

Abstract
Background:
Arteriosclerosis obliterans (ASO) is a major cause of adult limb loss worldwide. Autophagy of vascular endothelial cell (VEC) contributes to the ASO progression. However, the molecular mechanism that controls VEC autophagy remains unclear. In this study, we aimed to explore the role of the GRB2 associated binding protein 1 (GAB1) in regulating VEC autophagy.
Methods:
In vivo and in vitro studies were applied to determine the loss of adapt protein GAB1 in association with ASO progression. Histological GAB1 expression was measured in sclerotic vascular intima and normal vascular intima. Gain- and loss-of-function of GAB1 were applied in VEC to determine the effect and potential downstream signaling of GAB1.
Results:
The autophagy repressor p62 was significantly downregulated in ASO intima as compared to that in healthy donor (0.80 vs. 0.20, t = 6.43, P < 0.05). The expression level of GAB1 mRNA (1.00 vs. 0.24, t = 7.41, P < 0.05) and protein (0.72 vs. 0.21, t = 5.97, P < 0.05) was significantly decreased in ASO group as compared with the control group. Loss of GAB1 led to a remarkable decrease in LC3II (1.19 vs. 0.68, t = 5.99, P < 0.05), whereas overexpression of GAB1 significantly led to a decrease in LC3II level (0.41 vs. 0.93, t = 7.12, P < 0.05). Phosphorylation levels of JNK and p38 were significantly associated with gain- and loss-of-function of GAB1 protein.
Conclusion:
Loss of GAB1 promotes VEC autophagy which is associated with ASO. GAB1 and its downstream signaling might be potential therapeutic targets for ASO treatment.
Keywords: Arteriosclerosis obliterans, Autophagy, GRB2 associated binding protein 1, JNK pathway, p38 kinase pathway
Introduction
Arteriosclerosis obliterans (ASO) is one of the most prevalent peripheral arteriosclerotic vascular diseases.[1] The etiology of ASO has not been fully understood. Several individual risk factors have been reported to contribute to the pathogenesis of ASO including smoking, hypertension, hyperlipidemia, diabetes mellitus, and hyperhomocysteinemia.[2] Current therapeutic approaches include by-pass surgery and endovascular intervention. Even though initially effective, high restenosis rates post-treatment remains an obstacle for long-term recovery.[3]
Autophagy is a vital catabolic process that degrades cytoplasmic components within the lysosome, which has been observed in the majority types of human cells including vascular endothelial cells (VEC).[4] In addition to its role as a stress response pathway, autophagy also acts as a quality control system that promotes the basal turnover of long-lived proteins and organelles as well as selectively degrading damaged cellular components.[5] Therefore, dysregulation of autophagy is associated with various human diseases, such as neurodegenerative disease, cancer, and cardiovascular disease.[6–11]
The GRB2 associated binding protein 1 (GAB1) belongs to the insulin receptor substrate (IRS1)-like docking adapter protein family.[2,3] Like other IRS1 adapter proteins, GAB1 has a PH domain, that is, involved in assembling complexes downstream of cell-surface receptor tyrosine kinase (RTK). Activated RTK signaling controls a variety of critical cellular processes including cell-cycle progression, differentiation, metabolism, survival, adhesion, motility, and migration. In this study, we found that GAB1 which plays a critical role in the regulation of VEC autophagy was downregulated in ASO patients. In vitro experiments demonstrated that GAB1 inhibited VEC autophagy through JNK and p38 pathway. The association between a loss of GAB1 and excessive autophagy may provide a new direction for further drug development.
Methods
Ethical approval
The investigations were conducted according to the Declaration of Helsinki principles. The study was approved by the ethical committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University, Shanghai, China (No. RA-2020-071). The written informed consents were obtained from everyone who participates in this study.
Patients and healthy controls
We enrolled 11 patients diagnosed with ASO. The diagnosis of ASO of the lower extremity was made by history and physical examination. Ankle/brachial index and pulse wave velocity are widely used as complementary parameters to diagnose ASO. All ASO patients had the characteristic complaints of chronic limb ischemia, intermittent claudication, rest pain, or non-healing ischemic ulcers (Fontaine III, n = 2; Fontaine IV, n = 9) as confirmed by angiography. Individuals in our health examination center without any of the above symptoms were recruited as healthy controls. Sclerotic intima was collected, and the normal intima from amputation patients was used as control. The intima samples were stored at −80°C.
