

Abstract
Lysosomal storage diseases are a group of metabolic disorders caused by deficiencies of several components of lysosomal function. Most commonly affected are lysosomal hydrolases, which are involved in the breakdown and recycling of a variety of complex molecules and cellular structures. The understanding of lysosomal biology has progressively improved over time. Lysosomes are no longer viewed as organelles exclusively involved in catabolic pathways, but rather as highly dynamic elements of the autophagic‐lysosomal pathway, involved in multiple cellular functions, including signaling, and able to adapt to environmental stimuli. This refined vision of lysosomes has substantially impacted on our understanding of the pathophysiology of lysosomal disorders. It is now clear that substrate accumulation triggers complex pathogenetic cascades that are responsible for disease pathology, such as aberrant vesicle trafficking, impairment of autophagy, dysregulation of signaling pathways, abnormalities of calcium homeostasis, and mitochondrial dysfunction. Novel technologies, in most cases based on high‐throughput approaches, have significantly contributed to the characterization of lysosomal biology or lysosomal dysfunction and have the potential to facilitate diagnostic processes, and to enable the identification of new therapeutic targets.
Keywords: autophagy, lysosomal biology, lysosomal storage diseases, lysosomes
Subject Categories: Autophagy & Cell Death; Genetics, Gene Therapy & Genetic Disease;
In the last decade, our understanding of lysosomal storage diseases immensely improved. The current article comprehensively reviews recent advances in lysosomal storage diseases research and therapy development.

Glossary
- Autophagy
A multistep and regulated pathway that removes unnecessary or dysfunctional cellular components and allows for delivery of cargo materials to lysosomes, where they are degraded and recycled.
- High‐content imaging
Cell‐based technologies based on automated microscopy and complex image algorithms to extract multidimensional information on cell morphology, fluorescence intensity, or distribution of fluorescent markers within cells.
- Genome editing and CRISPR‐Cas9
An RNA‐guided targeted genome editing tool: This methodology which allows to introduce different specific genetic changes such as gene knock‐out, knock‐in, insertions, and deletions in cell lines and in vivo.
- Lysosomes
Membrane‐limited, ubiquitous, intracellular organelles involved in multiple cellular processes, such as catabolism and recycling of complex molecules and cellular components, signaling, and adaptation to environmental stimuli.
- Metabolome analyses
High‐throughput methodologies for the detection of multiple metabolites, mainly based on mass spectrometry or nuclear magnetic resonance‐based approaches.
- Next‐generation sequencing
A diagnostic tool based on technological platforms that allow for sequencing of millions of small fragments of DNA in parallel. Next‐generation sequencing can be used either for ”targeted” sequencing of selected gene panels or for “untargeted” approaches based on whole‐exome or genome analysis.
- MicroRNAs
Small non‐coding RNAs that that regulate gene expression by targeting messenger RNAs.
Introduction
Knowledge on lysosomal storage diseases (LSDs) has been evolving for more than a century (Fig 1). The first phenotypes and clinical entities were described in the 19th century (see Mehta et al, 2006 for review), long before the identification of lysosomes and the definition of their biochemistry and pathophysiology. At that time, the identification of these disorders was exclusively based on the characterization of clinical phenotypes and pathology.
Figure 1. The evolution of the knowledge on LSDs.
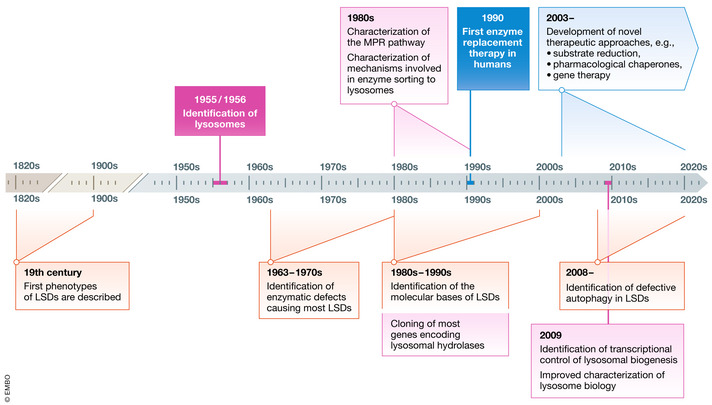
After the identification of the first clinical phenotypes during of the 19th century, the knowledge on LSDs evolved following the recognition of lysosomes in 1955/56 and the demonstration of the biochemical defects underlying LSDs, starting from 1963. Between the 1970s and 1990s, research in this field was focused on the mannose‐6‐phosphate receptor pathway and on the mechanisms underlying the sorting of lysosomal enzymes, on the identification of the molecular bases of LSDs, and on the development of tools and strategies to investigate lysosomal biology. The first attempts to treat these disorders by enzyme replacement therapy started in the 1990s. Current research is now focusing on the role of lysosomes as signaling platforms controlling cellular metabolism and on the development of new therapeutic approaches.
The biochemical and cellular bases of LSDs were elucidated much later, when Christian de Duve’s work, corroborated by Alex B. Novikoff's electron microscopy observations, led to the identification of lysosomes as cellular catabolic stations (de Duve et al, 1955; Novikoff et al, 1956), and when the biochemical defects underlying some of the previously described clinical entities were discovered. Pompe disease was the first disorder to be identified as an LSD in 1963, when Henri G. Hers demonstrated that this disease is due to the lack of an acidic α‐glucosidase, similar to rat liver lysosomal maltase (Hers, 1963), and that this deficiency is responsible for glycogen storage in tissues. He also suggested that other diseases, such as the mucopolysaccharide storage diseases, might be due to enzyme deficiencies. Between 1960 and the mid‐70s, the biochemistry of LSDs was further characterized with the identification of the primary storage materials for other LSDs and the recognition of the respective enzyme deficiencies (Van Hoof, 1974).
For decades, the biology and function of lysosomes remained associated with their catabolic function, and LSD pathophysiology was seen as a direct consequence of defective degradation and disposal of complex substrates (Vellodi, 2005; Heard et al, 2010).
Between the late 1970s and 1990s, research in this field progressed with studies that considerably expanded the knowledge on lysosome biology and on the pathophysiology of LSDs. These studies led to the characterization of the mechanism underlying the sorting of lysosomal enzymes (Sly & Fisher, 1982) and to the identification of the molecular bases of clinical variability of LSDs (Beck, 2001; Kroos et al, 2012).
Following the recognition of the mannose‐6‐phosphate pathway's role in lysosomal enzyme trafficking and the availability of new technologies for the purification and manufacturing of lysosomal enzymes, the early 1990s inaugurated the first attempts to treat these disorders by replacing the defective enzyme activity (Barton et al, 1990; Barton et al, 1991).
At the same time, the introduction of novel technologies had a critical role in the study of LSDs. Techniques for targeted gene disruption and generation of knock‐out animal models for human LSDs provided tools to better characterize the pathophysiology of these disorders and to develop innovative therapeutic strategies (Pastores et al, 2013). Mass spectrometry technology allowed for lysosomal proteome analyses (e.g., mannose‐6‐phosphate glycoproteome) (Sleat et al, 2005) that led to the identification of lysosomal proteins and novel molecular bases of some LSDs.
In the past decade, a number of studies have expanded our knowledge of lysosomal biology and provided new and important insights on LSD pathophysiology. These studies identified the lysosomes as highly dynamic organelles involved in multiple cellular functions, including signaling, and able to adapt to environmental stimuli. (Settembre et al, 2013; Perera & Zoncu, 2016; Ballabio & Bonifacino, 2020).
Significant advancements have also facilitated greater understanding on the molecular and metabolic mechanisms underlying LSDs and the development of new therapeutic strategies for these diseases (Parenti et al, 2015; Platt et al, 2018; Ren & Wang, 2020). This review will focus on new tools and technologies to study LSDs, on emerging aspects of lysosomal biology, and on recent discoveries on the cellular and organismal consequences of lysosomal dysfunction.
The biology of lysosomes, old concepts and new views
Lysosomes are membrane‐limited, ubiquitous, intracellular organelles involved in multiple cellular processes (Saftig & Klumperman, 2009; Ballabio & Bonifacino, 2020).
More than two hundred lysosomal‐resident proteins contribute to the biology and function of these organelles. Approximately 60 of them are acidic hydrolases (Lubke et al, 2009; Schroder et al, 2010). Most of them act as exoglycosidases or sulfatases and are localized to the lysosomal lumen. The others are localized at the lysosomal membrane and have multiple functions such as formation of a glycocalyx‐like layer, transport across the membrane, acidification, membrane stability, and mediating interaction between lysosomes and other cellular structures (Saftig & Klumperman, 2009; Ballabio & Bonifacino, 2020). In addition to lysosomal‐resident proteins, other proteins interact with the lysosome and participate to lysosomal function by being dynamically recruited to the lysosomal surface under certain conditions, for example, the transcription factor EB (TFEB), the mechanistic target of rapamycin complex 1 (mTORC1) (Perera & Zoncu, 2016; Ballabio & Bonifacino, 2020; Yim & Mizushima, 2020), the mTORC1 regulator tuberous sclerosis complex (TSC) (Dibble & Cantley, 2015), folliculin (FLCN) and FLCN‐interacting protein (FNIP) (Lawrence et al, 2019), the energy‐sensing complex AMP‐activated kinase (AMPK) (Zhang et al, 2014), and the signal transducer and activator of transcription‐3 (STAT3) (Liu et al, 2018).
The first function of normal lysosomes to be recognized is turnover of cellular constituents. Lysosomes are involved in the degradation of a broad variety of structurally diverse compounds, such as proteins, glycosaminoglycans, sphingolipids, oligosaccharides, glycogen, nucleic acids, and complex lipids. Cellular and extracellular materials and substrates destined for degradation reach lysosomes through different routes (endocytosis, phagocytosis, autophagy), or by direct transport. In this respect, lysosomes are part of a more complex pathway, referred to as the autophagy–lysosomal pathway (ALP). Autophagy plays a crucial role in cell homeostasis by controlling intracellular clearance and recycling of a variety of molecules and cellular components and also by sustaining cellular energy metabolism. Autophagy is a multistep pathway that involves autophagosome formation, cargo recruitment, and autophagosome–lysosome fusion. Importantly, autophagic function is entirely dependent on the ability of the lysosome to degrade and recycle autophagy substrates (Yim & Mizushima, 2020).
Much attention has been paid in recent years to the nutrient‐sensing function of lysosomes. Lysosomes are able to monitor the nutrient status of cells and to adjust their metabolism to changing energetic conditions. When nutrients are available mTORC1 is dynamically recruited to the lysosomal surface where it becomes active and promotes cellular anabolic processes (Saxton & Sabatini, 2017).
Recent studies have provided compelling evidence that lysosomal biogenesis and autophagy are controlled by the master transcriptional regulator transcription factor EB (TFEB) (Sardiello et al, 2009; Settembre et al, 2011). In addition to TFEB, another member of the MiT‐TFE family of transcription factors, TFE3, has a partially redundant function and is regulated in a similar manner (Martina et al, 2014; Raben & Puertollano, 2016).
TFEB subcellular localization and function is regulated by nutrient‐induced mTORC1‐mediated phosphorylation of specific serine residues (Settembre et al, 2012; Roczniak‐ Ferguson et al, 2012; Martina et al, 2012). On phosphorylation by mTORC1, TFEB is retained in the cytoplasm. A variety of stimuli leading to mTORC1 inactivation, such as starvation, induce TFEB dephosphorylation and nuclear translocation. Thus, the mTORC1‐TFEB regulatory axis enables lysosomes to adapt their function to environmental cues, such as nutrient availability (Ballabio & Bonifacino, 2020). Several additional conditions, such as infection, inflammation, physical exercise, endoplasmic reticulum stress, and mitochondrial damage, also promote TFEB nuclear translocation, highlighting the complexity of TFEB regulation (reviewed in Puertollano et al, 2018; Cinque et al, 2015).
Recent findings have added further complexity to the mechanisms by which mTORC1 phosphorylates TFEB (Napolitano et al, 2020). Unlike other substrates of mTORC1, TFEB is known to interact with RagGTPases (Martina & Puertollano, 2013). Due to this interaction, TFEB phosphorylation occurs through an mTORC1 substrate‐specific mechanism that is strictly dependent on the amino acid‐induced activation of RagC and RagD GTPases but is insensitive to Rheb activity induced by growth factors. This allows mTORC1 activity to be differentially regulated by different stimuli (Napolitano et al, 2020). This substrate‐specific regulation of TFEB by the mTORC1 pathway has a crucial role in Birt–Hogg–Dubé syndrome, a disorder characterized by benign skin tumors, lung, and kidney cysts and renal cell carcinoma (Kauffman et al, 2014; Calcagnì, et al, 2016) and caused by mutations in the lysosomal RagC/D activator folliculin (FLCN) (Napolitano et al, 2020).
Lysosomes are emerging as calcium (Ca2+) storage organelles. The concentration of free Ca2+ within the lysosome is around 500 µM, and therefore comparable to endoplasmic reticulum (ER) Ca2+ levels (Christensen et al, 2002). Ca2+ channels, such as transient receptor potential mucolipin‐1 (TRPML‐1, Mucolipin 1, MCOLN1) and the two‐pore channel (TPC), reside on the lysosomal membrane and have been shown to mediate local Ca2+ signals from intracellular compartments (e.g., mitochondria) (Xu et al, 2015; Xu & Ren, 2015). Lysosomal Ca2+ signaling participates in multiple cellular processes such as lysosomal acidification, the fusion of lysosomes with other cellular organelles, membrane trafficking and repair, autophagy, and formation of contact sites between the lysosome and the endoplasmic reticulum (Kilpatrick et al, 2013; Lloyd‐Evans & Waller‐Evans, 2019). Furthermore, lysosomal Ca2+ signaling is involved in the regulation of lysosomal biogenesis and autophagy through the activation of TFEB. Upon starvation, lysosomal Ca2+ release through TRPML1 activates the Ca2+‐dependent serine/threonine phosphatase calcineurin (CaN), which binds and dephosphorylates TFEB, thus promoting its nuclear translocation (Medina et al, 2015). TRPML1 also induces autophagic vesicle biogenesis through the generation of phosphatidylinositol 3‐phosphate (PI3P) and the recruitment of essential PI3P‐binding proteins to the nascent phagophore in a TFEB‐independent manner (Scotto‐Rosato et al, 2019).
The nosography of LSDs
LSDs are multisystem disorders that are associated with a broad range of clinical manifestations affecting multiple organs and systems and causing visceral, ocular, hematologic, skeletal, and neurological signs. These manifestations are often highly debilitating, causing progressive physical and neurological disabilities.
In general, LSD presentations show broad variability (Beck, 2001), ranging from early‐onset (in some cases neonatal), severe clinical forms that often result in premature death of patients, to late‐onset, attenuated phenotypes that have a lesser impact on patient health and lifespan. Albeit individually rare, their cumulative incidence is estimated in approximately 1 in 5,000–7,500 births, with higher rates in specific populations. It is noteworthy that newborn screening programs for LSDs, now active in some countries, may in the future significantly change these estimates and will likely provide a more precise figure of LSD incidence (Spada et al, 2006; Hopkins et al, 2018; Wasserstein et al, 2019).
The nosography of LSDs has evolved over time, reflecting the advancements in the knowledge of lysosomal function and the cellular consequences of its dysfunction. The traditional classification based on the classes of stored substrates (glycosaminoglycans in the mucopolysaccharidoses, glycosphingolipids in the glycosphingolipidoses, glycoproteins in the oligosaccharidosis, etc) largely reflects the vision of lysosomes as catabolic organelles and is centered on the disease biochemistry. Accurate and exhaustive information on LSD classification and nosography can be found elsewhere (Platt et al, 2018).
