

Abstract
Hydrogen/deuterium exchange with mass spectrometry (HDX-MS) is capable of providing unique insight into complex biological systems that are difficult to study by other techniques. Due to arduous sample handling requirements, automating HDX experimentation for higher throughput requires specialized equipment. While recent advances have enabled automation of sample preparation and analysis, several proteins of interest and types of HDX experiments remain incompatible with automated workflows and require manual sample preparation that greatly limits experimental throughput. To expand throughput and increase the precision of HDX-MS for systems requiring manual preparation, we have developed an inexpensive autosampler capable of thawing and injecting frozen HDX-MS samples in a highly reproducible manner.
Introduction
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) has recently seen a surge in popularity and evolving utilization across both academia and the biopharmaceutical industry.1 The versatility of HDX-MS has led to it becoming a routine tool for detailing protein–protein interactions, identifying allosteric effects, mapping folding pathways, and investigating mechanisms of protein structure and function across many challenging and otherwise inaccessible systems, including intrinsically disordered proteins.2 HDX-MS is also used within the biopharmaceutical industry to characterize antibody-based drugs and other protein therapies that continue to dominate the marketplace.3,4 Historically, HDX experimentation has suffered from low reproducibility along with low sample throughput, which can both be tied to back-exchange of the deuterium label.5−7 After each exchange, every sample must immediately be quenched to low pH, kept cold, and analyzed rapidly, all to slow the spontaneous loss of deuterium after labeling (“back-exchange”). In practice, samples are either analyzed directly after labeling via inline liquid chromatography–mass spectrometry (LC–MS) or flash frozen and kept at −80 °C for long-term storage and decoupled LC–MS analysis. An advantage of a decoupled approach is that samples are run in a continuous queue, rather than waiting as various time points are prepared, thus making the most efficient use of the MS instrument time. Recent commercial systems have been developed to automate nearly every step in HDX-MS analysis, including automated deuterium labeling with in-line injection, protease digestion, and LC–MS analysis.5,8−12 While this technology has greatly expanded the throughput and precision of HDX-MS, limitations still remain in terms of sampling different exchange conditions along with postquench sample manipulation(s). For example, current robotic systems are not well-suited for sampling rapid time scales (millisecond) or pulsed labeling experiments and are not equipped to perform complicated cleanup steps that are often required when working with complex systems, such as membrane-bound proteins.13−15
While some limitations for automated in-line HDX-MS are starting to be addressed, such as the recent development of filtration columns designed for postquench removal of lipids, there are still many types of samples and experimental parameters that cannot be automated and require manual sample preparation.16 While not as convenient, preparing samples manually does offer notable advantages: (1) all samples can be prepared in a short time window minimizing potential protein degradation; (2) exchanges can be performed on immobilized proteins, lyophilized proteins, or colloidal particles; (3) a wider range of exchange time points can be sampled; and (4) samples are amenable to complicated postquench cleanup steps. The major disadvantage, however, comes with having to minimize and maintain consistent levels of back-exchange. In a decoupled approach, samples remain frozen until the moment they are ready for LC–MS analysis, and every sample must be consistently thawed before injection. On top of this, reproducibility suffers when samples are analyzed on different days, meaning that every sample within a given experiment should be analyzed back-to-back for optimal results.3 Without the access to an autosampler capable of maintaining subfreezing temperatures (−80 °C), these constraints require users to be physically present for the manual thawing and injection of every sample within a given experiment.10,12 Here, we describe a simple and inexpensive sample manager capable of alleviating this inherent bottleneck with a decoupled HDX-MS pipeline.
Methods
HDX-MS Sample Preparation
Protein samples of 10 μL at concentrations of 0.1–1 mg/mL were diluted 10-fold into deuterated buffer (85% D2O final) for time points ranging from 3 s up to 20 h at room temperature. Samples were immediately quenched with an equal volume of ice-cold quench buffer (containing up to 8 M urea and 200 mM TCEP with 0.2% formic acid) to bring the pH to 2.5 and the final volume to 200 μL. For manual injections, samples were snap frozen in liquid nitrogen and stored at −80 °C until LC–MS analysis. Prior to manual injection, each sample was removed from a liquid nitrogen reservoir and allowed to sit on ice for 5 min. The sample was then placed on the countertop at room temperature for exactly 1 min to reach a slurry-like state and immediately injected with a 250 μL gastight syringe (Hamilton) wrapped in parafilm for insulation and precooled on ice. For automated injections, quenched samples were rapidly transferred to 1 mL autosampler glass vials (Total Recovery, Waters) prefrozen in a 54-vial sample tray within a bath of ethanol and dry ice. The bath was treated with the same precautions as used for cold traps employing organic solvents and dry ice, including necessary precautions for flammable solvents and the use of cryogenic gloves. Samples were capped with magnetic screw caps, and the entire 54-vial tray was stored at −80 °C until LC–MS analysis. Samples were thawed by moving the vials from the dry ice/ethanol compartment to a sample block held at 4 °C and waiting for a specified time before drawing the sample and injecting it into the injection port on the HDX system. Thaw times were in the range of 3–10 min dependent on the composition of the quenched solution. The full HDX-MS protocol including gradients, digestion conditions, and wash cycles are described in the Supporting Information.
