporal signal observed in SS14 lineage was not obtained by chance (Fig. S1). Therefore, we performed a temporal analysis using BEAST2 program with 102 genomes of SS14 lineage (Fig. 2a).
Figure 1.

Maximum likelihood phylogenetic analysis of 135 Treponema pallidum subspecies pallidum and 2 Treponema pallidum subspecies pertenue genomes. The boxes in the columns show (from the left) for each strain: its lineage, its country of origin, sexual orientation of the patient, and its macrolide resistance genetic profile. For macrolide resistance, if either the A2058G or A2059G mutation in the 23S rRNA genes was present, the strain was classified as resistant. If mixed alleles were detected in these sites, the strain was classified as ‘‘uncertain’’. The scale represents the number of substitutions per site.
Figure 2.


Bayesian maximum credibility phylogenetic analysis of 102 clinical Treponema pallidum subspecies pallidum genomes. (a) Time scaled phylogenetic tree of the TPA genomes in this study. (b) Expanded view of East Asian cluster-related node. The colored nodes represent the country of origin of the TPA genomes. The branch point nodes are shaded to indicate the posterior support (black ≥ 96%, dark grey ≥ 91%). The boxes on the right indicate the sexual orientation of the patient, the resistant mutations in the 23S rRNA genes, and the non-synonymous mutations in the penicillin-binding protein genes as shown in the legends. For the 23S rRNA genes, orange and green colors indicate the resistant-type and wild-type sites, respectively. For the penicillin-binding protein genes, pink and blue colors indicate the mutant and wild-type sites, respectively. The boxes with light-colored squares are single nucleotide polymorphism (SNP) sites that were not detected in the draft genome, and were determined by PCR-based Sanger sequencing with the primers shown in Table S2. The divergence estimates (95% HPD) of the time of each node are shown as follows: I, 1998 (1995–2002); II, 2002 (2000–2007); III, 2006 (2003–2009); IV, 2006 (2004–2010); V, 2007 (2006–2012); VI, 2008 (2006–2012); VII, 2009 (2008–2015); VIII, 2013 (2010–2016).
According to previous studies10,11, in addition to classification in the Nichols and SS14 lineages, as the SS14-lineage can be partitioned into two lineages (SS14Ω-A and SS14Ω-B) and these two lineages can be further partitioned into Sub-lineages 2–8 (SS14Ω-A) and Sub-lineages 1A/1B (SS14Ω-B), respectively, our results were in agreement with these classifications (Fig. 2a). The most recent common ancestor (MRCA) of the SS14-lineage was estimated to be extant between 1960 and 1980 (Fig. 2a). All of the SS14-lineage strains in Japan, except one, formed an East-Asian cluster (EAC), previously named Sub-lineage 1B11, consisting of 16 TPA strains detected in Japan and 11 TPA strains detected in China (Fig. 2a,b). The other SS14-lineage strain detected in Japan (strain 15A019HM, Fig. 2a) was in a sub-lineage that included TPA strains in European countries, although coming from a patient lacking information on nationality and a travel history.
Macrolide resistance and SNP analyses
With regard to the macrolide resistance mutations in Sub-lineage 1 genomes, unlike Sub-lineage 1A11 that contained four strains (UW186B, UW213B, UW228B, and UW254B) detected in the U.S, all of the macrolide-resistant strains in Sub-lineage 1B, the EAC, carried a single A2058G mutation in their 23S rRNA genes (Fig. 2b). In addition, 2 macrolide-sensitive strains were detected from MSM cases in Japan (derived from node V in Fig. 2b).