With the improved knowledge on the molecular and cellular bases of LSDs, an alternative way of classifying LSDs has been proposed, based on the process that is defective in the biogenesis of lysosomal enzymes, rather than on the stored substrate (Platt, 2018). The majority of these disorders is due to deficiencies of soluble hydrolases that are involved in the sequential degradation of a specific substrate. Other disorders are due to deficiencies in upstream processes, such as post‐translational modifications (multiple sulfatase deficiency, MSD, due to the lack of an enzyme converting a cysteine into a formylglycine residue in the catalytic site of sulfatases) (Cosma et al, 2002; Dierks et al, 2002), or to defective sorting of lysosomal enzymes to lysosomes (mucolipidoses—ML—II and III, with deficient generation of mannose‐6‐phosphate) (Hickman & Neufeld, 1972). Others are due to mutations of non‐enzymatic activator proteins (saposin activator protein, SAP, deficiencies) (Tamargo et al, 2012), of solute carriers (cystinosis, infantile sialic acid storage disease) (Gahl et al, 1982; Verheijen et al, 1999) and other lysosomal membrane proteins (Danon disease, due to LAMP2 defective function) (Nishino et al, 2000; Tanaka et al, 2000), or are the consequence of defects in assembly and stability of multienzymatic complexes (galactosialidosis, due to cathepsin A deficiency) (d’Azzo et al, 1982).
New technologies and cellular modeling to study lysosomal function in health and disease
Novel technologies have had significant impacts on the characterization of lysosome biology, the development of diagnostic tools for patients with a suspicion of LSD, and the identification and validation of new therapeutic targets (Fig 2) (Table 1). In several cases, these approaches are based on high‐throughput techniques combined with bioinformatic analysis of a large body of information (metabolomic, genomic, proteomic approaches). Novel approaches also include automated robotic‐based, miniaturized or cell‐based procedures (high‐content imaging) as well as innovative techniques that allow for manipulation of genetic information and generation of in vitro and in vivo models of disease.
Figure 2. New technologies to study lysosomal function and biology.

New technologies, in most cases based on high‐throughput techniques combined with bioinformatic analysis, have been exploited for the diagnostic work‐up in patients with a suspected LSD, for the identification of new disease genes, for the search of disease biomarkers, for the characterization of lysosome biology and disease mechanisms, and for the identification and validation of new therapeutic targets.
Table 1.
Examples of application of novel technologies for LSDs.
| Technology | Applications | Examples of successful applications |
|---|---|---|
| Genomic sequencing |
|
Identification of new LSDs caused by mutation of the VPS33A (Pavlova et al, 2019) and VPS16 (Steel et al, 2020) genes |
| Transcriptomic analysis |
|
Similarities between the microglia expression profiles of LSDs (mucolipidosis type IV mouse and Niemann‐Pick disease type C1) with common neurodegenerative disorders (Cougnoux et al, 2019) |
| Genome‐wide association studies |
|
Identification of a c.510C > T variant that may be predictive of clinical course and outcome in late‐onset Pompe disease patients (Bergsma et al, 2019) |
| microRNA sequencing |
|
Identification of differentially expressed microRNAs potentially predictive of disease severity in Pompe disease (Tarallo et al, 2019) |
| Biochemical and metabolomic analyses |
|
Identification of disease biomarkers for several LSDs (Boutin & Auray‐Blais, 2015; Reunert et al, 2015; Polo et al, 2019) Development of methods for simultaneous detection of multiple enzyme activities in dried blood spots suitable for newborn screening programs for several LSDs (Anderson, 2018; Donati et al, 2018; Kumar et al, 2019; Lukacs et al, 2019; Scott et al, 2020) |
| Cell‐based assays and high‐content imaging technologies |
|
Development of multiplex staining assays that allow screening of FDA‐approved compounds and identification of correctors for cellular phenotypes of LSDs (Pipalia et al, 2006; Pugach et al, 2018) |
| Targeted gene knock‐out and genome editing—iPSc |
|
CRISPR‐Cas9‐mediated generation of knock‐out models of LSDs, such as sphingolipidoses and Niemann‐Pick disease type C (Santos & Amaral, 2019) |
| Organellar omics |
|
Identification of lysosomal proteome and interactome (Sleat et al, 2005; Abu‐Remaileh et al, 2017; Thelen et al, 2017; Rabanal‐Ruiz & Korolchuk, 2018) |
Genomic techniques and next‐generation sequencing (NGS)
A major advancement, particularly in the diagnostic approach to LSDs, was introduced by NGS, a powerful diagnostic tool based on technological platforms that allow sequencing of millions of small fragments of DNA in parallel. This technology has been used both through a targeted strategy with gene panels and by untargeted approaches based on whole‐exome sequencing.
Given the difficulties in the diagnostic work‐up, and due to overlapping clinical phenotypes in LSDs, patients with a clinical suspicion of these disorders are excellent candidates for the application of an NGS‐based diagnosis. Before the introduction of NGS, the traditional approach for the diagnosis of LSDs was based on a step‐by‐step, progressive process starting with physical examination, proceeding to metabolite identification in biological fluids, and leading to the exact diagnosis through the demonstration of an enzymatic deficiency (or the deficiency of a lysosomal function) and the identification of mutations in a specific gene (Winchester, 2014).
NGS‐based approaches are substantially changing this stepwise process. The molecular analysis and search for mutations in LSD‐related genes can be performed immediately after the clinical suspicion of LSD, whereas the functional analysis of the deficient enzyme (or a non‐enzymatic protein) offers a complementary approach to definitively confirm disease diagnoses. Such a diagnostic process may prove to be cost‐effective and by‐pass the need for multiple biochemical analyses or repeated hospital admissions. Some examples of this strategy are already available in the literature, with the development of a gene panel specific for 891 genes involved in the ALP function (Di Fruscio et al, 2015), or the identification of Pompe disease patients in cohorts of unidentified limb‐girdle muscular dystrophies (Savarese et al, 2018).
NGS‐based analysis also has the potential to identify new genes involved in lysosomal disorders, thus expanding the list of LSDs. Indeed, two novel disorders associated with lysosomal abnormalities and impaired vesicle trafficking were recently recognized through whole‐exome sequencing. Mucopolysaccharidosis plus (MPS‐plus), characterized by typical manifestations of mucopolysaccharidoses such as coarse facial features, skeletal abnormalities, hepatosplenomegaly, respiratory problems, and by remarkable levels of glycosaminoglycans excretion in urines (Kondo et al, 2017), was associated with mutations in the VPS33A gene (Pavlova et al, 2019). Loss‐of‐function VPS16 gene mutations were found in patients with an early‐onset dystonia and with ultrastructural lysosomal abnormalities (Steel et al, 2020). Both VPS33A and VPS16 genes encode for subunits of the homotypic fusion and vacuole protein sorting (HOPS) complex that is essential for lysosome fusion with endosomes and autophagosomes (Wartosch et al, 2015).
Genomic approaches may also contribute to the understanding of the complexity and clinical variability of LSDs. The application of these approaches to the study of LSDs is a vast and rapidly expanding field that has been comprehensively reviewed in recent years (Hassan et al, 2017; Davidson et al, 2018).
Genome‐wide association studies have been performed to identify modifiers in some LSDs, for example, Gaucher disease and Pompe disease. In Gaucher disease, single nucleotide polymorphisms (SNPs) within the CLN8 gene locus were in linkage disequilibrium and associated with disease severity, possibly regulating sphingolipid sensing and/or in glycosphingolipid trafficking (Zhang et al, 2012). In Pompe disease, a c.510C > T variant was identified as a genetic modifier in late‐onset patients. This variant negatively influences pre‐mRNA splicing in patients carrying the c.‐32‐13T > G mutation, with significant correlations with residual alpha‐glucosidase activity and may be predictive of clinical course and outcome in late‐onset patients (Bergsma et al, 2019).
Transcriptomic analysis has been performed in several types of LSDs, such as Niemann‐Pick disease type C (Martin et al, 2019), mucopolysaccharidoses (Salvalaio et al, 2017; Peck et al, 2019), progranulin deficiency (Evers et al, 2017), Pompe disease (Turner et al, 2016), and Gaucher disease (Dasgupta et al, 2013), mucolipidosis type IV (Cougnoux et al, 2019). Although the animal models and the tissues differed in these analyses, it was possible to recognize a few common patterns in some diseases. In mucolipidosis type IV mouse microglia, the mixed neuroprotective/neurotoxic expression pattern showed similarities with that observed in Niemann‐Pick disease type C1 (Cougnoux et al, 2019). Of note, the changes observed in mucolipidosis type IV microglia overlapped with alterations found in common neurodegenerative disorders such as Alzheimer's, Parkinson's, and Huntington's diseases. The analysis of mid‐cervical cord in the mouse model of Pompe disease showed up‐regulation of pathways associated with cell death, proinflammatory signaling, and dysregulation of signal transduction pathways suggestive of impaired synaptic function and plasticity (Turner et al, 2016). Transcriptomic analysis in Gaucher disease mice revealed dysregulation of genes involved in cell growth and proliferation, cell cycle, heme metabolism, and mitochondrial dysfunction in liver, lung, and spleen (Dasgupta et al, 2013).
Albeit interesting, these data are largely affected by heterogeneity of the samples, indicating the need for integrated and systematic approaches in homogenous animal and cellular models.
In addition to disease mechanisms, NGS‐based analysis also has the potential to identify novel disease biomarkers. This approach has been used in recent studies that identified microRNAs as markers of Pompe disease, with correlations between disease phenotype and severity, and possibly with the response to enzyme replacement therapy (Cammarata et al, 2018; Tarallo et al, 2019; Carrasco‐Rozas et al, 2019). In Pompe disease, this analysis showed altered expression of microRNAs implicated in signaling pathways related to the pathophysiology of the disease like the mTOR and AMPK pathways, ubiquitin‐mediated proteolysis, cardiac hypertrophy, muscle atrophy, and regeneration, regulation of stem cells pluripotency and myogenesis. One of the differentially expressed microRNAs, miR‐133a, was identified as a potential marker of disease severity and response to therapy (Tarallo et al, 2019).
In Fabry disease, some of the dysregulated microRNAs are also related to the disease pathophysiology, such as miR‐199a‐5p and miR‐126‐3p that are known to be involved in endothelial dysfunction, and miR‐423‐5p and miR‐451a that are involved in myocardial remodeling (Cammarata et al, 2018).
Biochemical, metabolomic analyses, and newborn screening for LSDs
New technologies for high‐throughput analysis of multiple metabolites have also significantly contributed to the diagnostics and monitoring of LSDs. The availability of measurable and objective disease markers remains a major challenge for many LSDs.
Clinical measures are often non‐specific (such as the 6‐min walk test, respiratory function tests, quality of life assessments) or can be influenced by inter‐ and intra‐investigator variance. Biochemical markers may complement these clinical measures and provide accessory, quantitative tools to follow disease course and patient response to therapies. The search for biochemical markers has benefited from a number of modern methodologies such as mass spectrometry or nuclear magnetic resonance‐based approaches. Mass spectrometry has become the most widely used platform for inborn metabolic diseases because of its ability to analyze a wide range of molecules in different body fluids, with optimal dynamic range and great sensitivity (Costanzo et al, 2017). Metabolome analyses have been performed in some LSDs, such as some mucopolysaccharidoses (Fu et al, 2017; Tebani et al, 2019), Fabry disease (Boutin & Auray‐Blais, 2015), Pompe disease (Sato et al, 2017), Niemann‐Pick disease type C (Fan et al, 2013; Maekawa et al, 2015; Probert et al, 2017), neuronal ceroid lipofuscinoses (Sindelar et al, 2018), or in some sphingolipidoses (Polo et al, 2019).
This approach has allowed the identification of markers that may serve to facilitate early‐stage diagnosis and monitoring of response to therapy, for example, galabiosyl ceramide analogs in Fabry disease (Boutin & Auray‐Blais, 2015), oxysterols in Niemann‐Pick disease type C2 (Reunert et al, 2015), and various sphingolipids in some sphingolipidoses (Reunert et al, 2015). However, some issues need to be addressed in these studies, such as those related to the heterogeneity of samples used for the analyses and the number of patients required to be recruited in order to satisfy statistical significance (Percival et al, 2020).
Tandem mass spectrometry and digital microfluidic fluorimetry (DMF‐F) have found important applications in the context of neonatal screening programs (Burlina et al, 2018; Gelb et al, 2019).
The first trials of newborn screening for LSDs started about two decades ago with an immunoassay for the lysosomal marker lysosomal‐associated membrane protein‐1 (LAMP‐1) in dried blood spots (Ranierri et al, 1999). Other approaches followed, such as a multiplex immune‐quantification assay of lysosomal enzymes (Meikle et al, 2006), enzyme assays by DMF‐F, and tandem mass spectrometry (Gelb et al, 2019). Methods to test lysosomal enzyme activities in dried blood spots suitable for newborn screening programs have been developed for Fabry disease, Gaucher disease, Krabbe disease, Niemann‐Pick A/B, Pompe disease, and mucopolysaccharidoses, and for less prevalent disorders such as alpha‐mannosidosis, alpha‐fucosidosis, lysosomal acid lipase deficiency, and ceroid lipofuscinosis 1 and ceroid lipofuscinosis 2 (Anderson, 2018; Donati et al, 2018; Kumar et al, 2019; Lukacs et al, 2019; Scott et al, 2020). In most instances, these methods allow for simultaneous detection of multiple LSDs (Kumar et al, 2019; Lukacs et al, 2019; Hong et al, 2020).
These screening programs, already active in several countries (Schielen et al, 2017), have had a significant impact on the care of LSDs, allowing early diagnosis and timely access to therapies (Chien et al, 2009) and have changed the figure of LSD prevalence in different countries and populations (Spada et al, 2006).
Cell‐based assays and high‐content imaging technologies
Together with robotics, the development of cell‐based assays in combination with high‐content imaging technologies for screening has represented a major and fascinating advance for diseases that are associated with an evident cellular phenotype. Cell‐based high‐content imaging usually exploits automated microscopy and complex image algorithms to extract multidimensional information from hundreds of samples, such as cell morphology, fluorescence intensity, or distribution of fluorescent markers within cells (Bellomo et al, 2017). Results usually have very high statistical power, due to the high number of cells that can be analyzed allowing averaging of large amount of data. The major advantage, however, is represented by the possibility of multiplexing different assays simultaneously in integrated cell populations, cell subpopulations, individual cells, and subcellular structures within a given population (Zanella et al, 2010; Peravali et al, 2011). For these reasons, high‐content imaging is currently used to study disease mechanisms by loss‐of‐function and gain‐of‐function studies, as well as in preclinical drug discovery including target identification, lead optimization, assay validation, or primary and secondary screenings (Bellomo et al, 2017).
Several studies substantiate the potential of cell‐based screening approaches to identify candidate molecules for the treatment of LSDs, some of them exploiting high‐content imaging assays. For example, lysotracker and filipin fluorescence staining assays, or multiplex staining with dual markers filipin and anti‐LAMP1 have been exploited to screen FDA‐approved compounds and identify correctors of Niemann‐Pick disease type C1 (Pipalia et a, 2006; Pugach et al, 2018). One such drug is the antimicrobial alexidine dihydrochloride that appeared to promote increases in NPC1 transcript and mature protein and to be a potent cholesterol‐reducing drug. A cell‐based assay was also developed to detect arylsulfatase A residual activity in cells from patients with metachromatic leukodystrophy (Geng et al, 2011). Furthermore, a cell‐based assay was used to identify three compounds that enhance galactocerebrosidase activity (Jang et al, 2016). Another phenotypic‐based approach was used to identify modulators of autophagy in a murine neuronal cell model of CLN3 disease and led to the identification of compounds that normalized lysosomal positioning and promote clearance of storage material (Petcherski et al, 2019).
In summary, together these data, albeit preliminarly, suggest that cell‐based screening approaches may lead to the development of novel therapeutics for lysosomal storage diseases.