Cell culture and transfection
Human umbilical vein endothelial cells (HUVEC) were cultured within Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Life Technologies, Grand Island, NY, USA) as previously described.[12] For complementary DNA (cDNA) or small interfering RNA (siRNA) transfection, HUVEC cells were transiently transfected with plasmids or siRNAs using Lipofectamine® 2000 (Life Technologies) as previously described (Clonetics, San Diego, CA, USA).[4] The siRNA against GAB1 was purchased from Dharmacon (Lafayette, CO, USA).
Quantitative polymerase chain reaction
Total RNA was extracted using the RNeasy Mini kit (Qiagen, Valencia, CA, USA). cDNA was synthesized using the SuperScript III First-Strand Synthesis kit following the manufacturer's protocol (TaKaRa, Shiga, Japan). Quantitative polymerase chain reaction (PCR) targeting GAB1 was performed in a 20 μL reaction containing 1 μg of cDNA template, 1 μL of SYBR Green probe (TaKaRa). The PCR reactions were performed on a ViiA 7 Real-Time PCR System (Applied Biosystems, Waltham, Massachusetts, USA) with the following condition: 50°C, 2 min; 95°C, 10 min; 40 cycles of 95°C, 15 s and 60°C, 1 min. Samples were run in triplicate concurrently with a 10× serial dilution of sample cDNA as a standard.
Western blotting analysis
The vascular tissues were ground under liquid nitrogen and then suspended in lysis buffer. Western blotting was performed as previously described.[13] Anti-β-actin and anti-GAPDH antibodies were obtained from Cell signaling Technology (#4970,#5174, CST, USA). The anti-GAB1 antibody was purchased from Cell signaling Technology (#3232, CST, USA). Anti-p-ERK1/2, the antibody was acquired from Cell signaling Technology (#4370, CST, USA).
Immunofluorescence (IF)
Tissues were fixed with 4% paraformaldehyde, followed by permeabilization with 0.1% Triton X-100 and blocked with 5% bovine serum album plus Tween 20. Coverslips were then incubated with primary antibodies at 4°C overnight, followed by incubation with secondary antibodies at room temperature for about 1 h. After washing with PBS, the coverslips were counterstained with 4′,6-Diamidino-2-phenylindole (#H1200; Vector Laboratories, Burlington, ON, Canada). Images were visualized by using a Leica SP2 AOBS inverted confocal laser scanning microscope (Leica, Wetzlar, Germany).
Statistical analysis
Statistical analyses were performed using SPSS 23.0 (International Business Machines Corporation, IBM, USA). Normally distributed continuous data were expressed as mean ± standard deviation. T-test or Mann-Whitney U-test was used to test the differences between the two groups. All results presented are representative of at least three independent experiments. Statistical analysis was performed with an unpaired Student's t test to test the differences between the two groups. A P value of <0.05 was considered as statistically significant.
Results
Patient demographics
From 2013 to 2016, a total of 11 patients with an average age of 50.5 (35–76) years were enrolled in the clinical study [Table 1]. The occluded segments of each patient were located at different places including superficial artery, popliteal artery, or both. The risk factors are smoking, hypercoagulable states, diabetes mellitus, high cholesterol levels, and atrial fibrillation.
Table 1.
Clinical information of patients with arteriosclerosis obliterans.
| No. | Age (years) | Fontaine classification | Symptoms | Previous operation | Occluded level | Risk factors |
| 1 | 76 | IV | Toe gangrene and rest pain | PTA + STENT | SFA, ATA | Hypertension |
| 2 | 72 | IV | Toe ulcer and rest pain | PTA + STENT | PA | Hypertension, diabetes mellitus |
| 3 | 64 | IV | Toe ulcer and rest pain | PTA | PA, PTA | Hypertension, diabetes mellitus |
| 4 | 71 | IV | Foot bottom ulcer and rest pain | PTA + STENT | SFA, ATA | High cholesterol level, smoking |
| 5 | 75 | IV | Forefoot gangrene and rest pain | PTA + STENT | SFA, PA | Hypertension, smoking |
| 6 | 55 | III | Rest pain | None | SFA | Hypertension |
| 7 | 54 | III | Rest pain | None | ATA, PTA | Diabetes mellitus |
| 8 | 58 | IV | Toe ulcer and rest pain | PTA + STENT | SFA, PA | Hypertension, diabetes mellitus, smoking |
| 9 | 73 | IV | Toe ulcer and rest pain | PTA + STENT | EIA, SFA | Diabetes mellitus |
| 10 | 63 | IV | Toe gangrene and rest pain | PTA + STENT | EIA, ATA, PTA | Hypertension, diabetes mellitus, smoking |
| 11 | 61 | IV | Toe gangrene and rest pain | PTA + STENT | SFA, ATA | Diabetes mellitus |
ATA: Anterior tibial artery; EIA: External iliac artery; PAP: Popliteal artery; PTA: Percutaneous balloon angioplasty; PTA: posterior tibial artery; SFA: Superficial femoral artery.