Various amino acid changes in the penicillin binding protein (PBP) in the Treponema genome have been reported9,11,12. Especially, Sun et al. discussed on the probable relationships between these amino acid changes among the strains in China and drug pressure of penicillin9. Although there has been no evidence for the hypothesis that those amino acid changes have some effect on penicillin resistance, they might be useful as phylogenetic markers. This prompted us to investigate those changes among the strains in Japan. Therefore, based on previous studies9,11, we selected four PBP genes (pbp1 (TPANIC_0500), pbp2 (TPANIC_0760), mrcA (TPANIC_0705), and Tp47 (TPANIC_0574)) for this study. The mrcA A506V mutation was a unique feature in the genomes of EAC strains and was in all the genomes of TPA strains detected in Japan and China. However, the mrcA A506V mutation was not found in the genomes of other sub-lineage genomes, except strain CZ27 (Fig. 2a,b). The variants associated with pbp2 (TPANIC_0760) were only found in the genomes of TPA strains in China (Fig. 2a,b), and most (9 of 11) of these strains harbored pbp2 (TPANIC_0760) I415F or I415M mutation. Among the EAC strains detected in Japan, no genetic difference in the four genes (i.e., pbp1, pbp2, mrcA and Tp47) was observed. In addition, among the eight Nichols-lineage strains, only one strain detected in Japan (17A021MM) carried the pbp1 P564L mutation (Fig. 2a). This was the first example of a Nichols-lineage strain that carried this mutation.
The genetic diversifications of TPA strains and calculations of MRCA in China and Japan
The EAC MRCA was estimated to have been extant in 2006 (95% Highest Posterior Density (HPD): 2003–2009, node III in Fig. 2b), with the TPA strains in China and Japan subsequently forming separate clusters (Fig. 2b). The MRCA of the clusters of TPA strains in China and Japan may have emerged in about 2006 (95% HPD: 2003–2009, node III in Fig. 2b) and 2007 (95% HPD: 2006–2012, node V in Fig. 2b, respectively. The 11 TPA strains in China (from 7 men, 2 women, and 2 cases with missing data)8,9,14 appeared to have increased their genetic diversity after the late 2000s. All but one of the TPA strains detected in Japan (15A011MM in Fig. 2a,b) formed distinct sub-clusters between strains from MSM cases and strains from heterosexual cases. Noticeably, the strain 15A011MM is very closely related to the strain in China, X-4, which was apparently separated from other strains in China (Fig. 2b).
The MRCA of the 3 TPA strains from MSM cases was likely to be extant in 2009 (2008–2015, node VII in Fig. 2b), and that of the 12 TPA strains from heterosexual cases was likely to be extant in 2013 (2010–2016, node VIII in Fig. 2b). The genomes of the TPA strains in Japan expanded their genetic diversity mainly after the mid-2010s (Fig. 2b).
Figure 3 shows the epidemiological trends of the primary and secondary syphilis cases reported in Japan and China between 1995 and 2016. Considering the epidemiological data and the results of the Bayesian temporal analysis for the EAC genomes, the genomes of the TPA strains in China appeared to have expanded their genetic diversity after about 2005 (Fig. 2b), which corresponded to the time of the second wave of syphilis cases in China (Fig. 3). The increase in the number of syphilis cases in Japan was after 2013 (Fig. 3), which corresponded to the time of increasing genetic diversity in TPA strains in Japan (Fig. 2b).
Figure 3.
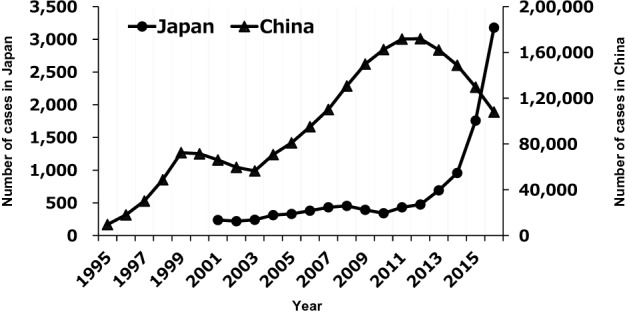
Number of primary and secondary syphilis cases reported in Japan and China, 1995–2016. The number of primary and secondary syphilis cases reported in Japan (plotted with circles, left scale) and in China (plotted with triangles, right scale). The original data were derived from The Japan National Tourism Organization (https://www.jnto.go.jp/jpn/statistics/visitor_trends/index.html).
Discussion
In this report, we have described the genomic and phylogenetic features of TPA strains detected in Japan compared to TPA strains detected in other countries, in particular in China. A significant feature of this study was that our analysis included information on the gender and sexual orientation of the syphilis patients from whom TPA strains had been detected.