Targeted gene knock‐out and genome editing techniques
Recently, the development of CRISPR/Cas9 approaches for genome editing and the technology for the generation of induced pluripotent stem cells (iPSCs) from skin or blood samples have opened a new era for the development of disease‐relevant cellular models of genetic diseases. These tools have found particular application for the generation of drug‐based screening systems and for the study of disease pathophysiology. iPSCs derived from patients are pluripotent and capable of differentiating into virtually any cell type, including disease‐relevant neuronal subtypes (Khurana et al, 2015) that display major common features of LSD pathology such as autophagic, lysosomal maturation, and mitochondrial defects (Lojewski et al, 2014). However, a major inconvenience of patient‐derived iPSCs is their genetic diversity. The use of (CRISPR)‐Cas9 gene editing to introduce specific genetic changes into a parental pluripotent line allows the production of isogenic lines representing selected mutations of LSD genes that can more readily be compared (Chaterji et al, 2017). CRISPR‐Cas9 is currently being exploited to generate knock‐out models for the study of LSDs. An exhaustive review about modeling of both cellular and in vivo modeling of rare sphingolipidoses and Niemann‐Pick disease type C, using this technology, has been published recently (Santos & Amaral, 2019).
Both CRISPR‐Cas9‐ and iPSC‐based approaches offer unprecedented opportunities for the generation of cellular models of LSDs. These can be used for the identification of new pathways involved in the physiology and pathogenesis of LSDs and for drug screenings to identify of new drugs for the therapy of these diseases. Additionally, CRISPR/Cas9 can be by itself an alternative therapeutic option for treating LSDs by correcting disease‐causing mutations, both in vitro and in vivo (Schwank et al, 2013; Wu et al, 2013; Xie et al, 2014).
Targeted gene knock‐out and knock‐in technologies have found important applications in the generation of in vivo models of LSDs. Compared to in vitro systems, animal models have major advantages as they provide opportunities to study pharmacokinetics, bioavailability, toxicity of therapeutic agents, and to evaluate critical endpoints such as metabolic responses in organs and tissues, and functional measures (Moro & Hanna‐Rose, 2020). Animal models have been developed for nearly all LSDs (Vaquer et al, 2013), the majority in small, prolific species such as mice and rats (Vaquer et al, 2013; Gurda & Vite, 2019). Knock‐in animal models have peculiar advantages as they allow characterization of the phenotypic, pathologic, and functional consequences of specific mutations found in patients (Praggastis et al, 2015) or for testing the response of such mutations to experimental treatments (Khanna et al, 2010).
Organellar omics
The characterization of the lysosomal proteome has been exploited as an important tool for the understanding of lysosomal dysfunction in human disease and of LSD pathophysiology. Proteomic analysis of lysosomes is mainly based on tandem mass spectrometry and has already proven to be a suitable strategy for the analysis of proteins interactions (Sleat et al, 2005), for the identification of biomarkers (Cologna et al, 2012; Matafora et al, 2015), for the identification of the molecular bases of LSDs, such as CLN2 (Sleat et al, 1997) and NPC2 (Naureckiene et al, 2000), and for the identification of novel potential lysosomal membrane transporters (Chapel et al, 2013).
Proteome analyses in LSD animal models and in selected samples from patients have been performed for Krabbe disease (Pellegrini et al, 2019), mucolipidosis III (Di Lorenzo et al, 2018) ceroid lipofuscinoses (Sleat et al, 2019), Niemann‐Pick type C1 (Pergande et al, 2019), in some mucopolysaccharidoses (Yuan et al, 2019), and others. In recent years, in light of the emerging role of lysosomes as signaling platforms controlling cellular metabolism, this approach has attracted renewed and growing interest. Depending on the studies, approximately 200 proteins have been identified, including bona fide lysosomal and lysosomal‐associated proteins. Interestingly, most of the lysosomal‐associated proteins correspond to members of the mTORC1 complex (Thelen et al, 2017; Rabanal‐Ruiz & Korolchuk, 2018). Searching for lysosomal terms in gene ontology and protein databases resulted in the identification of at least 500 proteins (Mi et al, 2019; The UniProt Consortium, 2019).
However, this approach is associated with important challenges related to the limited sensitivity of technologies in detecting low abundance proteins in lysosomes, lack of spatial information about the localization of the identified proteins in whole‐cell analysis, and interaction with other organelles. In recent years, methods to reduce the biomolecular complexity of a sample by isolation and purification of individual cellular organelles have been developed (Diettrich et al, 1998; Chen et al, 2005; Walker & Lloyd‐Evans, 2015; Tharkeshwar et al, 2017). These methods include density gradient centrifugation of cellular or tissue homogenates, use of magnetic iron oxide (FeO)‐coated high‐molecular‐weight dextran particles to purify lysosomes from mammalian cells (Diettrich et al, 1998; Chen et al, 2005), and delivery of superparamagnetic iron oxide nanoparticles (SPIONs) to lysosomes by endocytosis (Walker & Lloyd‐Evans, 2015). In this regard, it is important to mention the importance of the protein localization database based on careful subcellular localization studies combined with MS analysis (Prolocate, http://prolocate.cabm.rutgers.edu). More recent approaches are based on immunoaffinity enrichment of lysosomes from cells expressing a lysosomal transmembrane protein (i.e., TMEM192) fused to three tandem human influenza virus hemagglutinin (HA) epitopes using an antibody against HA conjugated to magnetic beads (Abu‐Remaileh et al, 2017; Wyant et al, 2018). This method is rapid at extracting highly pure lysosomes and may represent an important advance in the analysis of the lysosomal proteome and in the quantitative profiling of metabolites derived from the action of lysosomal enzymes or from the activity of lysosomal transporters under various cell states (Abu‐Remaileh et al, 2017).
Changes in the amounts of specific metabolites within the lysosome might uncover novel aspects involved in the pathophysiology of complex LSD and lead to the identification of aberrant accumulation of specific cargo or metabolites that could be used as biomarkers to test the efficacy of novel therapies.
How defects of lysosomal functions lead to disease
Historically, the pathology and the clinical manifestations of lysosomal disorders have been considered as direct consequences of the storage of inert substrates in tissues.
Indeed, manifestations such as visceromegaly, skin thickness, skeletal dysmorphisms, and ocular signs (corneal opacities, cherry‐red spot) might easily be viewed as the results of excessive undegraded substrates in cells and extracellular matrix.
This concept has been questioned by the new vision of lysosomal functions. Given the central role of lysosomes in cellular homeostasis and metabolism, it has been speculated that storage is just the “instigator” of a number of secondary events (Clarke, 2011) and that accumulation of undegraded substrates is able to prime complex pathogenetic cascades that are in fact responsible for LSD manifestations. Multiple and diverse events are now emerging as players in the pathogenesis of LSDs. Specifically, these events include storage of secondary substrates unrelated to the defective enzyme, abnormal composition of membranes and aberrant fusion and intracellular trafficking of vesicles, altered autophagic flux and accumulation of autophagic substrates, dysregulation of signaling pathways and activation of inflammation, abnormalities of calcium homeostasis, mitochondrial dysfunction, and oxidative stress (Ballabio & Gieselmann, 2009; Platt et al, 2018) (Fig 3).
Figure 3. The mechanisms involved in the pathophysiology of LSD.
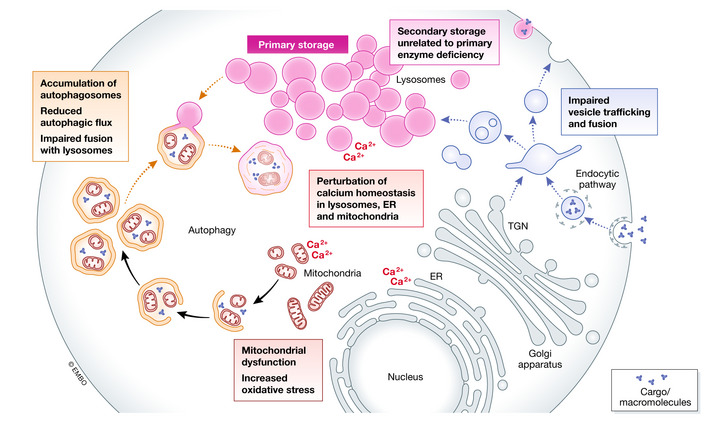
The accumulation of undegraded substrates triggers multiple events that are currently emerging as important players in the pathogenesis of LSDs. These events include storage of secondary substrates unrelated to the defective enzyme; abnormal composition of membranes and aberrant fusion and intracellular trafficking of vesicles; impairment of autophagy; perturbation of calcium homeostasis; and mitochondrial dysfunction and oxidative stress. In addition to the events shown in the figure, in several LSDs storage triggers dysregulation of signaling pathways and activation of inflammation.
Secondary storage
Secondary storage of unrelated and heterogeneous substrates has been extensively documented in several LSDs. For example, in mucopolysaccharidoses types I, II, IIIA, VI, and VII, characterized by primary storage of glycosaminoglycans, biochemical analysis of brain has shown that gangliosides GM2 and GM3 are also consistently and substantially elevated (Walkley & Vanier, 2009). Accumulation of GM2 and GM3 gangliosides has also been documented in a variety of other LSDs, including Niemann‐Pick type A and type C1 (Zervas, et al, 2001), mucolipidosis type IV (Micsenyi et al, 2009) neuronal ceroid lipofuscinoses (Jabs et al, 2008), and alpha‐mannosidosis (Goodman et al, 1991).
Secondary storage is thought to have a substantial role in the pathophysiology of LSDs. This idea is supported by the finding that secondary substrates localize in the brain areas that are most affected by the disease pathology (Tobias et al, 2019; Viana, et al, 2020) and that depletion of secondary substrates in experimental conditions (e.g., GM3 in the Niemann‐Pick type C1 mouse model) results in amelioration of neuropathology and disease manifestations (Lee et al, 2014). Secondary storage may also impair vesicle trafficking. Cholesterol and other lipids, for example, have been shown to impair the endo‐lysosomal system (Sobo et al, 2007; Walkley & Vanier, 2009), and the function of soluble N‐ethylmaleimide‐sensitive factor attachment protein receptors (SNAREs) (Fraldi et al, 2010; Sambri et al, 2017), which are critical for the fusion of cellular membranes (Lang et al, 2001).
These effects are not only limited to lysosomes but also implicated in the accumulation in other compartments of toxic storage materials, including multiple aggregate‐prone proteins, α‐synuclein, prion protein, Tau, amyloid β, and damaged mitochondria (Fraldi et al, 2016) that are known to be associated with common neurodegenerative disorders, such as Alzheimer's, Parkinson's, and Huntington's disease. Alpha‐synuclein accumulation has been found in several LSDs (Settembre et al, 2008a; Shachar et al, 2011; Di Malta et al, 2012) and suggests a link between alpha‐synuclein aggregation toxicity and neurodegeneration in LSDs. Extensive work has been done about beta‐glucocerebrosidase deficiency as a risk factor for the development of Parkinsonism (Aharon‐Peretz et al, 2004; Gan‐Or et al, 2008; Blanz & Saftig, 2016).
Impairment of autophagy
As lysosomes are the terminal compartment of the ALP, a general impairment of this pathway and of its critical functions in cell homeostasis is an obvious and expected finding in LSDs. A block of autophagy was initially recognized in a few of these disorders, namely in mucopolysaccharidosis type IIIA and in MSD (Settembre et al, 2008a), as well as in Pompe disease in which the accumulation of large pools of autophagic debris is a typical and consistent feature of skeletal muscle pathology (Fukuda et al, 2006a; Shea & Raben, 2009) and appears to be associated with dysregulation of mTORC1 and AMPK signaling (Lim et al, 2017).
Autophagic vesicle accumulation, together with increased poly‐ubiquitinated proteins and dysfunctional mitochondria, has now been reported also in several other LSDs, such as some mucopolysaccharidoses, sphingolipidoses (Gaucher and Fabry disease, Niemann‐Pick type C), mucolipidoses (type II, III and IV), Danon disease, and some neuronal ceroid lipofuscinoses (CLN 3, 10) (Liebermann et al, 2012).
The impairment of autophagy has important and deleterious consequences and is thought to contribute substantially to LSD pathophysiology. In Pompe disease, for example, the presence of autophagic accumulation was shown to impair the contractile function of muscles (Drost et al, 2005) and to affect the trafficking of recombinant enzymes used for enzyme replacement therapy (Fukuda et al, 2006b).
In neurons, a dysfunction of the ALP is thought to be involved in the pathophysiology of neurodegeneration (Hara et al, 2006; Komatsu et al, 2006; Monaco et al, 2020). The impairment of the ALP has also been shown to affect extracellular matrix formation and skeletal development and growth in chondrocytes from the mouse model of MPS VII (Bartolomeo et al, 2017; Settembre et al, 2018).
Mitochondrial dysfunction
One of the primary functions of autophagy is to execute mitochondrial turnover (Plotegher & Duchen, 2017; Wang & Wang, 2019). Thus, it is not surprising that mitochondrial dysfunction is emerging as an important player in the pathophysiology of LSDs. Substantial evidence supports the existence of a crosstalk and reciprocal functional relationships between mitochondria and lysosomes (Pshezhetsky, 2015). While defective autophagy affects mitochondrial quality control pathways and causes accumulation of damaged mitochondria, in turn mitochondrial dysfunction can impair lysosomal functions, such as acidification by the acidic pump V‐ATPase that relies on the ATP generated by mitochondria (Stepien et al, 2020).
Perturbations in mitochondrial function and homeostasis have been recognized in several LSDs, including sphingolipidoses (Gaucher disease, Niemann‐Pick disease type C, Krabbe disease), gangliosidoses, some mucopolysaccharidoses, multiple sulfatase deficiency, and neuronal ceroid lipofuscinoses (Plotegher & Duchen, 2017; Stepien et al, 2020) and have been proposed as one of the mechanisms underlying neurodegeneration (Martins et al, 2015; Saffari et al, 2017; Annunziata et al, 2018).
Multiple mitochondrial defects were found in the mouse model of Pompe disease, including mitochondrial calcium excess, increased reactive oxygen species, decreased mitochondrial membrane potential, and decreased oxygen consumption and ATP production (Lim et al, 2015). Increased oxidative stress, elevation of reactive oxygen species (ROS), and enhanced susceptibility of cells to mitochondria‐mediated apoptotic insults are obvious consequences of defects in mitophagy and mitochondrial dysfunction (Filomeni et al, 2015). Oxidative stress was observed in animal models of mucopolysaccharidoses type IIIB (Villani et al, 2009), type I (Donida et al, 2015), and type IIIA (Arfi et al, 2011) and in blood samples from patients affected by mucopolysaccharidoses types I (Pereira et al, 2008) and type II (Filippon et al, 2011).
Alteration of signaling pathways and inflammation
Non‐physiologic activation of signal transduction pathways by storage compounds is another consequence of storage in LSDs. Stored materials may interfere with normal ligand–receptor interactions, modify receptor responses, influence internalization and recycling of receptors, and lead to altered activation of signaling pathways involved in cellular transport and vesicle trafficking, calcium homeostasis, oxidative stress, morphogen signaling, inflammatory and innate immune responses, and cell death (von Zastrow & Sorkin, 2007; Ballabio & Gieselman, 2009; Fiorenza et al, 2018; Platt et al, 2018).