Loss of GAB1 is associated with VEC autophagy and ASO progression
It has been well-documented that GAB1 serves as a critical regulatory component of VEC migration and capillary formation, and loss of GAB1 accelerates vascular inflammation.[14] To determine whether GAB1 also plays a regulatory role in ASO progression, we compared the GAB1 protein level in the vascular endothelial lesion of ASO patients with control vascular endothelium using immunoblotting. The expression level of GAB1 was almost three fold lower in ASO groups as compared with the control group (0.72 vs. 0.21, t = 5.97, P < 0.05) [Figure 1A]. Furthermore, real time-qPCR results showed that the GAB1 mRNA level in the ASO group was significantly decreased compared to the control group (1.00 vs. 0.24, t = 7.41, P < 0.05), suggesting downregulation of GAB1 in the arteries was due to transcriptional regulation [Figure 1B]. Moreover, immunofluorescence (IF) analysis confirmed that the loss of GAB1 predominantly happened within the vascular intima in ASO arteries [Figure 1C]. Taken into consideration, these results indicated that loss of GAB1 within the vascular intima was highly associated with ASO progression.
Figure 1.
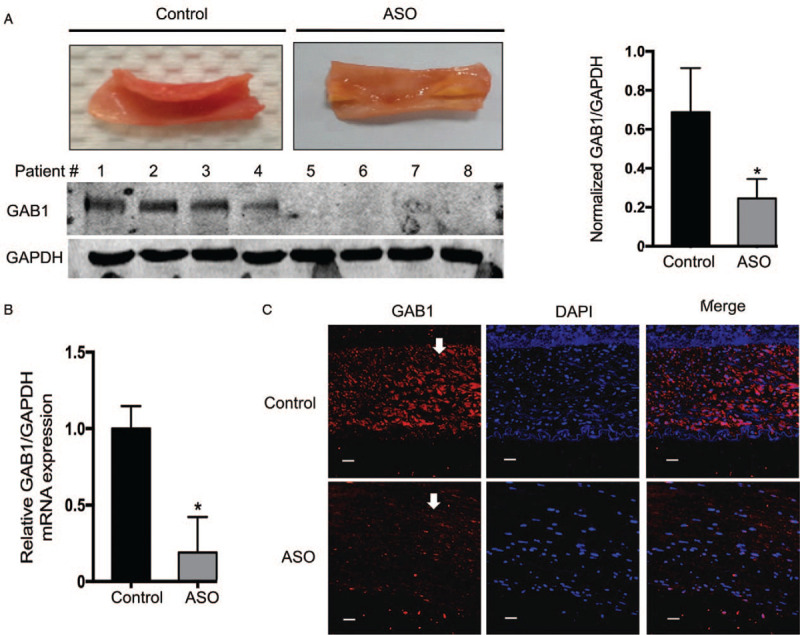
Expression of GAB1 in ASO patients. (A) Sclerotic intima was collected from four patients clinically diagnosed with ASO and the normal intima obtained from amputation patients was used as a control. The tissues were homogenized in RIPA buffer with a complete protease inhibitor. The lysates were processed for Western blotting for detecting the protein expression level of GAB1. The protein expression level of GAPDH was used as a loading control. (B) Protein levels of GAB1 were quantitated by means of densitometric analysis using Image J normalized to GAPDH and presented as fold changes compared with the control group (mean ± standard deviation, n = 4). ∗P < 0.05. (C) Immunocytochemical staining was conducted using anti-GAB1 (Red). Nuclei were counterstained by DAPI (Blue). Scale bars: 10 μm. ASO: Arteriosclerosis obliterans; DAPI: 4′,6-Diamidino-2-phenylindole; GAB1: GRB2 associated binding protein 1; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; RIPAA: Radioimmunoprecipitation assay.
Next, we questioned whether VEC autophagy altered within ASO intima. Surprisingly, we found that the autophagy repressor p62, also known as Sequestosome-1, was significantly downregulated in ASO intima as compared to control (0.80 vs. 0.20, t = 6.43, P < 0.05) [Figure 2A]. Furthermore, IF analysis demonstrated that LC3II, a component of autophagosome was significantly upregulated in the endothelium of ASO intima [Figure 2B]. These results suggesting that autophagy is significantly increased in VEC of ASO intima. Taken all together, in ASO intima, loss of GAB1 is associated with VEC autophagy and ASO progression.