The maximum likelihood phylogenetic analysis and the Bayesian temporal analysis of the genomes of global TPA strains in this study found similar results to previous reports in terms of lineages and sub-lineages. In those reports, most of the SS14-lineage strains in American and European countries were classified in lineages SS14Ω-A10,11.
The majority of TPA strains analyzed in this study in Japan (16 of 20) were classified in the SS14-lineage and formed an EAC, designated Sub-lineage 1B (in lineage SS14Ω-B) in a previous study11, that included strains in China. In addition to these strains, there were 3 Nichols-lineage strains and a strain belonging to another SS14 sub-lineage, previously designated as Sub-lineage 8, which contained strains in the U.S. and European countries11. WGS indicated an ongoing concurrent circulation of Nichols- and SS14-lineage strains in Japan, as has also been observed in several American and European countries10,11. Recently, four TPE strains from Japan were reported between 2014 and 201816. However, the definitions of subspecies of those strains were based on the sequencing of tp0548 and tp0856 genes, but were not based on WGS analyses16. Although we could not detect any TPE strain among the strains that passed our criteria described above and in “Methods”, we have to keep monitoring the emergence and spread of TPE strains in Japan.
The EAC was composed of SS14-lineage TPA strains in China and Japan, with most TPA strains in Japan (16/17) being subtype 14d/f, which is the dominant subtype among both heterosexual and MSM cases in Japan17. We could not evaluate the difference between subtype 14d/f strains from MSM and heterosexual cases in our previous molecular typing studies17,18. The WGS study in this report elucidated the separate phylogeny of the strains from MSM and heterosexuals belonging to this same subtype (Table 1, and Fig. 2b).
These results underlined the importance of having information on gender and sexual orientation in analyzing WGS studies of TPA to comprehend the detailed aspect of the circulations of strains in the respective communities of MSM and heterosexuals.
From the geographical point of view, the experimental results for strains from heterosexual cases collected in Tokyo and Osaka prefectures were mixed, showing that genetically similar strains were circulating among heterosexuals in these two prefectures that have the largest populations in Japan.
Based on the phylogenetic analyses with the strains collected since 2011 to 2018 (Table S1, strains in China and Japan), the MRCA of the EAC strains and of strains in China appeared to emerge in 2006 (node III in Fig. 2b), followed by the MRCA of strains in Japan in 2007 (node V in Fig. 2b). Therefore, the EAC has separated into Chinese and Japanese clusters since the mid 2000s. The estimated time of emergence of the MRCA of the Chinese cluster, determined in this study, was similar to that in a recent report11. The genomes of TPA strains in China and Japan then expanded their genetic diversity after the late 2000s and mid-2010s, respectively, which approximately corresponded to the time each country had an increasing number of primary and secondary syphilis cases (Fig. 3). This correspondence may be consistent with the fact that TPA is an obligate human pathogen and its accumulation of SNP sites increases with time. However, for the genomes of TPA strains in China, there was a discrepancy in that there was not an increase in their genetic diversity from the mid-1990s to the early 2000s (Fig. 2b), although the onset of the increase in the number of cases was in the late 1990s (Fig. 3). This fact may be attributed to detection bias owing to a limited number of samples included in this study. The simplest explanation is that genomes of TPA in China during the early stage of the syphilis outbreak had not been collected or analyzed systematically enough.
Since contagious pathogens can cross borders, the EAC may be an example of cross-border propagation of TPA strains between Japan and China. Although the cause of the current large syphilis outbreak in Japan may be uncertain, an increase in the number of travelers from China to Japan has been noted. The Japan National Tourism Organization (https://www.jnto.go.jp/jpn/statistics/visitor_trends/index.html) has reported that the number of Chinese travelers to Japan has increased rapidly since 2014, which corresponded with the onset of the recent syphilis outbreak (Fig. 3) mainly among heterosexuals in Japan. However, our results indicated that the MRCA of the TPA strains in the sub-cluster formed by the strains from heterosexuals in Japan was likely to be extant in 2013 (node VIII in Fig. 2b). In addition, based on their phylogeny, the MRCA of TPA strain 15A011MM and the strains from heterosexuals in Japan may have been extant in 2007 (node V in Fig. 2b). Therefore, we consider the hypothesis of a connection between Chinese tourists and the syphilis outbreak in Japan controversial, although our interpretation is based on a limited number of samples and on values with a wide (6–7 year) 95% HPD (Fig. 2b).