Neuroinflammation and bone involvement are paradigm examples of manifestations related to altered signaling. Neuroinflammation has been reported in a large variety of LSDs, such as mucopolysaccharidoses (Zalfa et al, 2016; Viana et al, 2020; Heon‐Roberts et al, 2020), sphingolipidoses (Potter & Petryniak, 2016; Fiorenza et al, 2018; Cougnoux et al, 2019), neuronal ceroid lipofuscinoses (Groh et al, 2016), and gangliosidoses (Utz et al, 2015). In some mucopolysaccharidoses, it has been shown that structurally anomalous glycosaminoglycans may mimic lipopolysaccharide, an endotoxin of gram‐negative bacteria, and activate the Toll‐like receptor 4 (TLR4) innate immune responses. As a consequence of TLR4 activation, secretion of proinflammatory cytokines increases together with the activation of tumor necrosis factor (TNF)‐alpha (Simonaro et al, 2010; Parker & Bigger, 2019). Furthermore, the activation of an atypical pattern of interferon downstream signaling, involving both interferon (IFN)‐gamma‐ and IFN‐alpha‐responsive genes, was detected in the cerebellum of the Niemann‐Pick disease type C1 mouse model, resulting in the elevation of IFN‐gamma‐responsive cytokines (Shin et al, 2019).
Interestingly, aberrant morphogen signaling has emerged as a possible mechanism that may explain some clinical features of LSDs, such as skeletal dysmorphisms, traditionally viewed as a direct consequence of substrate accumulation (Fiorenza et al, 2018). For example, excess of accumulated glycosaminoglycans and defective proteoglycan desulfation have been shown to alter fibroblast growth factors‐2 (FGF2)‐heparan sulfate interactions and the FGF2 signaling pathway in a murine model of MSD (Settembre et al, 2008b), and bone morphogenetic protein (BMP)‐4 signaling activity in mucopolysaccharidosis type I cells (Pan et al, 2005).
Altered FGF2 and Indian hedgehog distribution and impaired FGF2 signaling have been observed in growth plates from mucopolysaccharidosis type I mice (Kingma et al, 2016). In a zebrafish model of mucopolysaccharidosis type II, perturbations of glycosaminoglycan catabolism were associated with aberrant distribution and signaling of morphogens, such as sonic hedgehog (Shh), dysregulation of the Shh and Wnt/β‐catenin signaling, and aberrant heart development and atrioventricular valve formation (Costa et al, 2017).
Shh dysregulation, with severely disturbed subcellular localization of the Shh effectors Patched (Ptch) and Smoothened (Smo), and of ciliary proteins were found in Niemann‐Pick disease type C1 mice. Dysregulation of Shh signaling has been associated with shortening of primary cilium length and reduction in ciliated cells in animal brains and was proposed as a mechanism underlying abnormal cerebellum morphogenesis in this mouse model (Canterini et al, 2017; Formichi et al, 2018).
Abnormalities of Ca2+ signaling are thought to play an important role in LSDs (Lloyd‐Evans & Waller‐Evans, 2019; Liu & Lieberman, 2019). The importance of this signaling pathway for LSD pathophysiology was revealed by mucolipidosis type IV. This disorder is due to mutations in the mucolipin 1 (MCOLN1) gene that encodes the lysosomal Ca2+‐releasing channel TRPML1. Mucolipidosis IV pathology is the consequence of defective TRPML1 function and aberrant Ca2+ signaling, and is characterized by impaired vesicle trafficking and extensive storage of granular material and lamellar and concentric bodies (LaPlante et al, 2004). Ca2+ abnormalities have also been found in other LSDs and thought to participate in disease pathophysiology. In Niemann‐Pick type C1 mutant cells, for example, remarkable reduction in the acidic compartment calcium stores was observed, compared to wild‐type cells, likely due to sphingosine storage that induces calcium depletion in lysosomes, possibly through an inhibitory effect on Na+/Ca2+ exchangers (Lloyd‐Evans et al, 2008; Lloyd‐Evans & Platt, 2011).
Current and future therapies
Enzyme replacement therapy
Given that the majority of LSDs are due to the deficiency of a lysosomal hydrolase, the primary approach, and the first to be explored, was based on replacing the defective activity with a wild‐type functional enzyme. This strategy was pursued through hematopoietic stem cell transplantation and enzyme replacement therapy (ERT), which are based on the concept that lysosomal enzymes can be taken up by cells and correctly delivered to lysosomes through the mannose‐6‐phosphate pathway.
After the pioneering studies in Gaucher disease (Barton et al, 1990; Barton et al, 1991) that demonstrated the feasibility and the efficacy of ERT in the non‐neuronopathic forms of this disorder, this approach is currently considered the standard treatment for several LSDs, including the most prevalent of these disorders, such as Gaucher disease, Fabry disease, Pompe disease, some mucopolysaccharidoses, and a few ultra‐rare disorders (e.g., lysosomal acid lipase deficiency, mucopolysaccharidosis type VII) and is in clinical development for others (Platt et al, 2018; Poswar et al, 2019). However, despite remarkable success in treating some aspects of LSDs, we have learned from the experience of many years that ERT has limitations. Recombinant enzymes are immunogenic and may induce immune responses, particularly in cross‐reactive immunologic material (CRIM)‐negative patients (Berrier et al, 2015). Development of antibodies against the therapeutic enzyme may impact on the efficacy of therapies or cause immune‐mediated severe adverse reactions that require laborious and expensive protocols for induction of immune tolerance (Desai et al, 2019). ERT is based on periodic infusions, often requiring the use of intra‐venous devices that are associated with risk of life‐threatening infections. Thus, the impact on the quality of life of patients and their caregivers is substantial (Wyatt et al, 2012; Platt, 2018). In addition, costs of therapies are high and represent an economical burden for healthcare systems (Wyatt et al, 2012).
An additional and major issue is the insufficient biodistribution of the recombinant enzymes used for ERT, leading to inability to reach therapeutic concentrations in specific tissues. Recombinant enzymes are large molecules, often unable to cross anatomical and functional barriers. In several LSDs, the main target tissues, where the correction of enzyme activity is required to clear storage and improve pathology, are in fact the most difficult to reach (van Gelder et al, 2012). For these reasons, second‐generation recombinant enzymes with improved targeting properties or pharmacodynamics are currently under development, for example, avalglucosidase‐apha, a glycoengineered GAA, for the treatment of Pompe disease (Pena et al, 2019; Xu et al, 2019) and pegunigalsidase alfa, a PEGylated covalently cross‐linked alpha‐galactosidase A, for the treatment of Fabry disease (Schiffmann et al, 2019).
The inability of recombinant enzymes to cross the blood–brain barrier has major clinical relevance, as the majority of LSDs can be associated with neurological manifestations. Intrathecal or intraventricular delivery of enzyme has been proposed for some mucopolysaccharidoses (Giugliani et al, 2018), metachromatic leukodystrophy (I Dali et al, 2020), and is now an approved treatment for neuronal ceroid lipofuscinosis 2, in which intraventricular infusions of cerliponase‐alpha reduced disease progression (Schulz et al, 2018). The use of chimeric lysosomal enzymes, in which the therapeutic enzyme is conjugated with different peptides or antibody components that exploit interactions with specific receptors and mediate transport across the blood–brain barrier, is also a strategy that is currently being investigated (Sonoda et al, 2018; Pardridge et al, 2018; Yogalingam et al, 2019).
Pharmacological therapy
Indeed, LSDs have proven to be an extraordinary field of investigation with the development of multiple and diverse therapeutic strategies that hit different targets in the pathogenetic cascade of these disorders (Cox, 2015).
One of these strategies was aimed to restore the equilibrium of the so‐called “storage equation” (i.e., the balance between the amount of substrate that is delivered to lysosomes for degradation, and the amount of enzymes that are involved in its breakdown) by reducing flux of substrates to lysosomes with small‐molecule inhibitors of substrate synthesis (substrate reduction therapy—SRT). The first of such compounds to be used was the imino sugar Miglustat (N‐butyl‐deoxynojirimicin), a reversible inhibitor of glucosylceramide synthase, that catalyzes the formation of glucocerebroside and thereby initiates the glycosphingolipid biosynthetic pathway (Pastores & Barnett, 2003). Miglustat showed efficacy in correcting some clinical and biochemical parameters in Gaucher disease patients (Cox, 2005; Charrow & Scott, 2015). For its effect on this early step of the glycosphingolipid biosynthetic pathway, Miglustat has also been proposed for the treatment of GM1 and GM2 gangliosidoses, with a slowed disease progression (Poswar et al, 2019; Fischetto et al, 2020), and is approved for the treatment Niemann‐Pick disease type, in which gangliosides GM2, GM3 gangliosides, and other glycosphingolipids play a role in the pathogenesis of neurological manifestations (Zervas et al, 2001). Other substrate‐reducing molecules are now in clinical development or approved for the treatment of Gaucher and Fabry disease, such as eliglustat tartrate, venglustat, and lucerastat (Felis et al, 2019; Poswar et al, 2019). This approach is attractive, as substrate‐reducing drugs are small molecules that reach therapeutic concentrations in tissues, including those that are difficult to reach by ERT, and can be taken orally, with minimal impact on patient quality of life.
Newer approaches, based on different strategies, are now in a phase of advanced development. In most cases, they are also aimed at increasing or replacing the defective enzyme activity. Pharmacological chaperone therapy (PCT) is based on the use of small‐molecule drugs that enhance the stability of mutant enzyme proteins with residual activity, through specific non‐covalent interactions with the target enzymes, thus favoring their trafficking to lysosomes (Parenti et al, 2015).
PCT is now approved for the treatment of patients affected by Fabry disease. Migalastat (1‐deoxygalactonojirimycin), an active site‐directed imino sugar reversible inhibitor of alpha‐galactosidase A, was shown to paradoxically rescue the activity and the stability of this enzyme in cells from Fabry disease patients, opening the way to further development of this approach (Fan et al, 1999). This compound has shown clinical efficacy on different clinical manifestations of Fabry disease (Germain et al, 2016; Hughes et al, 2017; Lenders et al, 2020) and is now approved for the treatment of patients with amenable GLA gene mutations. PCT has evident advantages compared with ERT, as chaperones are small‐molecule drugs that can be taken orally by patients and are expected to penetrate across membranes and physiological barriers, thus reaching therapeutic concentrations in multiple tissues. On the other hand, a major limitation of this approach is the possibility to treat only patients carrying specific mutations (Benjamin et al, 2017). Concerns on the use of PCT have also been raised as chaperones interact with the catalytic sites and are thus reversible competitive inhibitors of their target enzymes. For the treatment of Fabry disease, this risk has been eluded using a protocol based on discontinuous, every other day administration (Germain et al, 2016), taking advantage of the short half‐life of the drug compared with that of alpha‐galactosidase A. New, allosteric drugs that interact with non‐catalytic domains of the enzyme and are thus non‐inhibitory may represent an alternative strategy to minimize the risk of unwanted enzyme inhibition (Porto et al, 2012; Parenti et al, 2015a).
The use of proteostasis modulators has also been proposed to rescue mutant, unstable lysosomal enzymes (Mu et al, 2008; Fog et al, 2018; Seemann et al, 2020). However, the drugs used for this approach are non‐specific and may be associated with significant adverse effects.
The demonstration of a synergy between PCT and ERT attracted further interest on this approach. This effect was first demonstrated in vitro and in vivo in cells from patients with Pompe disease and in the murine model of this disease (Porto et al, 2009; Khanna et al, 2012). This synergy was translated into clinical trials (Parenti et al, 2014; Kishnani et al, 2017) and is now under further clinical development (Data ref: ClinicalTrials.gov NCT04138277). The advantage of this synergy, compared with the “traditional” use of chaperones, is that the effect of the drug is directed toward the recombinant enzyme used for ERT, and not to the endogenous mutant enzyme. Thus, the effect of chaperones with this approach is mutation‐independent and can in principle be extended to all patients on ERT. Also, the administration of the chaperone is limited to the time of the ERT infusion (e.g., every other week in Pompe disease), with less risk of undesired effects of the drug.
Gene therapy
Gene therapy holds great promise and is under advanced clinical development for several LSDs. The approaches used so far for gene therapy of LSDs are based both on the use of adeno‐associated viral (AAV) vectors in vivo and of lentiviral vectors ex vivo. AAV‐mediated in vivo gene transfer is based on injection of a vector carrying the transgene under the control of ubiquitous or organ‐specific promoters. Ex vivo gene therapy is based on the correction of patient's cells, such as hematopoietic stem cells, followed by genetic modification in vitro and re‐implantation in the patients of the modified cells. Both approaches imply both local correction of a target tissue/organ and cross‐correction of distant tissues/organs by secreted enzymes that are internalized through the mannose‐6‐phosphate receptor pathway (Sands & Davidson, 2006).
Quite encouraging results have been obtained using the ex vivo approach in metachromatic leukodystrophy (Biffi et al, 2013; Sessa et al, 2016). Patients treated early showed improved course or complete prevention of disease manifestations, associated with improved brain MRI scores. In vivo AAV‐mediated gene therapy studies have been completed (Corti et al, 2017) or are in progress.
However, like for ERT, important challenges remain to be faced also by gene therapy, particularly the need for sustained expression of the therapeutic enzyme, and the need for correction of neuropathology. To address this latter issue, studies aimed at correcting brain involvement through direct intraparenchymal or intrathecal vector administration have been performed for the treatment of mucopolysaccharidose types IIIA and IIIB (Tardieu et al, 2014; Tardieu et al, 2017).
Other approaches based on gene editing for mucopolysaccharidosis types I and II (Data ref: ClinicalTrials.gov NCT03041324), inhibition of nonsense‐mediated decay and translational read‐through (Banning et al, 2018), and messenger RNA (mRNA) therapy (an approach based on biosynthetic mRNA transcripts to drive the synthesis of therapeutic proteins) (Zhu et al, 2019) are under evaluation.
Other strategies that are currently under investigation are based on the use of antisense oligonucleotides that allow for rescue of the normal splicing of transcripts. For example, this approach appears to particularly attractive for the treatment of late‐onset Pompe disease patients, in which a c.‐32‐13T > C mutation causes aberrant splicing and exon 2 partial or complete skipping with reduced synthesis of normal mRNA. This variant is highly prevalent (40–70% of alleles). Thus, a therapy for this mutation would have the advantage of being effective in a large fraction of patients. The characterization of splicing regulatory elements in GAA intron 1 and exon 2 and of the effects of the c.510C > T variant that modulates the effects of the c.‐32‐13T > C mutation (Bergsma et al, 2019) helped developing an antisense oligonucleotide‐based approach to promote exon 2 inclusion and enhanced GAA enzyme activity to levels above the disease threshold (van der Wal, 2017). Preliminary in vitro data show promising rescue of acid alfa glucosidase activity in cells from Pompe patients by using this approach (Goina et al, 2017; van der Wal, 2017).
Adjuvant therapies
In view of the emerging complexity of LSD pathophysiology, novel strategies are being explored that are based on an entirely different rationale and may represent adjunctive approaches to the treatment of LSDs. These approaches are not directed toward correction of the enzymatic defects and the causative gene mutations, or on modulation of the flux of substrates to lysosomes, but they are rather targeted to the manipulation of the pathways that are secondarily altered in LSDs.
For example, the abnormalities of the autophagic pathway are attractive targets. Indeed, manipulation of this pathway has been proposed as a therapeutic strategy for Pompe disease, providing some evidence of efficacy. In vitro and in vivo overexpression of TFEB also induced exocytosis, enhanced glycogen clearance, and resulted in some improvements in physical performance of Pompe disease mice (Spampanato et al, 2013; Gatto et al, 2017). In vitro overexpression of TFE3 triggered lysosomal exocytosis and resulted in efficient cellular clearance (Martina et al, 2014). This approach that has been explored so far through overexpression of master genes controlling the autophagic pathway may be difficult to translate into clinical applications. However, based on these proof‐of‐concept studies, alternative strategies based on the search for small‐molecule drugs modulating autophagy may be envisaged, with a better potential for clinical translation.