Data Analysis
Peptide assignments were made using MS/MS data obtained from undeuterated samples analyzed with Protein Prospector and Byonic (Protein Metrics). Deuterium uptake was analyzed and summarized using HDExaminer v2 software. All time points and replicates from available data sets were included unless the sample encountered a known technical problem (e.g., leaks or clogs). Variability statistics (standard deviations) were calculated from each discrete sample set using the deuterium uptake of a reporter peptide known to be fully deuterated by the earliest exchange time point (3 s).
Results and Discussion
System Design and Overview
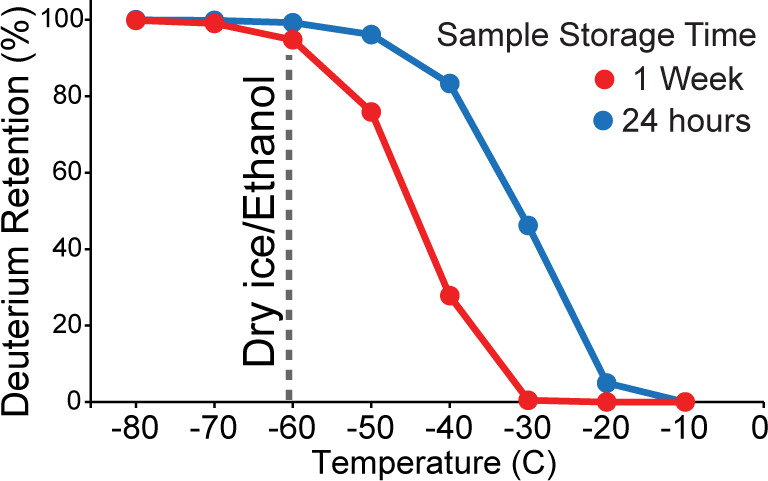
The influence that temperature, pH, and amino acid identity have on exchange rates are well-established and allow for accurate prediction of back-exchange under different conditions.17 These rates suggest that a temperature of −60 °C is sufficient for reducing deuterium loss in quenched samples (pH 2.5) to less than 1% over a period of 24 h (Figure 1). However, longer storage times at −60 °C would be detrimental, with more than 5% deuterium loss after 1 week. While these predictions overlook the possibility that solid-state exchange kinetics may significantly deviate from that predicted in solution, they do indicate that, while long-term storage of HDX-MS samples at −80 °C temperatures is warranted, samples may not necessarily need to be kept at such low temperatures to mitigate back-exchange for sample queues less than 24 h.
Figure 1.
Theoretical temperature dependence on back-exchange of frozen HDX samples. The deuterium retention for an average protein backbone amide is shown after 24 h (blue) or 1 week (red) when stored at different temperatures. Notably, −60 °C is sufficient for maintaining >99% of deuterium after 24 h (blue).
Baths composed of dry ice and ethanol are commonly utilized in chemistry and offer a simple and inexpensive way to achieve a stable temperature under −60 °C. We therefore devised a subfreezing sample manager capable of maintaining frozen HDX-MS samples using dry ice/ethanol for integration into a cost-effective automated LC–MS platform. The sample manager is an in-house fabricated unit designed to house a single 54-well sample tray (Figure 2). Dry ice is stored in a large peripheral reservoir, while the ethanol conducts heat to keep the sample tray at −65 °C, as measured by a calibrated K-type thermocouple. An insulated sliding lid above the sample tray allows for access to the sample manager via the integrated LEAP robot. A standard LEAP stack cooler is maintained at 4 °C and used for controlled thawing of frozen samples. The full description, schematics, and designs for the sample manager and 54-well sample tray, along with the detailed thaw/injection protocols, are included in the Supporting Information.
Figure 2.