The results of this study indicated several features about the genetic variants of some TPA genes. First, macrolide resistance mutations in TPA might be reversible, although there have been no data indicating reversion to the wild-type allele. As all the strains in Japan in Fig. 2b were phylogenetic branches from node V, it seems more likely that macrolide resistance was extant at the time node V formed, rather than that most of the strains in Fig. 2b independently mutated to macrolide resistance during the relatively short time after node V formed. Macrolide resistance mutations have been noted previously to have strong stability11,19, but existence of 2 macrolide-sensitive TPA strains in Japan (Fig. 2b) implied that there might have been a reverse mutation because of a fitness cost associated with carrying those mutations. However, a fitness cost- hypothesis contradicts the fact that most of the strains in Japan depicted in Fig. 2b still keep the resistance mutations. So, we could not exclude the possibility that the apparent ‘reverse mutations’ occurred independent of the putative fitness cost during the diversifications as other general SNPs did and have been kept under the absence of the drug. This scenario might meet the fact that azithromycin is no longer recommended for treatment to syphilis in most of the countries in the world.
Second, all the macrolide-resistant strains in Japan in this study carried the A2058G mutation in the 23S rRNA gene, but not the A2059G mutation. This was in agreement with the results of our previous study of over 100 strains in Japan17,18.
Third, in this study, the mrcA A506V mutation, which was considered to be unique to strains in China9,11, was also observed in EAC strains in Japan. However, some mutations in the pbp2 gene (TPANIC_0760) were not identified in any of the strains in Japan, while most of the strains (9 of 11) in China harbored at least 1 mutation in this gene. Of the 3 SNPs (i.e., A366T, I415F, and I415M) in the TPANIC_0760 gene, only I415F has been suggested to have a deleterious effect on the protein’s structural flexibility or its binding constant for substrate stability9. However, the effect of these SNPs on the possible generation of the penicillin-resistance is not known at present, because there has been no documentation of penicillin-resistant TPA strain so far, although penicillin has been used extensively to treat syphilis for more than 70 years.
Apart from that, in this study, the separation between the clusters of TPA strains in China and in Japan shown by the phylogenetic analyses was found, for most of the strains, in the SNP analyses of the TPANIC_0760 gene, although there were 2 exceptional strains in China that had no mutation in this gene (SMUTp_04 and X-4, Fig. 2b). In this context, it is noticeable that the strain X-4 was one of those 2 exceptional strains in China, and was phylogenetically separated from other strains in China (Fig. 2b). This strain was, rather, very closely related to a Japanese strain 15A011MM forming a small sub-cluster which was branch from node VI (Fig. 2b). These lines of information implied that this small sub-cluster might reflect the limited case(s) of direct international spread of TPA between China and Japan by the ‘Japanese type’ strain(s) (with TPANIC_0705 A506V mutation and without any mutation in TPANIC_0760 gene) in the recent year.
Finally, this study confirmed a Nichols-lineage strain carrying the pbp1 P564L mutation. This SNP has been commonly observed in, and limited to, the genomes of SS-14 strains9,11. The study reported here strongly suggested that TPA strains in any lineage could carry mutations in pbp1.
In conclusion, most of the TPA strains in Japan in this study had a close relationship to TPA strains in China, forming the EAC. The MRCA of the EAC is likely to have become extant about 2006. The TPA strains in China and Japan subsequently formed a separate cluster in each country in about 2007. The genomes of the TPA strains in each country then expanded their phylogenetic diversity during the time that country had an increasing number of syphilis cases. In addition, phylogenetic analysis showed that TPA strains from MSM cases in Japan clustered separately from strains from heterosexual cases. These findings, within the context of the recent global resurgence of syphilis, provide a better understanding of the phylogenetic features and transmission networks of syphilis, both domestic and global.