Aberrant activation inflammation is another potential therapeutic target. For example, as neuroinflammation has been documented in several LSDs, the pathways that are involved in the activation of the inflammasome are now being considered as additional potential therapeutic targets. Pentosan polysulfate, a mixture of semisynthetic sulfated polyanions, has been shown to have anti‐inflammatory effects in some LSDs, in particular targeting the activation TLR4 and the consequent secretion of proinflammatory cytokines and tumor necrosis factor (TNF)‐α. This drug has been tested in mucopolysaccharidosis type I and II patients and in animal models of mucopolysaccharidosis types I, IIIA, and VI (Simonaro et al, 2010; Simonaro et al, 2016; Orii et al, 2019), and in in vitro models of Fabry and Gaucher disease (Crivaro et al, 2019). Intraperitoneal high‐dose aspirin reduced neuroinflammation in mucopolysaccharidosis type IIIB mice, with significantly reduced transcript levels of MIP‐1α, IL‐1β, and GFAP (Arfi et al, 2011). A combination of Miglustat as a substrate‐reducing agent, the Ca2+‐modulator curcumin, and a non‐steroidal anti‐inflammatory drug to target neuroinflammation was evaluated in Niemann‐Pick disease type C1 mice and resulted into maintained body weight and motor function, reduced microglial activation, and delayed onset of Purkinje cell loss (Williams et al, 2014).
Other experimental therapeutic approaches have been directed toward correction of intralysosomal calcium levels in Niemann‐Pick disease type C1 (Lloyd‐Evans et al, 2008) and reduction in oxidative stress in Krabbe disease (Hawkins‐Salsbury et al, 2012), while stimulation of the cytoprotective effect of HSP70 with the small‐molecule arimoclomol in Niemann‐Pick disease type C1 is under clinical evaluation (Kirkegaard et al, 2016).
Although these approaches directed toward correction of the secondary abnormalities in LSDs are not expected to be curative, they may be of help in improving quality of life and slow disease progression. It is possible to speculate that correction of these abnormalities may synergize with existing therapies. For example, in Pompe disease the block of autophagy has been shown to impact on the lysosomal trafficking of the recombinant enzyme used for ERT (Fukuda et al, 2006b). It may be conceivable that improving the status of autophagic pathways may translate into improved lysosomal delivery of the therapeutic enzyme.
It is possible that other potential therapeutic targets will be identified thanks to the precise characterization of the pathogenetic cascade of LSDs and will open new avenues to the treatment of LSDs.
Conflict of interest
A. Ballabio is co‐founder of CASMA Therapeutics and of Next Generation Diagnostics (NGD).
For more information.
NORD—National Association for rare diseases: https://rarediseases.org/rare‐diseases/lysosomal‐storage‐disorders/
Lysosomal Disease Network Patients & Families: https://lysosomaldiseasenetwork.org/
National Gaucher Foundation: https://www.gaucherdisease.org/
National Fabry Disease Foundation: https://www.fabrydisease.org/
Sanfilippo Children Association: https://www.sanfilippo.org.au/
Acid Maltase Deficiency Association: https://amda‐pompe.org/
European Reference Network for Hereditary Metabolic Disorders: https://metab.ern‐net.eu/
Tigem—The Telethon Institute of Genetics and Medicine: https://www.tigem.it/
Telethon Foundation: https://www.telethon.it/
Pending Issues.
The current understanding of lysosome biology and function is still evolving. There is a need for further characterization of these aspects that may provide critical information on the pathophysiology of lysosomal storage diseases.
New methodologies should be exploited to improve our knowledge on lysosomal biology and on lysosomal disease pathophysiology. These methodologies may also have a major impact of patient care, with more efficient diagnostic pathways and availability of biomarkers to follow disease progression and effects of therapies.
Current therapies for the treatment of lysosomal storage disease have significant limitations. Particularly, biodistribution in target organs, such as brain, is a critical issue as many of these disorders are associated with central nervous system involvement.
The understanding of disease pathophysiology is critical as it has the potential to identify novel therapeutic targets and to indicate new strategies for the treatment of these disorders.
Acknowledgements
This work was supported by grants from the Italian Telethon Foundation (TGM16CB6); MIUR FIRB RBAP11Z3YA (A.B.); European Research Council Advanced Investigator no. 694282 (LYSOSOMICS) (A.B.); U.S. National Institutes of Health (R01‐NS078072) (A.B.); the Huffington Foundation (A.B.); the Associazione Italiana per la Ricerca sul Cancro (A.I.R.C.) (A.B.); European Regional Development Fund—POR Campania FESR 2014/2020; (IG 2015 Id17639) and (PPDF) (MCO 10000 and IG 18988 to PPDF); and the Italian Ministry of Health (PPDF) (RF‐2016‐02361540). This manuscript is dedicated to the memory of Thomas Dierks, Bielefeld University, Germany.
EMBO Mol Med (2021) 13: e12836.
See the Glossary for abbreviations used in this article.
References
- A Study to Assess the Long‐term Safety and Efficacy of ATB200/AT2221 in Adult Subjects with LOPD (2019) ClinicalTrials.Gov NCT04138277 (https://clinicaltrials.gov/ct2/show/NCT04138277) [DATASET]
- Abu‐Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, Sabatini DM (2017) Lysosomal metabolomics reveals V‐ATPase‐ and mTOR‐dependent regulation of amino acid efflux from lysosomes. Science 358: 807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aharon‐Peretz J, Rosenbaum H, Gershoni‐Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 351: 1972–1977 [DOI] [PubMed] [Google Scholar]
- Anderson S (2018) Newborn screening for lysosomal storage disorders. J Pediatr Health Care 32: 285–294 [DOI] [PubMed] [Google Scholar]
- Annunziata I, Sano R, d'Azzo A (2018) Mitochondria‐associated ER membranes (MAMs) and lysosomal storage diseases. Cell Death Dis 9: 328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfi A, Richard M, Gandolphe C, Bonnefont‐Rousselot D, Therond P, Scherman D (2011) Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: the positive effect of long‐term aspirin treatment. Mol Genet Metab 103: 18–25 [DOI] [PubMed] [Google Scholar]
- Ascending Dose Study of Genome Editing by the Zinc Finger Nuclease (ZFN) Therapeutic SB‐913 in subjects with MPS II (2017) ClinicalTrials.gov NCT03041324 (https://clinicaltrials.gov/ct2/show/NCT03041324) [DATASET]
- Ballabio A, Gieselmann V (2009) Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 179: 684–696 [DOI] [PubMed] [Google Scholar]
- Ballabio A, Bonifacino JS (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21: 101–118 [DOI] [PubMed] [Google Scholar]
- Banning A, Schiff M, Tikkanen R (2018) Amlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria. Biochim Biophys Acta Mol Basis Dis 1864: 668–675 [DOI] [PubMed] [Google Scholar]
- Bartolomeo R, Cinque L, De Leonibus C, Forrester A, Salzano AC, Monfregola J, De Gennaro E, Nusco E, Azario I, Lanzara C et al (2017) mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest 127: 3717–3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO (1990) Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci USA 87: 1913–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE et al (1991) Replacement therapy for inherited enzyme deficiency–macrophage‐targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 324: 1464–1470 [DOI] [PubMed] [Google Scholar]
- Beck M (2001) Variable clinical presentation in lysosomal storage disorders. J Inherit Metab Dis. 24(Suppl 2): 47–51 [DOI] [PubMed] [Google Scholar]
- Bellomo F, Medina DL, De Leo E, Panarella A, Emma F (2017) High‐content drug screening for rare diseases. J Inherit Metab Dis 40: 601–607 [DOI] [PubMed] [Google Scholar]
- Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, Bronfin B, Williams H, Yu J, Bichet DG et al (2017) The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 19: 430–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsma AJ, In't Groen SLM, van den Dorpel JJA, van den Hout HJMP, van der Beek NAME, Schoser B, Toscano A, Musumeci O, Bembi B, Dardis A et al (2019) A genetic modifier of symptom onset in Pompe disease. EBioMedicine 43: 553–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrier KL, Kazi ZB, Prater SN, Bali DS, Goldstein J, Stefanescu MC, Rehder CW, Botha EG, Ellaway C, Bhattacharya K et al (2015) CRIM‐negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med 17: 912–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S et al (2013) Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 341: 1233158 [DOI] [PubMed] [Google Scholar]
- Blanz J, Saftig P (2016) Parkinson's disease: acid‐glucocerebrosidase activity and alpha‐synuclein clearance. J Neurochem 139(Suppl 1): 198–215 [DOI] [PubMed] [Google Scholar]
- Boutin M, Auray‐Blais C (2015) Metabolomic discovery of novel urinary galabiosylceramide analogs as Fabry disease biomarkers. J Am Soc Mass Spectrom. 26: 499–510 [DOI] [PubMed] [Google Scholar]
- Burlina AB, Polo G, Salviati L, Duro G, Zizzo C, Dardis A, Bembi B, Cazzorla C, Rubert L, Zorden X et al (2018) Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J Inherit Metab Dis 41: 209–219 [DOI] [PubMed] [Google Scholar]
- Calcagnì A, Kors L, Verschuren E, De Cegli R, Zampelli N, Nusco E, Confalonieri S, Bertalot G, Pece S, Settembre C et al (2016) Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. Elife 5: e17047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarata G, Scalia S, Colomba P, Zizzo C, Pisani A, Riccio E, Montalbano M, Alessandro R, Giordano A, Duro G (2018) A pilot study of circulating microRNAs as potential biomarkers of Fabry disease. Oncotarget 9: 27333–27345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canterini S, Dragotto J, Dardis A, Zampieri S, De Stefano ME, Mangia F, Erickson RP, Fiorenza MT (2017) Shortened primary cilium length and dysregulated Sonic hedgehog signaling in Niemann‐Pick C1 disease. Hum Mol Genet. 26: 2277–2289 [DOI] [PubMed] [Google Scholar]
- Carrasco‐Rozas A, Fernández‐Simón E, Lleixà MC, Belmonte I, Pedrosa‐Hernandez I, Montiel‐Morillo E, Nuñez‐Peralta C, Llauger Rossello J, Segovia S, De Luna N et al (2019) Identification of serum microRNAs as potential biomarkers in Pompe disease. Ann Clin Transl Neurol 6: 1214–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapel A, Kieffer‐Jaquinod S, Sagné C, Verdon Q, Ivaldi C, Mellal M, Thirion J, Jadot M, Bruley C, Garin J et al (2013) An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 12: 1572–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrow J, Scott CR (2015) Long‐term treatment outcomes in Gaucher disease. Am J Hematol 90(Suppl 1): S19–24 [DOI] [PubMed] [Google Scholar]
- Chaterji S, Ahn EH, Kim DH (2017) CRISPR genome engineering for human pluripotent stem cell research. Theranostics. 7: 4445–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FW, Gordon RE, Ioannou YA (2005) NPC1 late endosomes contain elevated levels of non‐esterified ('free') fatty acids and an abnormally glycosylated form of the NPC2 protein. Biochem J 390: 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Lee NC, Thurberg BL, Chiang SC, Zhang XK, Keutzer J, Huang AC, Wu MH, Huang PH, Tsai FJ et al (2009) Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 124: e1116–e1125 [DOI] [PubMed] [Google Scholar]
- Christensen KA, Myers JT, Swanson JA (2002) pH‐dependent regulation of lysosomal calcium in macrophages. J Cell Sci 115: 599–607 [DOI] [PubMed] [Google Scholar]
- Cinque L, Forrester A, Bartolomeo R, Svelto M, Venditti R, Montefusco S, Polishchuk E, Nusco E, Rossi A, Medina DL et al (2015) FGF signalling regulates bone growth through autophagy. Nature 528: 272–275 [DOI] [PubMed] [Google Scholar]
- Clarke LA (2011) Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology 50(Suppl 5): v13–v18 [DOI] [PubMed] [Google Scholar]
- Cologna SM, Jiang XS, Backlund PS, Cluzeau CV, Dail MK, Yanjanin NM, Siebel S, Toth CL, Jun HS, Wassif CA et al (2012) Quantitative proteomic analysis of Niemann‐Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS One 7: e47845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti M, Liberati C, Smith BK, Lawson LA, Tuna IS, Conlon TJ, Coleman KE, Islam S, Herzog RW, Fuller DD et al (2017) Safety of intradiaphragmatic delivery of adeno‐associated virus‐mediated alpha‐glucosidase (rAAV1‐CMV‐hGAA) gene therapy in children affected by pompe disease. Hum Gene Ther Clin Dev 28: 208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A (2003) The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113: 445–456 [DOI] [PubMed] [Google Scholar]
- Costa R, Urbani A, Salvalaio M, Bellesso S, Cieri D, Zancan I, Filocamo M, Bonaldo P, Szabo I, Tomanin R et al (2017) Perturbations in cell signaling elicit early cardiac defects in mucopolysaccharidosis type II. Hum Mol Genet 26: 1643–1655 [DOI] [PubMed] [Google Scholar]
- Costanzo M, Zacchia M, Bruno G, Crisci D, Caterino M, Ruoppolo M (2017) Integration of proteomics and metabolomics in exploring genetic and rare metabolic diseases. Kidney Dis 3: 66–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougnoux A, Drummond RA, Fellmeth M, Navid F, Collar AL, Iben J, Kulkarni AB, Pickel J, Schiffmann R, Wassif CA et al (2019) Unique molecular signature in mucolipidosis type IV microglia. J Neuroinflammation 16: 276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TM (2005) Substrate reduction therapy for lysosomal storage diseases. Acta Paediatr Suppl 94: 57–69 [DOI] [PubMed] [Google Scholar]
- Cox TM (2015) Innovative treatments for lysosomal diseases. Best Pract Res Clin Endocrinol Metab 29: 275–311 [DOI] [PubMed] [Google Scholar]
- Crivaro AN, Mucci JM, Bondar CM, Ormazabal ME, Ceci R, Simonaro C, Rozenfeld PA (2019) Efficacy of pentosan polysulfate in in vitro models of lysosomal storage disorders: Fabry and Gaucher disease. PLoS One 14: e0217780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H (1982) Molecular defect in combined beta‐galactosidase and neuraminidase deficiency in man. Proc Natl Acad Sci USA 79: 4535–4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dali IC, Sevin C, Krägeloh‐Mann I, Giugliani R, Sakai N, Wu J, Wasilewski M(2020) Safety of intrathecal delivery of recombinant human arylsulfatase A in children with metachromatic leukodystrophy: results from a phase 1/2 clinical trial. Mol Genet Metab 16: 30152–30159 [DOI] [PubMed] [Google Scholar]
- Dasgupta N, Xu YH, Oh S, Sun Y, Jia L, Keddache M, Grabowski GA (2013) Gaucher disease: transcriptome analyses using microarray or mRNA sequencing in a Gba1 mutant mouse model treated with velaglucerase alfa or imiglucerase. PLoS One 8: e74912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson BA, Hassan S, Garcia EJ, Tayebi N, Sidransky E (2018) Exploring genetic modifiers of Gaucher disease: the next horizon. Hum Mutat. 39: 1739–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F (1955) Tissue fractionation studies. Intracellular distribution patterns of enzymes in rat‐liver tissue. Biochem J 60: 604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai AK, Li C, Rosenberg AS, Kishnani PS (2019) Immunological challenges and approaches to immunomodulation in Pompe disease: a literature review. Ann Transl Med. 7: 285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibble CC, Cantley LC (2015) Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 25: 545–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K (2003) Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)‐formylglycine generating enzyme. Cell 113: 435–444 [DOI] [PubMed] [Google Scholar]
- Diettrich O, Mills K, Johnson AW, Hasilik A, Winchester BG (1998) Application of magnetic chromatography to the isolation of lysosomes from fibroblasts of patients with lysosomal storage disorders. FEBS Lett 441: 369–372 [DOI] [PubMed] [Google Scholar]