Figure 2.
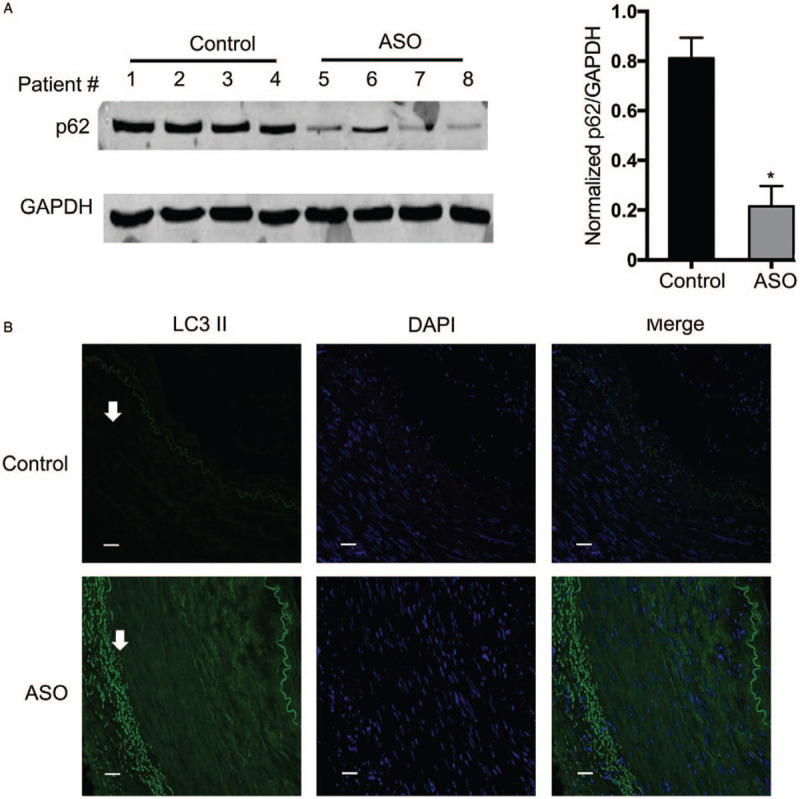
Autophagy is upregulated in ASO patients. (A) The same cell lysates were used as described in Figure 1 and processed for Western blotting for detecting the protein expression level of p62. The protein expression level of GAPDH was used as a loading control. Protein levels of p62 were quantitated by densitometric analysis using Image J normalized to GAPDH and presented as fold changes compared with the control group (mean ± standard deviation, n = 4). ∗P < 0.05. (B) Immunocytochemical staining was conducted using anti-LC3 II (Green). Nuclei were counterstained by DAPI (Blue). Scale bars: 10 μm. ASO: Arteriosclerosis obliterans; DAPI: 4′,6-Diamidino-2-phenylindole; GAB1: GRB2 associated binding protein 1; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase.
GAB1 negatively regulates autophagy in VEC
Autophagy dysregulation has been found to promote atherosclerosis in part through inflammasome hyperactivation.[15,16] It is also reported that autophagy can be regulated by insulin growth factor 1 receptor (IGF-1R) signaling.[17]GAB1 is a key downstream adapt protein of IGF-1R signalling; so the loss of GAB1 may contribute to arteriosclerosis through inducing VEC autophagy. To validate this hypothesis, we knocked down GAB1 in HUVECs and found that loss of GAB1 led to a remarkable increase of LC3II, and this effect cannot be reversed by the autophagy inducer Hank balanced salt solution (HBSS) (1.19 vs. 0.68, 0.46 vs. 0.14, and 0.54 vs. 0.40) [Figures 3A and 3C]. Correspondingly, overexpression of GAB1 significantly decreased LC3II level (0.41 vs. 0.93, 0.17 vs. 0.34, and 0.21 vs. 0.60) [Figures 3B and 3D]. These results suggested that GAB1 plays a negative role in regulating VEC autophagy.
Figure 3.