Methods
Clinical samples
Samples were collected from patients with suspected syphilis between 2014 and 2018 in four clinics and one hospital in Tokyo and Osaka prefectures, along with information on each patient’s gender and self-reported sexual orientation. These information were collected voluntarily. Other information of the patients was not systematically collected. It was suggested that most of the patients were in the primary or secondary stage, although not necessarily specified. This study was approved by the institutional review board of the National Institute of Infectious Diseases (approval numbers 508 and 705), and all the experiments were performed in accordance with the directions from the review board above. The informed consent was obtained from all participants.
Detection, DNA extraction, and molecular typing of treponemal DNA
Tris–EDTA (TE) buffer-suspensions (swab samples from genital, anal, and oral lesions) and DNA extracts (the other samples) prepared by a DNeasy Blood & Tissue Kit (Qiagen, Inc., Valencia, CA) were used as the PCR templates. The TPA polA and tp47 genes-targeted PCR was performed as described previously17,18. When at least one amplicon of the two genes was generated, a sample was considered to contain TPA-DNA. Molecular typing according to the ECDCT system4 and determining the presence of the A2058G and/or A2059G mutations in one or both of the TPA 23S rRNA genes were performed by short-read mapping as described below. For the Japanese strains which showed less than genome × 50 coverage in 23S rRNA genes (Table S1), Sanger sequencing was performed and no discrepancies between the two methods were confirmed.
Whole-genome sequence and phylogenetic analyses
For WGS analysis, we selected 49 TPA strains with complete typing results in the ECDCT system4, which had been previously detected in Japan. Extracted DNA of the clinical samples of these strains was amplified evenly using the Illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare UK Ltd., Buckinghamshire, UK) by the functions of random hexamer nucleotides and the bacteriophage phi-29 DNA polymerase. The amplified DNA was served for fragmentation and simultaneous transfer of the adaptor tags by transposase, followed by selective concentration of TPA genomic DNA using a SureSelect QXT Target Enrichment Kit and Custom Capture Library RNA (Agilent Technologies, Ceder Creek, TX, USA) for the TPA strain Nichols genome sequence by the hybridization to Library RNA in the tube. These procedures were all performed following the manufacturer’s directions. The pooled libraries were analyzed by multiplex paired-end sequencing (300-mer × 2) using the MiSeq System (Illumina, San Diego, CA, USA). The short-reads were mapped to the complete genome of TPA strain Nichols (NCBI accession no. NC_021490.2). The genomes of 20 of the 49 TPA strains met our criteria of at least 90% coverage of the Nichols genome and a minimum of 10 reads. In addition, published treponemal genomes that met our criteria (Table S1) were included in our analyses. These short-reads were mapped to the Nichols genome using bwasw function of bwa v.0.7.1720 and SNPs were identified using samtools v.1.921 and VarScan v.2.4.322 as previously described23. The default parameters were used for SNP identification except that the minimum coverage was set to ten. Repeat and recombinogenic regions were detected by MUMmer v.3.225924 and gubbins25, respectively, and were removed from further analyses. Phylogenetic relationships were determined by reconstructing a phylogenetic tree by the maximum likelihood method using IQ-TREE with 1000 ultrafast bootstrap replicates26.
For temporal analyses, the Bayesian Evolutionary Analysis Sampling Tree (BEAST) v. 2.6.1 program27 was used. Whole genome sequences of all the TPA strains detected in Japan in this study and TPA genomes in the database with no or limited passages (Table S1) were used for this analysis as previously described11. Briefly, a recombination-masked SNP alignment was analyzed using a GTR4 substitution model and a strict clock model with a burnin of 10 million cycles followed by 100 million MCMC cycles. The starting molecular clock rate was set to 3.6 × 10–4 as described previously11. Maximum likelihood and BEAST trees were plotted using a ggtree and a ggplot package of R v. 3.6.228. To confirm the temporal signal in our tree, we used TempEst v.1.5.3 for root-to-tip analysis and the TIPDATINGBEAST package29 in R for date-randomization test as previously described11. Twenty new datasets with randomly assigned dates were analyzed by BEAST and the results showed no evidence of temporal signal in these replicates, suggesting that the signal in our tree was not found by chance.