- Di Fruscio G, Schulz A, De Cegli R, Savarese M, Mutarelli M, Parenti G, Banfi S, Braulke T, Nigro V, Ballabio A (2015) Lysoplex: an efficient toolkit to detect DNA sequence variations in the autophagy‐lysosomal pathway. Autophagy 11: 9289–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo G, Velho RV, Winter D, Thelen M, Ahmadi S, Schweizer M, De Pace R, Cornils K, Yorgan TA, Grüb S et al (2018) Lysosomal proteome and secretome analysis identifies missorted enzymes and their nondegraded substrates in mucolipidosis III mouse cells. Mol Cell Proteomics. 17: 1612–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Malta C, Fryer JD, Settembre C, Ballabio A (2012) Autophagy in astrocytes: a novel culprit in lysosomal storage disorders. Autophagy 8: 1871–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati MA, Pasquini E, Spada M, Polo G, Burlina A (2018) Newborn screening in mucopolysaccharidoses. Ital J Pediatr 44(Suppl 2): 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donida B, Marchetti DP, Biancini GB, Deon M, Manini PR, da Rosa HT, Moura DJ, Saffi J, Bender F, Burin MG et al (2015) Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim Biophys Acta 1852: 1012–1019 [DOI] [PubMed] [Google Scholar]
- Drost MR, Hesselink RP, Oomens CW, van der Vusse GJ (2005) Effects of non‐contractile inclusions on mechanical performance of skeletal muscle. J Biomech 38: 1035–1043 [DOI] [PubMed] [Google Scholar]
- Evers BM, Rodriguez‐Navas C, Tesla RJ, Prange‐Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F et al (2017) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep 20: 2565–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JQ, Ishii S, Asano N, Suzuki Y (1999) Accelerated transport and maturation of lysosomal alpha‐galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med 5: 112–115 [DOI] [PubMed] [Google Scholar]
- Fan M, Sidhu R, Fujiwara H, Tortelli B, Zhang J, Davidson C, Walkley SU, Bagel JH, Vite C, Yanjanin NM et al (2013) Identification of Niemann‐Pick C1 disease biomarkers through sphingolipid profiling. J Lipid Res 54: 2800–2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felis A, Whitlow M, Kraus A, Warnock DG, Wallace E (2019) Current and investigational therapeutics for fabry disease. Kidney Int Rep 5: 407–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippon L, Vanzin CS, Biancini GB, Pereira IN, Manfredini V, Sitta A, Peralba Mdo C, Schwartz IV, Giugliani R, Vargas CR (2011) Oxidative stress in patients with mucopolysaccharidosis type II before and during enzyme replacement therapy. Mol Genet Metab 103: 121–127 [DOI] [PubMed] [Google Scholar]
- Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22: 377–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorenza MT, Moro E, Erickson RP (2018) The pathogenesis of lysosomal storage disorders: beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum Mol Genet 27: R119–R129 [DOI] [PubMed] [Google Scholar]
- Fischetto R, Palladino V, Mancardi MM, Giacomini T, Palladino S, Gaeta A, Di Rocco M, Zampini L, Lassandro G, Favia V et al (2020) Substrate reduction therapy with Miglustat in pediatric patients with GM1 type 2 gangliosidosis delays neurological involvement: a multicenter experience. Mol Genet Genomic Med 8: e1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fog CK, Zago P, Malini E, Solanko LM, Peruzzo P, Bornaes C, Magnoni R, Mehmedbasic A, Petersen NHT, Bembi B et al (2018) The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 38: 142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formichi P, Battisti C, De Santi MM, Guazzo R, Tripodi SA, Radi E, Rossi B, Tarquini E, Federico A (2018) Primary cilium alterations and expression changes of Patched1 proteins in niemann‐pick type C disease. J Cell Physiol 233: 663–672 [DOI] [PubMed] [Google Scholar]
- Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K et al (2010) Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J 29: 3607–3620 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Fraldi A, Klein AD, Medina DL, Settembre C (2016) Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci 39: 277–295 [DOI] [PubMed] [Google Scholar]
- Fu H, Meadows AS, Pineda RJ, Mohney RP, Stirdivant S, McCarty DM (2017) Serum global metabolomics profiling reveals profound metabolic impairments in patients with MPS IIIA and MPS IIIB. Metab Brain Dis 32: 1403–1415 [DOI] [PubMed] [Google Scholar]
- Fukuda T, Roberts A, Ahearn M, Zaal K, Ralston E, Plotz PH, Raben N (2006a) Autophagy and lysosomes in Pompe disease. Autophagy 2: 318–320 [DOI] [PubMed] [Google Scholar]
- Fukuda T, Ahearn M, Roberts A, Mattaliano RJ, Zaal K, Ralston E, Plotz PH, Raben N (2006b) Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol Ther 14: 831–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahl WA, Bashan N, Tietze F, Bernardini I, Schulman JD (1982) Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217: 1263–1265 [DOI] [PubMed] [Google Scholar]
- Gan‐Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, Bar‐Shira A, Orr‐Urtreger A (2008) Genotype‐phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70: 2277–2283 [DOI] [PubMed] [Google Scholar]
- Gatto F, Rossi B, Tarallo A, Polishchuk E, Polishchuk R, Carrella A, Nusco E, Alvino FG, Iacobellis F, De Leonibus E et al (2017) AAV‐mediated transcription factor EB (TFEB) gene delivery ameliorates muscle pathology and function in the murine model of Pompe Disease. Sci Rep 7: 15089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelb MH, Lukacs Z, Ranieri E, Schielen PCJI (2019) Newborn screening for lysosomal storage disorders: methodologies for measurement of enzymatic activities in dried blood spots. Int J Neonatal Screen 5: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng H, Whiteley G, Ribbens J, Zheng W, Southall N, Hu X, Marugan JJ, Ferrer M, Maegawa GH (2011) Novel patient cell‐based HTS assay for identification of small molecules for a lysosomal storage disease. PLoS One 6: e29504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, Feliciani C, Shankar SP, Ezgu F, Amartino H et al (2016) Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 375: 545–555 [DOI] [PubMed] [Google Scholar]
- Giugliani R, Vairo F, Kubaski F, Poswar F, Riegel M, Baldo G, Saute JA (2018) Neurological manifestations of lysosomal disorders and emerging therapies targeting the CNS. Lancet Child Adolesc Health 2: 56–68 [DOI] [PubMed] [Google Scholar]
- Goina E, Peruzzo P, Bembi B, Dardis A, Buratti E (2017) Glycogen reduction in myotubes of late‐onset pompe disease patients using antisense technology. Mol Ther 25: 2117–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LA, Livingston PO, Walkley SU (1991) Ectopic dendrites occur only on cortical pyramidal cells containing elevated GM2 ganglioside in alpha‐mannosidosis. Proc Natl Acad Sci USA 88: 11330–11334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh J, Ribechini E, Stadler D, Schilling T, Lutz MB, Martini R (2016) Sialoadhesin promotes neuroinflammation‐related disease progression in two mouse models of CLN disease. Glia 64: 792–809 [DOI] [PubMed] [Google Scholar]
- Gurda BL, Vite CH (2019) Large animal models contribute to the development of therapies for central and peripheral nervous system dysfunction in patients with lysosomal storage diseases. Hum Mol Genet 28: R119–R131 [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki‐Migishima R, Yokoyama M, Mishima K, Saito I, Okano H et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: 885–889 [DOI] [PubMed] [Google Scholar]
- Hassan S, Sidransky E, Tayebi N (2017) The role of epigenetics in lysosomal storage disorders: uncharted territory. Mol Genet Metab 122: 10–18 [DOI] [PubMed] [Google Scholar]
- Hawkins‐Salsbury JA, Qin EY, Reddy AS, Vogler CA, Sands MS (2012) Oxidative stress as a therapeutic target in globoid cell leukodystrophy. Exp Neurol 237: 444–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard JM, Bruyère J, Roy E, Bigou S, Ausseil J, Vitry S (2010) Storage problems in lysosomal diseases. Biochem Soc Trans 38: 1442–1447 [DOI] [PubMed] [Google Scholar]
- Heon‐Roberts R, Nguyen ALA, Pshezhetsky AV (2020) Molecular bases of neurodegeneration and cognitive decline, the major burden of sanfilippo disease. J Clin Med 9: 344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hers HG (1963) alpha‐Glucosidase deficiency in generalized glycogen‐storage disease (Pompe's disease). Biochem J. 86: 11–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Neufeld EF (1972) A hypothesis for I‐cell disease: defective hydrolases that do not enter lysosomes. Biochem Biophys Res Commun 49: 992–999 [DOI] [PubMed] [Google Scholar]
- Hong X, Sadilek M, Gelb MH (2020) A highly multiplexed biochemical assay for analytes in dried blood spots: application to newborn screening and diagnosis of lysosomal storage disorders and other inborn errors of metabolism. Genet Med 22: 1262–1268 [DOI] [PubMed] [Google Scholar]
- Hopkins PV, Klug T, Vermette L, Raburn‐Miller J, Kiesling J, Rogers S (2018) Incidence of 4 lysosomal storage disorders from 4 years of newborn screening. JAMA Pediatr. 172: 696–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DA, Nicholls K, Shankar SP, Sunder‐Plassmann G, Koeller D, Nedd K, Vockley G, Hamazaki T, Lachmann R et al (2017) Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18‐month results from the randomised phase III ATTRACT study. J Med Genet 54: 288–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs S, Quitsch A, Käkelä R, Koch B, Tyynelä J, Brade H, Glatzel M, Walkley S, Saftig P, Vanier MT et al (2008) Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J Neurochem 106: 1415–1425 [DOI] [PubMed] [Google Scholar]
- Jang DS, Ye W, Guimei T, Solomon M, Southall N, Hu X, Marugan J, Ferrer M, Maegawa GH (2016) Cell‐based high‐throughput screening identifies galactocerebrosidase enhancers as potential small‐molecule therapies for Krabbe's disease. J Neurosci Res 94: 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman EC, Ricketts CJ, Rais‐Bahrami S, Yang Y, Merino MJ, Bottaro DP, Srinivasan R, Linehan WM (2014) Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol 11: 465–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S (2013) Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci 126: 60–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma SDK, Wagemans T, IJlst L, Bronckers ALJJ, van Kuppevelt TH, Everts V, Wijburg FA, van Vlies N(2016) Altered interaction and distribution of glycosaminoglycans and growth factors in mucopolysaccharidosis type I bone disease. Bone 88: 92–100 [DOI] [PubMed] [Google Scholar]
- Kirkegaard T, Gray J, Priestman DA, Wallom KL, Atkins J, Olsen OD, Klein A, Drndarski S, Petersen NH, Ingemann L et al (2016) Heat shock protein‐based therapy as a potential candidate for treating the sphingolipidoses. Sci Transl Med 8: 355ra118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani P, Tarnopolsky M, Roberts M, Sivakumar K, Dasouki M, Dimachkie MM, Finanger E, Goker‐Alpan O, Guter KA, Mozaffar T et al (2017) Duvoglustat HCl increases systemic and tissue exposure of active acid alpha‐glucosidase in pompe patients co‐administered with alglucosidase alpha. Mol Ther 25: 1199–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Soska R, Lun Y, Feng J, Frascella M, Young B, Brignol N, Pellegrino L, Sitaraman SA, Desnick RJ et al (2010) The pharmacological chaperone 1‐deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol Ther 18: 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Flanagan JJ, Feng J, Soska R, Frascella M, Pellegrino LJ, Lun Y, Guillen D, Lockhart DJ, Valenzano KJ (2012) The pharmacological chaperone AT2220 increases recombinant human acid α‐glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS One 7: e40776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Ichiyama Y, Kominami E et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884 [DOI] [PubMed] [Google Scholar]
- Kondo H, Maksimova N, Otomo T, Kato H, Imai A, Asano Y, Kobayashi K, Nojima S, Nakaya A, Hamada Y et al (2017) Mutation in VPS33A affects metabolism of glycosaminoglycans: a new type of mucopolysaccharidosis with severe systemic symptoms. Hum Mol Genet 26: 173–183 [DOI] [PubMed] [Google Scholar]
- Kroos M, Hoogeveen‐Westerveld M, van der Ploeg A, Reuser AJ (2012) The genotype‐phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet. 160: 59–68 [DOI] [PubMed] [Google Scholar]
- Kumar AB, Hong X, Yi F, Wood T, Gelb MH (2019) Tandem mass spectrometry‐based multiplex assays for α‐mannosidosis and fucosidosis. Mol Genet Metab 127: 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Bruns D, Wenzel D, Riedel D, Holroyd P, Thiele R, Jahn R (2001) SNAREs are concentrated in cholesterol‐dependent clusters that define docking and fusion sites for exocytosis. EMBO J 20: 2202–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPlante JM, Ye CP, Quinn SJ, Goldin E, Brown EM, Slaugenhaupt SA, Vassilev PM (2004) Functional links between mucolipin‐1 and Ca2+‐dependent membrane trafficking in mucolipidosis IV. Biochem Biophys Res Commun 322: 1384–1391 [DOI] [PubMed] [Google Scholar]
- Lawrence RE, Fromm SA, Fu Y, Yokom AL, Kim DJ, Thelen AM, Young LN, Lim CY, Samelson AJ, Hurley JH et al (2019) Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 366: 971–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lee JK, Bae YC, Yang SH, Okino N, Schuchman EH, Yamashita T, Bae JS, Jin HK (2014) Inhibition of GM3 synthase attenuates neuropathology of Niemann‐Pick disease Type C. by affecting sphingolipid metabolism. Mol Cells 37: 161–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenders M, Nordbeck P, Kurschat C, Karabul N, Kaufeld J, Hennermann JB, Patten M, Cybulla M, Müntze J, Üçeyler N et al (2020) Treatment of Fabry's disease with migalastat: outcome from a prospective observational multicenter study (FAMOUS). Clin Pharmacol Ther 108: 326–337 [DOI] [PubMed] [Google Scholar]
- Lim JA, Li L, Kakhlon O, Myerowitz R, Raben N (2015) Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 11: 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim JA, Li L, Shirihai OS, Trudeau KM, Puertollano R, Raben N (2017) Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO Mol Med 9: 353–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lojewski X, Staropoli JF, Biswas‐Legrand S, Simas AM, Haliw L, Selig MK, Coppel SH, Goss KA, Petcherski A, Chandrachud U et al (2014) Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet 23: 2005–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd‐Evans E, Platt FM (2011) Lysosomal Ca(2+) homeostasis: role in pathogenesis of lysosomal storage diseases. Cell Calcium 50: 200–205 [DOI] [PubMed] [Google Scholar]
- Lloyd‐Evans E, Morgan AJ, He X, Smith DA, Elliot‐Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM (2008) Niemann‐Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med 14: 1247–1255 [DOI] [PubMed] [Google Scholar]
- Lloyd‐Evans E, Waller‐Evans H (2019) Lysosomal Ca(2+) Homeostasis and Signaling in Health and Disease. Cold Spring Harb Perspect Biol a035311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Tardiff DF, Chung CY, Lindquist S (2015) Toward stem cell‐based phenotypic screens for neurodegenerative diseases. Nat Rev Neurol 11: 339–350 [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A (2012) Autophagy in lysosomal storage disorders. Autophagy 8: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Palmfeldt J, Lin L, Colaço A, Clemmensen KKB, Huang J, Xu F, Liu X, Maeda K, Luo Y et al (2018) STAT3 associates with vacuolar H+‐ATPase and regulates cytosolic and lysosomal pH. Cell Res 28: 996–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu EA, Lieberman AP (2019) The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neurosci Lett 697: 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübke T, Lobel P, Sleat DE (2009) Proteomics of the lysosome. Biochim Biophys Acta 1793: 625–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacs Z, Nickel M, Murko S, Nieves Cobos P, Schulz A, Santer R, Kohlschütter A (2019) Validity of a rapid and simple fluorometric tripeptidyl peptidase 1 (TPP1) assay using dried blood specimens to diagnose CLN2 disease. Clin Chim Acta 492: 69–71 [DOI] [PubMed] [Google Scholar]