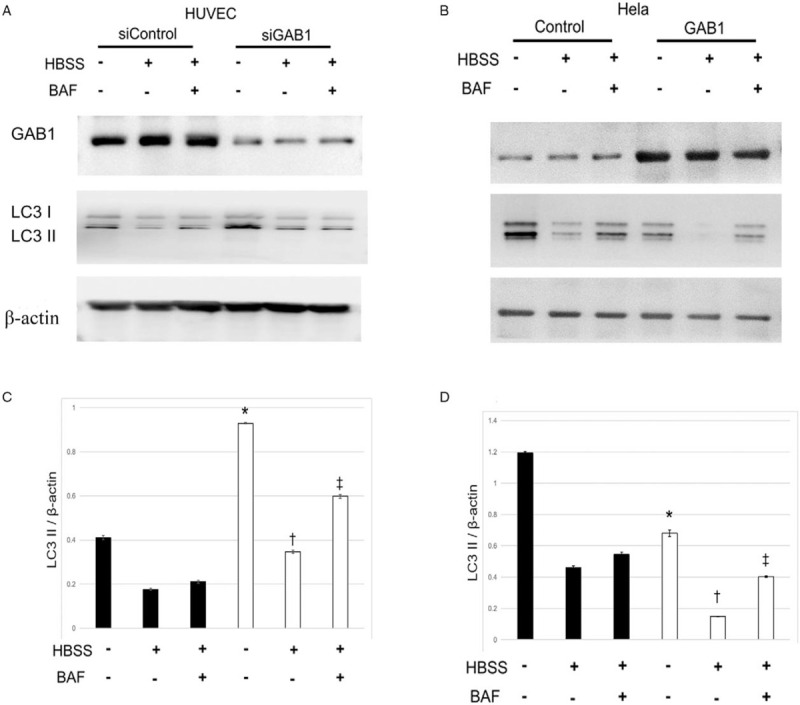
GAB1 natively regulates autophagy. (A) HUVECs were treated with either control siRNA (siControl) or GAB1-targeting siRNA (siGAB1) for 48 h, followed by treatment of HBSS for 6 h for the induction of autophagy presented with or without BAF. Cell lysates were harvested to examine the protein levels of GAB1 (using anti-GAB1 antibody), LC3I/II, and β-actin by Western blotting. (B) HeLa cells were transiently transfected with control or GAB1 expression vector control for 24 h, followed by the treatment as mentioned above. Western blotting was performed and analyzed as mentioned above. (C) Protein levels of LC3II were quantitated by densitometric analysis using Image J normalized to β-actin and presented as fold changes compared with the control group. ∗siControl vs. siGAB1 in HBSS− and BAF−; †siControl vs. siGAB1 in HBSS+ and BAF−; ‡siControl vs. siGAB1 in HBSS+ and BAF+. (D) Protein levels of LC3II were quantitated by densitometric analysis using Image J normalized to β-actin and presented as fold changes compared with the control group. ∗Control vs. GAB1 in HBSS− and BAF−; †Control vs. GAB1 in HBSS+ and BAF−; ‡Control vs. GAB1 in HBSS+ and BAF+. ASO: Arteriosclerosis obliterans; BAF: Bafilomycin A1; DAPI: 4′,6-Diamidino-2-phenylindole; GAB1: GRB2 associated binding protein 1; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; HBSS: Hank balanced salt solution; HUVEC: Human umbilical vein endothelial cells.
GAB1 regulates autophagy through stress-activated protein kinase pathway
Autophagy is upregulated in response to extra- or intracellular stress, such as starvation, growth factor deprivation, ER stress, and pathogen infection. Several stress-activated protein kinase pathways, such as mTOR1, JNK, Akt, and Erk have been demonstrated to play a key role in regulating autophagy upon those stimulations.[17–20] As an adapt protein, GAB1 served as an upstream of those signaling. Here, we demonstrated that the phosphorylation levels of JNK and p38 were significantly increased in ASO intima compared with control (0.54 vs. 2.67, t = 16.3, P < 0.05) [Figure 4A]. Meanwhile, the phosphorylation levels of Akt and ERK1/2 remained unchanged. Furthermore, we found that the p-p38 and p-JNK levels were significantly associated with gain- and loss-of-function of GAB1 protein [Figures 4B and 4C]. The p38 inhibitor (SB203580) attenuated host autophagy at basal condition by demonstrating the up-regulation of p62. However, the autophagic flux did not affect by the reduction of p62 in the Bafilomycin (BAF) + HBSS group, suggesting inactivation of p38 may only play a role in attenuating autophagy under normal conditions (0.70 vs. 1.38, 0.61 vs. 0.58, and 0.94 vs. 0.92) [Figures 4E and 4G]. On the other hand, we found that JNK inhibitor (SP600215) led to an up-regulation of p62 in normal condition, HBSS group and BAF+HBSS group (0.72 vs. 1.01, 0.56 vs. 0.73, and 0.71 vs. 0.82) [Figures 4D and 4F], indicating that JNK can positively activate basal autophagy and autophagic flux. Taken all together, our results suggest that the JNK pathway may play an important role in the loss of GAB1 induced VEC autophagy.