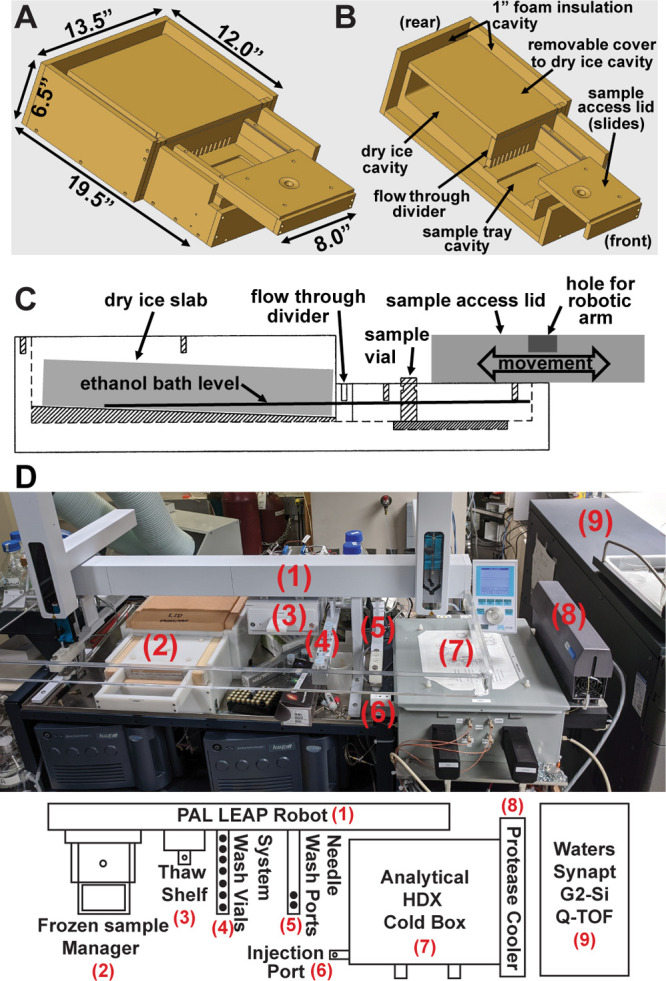
Design of the dry ice/ethanol sample manager and overview of automated HDX platform. (A) Size dimensions and (B) cut-away view of internal compartments and features of the sample manager. (C) Sample manager utilizes a dry ice/ethanol bath housed in a slanted chamber with a flow-through divider to ensure that HDX samples are kept frozen as the dry ice reservoir depletes over time. (D) Overview of the automated HDX LC–MS system with major system components labeled.
Increasing Throughput
The constraints of HDX-MS sample handling have greatly restricted the throughput of LC–MS analysis. Accounting for the time required for wash steps to minimize carryover, we can only achieve an average sampling rate on the order of two samples per hour, or 24 samples within a 12 h shift. Furthermore, to minimize variability, every sample within a given data set should be collected by the same analyst within a single continual session, making the manual LC–MS analysis of HDX samples a long and tedious process. By creating a sample manager capable of being accessed robotically, we aimed to improve throughput by directly lifting the burden of a manual thaw/inject approach.
To assess our viable range of sample storage time, we setup an extended queue of HDX-MS samples that contained fully deuterated bradykinin peptide to serve as an internal reporter for back-exchange. After filling the dry ice reservoir, the queue was initiated and data were collected every 40 min over the course of 20 continuous hours without any manual intervention or resupply of dry ice. Consistent with our initial estimation of back-exchange at low temperatures, bradykinin deuteration levels showed no systematic change throughout the course of 18 h, after which a sharp drop in deuterium retention was observed (Figure 3A). This drop in deuterium level is a direct consequence of accelerated back-exchange, occurring as the sample manager begins to warm after depletion of the dry ice reservoir. The design of our sample manager addresses this issue by allowing for the resupply of dry ice at any point without halting data collection. As a queue nears completion, the entire 54-well sample tray may be quickly swapped with another tray for a seamless and virtually unlimited continuation of LC–MS analysis with little manual interaction required.
Figure 3.