- Maekawa M, Shimada M, Ohno K, Togawa M, Nittono H, Iida T, Hofmann AF, Goto J, Yamaguchi H, Mano N (2015) Focused metabolomics using liquid chromatography/electrospray ionization tandem mass spectrometry for analysis of urinary conjugated cholesterol metabolites from patients with Niemann‐Pick disease type C and 3β‐hydroxysteroid dehydrogenase deficiency. Ann Clin Biochem. 52: 576–587 [DOI] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2013) RRAG GTPases link nutrient availability to gene expression, autophagy and lysosomal biogenesis. Autophagy 9: 928–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Lishu L, Jeong‐A L, Patange S, Raben N, Puertollano R (2014) The nutrient‐responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal 7: ra9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin KB, Williams IM, Cluzeau CV, Cougnoux A, Dale RK, Iben JR, Cawley NX, Wassif CA, Porter FD (2019) Identification of novel pathways associated with patterned cerebellar purkinje neuron degeneration in niemann‐pick disease, type C1. Int J Mol Sci 21: 292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins C, Hůlková H, Dridi L, Dormoy‐Raclet V, Grigoryeva L, Choi Y, Langford‐Smith A, Wilkinson FL, Ohmi K, DiCristo G et al (2015) Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 138: 336–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matafora V, Cuccurullo M, Beneduci A, Petrazzuolo O, Simeone A, Anastasio P, Mignani R, Feriozzi S, Pisani A, Comotti C et al (2015) Early markers of Fabry disease revealed by proteomics. Mol Biosyst 11: 1543–1551 [DOI] [PubMed] [Google Scholar]
- Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto‐Rosato A, Prezioso C, Forrester A et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17: 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle PJ, Grasby DJ, Dean CJ, Lang DL, Bockmann M, Whittle AM, Fietz MJ, Simonsen H, Fuller M, Brooks DA et al (2006) Newborn screening for lysosomal disease. Mol Genet Metab 88: 307–314 [DOI] [PubMed] [Google Scholar]
- Mehta A, Beck M, Linhart A, Sunder‐Plassmann G, Widmer U (2006) History of lysosomal storage diseases: an overview In: Mehta A, Beck M, Sunder‐Plassmann G (eds). Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD (2019) Protocol Update for large‐scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14: 703–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsenyi MC, Dobrenis K, Stephney G, Pickel J, Vanier MT, Slaugenhaupt SA, Walkley SU (2009) Neuropathology of the Mcoln1(‐/‐) knockout mouse model of mucolipidosis type IV. J Neuropathol Exp Neurol 68: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco A, Maffia V, Sorrentino NC, Sambri I, Ezhova Y, Giuliano T, Cacace V, Nusco E, De Risi M, De Leonibus E et al (2020) The amyloid inhibitor CLR01 relieves autophagy and ameliorates neuropathology in a severe lysosomal storage disease. Mol Ther 28: 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro CA, Hanna‐Rose W (2020) Animal model contributions to congenital metabolic disease. Adv Exp Med Biol 1236: 225–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, Kelly JW (2008) Chemical and biological approaches synergize to ameliorate protein‐folding diseases. Cell 134: 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Di Malta C, Esposito A, de Araujo MEG, Pece S, Bertalot G, Matarese M, Benedetti V, Zampelli A, Stasyk T et al (2020) A substrate‐specific mTORC1 pathway underlies Birt‐Hogg‐Dube’ syndrome. Nature 585: 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P (2000) Identification of HE1 as the second gene of Niemann‐Pick C disease. Science 290: 2298–2301 [DOI] [PubMed] [Google Scholar]
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y et al (2000) Primary LAMP‐2 deficiency causes X‐linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406: 906–910 [DOI] [PubMed] [Google Scholar]
- Novikoff AB, Beaufay AB, de Duve C (1956) Electron microscopy of lysosome rich fractions from rat liver. J Biophys Biochem Cytol. 2: 179–184 [PMC free article] [PubMed] [Google Scholar]
- Orii K, Lim A, Tomatsu S, Stapleton M, Suzuki Y, Simonaro CM, Schuchman EH, Fukao T, Matsumoto T (2019) Safety study of sodium pentosan polysulfate for adult patients with mucopolysaccharidosis type II. Diagnostics 9: 226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C, Nelson MS, Reyes M, Koodie L, Brazil JJ, Stephenson EJ, Zhao RC, Peters C, Selleck SB, Stringer SE et al (2005) Functional abnormalities of heparan sulfate in mucopolysaccharidosis‐I are associated with defective biologic activity of FGF‐2 on human multipotent progenitor cells. Blood 106: 1956–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardridge WM, Boado RJ, Giugliani R, Schmidt M (2018) Plasma pharmacokinetics of valanafusp alpha, a human insulin receptor antibody‐iduronidase fusion protein, in patients with mucopolysaccharidosis type I. BioDrugs 32: 169–176 [DOI] [PubMed] [Google Scholar]
- Parenti G, Fecarotta S, la Marca G, Rossi B, Ascione S, Donati MA, Morandi LO, Ravaglia S, Pichiecchio A, Ombrone D et al (2014) A chaperone enhances blood α‐glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol Ther 22: 2004–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G, Andria G, Ballabio A (2015a) Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 66: 471–486 [DOI] [PubMed] [Google Scholar]
- Parenti G, Andria G, Valenzano KJ (2015b) Pharmacological chaperone therapy: preclinical development, clinical translation, and prospects for the treatment of lysosomal storage disorders. Mol Ther. 23: 1138–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker H, Bigger BW (2019) The role of innate immunity in mucopolysaccharide diseases. J Neurochem 148: 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastores GM, Torres PA, Zeng BJ (2013) Animal models for lysosomal storage disorders. Biochemistry 78: 721–725 [DOI] [PubMed] [Google Scholar]
- Pastores GM, Barnett NL (2003) Substrate reduction therapy: miglustat as a remedy for symptomatic patients with Gaucher disease type 1. Expert Opin Invest Drugs 12: 273–281 [DOI] [PubMed] [Google Scholar]
- Pavlova EV, Shatunov A, Wartosch L, Moskvina AI, Nikolaeva LE, Bright NA, Tylee KL, Church HJ, Ballabio A, Luzio JP et al (2019) The lysosomal disease caused by mutant VPS33A. Hum Mol Genet 28: 2514–2530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck SH, Tobias JW, Shore EM, Malhotra NR, Haskins ME, Casal ML, Smith LJ (2019) Molecular profiling of failed endochondral ossification in mucopolysaccharidosis VII. Bone 128: 115042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini D, Del Grosso A, Angella L, Giordano N, Dilillo M, Tonazzini I, Caleo M, Cecchini M, McDonnell LA (2019) Quantitative microproteomics based characterization of the central and peripheral nervous system of a mouse model of krabbe disease. Mol Cell Proteomics 18: 1227–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena LDM, Barohn RJ, Byrne BJ, Desnuelle C, Goker‐Alpan O, Ladha S, Laforêt P, Mengel KE, Pestronk A, Pouget J et al (2019) Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment‐naïve and alglucosidase alfa‐treated patients with late‐onset Pompe disease: A phase 1, open‐label, multicenter, multinational, ascending dose study. Neuromuscul Disord 29: 167–186 [DOI] [PubMed] [Google Scholar]
- Pereira VG, Martins AM, Micheletti C, D'Almeida V (2008) Mutational and oxidative stress analysis in patients with mucopolysaccharidosis type I undergoing enzyme replacement therapy. Clin Chim Acta 387: 75–79 [DOI] [PubMed] [Google Scholar]
- Pergande MR, Nguyen TTA, Haney‐Ball C, Davidson CD, Cologna SM (2019) Quantitative, label‐free proteomics in the symptomatic niemann‐pick, type C1 mouse model using standard flow liquid chromatography and thermal focusing electrospray ionization. Proteomics 19: e1800432 [DOI] [PubMed] [Google Scholar]
- Peravali R, Gehrig J, Giselbrecht S, Lütjohann DS, Hadzhiev Y, Müller F, Liebel U (2011) Automated feature detection and imaging for high‐resolution screening of zebrafish embryos. Biotechniques 50: 319–324 [DOI] [PubMed] [Google Scholar]
- Percival BC, Gibson M, Wilson PB, Platt FM, Grootveld M (2020) Metabolomic studies of lipid storage disorders, with special reference to niemann‐pick type C disease: a critical review with future perspectives. Int J Mol Sci 21: 2533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Zoncu R (2016) The lysosome as a regulatory hub. Annu Rev Cell Dev Biol 32: 223–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petcherski A, Chandrachud U, Butz ES, Klein MC, Zhao WN, Reis SA, Haggarty SJ, Ruonala MO, Cotman SL (2019) An autophagy modifier screen identifies small molecules capable of reducing autophagosome accumulation in a model of CLN3‐mediated neurodegeneration. Cells 8: 1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pshezhetsky AV (2015) Crosstalk between 2 organelles: lysosomal storage of heparan sulfate causes mitochondrial defects and neuronal death in mucopolysaccharidosis III type C. Rare Dis 3(1): e1049793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipalia NH, Huang A, Ralph H, Rujoi M, Maxfield FR (2006) Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann‐Pick C cells. J Lipid Res 47: 284–301 [DOI] [PubMed] [Google Scholar]
- Platt FM (2018) Emptying the stores: lysosomal diseases and therapeutic strategies. Nat Rev Drug Discov 17: 133–150 [DOI] [PubMed] [Google Scholar]
- Platt FM, d'Azzo A, Davidson BL, Neufeld EF, Tifft CJ (2018) Lysosomal storage diseases. Nat Rev Dis Primers 4: 27 [DOI] [PubMed] [Google Scholar]
- Plotegher N, Duchen MR (2017) Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol Med 23: 116–134 [DOI] [PubMed] [Google Scholar]
- Polo G, Burlina AP, Ranieri E, Colucci F, Rubert L, Pascarella A, Duro G, Tummolo A, Padoan A, Plebani M et al (2019) Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study. Clin Chem Lab Med 57: 1863–1874 [DOI] [PubMed] [Google Scholar]
- Porto C, Cardone M, Fontana F, Rossi B, Tuzzi MR, Tarallo A, Barone MV, Andria G, Parenti G (2009) The pharmacological chaperone N‐butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol Ther 17: 964–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto C, Ferrara MC, Meli M, Acampora E, Avolio V, Rosa M, Cobucci‐Ponzano B, Colombo G, Moracci M, Andria G et al (2012) Pharmacological enhancement of α‐glucosidase by the allosteric chaperone N‐acetylcysteine. Mol Ther. 20: 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poswar FO, Vairo F, Burin M, Michelin‐Tirelli K, Brusius‐Facchin AC, Kubaski F, Souza CFM, Baldo G, Giugliani R (2019) Lysosomal diseases: Overview on current diagnosis and treatment. Genet Mol Biol 42(suppl 1): 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter GB, Petryniak MA (2016) Neuroimmune mechanisms in Krabbe's disease. J Neurosci Res 94: 1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praggastis M, Tortelli B, Zhang J, Fujiwara H, Sidhu R, Chacko A, Chen Z, Chung C, Lieberman AP, Sikora J et al (2015) A murine Niemann‐Pick C1 I1061T knock‐in model recapitulates the pathological features of the most prevalent human disease allele. J Neurosci 35: 8091–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probert F, Ruiz‐Rodado V, Vruchte DT, Nicoli ER, Claridge TDW, Wassif CA, Farhat N, Porter FD, Platt FM, Grootveld M (2017) NMR analysis reveals significant differences in the plasma metabolic profiles of Niemann Pick C1 patients, heterozygous carriers, and healthy controls. Sci Rep 7: 6320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puertollano R, Ferguson SM, Brugarolas J, Ballabio A (2018) The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 37: e98804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugach EK, Feltes M, Kaufman RJ, Ory DS, Bang AG (2018) High‐content screen for modifiers of Niemann‐Pick type C disease in patient cells. Hum Mol Genet 27: 2101–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal‐Ruiz Y, Korolchuk VI (2018) mTORC1 and nutrient homeostasis: the central role of the lysosome. Int J Mol Sci 19: 818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Puertollano R (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol 32: 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranierri E, Gerace RL, Ravenscroft EM, Hopwood JJ, Meikle PJ (1999) Pilot neonatal screening program for lysosomal storage disorders, using lamp‐1. Southeast Asian. J Trop Med Public Health 30: 111–113 [PubMed] [Google Scholar]
- Ren H, Wang G (2020) Autophagy and lysosome storage disorders. Adv Exp Med Biol 1207: 87–102 [DOI] [PubMed] [Google Scholar]
- Reunert J, Lotz‐Havla AS, Polo G, Kannenberg F, Fobker M, Griese M, Mengel E, Muntau AC, Schnabel P, Sommerburg O et al (2015) Niemann‐Pick Type C‐2 disease: identification by analysis of plasma cholestane‐3β,5α,6β‐triol and further insight into the clinical phenotype. JIMD Rep 23: 17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffari A, Kölker S, Hoffmann GF, Ebrahimi‐Fakhari D (2017) Linking mitochondrial dysfunction to neurodegeneration in lysosomal storage diseases. J Inherit Metab Dis 40: 631–640 [DOI] [PubMed] [Google Scholar]
- Saftig P, Klumperman J (2009) Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 10: 623–635 [DOI] [PubMed] [Google Scholar]
- Salvalaio M, D'Avanzo F, Rigon L, Zanetti A, D'Angelo M, Valle G, Scarpa M, Tomanin R (2017) Brain RNA‐Seq profiling of the mucopolysaccharidosis type II mouse model. Int J Mol Sci 18: 1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambri I, D'Alessio R, Ezhova Y, Giuliano T, Sorrentino NC, Cacace V, De Risi M, Cataldi M, Annunziato L, De Leonibus E et al (2017) Lysosomal dysfunction disrupts presynaptic maintenance and restoration of presynaptic function prevents neurodegeneration in lysosomal storage diseases. EMBO Mol Med 9: 112–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands MS, Davidson BL (2006) Gene therapy for lysosomal storage diseases. Mol Ther 13: 839–849 [DOI] [PubMed] [Google Scholar]
- Santos R, Amaral O (2019) Advances in sphingolipidoses: CRISPR‐Cas9 editing as an option for modelling and therapy. Int J Mol Sci 20: 5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS et al (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- Sato Y, Kobayashi H, Higuchi T, Shimada Y, Ida H, Ohashi T (2017) Metabolomic profiling of Pompe disease‐induced pluripotent stem cell‐derived cardiomyocytes reveals that oxidative stress is associated with cardiac and skeletal muscle pathology. Stem Cells Transl Med. 6: 31–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarese M, Torella A, Musumeci O, Angelini C, Astrea G, Bello L, Bruno C, Comi GP, Di Fruscio G, Piluso G et al (2018) Targeted gene panel screening is an effective tool to identify undiagnosed late onset Pompe disease. Neuromuscul Disord 28: 586–591 [DOI] [PubMed] [Google Scholar]
- Saxton RA, Sabatini DM (2017) mTOR signaling in growth, metabolism, and disease. Cell 168: 960–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schielen PCJI, Kemper EA, Gelb MH (2017) Newborn screening for lysosomal storage diseases: a concise review of the literature on screening methods, therapeutic possibilities and regional programs. Int J Neonatal Screen 3: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R, Goker‐Alpan O, Holida M, Giraldo P, Barisoni L, Colvin RB, Jennette CJ, Maegawa G, Boyadjiev SA, Gonzalez D et al (2019) Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1‐year phase 1/2 clinical trial. J Inherit Metab Dis 42: 534–544 [DOI] [PubMed] [Google Scholar]