Figure 4.
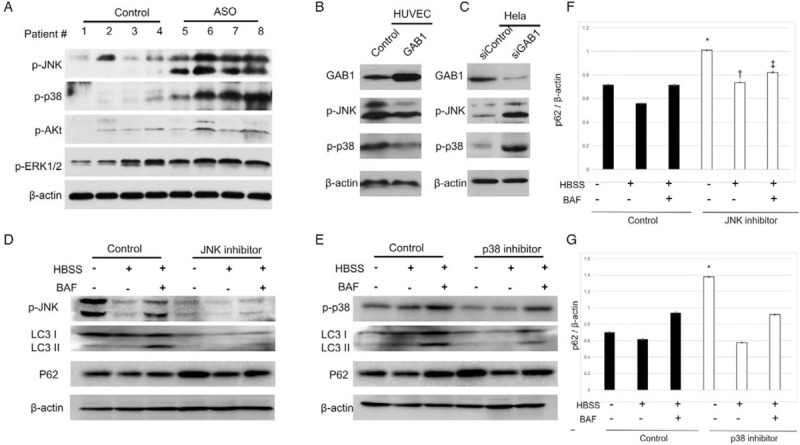
Stress-activated protein kinase pathway was involved in GAB1-mediated autophagy. (A) The tissue lysates were harvested as mentioned above to examine the protein levels of p-JNK, p-p38, p-Akt, p-ERK1/2, and β-actin by Western blotting. (B) HUVECs were treated with either control siRNA (siControl) or GAB1-targeting siRNA (siGAB1) for 48 h. (C) HeLa cells were transiently transfected with control or GAB1 expression vector control for 24 h, cell lysates were harvested to examine the protein levels of GAB1, p-p38, p-JNK, and β-actin by Western blotting. HUVEC with either (D) JNK inhibitor (SP600215) or (E) p38 inhibitor (SB203580) for 24 h, followed by treatment of HBSS for 6 h for the induction of autophagy presented with or without BAF. Cell lysates were harvested to examine the protein levels of GAB1, LC3I/II, and β-actin by Western blotting. (F) Protein levels of p62 were quantitated by densitometric analysis using Image J normalized to β-actin and presented as fold changes compared with the control group. ∗control vs. p38 inhibitor in HBSS− and BAF−; †control vs. p38 inhibitor in HBSS+ and BAF−; ‡control vs. p38 inhibitor in HBSS+ and BAF+. (G, F) Protein levels of p62 were quantitated by densitometric analysis using Image J normalized to β-actin and presented as fold changes compared with the control group. ∗control vs. JNK inhibitor in HBSS− and BAF−; †control vs. JNK inhibitor in HBSS+ and BAF−; ‡control vs. JNK inhibitor in HBSS+ and BAF+. ASO: Arteriosclerosis obliterans; BAF: Bafilomycin A1; DAPI: 4′,6-Diamidino-2-phenylindole; GAB1: GRB2 associated binding protein 1; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; HBSS: Hank's balanced salt solution; HUVEC: Human umbilical vein endothelial cells.
Discussion
As a docking protein, GAB1 has been well-documented in regulating cellular processes including cell proliferation, differentiation, apoptosis, and stress responses.[21] The depletion of GAB1 has been proved to result in embryonic lethality due to severe organ development defects.[22] As GAB1 was involved in multiple intracellular signaling pathways, the dysregulation of GAB1 was reported to be associated with different human diseases.[23] For instance, selective deletion of hepatic GAB1 showed enhanced hepatic insulin sensitivity with reduced glycemic and improved glucose tolerance.[24] Endothelial GAB1 deletion accelerates AngII-dependent vascular inflammation and atherosclerosis.[25] Atherosclerosis is a complex inflammatory process involving several signaling pathways, such as transforming growth factor-β (TGF-β) and CD40/CD40L.[26–28] In this study, we found that both JNK and p38 pathways were involved in ASO. It has been reported that the activation of JNK is involved in the induction of autophagy during oxidative stress and persistently hyper-expressed and activated SAPK/JNK in lesions plays a key role in mediating cell differentiation and apoptosis during the development of atherosclerosis.[17] The p38 pathway was previously reported to play a critical role in the tight control of the early development of atherosclerosis.[29] In this study, we found loss of GAB1 repressed JNK and p38 signaling which contributes to VEC autophagy o and eventually promotes ASO progression.