Macrolide resistance SNPs were determined by mapping the short reads to the Nichols genome. For identification of non-synonymous variants of penicillin binding protein (PBP) genes pbp1 (TPANIC_0500), pbp2 (TPANIC_0760), mrcA (TPANIC_0705) and Tp47 (TPANIC_0574), the short-reads were assembled using the SPAdes v. 3.13.130. The PBP gene sequences were extracted from the draft genomes. In the cases of those variants could not be determined from draft genomes, the variants were determined by PCR-based Sanger sequencing of the initial DNA samples with the primers shown in Table S2.
Epidemiological data on syphilis cases in Japan and China
Information on syphilis cases in Japan between 2001 and 2016 was obtained from public annual reports provided by the National Institute of Infectious Diseases website31. Information on syphilis cases in China between 1995 and 2016 was obtained from a published article32. Using these data, we determined the number of primary and secondary syphilis cases, which generally indicate more recent infections and can be a more timely indication of the ongoing spread of syphilis at that time than latent syphilis or asymptomatic cases.
Supplementary Information
Acknowledgements
This work was supported by grants from Ministry of Health, Labour, and Welfare [grant numbers H28-Shinkou-Ippan-008, H30-Shinkou-Ippan-004], and the Ministry of Education, Culture, Sports, Science and Technology of Japan [grant number 18K09184]. We thank Hiroshi Kameoka, Ichiro Itoda, Igen Hongo, Masayuki Sawamura, and Takashi Hamada, who generously and continually provided us with specimens from clinically suspected syphilis cases. We also thank Kimiko Matsumoto and Ai Yoshida for their technical support in the detection and typing of T. pallidum subsp. pallidum, including the PCR procedures and Yu Takizawa for support in the genome analyses.
Author contributions
S.N., K.L., M.K., and S.I.N. designed this study, set up the operational aspects of this study, performed the laboratory work, and analyzed the data. M.O. contributed to the design of this study and supervised this project. All authors contributed to the writing and reviewed this manuscript. All authors read and approved the final manuscript.
Data availability
Accession numbers Short read data have been deposited in GenBank under accession number DRA009733.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shingo Nishiki and Kenichi Lee.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-82337-7.
References
- 1.Newman L, et al. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS ONE. 2015;10:e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fenton KA, et al. Infectious syphilis in high-income settings in the 21st century. Lancet Infect. Dis. 2008;8:244–253. doi: 10.1016/S1473-3099(08)70065-3. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi T, et al. Rapid increase in reports of syphilis associated with men who have sex with women and women who have sex with men, Japan, 2012 to 2016. Sex. Transm. Dis. 2018;45:139–143. doi: 10.1097/OLQ.0000000000000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marra C, et al. Enhanced molecular typing of Treponema pallidum: Geographical distribution of strain types and association with neurosyphilis. J. Infect. Dis. 2010;202:1380–1388. doi: 10.1086/656533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grillová L, et al. Molecular characterization of Treponema pallidum subsp. pallidum in Switzerland and France with a new multilocus sequence typing scheme. PLoS ONE. 2018;13:e0200773. doi: 10.1371/journal.pone.0200773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tipple C, Taylor GP. Syphilis testing, typing, and treatment follow-up: A new era for an old disease. Curr. Opin. Infect. Dis. 2015;28:53–60. doi: 10.1097/QCO.0000000000000124. [DOI] [PubMed] [Google Scholar]
- 7.Pětrošová H, et al. Resequencing of Treponema pallidum ssp. pallidum strains Nichols and SS14: Correction of sequencing errors resulted in increased separation of syphilis treponeme subclusters. PLoS ONE. 2013;8:e74319. doi: 10.1371/journal.pone.0074319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tong ML, et al. Whole genome sequence of the Treponema pallidum subsp. pallidum strain Amoy: An Asian isolate highly similar to SS14. PLoS ONE. 2017;12:e0182768. doi: 10.1371/journal.pone.0182768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun J, et al. Tracing the origin of Treponema pallidum in China using next-generation sequencing. Oncotarget. 2016;7:42904–42918. doi: 10.18632/oncotarget.10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arora N, et al. Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster. Nat. Microbiol. 2016;2:16245. doi: 10.1038/nmicrobiol.2016.245. [DOI] [PubMed] [Google Scholar]