- Schröder BA, Wrocklage C, Hasilik A, Saftig P (2010) The proteome of lysosomes. Proteomics 10: 4053–4076 [DOI] [PubMed] [Google Scholar]
- Schulz A, Ajayi T, Specchio N, de Los RE, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D et al (2018) Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med 378: 1898–1907 [DOI] [PubMed] [Google Scholar]
- Scott CR, Elliott S, Hong X, Huang JY, Kumar AB, Yi F, Pendem N, Chennamaneni NK, Gelb MH (2020) Newborn screening for mucopolysaccharidoses: results of a pilot study with 100 000 dried blood spots. J Pediatr 216: 204–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto Rosato A, Montefusco S, Soldati C, Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A, Grimm C, Lombardo A et al (2019) TRPML1 links lysosomal calcium to autophagosome biogenesis through the activation of the CaMKKβ/VPS34 pathway. Nat Commun 10: 630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann S, Ernst M, Cimmaruta C, Struckmann S, Cozma C, Koczan D, Knospe AM, Haake LR, Citro V, Bräuer AU et al (2020) Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in Fabry disease. Biochem J. 477: 359–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, Canale S, Lopez ID, Morena F, Calabria A et al (2016) Lentiviral haemopoietic stem‐cell gene therapy in early‐onset metachromatic leukodystrophy: an ad‐hoc analysis of a non‐randomised, open‐label, phase 1/2 trial. Lancet 388: 476–487 [DOI] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Jahreiss L, Spampanato C, Venturi C, Medina D, de Pablo R, Tacchetti C, Rubinsztein DC, Ballabio A (2008a) A block of autophagy in lysosomal storage disorders. Hum Mol Genet 17: 119–129 [DOI] [PubMed] [Google Scholar]
- Settembre C, Arteaga‐Solis E, McKee MD, de Pablo R, Al Awqati Q, Ballabio A, Karsenty G (2008b) Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes Dev 22: 2645–2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P et al (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin SU, Huynh T, Ferron M, Karsenty G, Vellard MC et al (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A (2013) Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 14: 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Cinque L, Bartolomeo R, Di Malta C, De Leonibus C, Forrester A (2018) Defective collagen proteostasis and matrix formation in the pathogenesis of lysosomal storage disorders. Matrix Biol 71–72: 283–293 [DOI] [PubMed] [Google Scholar]
- Shea L, Raben N (2009) Autophagy in skeletal muscle: implications for Pompe disease. Int J Clin Pharmacol Ther 47(Suppl 1): S42–S47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas‐Rothschild A, Futerman AH (2011) Lysosomal storage disorders and Parkinson's disease: Gaucher disease and beyond. Mov Disord 26: 1593–1604 [DOI] [PubMed] [Google Scholar]
- Shin SD, Shin A, Mayagoitia K, Wilson CG, Bellinger DL, Soriano S (2019) Interferon downstream signaling is activated early in pre‐symptomatic Niemann‐Pick disease type C. Neurosci Lett. 27(706): 43–50 [DOI] [PubMed] [Google Scholar]
- Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH (2010) Involvement of the Toll‐like receptor 4 pathway and use of TNF‐alpha antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci USA 107: 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonaro CM, Tomatsu S, Sikora T, Kubaski F, Frohbergh M, Guevara JM, Wang RY, Vera M, Kang JL, Smith LJ et al (2016) Pentosan polysulfate: oral versus subcutaneous injection in mucopolysaccharidosis type I dogs. PLoS One 11: e0153136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindelar M, Dyke JP, Deeb RS, Sondhi D, Kaminsky SM, Kosofsky BE, Ballon DJ, Crystal RG, Gross SS (2018) Untargeted metabolite profiling of cerebrospinal fluid uncovers biomarkers for severity of late infantile neuronal ceroid lipofuscinosis (CLN2, Batten Disease). Sci Rep 8: 15229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P (1997) Association of mutations in a lysosomal protein with classical late‐infantile neuronal ceroid lipofuscinosis. Science 277: 1802–1805 [DOI] [PubMed] [Google Scholar]
- Sleat DE, Lackland H, Wang Y, Sohar I, Xiao G, Li H, Lobel P (2005) The human brain mannose 6‐phosphate glycoproteome: a complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics 5: 1520–1532 [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El‐Banna M, Zheng H, Zhao C, Soherwardy A, Moore DF, Lobel P (2019) Analysis of brain and cerebrospinal fluid from mouse models of the three major forms of neuronal ceroid lipofuscinosis reveals changes in the lysosomal proteome. Mol Cell Proteomics. 18: 2244–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly WS, Fischer HD (1982) The phosphomannosyl recognition system for intracellular and intercellular transport of lysosomal enzymes. J Cell Biochem 18: 67–85 [DOI] [PubMed] [Google Scholar]
- Sobo K, Le Blanc I, Luyet PP, Fivaz M, Ferguson C, Parton RG, Gruenberg J, van der Goot FG (2007) Late endosomal cholesterol accumulation leads to impaired intra‐endosomal trafficking. PLoS One 2: e851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda H, Morimoto H, Yoden E, Koshimura Y, Kinoshita M, Golovina G, Takagi H, Yamamoto R, Minami K, Mizoguchi A et al (2018) A blood‐brain‐barrier‐penetrating anti‐human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Mol Ther 26: 1366–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ (2006) High incidence of later‐onset fabry disease revealed by newborn screening. Am J Hum Genet 79: 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G et al (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5: 691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel D, Zech M, Zhao C, Barwick KES, Burke D, Demailly D, Kumar KR, Zorzi G, Nardocci N, Kaiyrzhanov R et al (2020) Loss‐of‐function variants in HOPS complex genes VPS16 and VPS41 cause early onset dystonia associated with lysosomal abnormalities. Ann Neurol 88: 867–877 [DOI] [PubMed] [Google Scholar]
- Stepien KM, Roncaroli F, Turton N, Hendriksz CJ, Roberts M, Heaton RA, Hargreaves I (2020) Mechanisms of mitochondrial dysfunction in lysosomal storage disorders: a review. J Clin Med. 9: 2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CR et al (2013) Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13: 653–658 [DOI] [PubMed] [Google Scholar]
- Tamargo RJ, Velayati A, Goldin E, Sidransky E (2012) The role of saposin C in Gaucher disease. Mol Genet Metab 106: 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann‐Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P (2000) Accumulation of autophagic vacuoles and cardiomyopathy in LAMP‐2‐deficient mice. Nature 406: 902–906 [DOI] [PubMed] [Google Scholar]
- Tharkeshwar AK, Trekker J, Vermeire W, Pauwels J, Sannerud R, Priestman DA, Te Vruchte D, Vints K, Baatsen P, Decuypere JP et al (2017) A novel approach to analyze lysosomal dysfunctions through subcellular proteomics and lipidomics: the case of NPC1 deficiency. Sci Rep 7: 41408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarallo A, Carissimo A, Gatto F, Nusco E, Toscano A, Musumeci O, Coletta M, Karali M, Acampora E, Damiano C et al (2019) microRNAs as biomarkers in Pompe disease. Genet Med 21: 591–600 [DOI] [PubMed] [Google Scholar]
- Tardieu M, Zérah M, Husson B, de Bournonville S, Deiva K, Adamsbaum C, Vincent F, Hocquemiller M, Broissand C, Furlan V et al (2014) Intracerebral administration of adeno‐associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther 25: 506–516 [DOI] [PubMed] [Google Scholar]
- Tardieu M, Zérah M, Gougeon ML, Ausseil J, de Bournonville S, Husson B, Zafeiriou D, Parenti G, Bourget P, Poirier B et al (2017) Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol 16: 712–720 [DOI] [PubMed] [Google Scholar]
- Tebani A, Abily‐Donval L, Schmitz‐Afonso I, Piraud M, Ausseil J, Zerimech F, Pilon C, Pereira T, Marret S, Afonso C et al (2019) Analysis of mucopolysaccharidosis type VI through integrative functional metabolomics. Int J Mol Sci 20: 446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen M, Winter D, Braulke T, Gieselmann V (2017) SILACBased comparative proteomic analysis of lysosomes from mammalian cells using LC‐MS/MS. Methods Mol Biol 1594: 1–18 [DOI] [PubMed] [Google Scholar]
- Tobias F, Pathmasiri KC, Cologna SM (2019) Mass spectrometry imaging reveals ganglioside and ceramide localization patterns during cerebellar degeneration in the Npc1−/− mouse model. Analyt Bioanalyt Chem 411: 5659–5668 [DOI] [PubMed] [Google Scholar]
- Turner SMF, Falk DJ, Byrne BJ, Fuller DD (2016) Transcriptome assessment of the Pompe (Gaa‐/‐) mouse spinal cord indicates widespread neuropathology. Physiol Genomics 48: 785–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utz JR, Crutcher T, Schneider J, Sorgen P, Whitley CB (2015) Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol Genet Metab 114: 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wal E, Bergsma AJ, Pijnenburg JM, van der Ploeg AT, Pijnappel WWMP (2017) Antisense oligonucleotides promote exon inclusion and correct the common c.‐32‐13T>G GAA splicing variant in Pompe disease. Mol Ther Nucleic Acids 7: 90–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gelder CM, Vollebregt AA, Plug I, van der Ploeg AT, Reuser AJ (2012) Treatment options for lysosomal storage disorders: developing insights. Expert Opin Pharmacother 13: 2281–2299 [DOI] [PubMed] [Google Scholar]
- Van Hoof F (1974) Mucopolysaccharidoses and mucolipidoses. J Clin Pathol Suppl. 8: 64–93 [PMC free article] [PubMed] [Google Scholar]
- Vaquer G, Rivière F, Mavris M, Bignami F, Llinares‐Garcia J, Westermark K, Sepodes B (2013) Animal models for metabolic, neuromuscular and ophthalmological rare diseases. Nat Rev Drug Discov 12: 287–305 [DOI] [PubMed] [Google Scholar]
- Vellodi A (2005) Lysosomal storage disorders. Br J Haematol. 128: 413–431 [DOI] [PubMed] [Google Scholar]
- Verheijen FW, Verbeek E, Aula N, Beerens CE, Havelaar AC, Joosse M, Peltonen L, Aula P, Galjaard H, van der Spek PJ et al (1999) A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet 23: 462–465 [DOI] [PubMed] [Google Scholar]
- Viana GM, Priestman DA, Platt FMK, Tomatsu S, Pshezhetsky AV(2020) Brain pathology in mucopolysaccharidoses (MPS) patients with neurological forms. J Clin Med 9: 396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani GR, Di Domenico C, Musella A, Cecere F, Di Napoli D, Di Natale P (2009) Mucopolysaccharidosis IIIB: oxidative damage and cytotoxic cell involvement in the neuronal pathogenesis. Brain Res 1279: 99–108 [DOI] [PubMed] [Google Scholar]
- von Zastrow M, Sorkin A (2007) Signaling on the endocytic pathway. Curr Opin Cell Biol 19: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MW, Lloyd‐Evans E (2015) A rapid method for the preparation of ultrapure, functional lysosomes using functionalized superparamagnetic iron oxide nanoparticles. Methods Cell Biol 126: 21–43 [DOI] [PubMed] [Google Scholar]
- Walkley SU, Vanier MT (2009) Secondary lipid accumulation in lysosomal disease. Biochim Biophys Acta 1793: 726–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Wang G (2019) Autophagy in mitochondrial quality control. Adv Exp Med Biol 1206: 421–434 [DOI] [PubMed] [Google Scholar]
- Wartosch L, Günesdogan U, Graham SC, Luzio JP (2015) Recruitment of VPS33A to HOPS by VPS16 Is required for lysosome fusion with endosomes and autophagosomes. Traffic 16:727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein MP, Caggana M, Bailey SM, Desnick RJ, Edelmann L, Estrella L, Holzman I, Kelly NR, Kornreich R, Kupchik SG et al (2019) The New York pilot newborn screening program for lysosomal storage diseases: Report of the First 65,000 Infants. Genet Med 21: 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams IM, Wallom KL, Smith DA, Al Eisa N, Smith C, Platt FM (2014) Improved neuroprotection using miglustat, curcumin and ibuprofen as a triple combination therapy in Niemann‐Pick disease type C1 mice. Neurobiol Dis 67: 9–17 [DOI] [PubMed] [Google Scholar]
- Winchester B (2014) Lysosomal diseases: diagnostic update. J Inherit Metab Dis 37: 599–608 [DOI] [PubMed] [Google Scholar]
- Wyant GA, Abu‐Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A, Sabatini DM (2018) NUFIP1 is a ribosome receptor for starvation‐induced ribophagy. Science 360: 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt K, Henley W, Anderson L, Anderson R, Nikolaou V, Stein K, Klinger L, Hughes D, Waldek S, Lachmann R et al (2012) The effectiveness and cost‐effectiveness of enzyme and substrate replacement therapies: a longitudinal cohort study of people with lysosomal storage disorders. Health Technol Assess 16: 1–543 [DOI] [PubMed] [Google Scholar]
- Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J (2013) Correction of a genetic disease in mouse via use of CRISPR/Cas9. Cell Stem Cell 13: 659–662 [DOI] [PubMed] [Google Scholar]
- Yim WW, Mizushima N (2020) Lysosome biology in autophagy. Cell Discov 6: 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogalingam G, Luu AR, Prill H, Lo MJ, Yip B, Holtzinger J, Christianson T, Aoyagi‐Scharber M, Lawrence R, Crawford BE et al (2019) BMN 250, a fusion of lysosomal alpha‐N‐acetylglucosaminidase with IGF2, exhibits different patterns of cellular uptake into critical cell types of Sanfilippo syndrome B disease pathogenesis. PLoS One 14: e0207836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Meng Y, Chen C, Liang S, Ma Y, Jiang W, Duan J, Wang C (2019) Proteomic approaches in the discovery of potential urinary biomarkers of mucopolysaccharidosis type II. Clin Chim Acta 499: 34–40 [DOI] [PubMed] [Google Scholar]
- Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW (2014) Seamless gene correction of beta‐thalassemia mutations in patient specific iPSCs using CRISPR/Cas9 and piggybac. Genome Res 24: 1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Martinoia E, Szabo I (2015) Organellar channels and transporters. Cell Calcium 58: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ren D (2015) Lysosomal physiology. Annu Rev Physiol 77: 57–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Lun Y, Frascella M, Frascella M, Garcia A, Soska R, Nair A, Ponery AS, Schilling A, Feng J et al (2019) Improved efficacy of a next‐generation ERT in murine Pompe disease. JCI Insight 4: e125358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalfa C, Verpelli C, D'Avanzo F, Tomanin R, Vicidomini C, Cajola L, Manara R, Sala C, Scarpa M, Vescovi AL et al (2016) Glial degeneration with oxidative damage drives neuronal demise in MPSII disease. Cell Death Dis 7: e2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella F, Lorens JB, Link W (2010) High content screening: seeing is believing. Trends Biotechnol 28: 237–245 [DOI] [PubMed] [Google Scholar]
- Zhang CK, Stein PB, Liu J, Wang Z, Yang R, Cho JH, Gregersen PK, Aerts JM, Zhao H, Pastores GM et al (2012) Genome‐wide association study of N370S homozygous Gaucher disease reveals the candidacy of CLN8 gene as a genetic modifier contributing to extreme phenotypic variation. Am J Hematol 87: 377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CS, Jiang B, Li M, Zhu M, Peng Y, Zhang YL, Wu YQ, Li TY, Liang Y, Lu Z et al (2014) The lysosomal v‐ATPase‐Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20: 526–540 [DOI] [PubMed] [Google Scholar]
- Zervas M, Dobrenis K, Walkley SUJ (2001) Neurons in Niemann‐Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol 60: 49–64 [DOI] [PubMed] [Google Scholar]
- Zhu X, Yin L, Theisen M, Siddiqui S, Levy B, Presnyak V, Frassetto A, Milton J, Salerno T, Benenato KE et al (2019) Systemic mRNA therapy for the treatment of fabry disease: preclinical studies in wild‐type mice, fabry mouse model, and wild‐type non‐human primates. Am J Hum Genet 104: 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]