Previous studies showed that blocking autophagy promotes plaque necrosis in the atherosclerosis mouse model.[30] Moreover, autophagy has been reported to be involved in the regulation of atherosclerosis plaque stability.[31] In this study, we found that loss of GAB1 induced VEC autophagy and this can be attenuated by blockage of JNK and p38 signaling. It has been reported JNK engaged activator protein-1 bind to the p62 promoter. Meanwhile, activation of p38 can promote autophagy in an mTOR-dependent manner. These may explain how GAB1 regulated autophagy by JNK. Even though the detailed mechanism by which GAB1 controls JNK and p38 remains unclear, our results suggest targeting downstream signaling of GAB1 can significantly repress the VEC autophagy.
The mechanisms that control GAB1 transcription in VEC remain unknown. No methylation in the GAB1 promoter region of these ASO patients was found (data not shown). However, we cannot exclude other genomic alterations that may indirectly induce GAB1 repression. A recent study reported that microRNA is associated with GAB1 expression in colorectal cancer.[32] Whether such alterations regulate GAB1 expression warrants further investigation. Autophagy is a complex process in a variety of physiological activities. The regulation of autophagy under different physiological activities may vary from each other. The arteries consist of three different structures including intima, tunica media, and adventitia. In this study, we focused on the intima autophagy, which represents the initiation of the arteriosclerosis. Further study will focus on the mechanism by which GAB1 regulates host autophagy in mice models.
In this study, we did not profile all signaling upon GAB1 alteration. As an adapt protein, while it cannot be excluded that GAB1 may also exert its autophagy regulating function through other pathways, we demonstrated that the p38 and JNK inhibitor significantly antagonized HUVEC autophagy, suggesting that the p38 and JNK may not be essential for the loss of GAB1 induced cell autophagy. We also cannot rule out the possibility that GAB1 may be associated with diabetes as 7 of 11 patients included in this study have diabetes. However, this does not impede the conclusion that GAB1 is associated with VEC autophagy in ASO patients.
However, taken all together, our study reported that loss of GAB1 in ASO patients. Moreover, we found that GAB1 plays a negative role in promoting autophagy in HUVECs. This finding suggests a new mechanism to explain the development of the pathogenesis of ASO, which provides a new target for future drug development.
Funding
This work was supported by the grants from the Shanghai Municipal Science and Technology Commission (No. 14430721400), National Natural Science Foundation (Nos. 81700421 and 81670442), and Clinical innovative research funding of Shanghai Jiaotong University School of Medicine (No. PY2018-IIC-05).
Conflicts of interest
None.
Footnotes
How to cite this article: Ye M, Guo XJ, Kan KJ, Ni QH, Chen JQ, Wang H, Qian X, Xue GH, Deng HY, Zhang L. Loss of GRB2 associated binding protein 1 in arteriosclerosis obliterans promotes host autophagy. Chin Med J 2021;134:73–80. doi: 10.1097/CM9.0000000000001255
References
- 1.Horie T, Kimura T, Ono K. Emerging novel biomarkers for arteriosclerosis obliterans. J Atheroscler Thromb 2016; 23:171–172. doi: 10.5551/jat.ED028. [DOI] [PubMed] [Google Scholar]
- 2.Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013; 382:1329–1340. doi: 10.1016/S0140-6736(13)61249-0. [DOI] [PubMed] [Google Scholar]
- 3.Hoshino J, Fujimoto Y, Naruse Y, Hasegawa E, Suwabe T, Sawa N, et al. Characteristics of revascularization treatment for arteriosclerosis obliterans in patients with and without hemodialysis. Circ J 2010; 74:2426–2433. doi: 10.1253/circj.CJ-09-0910. [DOI] [PubMed] [Google Scholar]
- 4.Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, et al. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E(−)/(−) mice. Sci Rep 2014; 4:5519.doi: 10.1038/srep05519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arias E, Cuervo A. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol 2010; 23:184–189. doi: 10.1016/j.ceb.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nixon R. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 7.Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther 2011; 10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 2013; 13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol 2011; 31:2787–2791. doi: 10.1161/atvbaha.111.224899. [DOI] [PubMed] [Google Scholar]