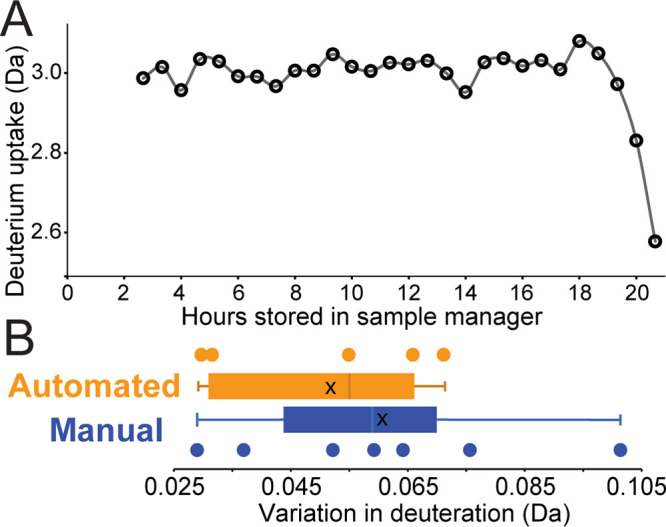
Performance assessment of the sample manager and automated HDX platform. (A) Deuteration levels of internal standard Bradykinin remain consistent for each sample stored in the dry ice/ethanol sample manager until total depletion of the dry ice reservoir occurs after 18 continuous hours. (B) Collection of discrete data sets was analyzed to compare variability between automated and manual thaw/inject methods. Within each data set, a fully deuterated reporter peptide was used to assess variability in back-exchange. The average deviation (x) for the automated and manual method was determined to be 0.051 and 0.060 Da, respectively. Details are shown in Table S1.
Reducing Variability
To gauge the precision of our automated system, we compared an extensive series of decoupled HDX-MS data sets to assess the general variability between manual and automated thaw/inject methods. Using a fully deuterated peptide, we determined that the automated platform reduces overall variability by more than 15% (Figure 3B). While the study included roughly the same number of discrete sample sets between the two methods, we note that the automated sample sets are considerably larger due to the increased throughput afforded by the system (Table S1). While sample preparation (labeling and quenching) remains a large contributor to variance within any given experiment, our system shows an overall reduction in variability due to the precise control of timing and temperature throughout every thaw/inject cycle.
Conclusion
We present an inexpensive sample manager integrated with a robotic autosampler for high-throughput decoupled HDX-MS. The platform enables collection of frozen HDX-MS samples with superior precision over manual injections and enables collection of large data sets for complex systems that are currently unattainable by fully automated HDX-MS systems.
Acknowledgments
The authors wish to thank Dale Whittington and J. Scott Edgar for assistance with data collection. This work was supported by the Global Health Vaccine Accelerator Platform (GH-VAP #OPP1126258) through the Bill and Melinda Gates Foundation along with support from the UW Molecular Biophysics Training Program (MBTG #T32GM008268).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jasms.0c00341.
Detailed description of sample chiller components and sample handling protocols; design of the vial trays; table of variation in back-exchange for automated vs manually injected samples (PDF)
Full sequence robot sample thaw video (MP4)
Sample tray, frame slider, and sample manager .pdf files (ZIP)
Chiller solidworks files (ZIP)
Modified 54-vial HDX tray .stl files (ZIP)
Chiller .dxf files (ZIP)
Chiller .step files (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Hodge E. A.; Benhaim M. A.; Lee K. K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 2020, 29, 843–855. 10.1002/pro.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson G. R.; Burke J. E.; Ahn N. G.; Anand G. S.; Borchers C.; Brier S.; Bou-Assaf G. M.; Engen J. R.; Englander S. W.; Faber J.; Garlish R.; Griffin P. R.; Gross M. L.; Guttman M.; Hamuro Y.; Heck A. J. R.; Houde D.; Iacob R. E.; Jørgensen T. J. D.; Kaltashov I. A.; Klinman J. P.; Konermann L.; Man P.; Mayne L.; Pascal B. D.; Reichmann D.; Skehel M.; Snijder J.; Strutzenberg T. S.; Underbakke E. S.; Wagner C.; Wales T. E.; Walters B. T.; Weis D. D.; Wilson D. J.; Wintrode P. L.; Zhang Z.; Zheng J.; Schriemer D. C.; Rand K. D. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. 10.1038/s41592-019-0459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D.; Berkowitz S. A.; Engen J. R. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011, 100, 2071–2086. 10.1002/jps.22432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Mo J.; Tao L.; Russell R. J.; Tymiak A. A.; Chen G.; Iacob R. E.; Engen J. R. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: methodology and applications. Drug Discovery Today 2014, 19, 95–102. 10.1016/j.drudis.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins D. J.; Espada A.; Novick S. J.; Molina-Martin M.; Stites R. E.; Espinosa J. F.; Broughton H.; Goswami D.; Pascal B. D.; Dodge J. A.; Chalmers M. J.; Griffin P. R. Two-Site Evaluation of the Repeatability and Precision of an Automated Dual-Column Hydrogen/Deuterium Exchange Mass Spectrometry Platform. Anal. Chem. 2016, 88, 6607–6614. 10.1021/acs.analchem.6b01650. [DOI] [PubMed] [Google Scholar]