- 11.Beale MA, et al. Genomic epidemiology of syphilis reveals independent emergence of macrolide resistance across multiple circulating lineages. Nat. Commun. 2019;10:3255. doi: 10.1038/s41467-019-11216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto M, et al. Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation. Nat. Microbiol. 2016;2:16190. doi: 10.1038/nmicrobiol.2016.190. [DOI] [PubMed] [Google Scholar]
- 13.Grillova L, et al. Directly sequenced genomes of contemporary strains of syphilis reveal recombination-driven diversity in genes encoding predicted surface-exposed antigens. Front. Microbiol. 2019 doi: 10.3389/fmib.2019.0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen W, et al. Analysis of Treponema pallidum strains from China using improved methods for whole-genome sequencing from primary syphilis chancres. J. Infect. Dis. 2020 doi: 10.1093/infdis/jiaa449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujikura H, et al. Syphilis notification rate in Japan, 2004–2018. Jpn. J. Sex. Transm. Infect. 2019;30:1–7. [Google Scholar]
- 16.Kawahata T, et al. Bejel, a nonveneral Treponematosis, among men who have sex with men, Japan. Emerg. Infect. Dis. 2019;25:1581–1582. doi: 10.3201/eid2508.181690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai M, et al. Molecular typing and macrolide resistance analyses of Treponema pallidum in heterosexuals and men who have sex with men in Japan, 2017. J. Clin. Microbiol. 2019;57:e01167–e1218. doi: 10.1128/JCM.01167-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nishiki S, et al. Epidemiology, molecular strain types, and macrolide resistance of Treponema pallidum in Japan, 2017–2018. J. Infect. Chemother. 2020;26:1042–1047. doi: 10.1016/j.jiac.2020.05.022. [DOI] [PubMed] [Google Scholar]
- 19.Stamm LV. Syphilis: Antibiotic treatment and resistance. Epidemiol. Infect. 2015;143:1567–1574. doi: 10.1017/S0950268814002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;2011(27):2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koboldt DC, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee K, Izumiya H, Iyoda S, Ohnishi M. Effective surveillance using multilocus variable-number tandem-repeat analysis and whole-genome sequencing for enterohemorrhagic Escherichia coli O157. Appl. Environ. Microbiol. 2019;85:e00728–e819. doi: 10.1128/AEM.00728-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Croucher NJ, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole-genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouckaert R, et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019;15:e1006650. doi: 10.1371/journal.pcbi.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Core Team. 2019. R: a language and environment for statistical computing (R Foundation for Statistical Computing, Vienna) http://www.R-project.org/, Accessed 14 Feb 2020.
- 29.Rieux A, Khatchikian CE. tipdatingbeast: An r package to assist the implementation of phylogenetic tip-dating tests using beast. Mol. Ecol. Resour. 2017;17:608–613. doi: 10.1111/1755-0998.12603. [DOI] [PubMed] [Google Scholar]
- 30.Bankevich A, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Infectious Disease Surveillance Center, National Institute of Infectious Diseases and Tuberculosis and Infectious Diseases Control Division, Ministry of Health, Labour and Welfare. National Epidemiological Surveillance of Infectious Diseases (NESID) Program annual reports, 2001–2016. https://www.niid.go.jp/niid/ja/allarticles/surveillance/2270-idwr/nenpou/8563-kako2017.html, Accessed 14 Feb 2020.
- 32.Tao Y, et al. A nationwide spatiotemporal analysis of syphilis over 21 years and implications for prevention and control in China. Clin. Infect. Dis. 2020;70:136–139. doi: 10.1093/cid/ciz331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers Short read data have been deposited in GenBank under accession number DRA009733.