- 10.Luo XY, Yuan JL, Liu J, Luo CN, Yang MH, Wei Q, et al. Increased macroautophagy in interferon-gamma-producing T cells from patients with newly diagnosed systemic lupus erythematosus. Chin Med J 2018; 131:1527–1532. doi: 10.4103/0366-6999.235110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu HZ, Li L, Chen SL, Wei JR, Zhang JX, Liu J, et al. Low shear stress regulating autophagy mediated by the p38 mitogen activated protein kinase and p53 pathways in human umbilical vein endothelial cells. Chin Med J 2018; 131:1132–1133. doi: 10.4103/0366-6999.230724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan GQ, Qin RR, Li YH, Song DJ, Chen TS, Zhang W, et al. Endothelial cells microparticle-associated protein disulfide isomerase promotes platelet activation in metabolic syndrome. Oncotarget 2016; 7:83231–83240. doi: 10.18632/oncotarget.13081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng H, Fung G, Shi J, Xu S, Wang C, Yin M, et al. Enhanced enteroviral infectivity via viral protease-mediated cleavage of Grb2-associated binder 1. FASEB J 2015; 29:4523–4531. doi: 10.1096/fj.15-274829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Xu S, Yin M, Jin ZG. Essential roles of Gab1 tyrosine phosphorylation in growth factor-mediated signaling and angiogenesis. Int J Cardiol 2015; 181:180–184. doi: 10.1016/j.ijcard.2014.10.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 2012; 15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res 2012; 93:320–329. doi: 10.1093/cvr/cvr321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haberzettl P, Hill BG. Oxidized lipids activate autophagy in a JNK-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biol 2013; 1:56–64. doi: 10.1016/j.redox.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moscat J, Diaz Meco M. Feedback on Fat: p62-mTORC1-autophagy connections. Cell 2011; 147:724–727. doi: 10.1016/j.cell.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu P, Lai D, Lu P, Gao J, He H. ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med 2012; 29:613–618. doi: 10.3892/ijmm.2012.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun 2013; 4:2799.doi: 10.1038/ncomms3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lemarié CA, Lehoux S. The gift of Gab1 (Grb-2-associated binder 1). Arterioscler Thromb Vasc Biol 2011; 31:956–957. doi: 10.1161/ATVBAHA.111.225987. [DOI] [PubMed] [Google Scholar]
- 22.Gu H, Neel BG. The “Gab” in signal transduction. Trends Cell Biol 2003; 13:122–130. doi: 10.1016/s0962-8924(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 23.Nakaoka Y, Komuro I. Gab docking proteins in cardiovascular disease, cancer, and inflammation. Int J Inflam 2013; 2013:141068.doi: 10.1155/2013/141068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bard-Chapeau EA, Hevener AL, Long S, Zhang EE, Olefsky JM, Feng G-S. Deletion of Gab1 in the liver leads to enhanced glucose tolerance and improved hepatic insulin action. Nat Med 2005; 11:567–571. doi: 10.1038/nm1227. [DOI] [PubMed] [Google Scholar]
- 25.Higuchi K, Nakaoka Y, Shioyama W, Arita Y, Hashimoto T, Yasui T, et al. Endothelial Gab1 deletion accelerates angiotensin II-dependent vascular inflammation and atherosclerosis in apolipoprotein E knockout mice. Circ J 2012; 76:2031–2040. doi: 10.1253/circj.cj-11-1507. [DOI] [PubMed] [Google Scholar]
- 26.Masters SL, Latz E, O’Neill LA. The inflammasome in atherosclerosis and type 2 diabetes. Sci Transl Med 2011; 3:81ps17.doi: 10.1126/scitranslmed.3001902. [DOI] [PubMed] [Google Scholar]
- 27.Bobik A, Agrotis A, Kanellakis P, Dilley R, Krushinsky A, Smirnov V, et al. Distinct patterns of transforming growth factor-beta isoform and receptor expression in human atherosclerotic lesions. Colocalization implicates TGF-beta in fibrofatty lesion development. Circulation 1999; 99:2883–2891. doi: 10.1161/01.cir.99.22.2883. [DOI] [PubMed] [Google Scholar]
- 28.Mach F, Schönbeck U, Sukhova GK, Bourcier T, Bonnefoy J-Y, Pober JS, et al. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for CD40-CD40 ligand signaling in atherosclerosis. Proc Natl Acad Sci U S A 1997; 94:1931.doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun C, Liang C, Ren Y, Zhen Y, He Z, Wang H, et al. Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways. Basic Res Cardiol 2009; 104:42–49. doi: 10.1007/s00395-008-0738-8. [DOI] [PubMed] [Google Scholar]
- 30.Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007; 450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 31.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 2006; 84:448–454. doi: 10.1111/j.1440-1711.2006.01454.x. [DOI] [PubMed] [Google Scholar]
- 32.Bai R, Weng C, Dong H, Li S, Chen G, Xu Z. MicroRNA-409-3p suppresses colorectal cancer invasion and metastasis partly by targeting GAB1 expression. Int J Cancer 2015; 137:2310–2322. doi: 10.1002/ijc.29607. [DOI] [PubMed] [Google Scholar]