- Hudgens J. W.; Gallagher E. S.; Karageorgos I.; Anderson K. W.; Filliben J. J.; Huang R. Y. C.; Chen G.; Bou-Assaf G. M.; Espada A.; Chalmers M. J.; Harguindey E.; Zhang H. M.; Walters B. T.; Zhang J.; Venable J.; Steckler C.; Park I.; Brock A.; Lu X.; Pandey R.; Chandramohan A.; Anand G. S.; Nirudodhi S. N.; Sperry J. B.; Rouse J. C.; Carroll J. A.; Rand K. D.; Leurs U.; Weis D. D.; Al-Naqshabandi M. A.; Hageman T. S.; Deredge D.; Wintrode P. L.; Papanastasiou M.; Lambris J. D.; Li S.; Urata S. Interlaboratory Comparison of Hydrogen-Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Anal. Chem. 2019, 91, 7336–7345. 10.1021/acs.analchem.9b01100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkitt W.; O’Connor G. Assessment of the repeatability and reproducibility of hydrogen/deuterium exchange mass spectrometry measurements. Rapid Commun. Mass Spectrom. 2008, 22, 3893–3901. 10.1002/rcm.3794. [DOI] [PubMed] [Google Scholar]
- Keppel T. R.; Jacques M. E.; Young R. W.; Ratzlaff K. L.; Weis D. D. An efficient and inexpensive refrigerated LC system for H/D exchange mass spectrometry. J. Am. Soc. Mass Spectrom. 2011, 22, 1472–1476. 10.1007/s13361-011-0152-6. [DOI] [PubMed] [Google Scholar]
- Chalmers M. J.; Busby S. A.; Pascal B. D.; He Y.; Hendrickson C. L.; Marshall A. G.; Griffin P. R. Probing protein ligand interactions by automated hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2006, 78, 1005–1014. 10.1021/ac051294f. [DOI] [PubMed] [Google Scholar]
- Woods V. L.; Hamuro Y. High resolution, high-throughput amide Deuterium Exchange-Mass Spectrometry (DXMS) determination of protein binding site structure and dynamics: Utility in pharmaceutical design. J. Cell. Biochem. 2001, 84, 89–98. 10.1002/jcb.10069. [DOI] [PubMed] [Google Scholar]
- Englander J. J.; Del Mar C.; Li W.; Englander S. W.; Kim J. S.; Stranz D. D.; Hamuro Y.; Woods V. L. Protein structure change studied by hydrogen-deuterium exchange, functional labeling, and mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 7057–7062. 10.1073/pnas.1232301100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada A.; Haro R.; Castañon J.; Sayago C.; Perez-Cozar F.; Cano L.; Redero P.; Molina-Martin M.; Broughton H.; Stites R. E.; Pascal B. D.; Griffin P. R.; Dodge J. A.; Chalmers M. J. A Decoupled Automation Platform for Hydrogen/Deuterium Exchange Mass Spectrometry Experiments. J. Am. Soc. Mass Spectrom. 2019, 30, 2580–2583. 10.1007/s13361-019-02331-2. [DOI] [PubMed] [Google Scholar]
- Keppel T. R.; Weis D. D. Analysis of disordered proteins using a simple apparatus for millisecond quench-flow H/D exchange. Anal. Chem. 2013, 85, 5161–5168. 10.1021/ac4004979. [DOI] [PubMed] [Google Scholar]
- Walters B. T.; Mayne L.; Hinshaw J. R.; Sosnick T. R.; Englander S. W. Folding of a large protein at high structural resolution. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18898–18903. 10.1073/pnas.1319482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astorga-Wells J.; Landreh M.; Johansson J.; Bergman T.; Jörnvall H. A membrane cell for on-line hydrogen/deuterium exchange to study protein folding and protein-protein interactions by mass spectrometry. Mol. Cell. Proteomics 2011, 10, M110.006510. 10.1074/mcp.M110.006510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. W.; Gallagher E. S.; Hudgens J. W. Automated Removal of Phospholipids from Membrane Proteins for H/D Exchange Mass Spectrometry Workflows. Anal. Chem. 2018, 90, 6409–6412. 10.1021/acs.analchem.8b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Milne J. S.; Mayne L.; Englander S. W. Primary structure effects on peptide group hydrogen exchange. Proteins: Struct., Funct., Genet. 1993, 17, 75–86. 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.